

Green blood, a remarkable physiological trait, evolved multiple times in lizards.
Abstract
Several species of lizards from the megadiverse island of New Guinea have evolved green blood. An unusually high concentration of the green bile pigment biliverdin in the circulatory system of these lizards makes the blood, muscles, bones, tongue, and mucosal tissues bright green in color, eclipsing the crimson color from their red blood cells. This is a remarkable physiological feature because bile pigments are toxic physiological waste products of red blood cell catabolism and, when chronically elevated, cause jaundice in humans and all other vertebrates. Although these lizards offer a promising system to examine the evolution of extraordinary physiological characteristics, little is known about the phylogenetic relationships of green-blooded lizards or the evolutionary origins of green blood. We present the first extensive phylogeny for green-blooded lizards and closely related Australasian lizards using thousands of genomic regions to examine the evolutionary history of this unusual trait. Maximum likelihood ancestral character state reconstruction supports four independent origins of green blood. Our results lay the phylogenetic foundation necessary to determine the role, if any, of natural selection in shaping this enigmatic physiological trait as well as understanding the genetic, proteomic, and biochemical basis for the lack of jaundice in those species that have independently evolved green blood.
INTRODUCTION
Understanding the evolutionary history of physiological adaptations is of critical importance to biology. In particular, unusual or outlier physiologies may provide important evolutionary insight into the breadth, tempo, and mode of key innovations. One of the most unusual amniote physiological traits is green blood that has evolved in several ecologically diverse species of New Guinea lizards (1, 2). Whereas most vertebrates have crimson red blood derived from the ferrous form of heme, these lizards have lime green–colored blood, resulting in bright green coloration of their muscles, bones, tongue, and mucosal tissues (1). The green coloration is due to exceptionally high concentrations of the bile pigment biliverdin (714 to 1020 μM) in the circulatory system, which eclipses the vivid red color of their nucleated red blood cells (2). In other vertebrates, bile pigments (biliverdin and bilirubin) remain generally undetectable in the circulatory system because their accumulation is known to be genotoxic, cytotoxic, neurotoxic, and causes jaundice (3–6). Bile pigments are normal degradation products of heme catabolism. As red blood cells senesce, degradation of the heme constituent of red blood cell hemoglobin is catalyzed by the enzyme heme oxygenase, resulting in biliverdin and the recycling of iron and carbon monoxide. In addition to immediate cellular damage, sustained biliverdin accumulation is associated with permanent nerve damage and developmental disorders (7, 8). Furthermore, recent research has shown that babies treated for jaundice are more likely to develop autism (9). Bile pigment accumulation is commonly seen in individuals with impaired metabolic and liver function, such as newborn babies or patients with the life-threatening Gilbert’s and Crigler-Najjar syndromes (10–12). Nevertheless, several New Guinea lizards have the highest concentration of blood biliverdin measured for any animal (2) without being jaundiced. In contrast, the highest recorded level of biliverdin measured in humans, 50 μM, resulted in death (13). We use the term hyperbiliverdinism for this nonpathological biliverdin accumulation. The unusual physiology found in these lizards offers a promising system to examine the underlying genomic and proteomic changes that allow these lizards to remain jaundice-free. However, the lack of a well-resolved phylogeny for these lizards and close relatives has prevented an understanding of the evolutionary history of hyperbiliverdinism.
Five described species of lizards with green blood are currently grouped into the same genus (Prasinohaema) based solely on blood coloration despite the fact that the five species are morphologically and ecologically divergent. We use phylogenomic data from thousands of genome-wide loci to establish a robust phylogeny for Prasinohaema and related Australasian lizards with normal blood to determine the evolutionary history of green blood. Our phylogenetic and ancestral character state reconstruction results produce a highly robust phylogeny that supports four independent origins of green blood, demonstrating surprising evolutionary dynamism of green blood. Our findings lay the foundation for determining the role of natural selection, if any, in shaping this enigmatic physiological feature in these lizards as well as understanding the genetic and biochemical basis for hyperbiliverdinism. Understanding how these lizards avoid jaundice with such high bile pigment levels may translate into nontraditional approaches to addressing specific practical health problems, providing valuable benefits to society.
RESULTS
Phylogenetic analyses
We investigated the phylogenetic relationships among 51 species (119 individuals) of Australasian skinks including 6 species with green blood, 2 of which are undescribed new species (table S1). We targeted 5060 ultraconserved elements (UCEs) for phylogenomic analyses and recovered an average of 2690 loci (100 to 4241) per individual with a mean length of 586 base pairs (bp) (224 to 5494) per locus (Table 1). We subsampled our full data set to examine the effects of number of loci on phylogeny estimation (our “1K” data set), to explore the influence of loci versus taxonomic sampling on phylogeny estimation (our “TWO” data set), and to examine the effects of uneven taxonomic sampling (our “ONE” data set). The phylogeny presented in the main text, from the ONE data set, was trimmed to include only one representative per species, resulting in 4333 loci shared among more than 50% of taxa. Concatenation and coalescent approaches to species tree inference recovered a well-supported topology (Fig. 1). Overall, highly supported phylogenetic relationships were concordant among our data sets, and any incongruencies had low bootstrap support (figs. S1 to S7). Our data were also robust to biases caused by missing data because analysis of multiple data sets with varying levels of matrix completeness and information content recovered highly concordant topologies within each data set (figs. S1 to S7).
Table 1. Summary of UCE alignments.
Ten samples with fewer than 499 (10%) UCE loci or missing voucher information were removed from our “ALL” data set. Our 1K data set removed all individuals with fewer than 1000 UCE loci, the TWO data set trimmed sampling to no more than two individuals per species, and the ONE data set removed all but one individual per species.
| Data set | Taxa | Total length (bp) | No. UCE loci in % taxa | |||
| 50% | 60% | 70% | 80% | |||
| ALL | 109 | 1,545,997 | 4088 | 3331 | 1931 | 542 |
| 1K | 98 | 1,641,106 | 4354 | 3952 | 3195 | 1624 |
| TWO | 63 | 1,748,774 | 4333 | 3946 | 3137 | 1606 |
| ONE | 43 | 1,838,540 | 4368 | 3986 | 3220 | 1910 |
Fig. 1. Ancestral reconstruction indicates four origins of green blood.
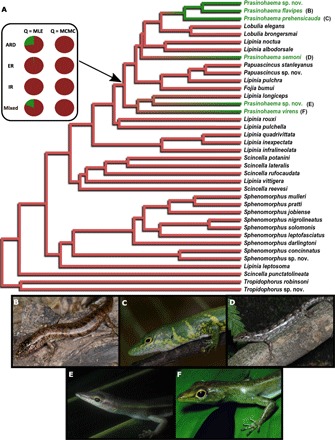
(A) Transitions between red blood and green blood in Australasian lizards were summarized from 1000 stochastic character simulations on a fixed species topology. The phylogeny was estimated from a 70% complete sequence matrix from our ONE data set using an unpartitioned concatenated analysis in RAxML. Bootstrap values for all nodes are 100. Branches are colored continuously according to posterior probability (PP) support for red (PP = 1.0) or green (PP = 0.0) blood under our “mixed” model. Pie charts indicate proportion of character histories that reconstructed red or green blood as the ancestral state for the most recent common ancestor of all Prasinohaema species under each transition rate model: all rates different (ARD), equal rates (ER), irreversible (IR), or mixed. Values for the character rate matrix were either fixed at their maximum likelihood estimates (MLE) or sampled from a posterior distribution of rate matrices (MCMC). Pictures show the following green-blooded species: (B) Prasinohaema flavipes, (C) P. prehensicauda, (D) Prasinohaema semoni, (E) Prasinohaema sp. nov., and (F) Prasinohaema virens.
Monophyly assessment
Surprisingly, our phylogenomic analysis strongly supports the nonmonophyly of green-blooded lizards; in addition, two other genera (Lipinia and Scincella) are not monophyletic. Our sampling included many Australasian taxa; however, the clade containing the most recent common ancestor for all green-blooded lizards is composed only of New Guinea species, indicating a Papuan origin for this radiation. We recover a robust fully resolved phylogeny with short internal branches that are likely the result of a rapid radiation. Given the large number of taxa and loci in our data set and the absence of particular loci in different taxa, Bayesian concordance analyses were not computationally feasible; however, statistical tests for monophyly corroborate the polyphyletic origins of the six Prasinohaema species. Shimodaira-Hasegawa (SH) tests on our ONE and TWO concatenated data sets supported the best unconstrained maximum likelihood (ML) tree over any of the constraint trees that forced either partial or complete Prasinohaema monophyly (14). We explored the possibility of hemiplasy by searching for gene tree histories that support fewer than four lineages with green blood and found no support for Prasinohaema monophyly among more than 4000 individually estimated gene trees (tables S2 and S3) (15). In addition, modeling for incomplete lineage sorting using a coalescent approach (ASTRAL) yielded topologies that were concordant with our unpartitioned ML approach (RAxML), indicating that our data were robust to problems of incomplete lineage sorting. Despite strong support for four green-blood lineages (Fig. 1), the number of evolutionary transitions between red and green blood is still unresolved: The evolution of green blood in lizards could be explained by four independent origins, a single origin with multiple reversals, or a complex pattern involving multiple gains and multiple losses.
Ancestral state reconstruction
We conducted an ML ancestral state reconstruction and stochastic character mapping of green blood on the unpartitioned concatenated ML species trees to investigate the most likely character state for blood physiology in the ancestor of all species with green blood. Model fitting for both ML reconstructions and character mappings resulted in similar support for alternative models (table S4). When the transition rate matrix was fixed at its empirical ML value, each model reconstructed red blood as the ancestral state with varying levels of support (27, 99, 100, and 84% probability) for both marginal reconstructions and stochastic mappings. Although the transition rates and mean number of reversals varied considerably between models, all reconstructions recovered an average of 4 (3.9 to 4.2) transitions from red to green blood (Table 2). In contrast, sampling a new transition rate matrix for each character history from a model’s posterior distribution of 1000 simulations using MCMC unequivocally supported (100% probability) red blood as the ancestral state and four independent origins of green blood. Model choice did not significantly change the number or placement of transitions using the MCMC approach but did vary the results of the ML approach, likely due to the use of a single rate matrix for all reconstructions under a given model (Fig. 1 and Table 2). All reconstructions presented in the main text were performed using our “ONE-p70” phylogeny as input, but we recovered similar results using all subsets of our ONE and TWO data sets (table S5). Furthermore, stochastic character mapping across randomly sampled subsets of posterior probability trees and gene trees never yielded less than four transitions from red to green blood for any given character history (table S6). These results suggest that our ancestral reconstruction and character mapping results are robust to hemiplasy caused by phylogenetic uncertainty. Together, our analyses indicate that hyperbiliverdinism is likely the result of repeated evolution.
Table 2. Integrating over multiple models for stochastic character mapping.
Three transition rate models (ARD, ER, and IR) and one mixed model were considered for stochastic character mapping. The number of simulations (Nsim) for each model was normalized using Akaike information criterion weights (wAIC) for that model and the mixed model sampled character histories from all three models. The transition rate matrix for a given model was either fixed at its empirical MLE or sampled using MCMC from its posterior distribution (MCMC). Rates (q), AIC scores, Likelihood (LnL), mean number of forward and reverse transitions, and PP of red blood reconstructed for the most recent common ancestor (MRCA) for all Prasinohaema are shown for the ONE-p70 tree. Models are described in the main text.
| Transition rate matrix | Model | Model summary | Mean no. of changes | PP of red-blooded MRCA | ||||||
| qRG | qGR | AIC | wAIC | ln (L) | Nsim | Red→Green | Red←Green | |||
| MLE (fixed) | ARD | 11.7 | 137.2 | 32.2 | 0.386 | −14.1 | 386 | 4.0 | 9.0 | 27.1 |
| ER | 11.8 | 11.8 | 32.5 | 0.332 | −15.3 | 332 | 4.2 | 0.5 | 99.9 | |
| IR | 10.6 | 0.0 | 32.8 | 0.282 | −15.4 | 282 | 4.0 | 0.0 | 100 | |
| Mixed | — | — | — | — | — | 1000 | 3.9 | 2.2 | 81.4 | |
| MCMC | ARD | 3.5 | 1.0 | 36.3 | 0.156 | −16.1 | 156 | 3.9 | 0.1 | 100 |
| ER | 3.8 | 3.8 | 33.5 | 0.622 | −15.7 | 622 | 3.9 | 0.1 | 100 | |
| IR | 3.9 | 0.0 | 35.5 | 0.222 | −16.7 | 222 | 4.0 | 0.0 | 100 | |
| Mixed | — | — | — | — | — | 1000 | 3.9 | 0.1 | 100 | |
DISCUSSION
Surprisingly, we find robust support for the nonmonophyly of green-blooded lizards, revealing a complex and dynamic evolutionary history of this unusual physiological trait. This conclusion is supported by extensive phylogenomic analyses of 5060 UCE loci and tests for monophyly. Considering the pathological consequences of elevated concentrations of bile pigments in most animals, green blood was assumed to have originated only once in amniotes. However, we reconstructed red blood as the most likely ancestral state for all Prasinohaema lineages with strong support for four separate origins of green blood. Although these lizards are the only amniotes to have hyperbiliverdinism, several other vertebrates and invertebrates have been shown to have moderately elevated levels of bile pigments. Elevated levels of biliverdin have been documented in the blood and scales of several unrelated species of fishes (16–19) and insects (20, 21), and biliverdin is deposited in eggshells of some birds (22). Biliverdin has been suggested as the cause of green bones, skin, and blood in several unrelated frogs (23, 24). The rare but widespread occurrence of moderately elevated levels of bile pigments in animals suggests that there may be some adaptive value of elevated biliverdin.
How cytotoxicity of bile pigments might be selected for is unclear; however, bile pigments have been shown to reduce pathogen activity (25–28), so elevated levels of bile pigments may preclude or reduce infection. Because lizards are the only amniotes that have this unusual physiology and remain healthy, further investigation will inform us how these lizards are not jaundiced. In fish with green blood plasma, excess biliverdin is tightly bound to albumin transport proteins, and this complex is neither metabolized nor excreted by the liver but remains in the bloodstream, which may obviate damage from biliverdin (29). This mechanism for biliverdin retention and tolerance in unrelated fish is the result of convergent changes in the serum albumin protein, lending support to the possibility of green blood being an advantageous trait, shaped by natural selection (19). Convergent and parallel evolution of unusual phenotypes may be more frequent than previously thought due to shared constraints in developmental patterns, genomic architecture, and body form (30).
In contrast to the toxicity aspect of bile pigments, recent data on biosynthetic pathways indicate possible positive functional roles for bile pigments, but at low concentrations (31). Excess biliverdin appears to have antioxidant and cytoprotective properties in vitro (32, 33), predominantly due to their ability to scavenge free radical species (4, 34–38). Bile pigments also appear to have a beneficial role in oxidative stress defense in animals (39) and have been identified as having significant neuroprotective antioxidant and antimutagenic properties (40, 41).
Our analyses indicate four independent origins of green blood from a normal, red-blooded ancestor with no losses. The primary alternative hypothesis is that there was only a single origin of green blood followed by four or more losses, an evolutionary process presumably driven by natural selection. To fully tease apart these hypotheses, our phylogenetic results lay the groundwork for future functional genomic and proteomic approaches to identify the genes responsible for the origins, and possible loss, of hyperbiliverdinism. We predict that independent gains will be identified by four unique genomic changes whereas a single origin will be identified by a shared genomic change and that we will be able to track the signature of independent reversals to red blood, if any, from genomic data. The exciting future challenges of this nonmodel system are to address the potential convergent and parallel evolution of green blood in these lizards, determine whether bile pigment retention confers any major selective advantages for cells in vivo, and examine potential proteomic or biochemical innovations associated with nonpathological biliverdin accumulation.
METHODS
DNA sampling and sequencing
We took a genome-wide next-generation sequencing approach and targeted 5060 UCE loci using an in-solution sequence capture protocol (42). Our sampling included 24 lizards with green blood and 95 closely related red-blooded individuals. Genomic DNA was extracted from tissue preserved in ethanol using a standard DNA salt extraction protocol (43) and sonicated into sizes between 400 and 600 bp, with a target of 400 bp using an EpiSonic 1000 (EpiGentek). The Kapa Hyper Prep Kit (Kapa Biosystems Inc.) was used to prepare libraries for target enrichment and sequencing, including ligation of custom dual-indexed adapters with unique barcode sequencing for each individual. Prior to sequence capture, we pooled indexed libraries into groups of 8 in equimolar ratios. For sequence capture, we used the Tetrapods-UCE-5Kv1 probe set (myBaits, MYcroarray) targeting 5060 UCE loci following a freely available open-source protocol. This approach has been successfully used in lizards and other vertebrates (44, 45). Following enrichment and limited-cycle polymerase chain reaction, pools of equimolar reactions were combined and sent to Oklahoma Medical Research Foundation (Oklahoma City, OK) for 150-bp paired-end sequencing on an Illumina HiSeq3000.
Data processing
To process demultiplexed raw reads for phylogenomic analyses, we followed the standard PHYLUCE pipeline (46). Illumiprocessor, in combination with Trimmomatic, was used to filter low-quality reads and remove adapters (47, 48), yielding an average of 8,504,473 reads (302,468 to 119,426,985) and 1,141,548,626 bp (42,543,058 to 17,007,264,440) per sample. Filtered reads were assembled into contigs using Trinity (49). To check for contamination, all contigs were aligned to the nonredundant protein database by BLASTP with an e value cutoff of 1 × 10−6 and checked for best hits to squamate reptiles. We also performed an all-versus-all blast search on the assemblies and removed six individuals that had reciprocal best hits to a different species, likely the result of cross contamination. The resulting assemblies were screened for matches to the UCE probes, and nontargets and paralogs were removed in PHYLUCE. The resulting regions were aligned using MAFFT (50), and quality-trimmed using a parallelized wrapper around Gblocks (51). We created three data sets from our “all taxa” data set. First, to examine the effects of number of loci in phylogeny estimation, we removed 21 individuals with <1000 UCE loci to create a 98-taxon 1K data set. Second, to explore the influence of number of loci versus taxonomic sampling, we created a TWO data set, which restricted sampling to no more than two individuals per species, independent of loci number. Third, to examine the effects uneven taxonomic sampling, we constructed a ONE data set, for which we removed individuals with less than 1000 UCEs and limited taxon sampling to one representative per species. To assess the sensitivity of phylogenetic inference to missing data, we subsampled each data set and generated alignments that allowed up to 20, 30, 40, or 50% missing data (data sets are appended with p80, p70, p60, p50, respectively). Finally, we generated four additional alignments for each data set containing only the top 100, 250, 500, or 750 loci with the most parsimonious informative sites (data sets are appended with i100, i250, i500, or i750, respectively). In summary, each of our data sets was subsampled into eight alignments using two metrics: missing data and information content.
Phylogenetic inference
We performed unpartitioned concatenated ML analysis of each alignment and data set in RAxML v8.2 (52), using 20 searches with GTR GAMMA model of nucleotide substitution for the best ML tree. Nodal support for the best ML topology was assessed using nonparametric bootstrapping with the autoMRE option in RAxML, and we reconciled the best ML tree with the bootstrap replicates in RAxML. Species-tree methods have been shown to be more accurate than concatenated approaches for multilocus data under a variety of conditions (53). Because of the large number of gene trees that are typically produced from phylogenomic data sets, species trees were estimated using a summary coalescent approach. Individual gene trees were first estimated in an ML-like framework implemented in RAxML v8.2. Next, multilocus bootstraps were generated by sampling loci with replacement to create 100 resampled data sets, and we performed nonparametric bootstrapping by sites of each locus in each of the resampled data sets using RAxML. Then, bootstrapped gene trees were sorted to ensure that each resampled data set was resampled by loci and by sites (54). The 100 resampled data sets were used to infer species trees in a summary coalescent framework, implemented in ASTRAL-II 4.7.8 (55) that allows unrooted, multifurcating trees as input and has been shown to be robust to effects of both incorrectly estimated gene trees and gene tree heterogeneity (56). Extended majority-rule consensus (MRE) trees were estimated using sumtrees in DendroPy v3.2 (57). After visually confirming species monophyly and topological concordance among data sets, we elected to proceed with the ONE and TWO data sets for all subsequent analyses because they had even taxonomic sampling, which resulted in a higher number of shared loci between species. For the main text, we presented the results from our 70% complete matrix which was trimmed to one representative per species (ONE-p70), because it was the most complete matrix without a significant reduction in the number of represented species or loci per individual (Table 1).
Monophyly assessment
To assess support for the nonmonophyly of lizards with green blood, we took two approaches. First, we conducted SH tests (14) on each ML estimate, or the “best tree,” for each concatenated data set in RAxML. This was accomplished by running 20 ML searches for the best “constrained” tree by passing RAxML a constraint file with the -g option, then comparing likelihood scores using the -f h option, and passing RAxML the best constrained tree with -z and the best tree with -t. Second, we searched for both partial and full green blood monophyly in the individually estimated gene trees, excluding the Prasinohaema flavipes + Prasinohaema prehensicauda clade. For this, we used the is.monophyletic option in the R package ape and iterated over more than 4000 gene trees in our ONE and TWO data sets. Our results are summarized in tables S2 and S3.
Ancestral state reconstruction
We used ML ancestral state reconstruction and stochastic character mapping to assess whether hyperbiliverdinism can best be explained by multiple independent origins or a single origin with multiple reversals. Because branch length estimates are important for ancestral state reconstruction, all reconstructions were performed on species trees inferred from the unpartitioned concatenated alignments or on individual ML gene trees for each data set. We reconstructed ancestral states for green blood in an ML framework using the ace command in the R package APE v3.4 (58). Three transition rate matrices were considered: (i) a two-parameter model that had different rates for forward and reverse transitions (ARD), (ii) a two-parameter model that assumed once green blood is gained there were no reversals to red blood (IR) model, and (iii) a single-parameter model that assumed forward and reverse transition rates were equal (ER). To determine the model fit, we examined the AIC for each model using the fitDiscrete command from the package geiger v2.0.6 (59). Because the three models gave comparable support (table S4), we applied a fourth mixed model for which we integrated stochastic character maps across all three transition models. For the mixed model, we calculated normalized Akaike weights from the log likelihood of the other three models and sampled a number of character histories in proportion to the weight of evidence in support for each model. For example, the Akaike weights for the ONE-p70 data set were 0.386, 0.332, and 0.282, for the ARD, ER, and IR models, respectively. Thus, we simulated 386 character histories under the ARD model, 332 character histories under the ER model, and 282 character histories under the IR model. For all four models, we used stochastic character mapping (60), implemented in phytools (61), to summarize the results of multiple trait mappings onto our phylogeny using a continuous-time Markov process. The transition rate matrix estimated for each model was applied to these simulations using two approaches. First, we fixed the transition matrix at its empirical ML value estimated for a given model and generated multiple character histories. Second, we sampled a new transition matrix for each character history from the posterior distribution of a given model using MCMC. For the mixed-model MCMC approach, a new transition rate matrix was sampled from the posterior distribution of each model in proportion to the calculated Akaike weights. We generated 1000 character histories for each model using the make.simmap command and summarized the results using describe.simmap to determine the posterior probabilities that each internal node is at each state, the proportion of time spent in each state, and the number of transitions across the phylogeny. Results for the ONE-p70 topology are shown in Fig. 1 and Table 2. Results for other input trees are summarized in table S5.
Supplementary Material
Acknowledgments
We thank the many villagers on whose land we conducted fieldwork. We thank B. Wilmot from the Papua New Guinea (PNG) Department of Environment and Conservation for permit support and J. Animiato and B. Iova from the PNG National Museum and Art Gallery for field assistance. All research was approved under Louisiana State University (LSU) Institutional Animal Care and Use Committee protocol 06-071. We thank R. Brown (University of Kansas), S. Donnellan (South Australian Museum), J. McGuire (University of California, Berkeley), A. Resetar (Field Museum of Natural History), and J. Vindum (California Academy of Sciences) for tissue loans. This research was expedited by use of the high-performance computing resources provided by LSU (www.hpc.lsu.edu). Funding: Z.B.R. acknowledges support from the National Science Foundation Graduate Research Fellowship Program under grant no. DGE-1247192 and Louis Stokes Alliances for Minority Participation Bridge to the Doctorate activity (HRD-1301957). This work was funded by the NSF grant DEB-1146033 to S.L.P. and C.C.A. Author contributions: C.C.A. conducted fieldwork and collected samples. C.C.A. and S.L.P. conceived of the project idea. Z.B.R. collected and analyzed the genomic data. Z.B.R., S.L.P., and C.C.A. wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Raw sequencing reads are available from the NCBI (National Center for Biotechnology Information) Sequence Read Archive (SRA BioProject PRJNA420910). Sequence alignments, command scripts, phylogenies, and other supplementary data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/5/eaao5017/DC1
fig. S1. ML tree for ONE-i100.
fig. S2. Species tree for ONE-i100.
fig. S3. ML tree for ONE-p70.
fig. S4. Species tree for ONE-p70.
fig. S5. ML tree for TWO-i100.
fig. S6. Species tree for TWO-i100.
fig. S7. Species tree for TWO-p70 data set.
table S1. Sampling information and data processing results.
table S2. SH test results from the SH tests for monophyly of Prasinohaema.
table S3. Partial and full monophyly of Prasinohaema.
table S4. Model test results for the ONE-p70 and TWO-p70 data sets.
table S5. Results from marginal ancestral state reconstructions and stochastic character mapping using the ONE and TWO data set as input.
table S6. Results from stochastic mapping across gene trees and posterior trees.
REFERENCES AND NOTES
- 1.Greer A. E., Raizes G., Green blood pigment in lizards. Science 166, 392 (1969). [DOI] [PubMed] [Google Scholar]
- 2.Austin C. C., Jessing K. W., Green-blood pigmentation in lizards. Comp. Biochem. Physiol. A Physiol. 109, 619–626 (1994). [Google Scholar]
- 3.Hawkins W. B., Whipple G. H., The life cycle of the red blood cell in the dog. Am. J. Physiol. 122, 418–427 (1938). [Google Scholar]
- 4.Asad S. F., Singh S., Ahmad A., Khan N. U., Hadi S. M., Prooxidant and antioxidant activities of bilirubin and its metabolic precursor biliverdin: A structure-activity study. Chem. Biol. Interact. 137, 59–74 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Asad S. F., Singh S., Ahmad A., Hadi S. M., Bilirubin/biliverdin–Cu(II) induced DNA breakage; reaction mechanism and biological significance. Toxicol. Lett. 131, 181–189 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Keshavan P., Schwemberger S. J., Smith D. L. H., Babcock G. F., Zucker S. D., Unconjugated bilirubin induces apoptosis in colon cancer cells by triggering mitochondrial depolarization. Int. J. Cancer 112, 433–445 (2004). [DOI] [PubMed] [Google Scholar]
- 7.S. Schenker, A. M. Hoyumpa, D. W. McCandless, in Bile Pigments and Jaundice: Molecular, Metabolic, and Medical Aspects, J. D. Ostrow, Ed. (Marcel Dekker Inc., New York, 1986), pp. 395–419. [Google Scholar]
- 8.Liu X.-m., Chapman G. B., Wang H., Durante W., Adenovirus-mediated heme oxygenase-1 gene expression stimulates apoptosis in vascular smooth muscle cells. Circulation 105, 79–84 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Maimburg R. D., Bech B. H., Væth M., Møller-Madsen B., Olsen J., Neonatal jaundice, autism, and other disorders of psychological development. Pediatrics 126, 872–878 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Ritter J. K., Yeatman M. T., Ferreira P., Owens I. S., Identification of a genetic alteration in the code for bilirubin UDP-glucuronosyltransferase in the UGT1 gene complex of a Crigler-Najjar type I patient. J. Clin. Invest. 90, 150–155 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McDonagh A. F., Phototherapy: From ancient Egypt to the new millennium. J. Perinatol. 21 (Suppl. 1), S7–S12 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Strassburg C. P., Pharmacogenetics of Gilbert’s syndrome. Pharmacogenomics 9, 703–715 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Greenberg A. J., Bossenmaier I., Schwartz S., Green jaundice. A study of serum biliverdin, mesobiliverdin and other green pigments. Am. J. Dig. Dis. 16, 873–880 (1971). [DOI] [PubMed] [Google Scholar]
- 14.Shimodaira H., Hasegawa M., Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol. Biol. Evol. 16, 1114–1116 (1999). [Google Scholar]
- 15.Avise J. C., Robinson T. J., Hemiplasy: A new term in the lexicon of phylogenetics. Syst. Biol. 57, 503–507 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Low P. S., Bada J. L., Bile pigments in the blood serum of fish from the family Cottidae. Comp. Biochem. Physiol. A Comp. Physiol. 47, 411–418 (1974). [DOI] [PubMed] [Google Scholar]
- 17.Mudge S. M., Davenport J., Serum pigmentation in Cyclopterus lumpus L. J. Fish Biol. 29, 737–745 (1986). [Google Scholar]
- 18.Makos B. K., Youson J. H., Serum levels of bilirubin and biliverdin in the sea lamprey, Petromyzon marinus L., before and after their biliary atresia. Comp. Biochem. Physiol. A Comp. Physiol. 87, 761–764 (1987). [DOI] [PubMed] [Google Scholar]
- 19.Fang L.-S., Bada J. L., The blue-green blood plasma of marine fish. Comp. Biochem. Physiol. B 97, 37–45 (1990). [DOI] [PubMed] [Google Scholar]
- 20.Goodman W. G., Adams B., Trost J. T., Purification and characterization of a biliverdin-associated protein from the hemolymph of Manduca sexta. Biochemistry 24, 1168–1175 (1985). [DOI] [PubMed] [Google Scholar]
- 21.Law J. H., Wells M. A., Insects as biochemical models. J. Biol. Chem. 264, 16335–16338 (1989). [PubMed] [Google Scholar]
- 22.Kennedy G. Y., Vevers H. G., A survey of avian eggshell pigments. Comp. Biochem. Physiol. B 55, 117–123 (1976). [DOI] [PubMed] [Google Scholar]
- 23.Marinetti G. V., Bagnara J. T., Yolk pigments of the Mexican leaf frog. Science 219, 985–987 (1983). [DOI] [PubMed] [Google Scholar]
- 24.Lee Grismer L., Thy N., Chav T., Holden J., A new species of Chiromantis Peters 1854 (Anura: Rhacophoridae) from Phnom Samkos in the northwestern Cardamom Mountains, Cambodia. Herpetologica 63, 392–400 (2007). [Google Scholar]
- 25.Kumar S., Guha M., Choubey V., Maity P., Srivastava K., Puri S. K., Bandyopadhyay U., Bilirubin inhibits Plasmodium falciparum growth through the generation of reactive oxygen species. Free Radic. Biol. Med. 44, 602–613 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Zhu Z., Wilson A. T., Luxon B. A., Brown K. E., Meleah Mathahs M., Bandyopadhyay S., McCaffrey A. P., Schmidt W. N., Biliverdin inhibits hepatitis C virus nonstructural 3/4A protease activity: Mechanism for the antiviral effects of heme oxygenase? Hepatology 52, 1897–1905 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gutiérrez-Grobe Y., Vitek L., Tiribelli C., Kobashi-Margáin R. A., Uribe M., Méndez-Sánchez N., Biliverdin and heme oxygenase antiviral activity against hepatitis C virus. Ann. Hepatol. 10, 105–107 (2011). [PubMed] [Google Scholar]
- 28.Alves E., Maluf F. V., Bueno V. B., Guido R. V. C., Oliva G., Singh M., Scarpelli P., Costa F., Sartorello R., Catalani L. H., Brady D., Tewari R., Garcia C. R. S., Biliverdin targets enolase and eukaryotic initiation factor 2 (eIF2α) to reduce the growth of intraerythrocytic development of the malaria parasite Plasmodium falciparum. Sci. Rep. 6, 22093 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fang L.-S., Bada J. L., A special pattern of haem catabolism in a marine fish, Clinocottus analis, with green blood plasma. J. Fish Biol. 33, 775–780 (1988). [Google Scholar]
- 30.Losos J. B., Convergence, adaptation, and constraint. Evolution 65, 1827–1840 (2011). [DOI] [PubMed] [Google Scholar]
- 31.McDonagh A. F., Turning green to gold. Nat. Struct. Biol. 8, 198–200 (2001). [DOI] [PubMed] [Google Scholar]
- 32.Stocker R., Maghzal G., Reply to Sedlak and Snyder: The little bighorn of the biliverdin reductase amplification cycle. J. Biol. Chem. 284, le12 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maghzal G. J., Leck M.-C., Collinson E., Li C., Stocker R., Limited role for the bilirubin-biliverdin redox amplification cycle in the cellular antioxidant protection by biliverdin reductase. J. Biol. Chem. 284, 29251–29259 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Farrera J.-A., Jaumà A., Ribó J. M., Peiré M. A., Parellada P. P., Roques-Choua S., Bienvenue E., Seta P., The antioxidant role of bile pigments evaluated by chemical tests. Bioorg. Med. Chem. 2, 181–185 (1994). [DOI] [PubMed] [Google Scholar]
- 35.Kaur H., Hughes M. N., Green C. J., Naughton P., Foresti R., Motterlini R., Interaction of bilirubin and biliverdin with reactive nitrogen species. FEBS Lett. 543, 113–119 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Kumar S., Bandyopadhyay U., Free heme toxicity and its detoxification systems in human. Toxicol. Lett. 157, 175–188 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Jansen T., Daiber A., Direct antioxidant properties of bilirubin and biliverdin. Is there a role for biliverdin reductase? Front. Pharmacol. 3, 30 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bai B., Liu Y., You Y., Li Y., Ma L., Intraperitoneally administered biliverdin protects against UVB-induced skin photo-damage in hairless mice. J. Photochem. Photobiol. B 144, 35–41 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Beale S. I., Yeh J. I., Deconstructing heme. Nat. Struct. Biol. 6, 903–905 (1999). [DOI] [PubMed] [Google Scholar]
- 40.Doré S., Takahashi M., Ferris C. D., Hester L. D., Guastella D., Snyder S. H., Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc. Natl. Acad. Sci. U.S.A. 96, 2445–2450 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bulmer A. C., Ried K., Blanchfield J. T., Wagner K.-H., The anti-mutagenic properties of bile pigments. Mutat. Res. 658, 28–41 (2008). [DOI] [PubMed] [Google Scholar]
- 42.Faircloth B. C., McCormack J. E., Crawford N. G., Harvey M. G., Brumfield R. T., Glenn T. C., Ultraconserved elements anchor thousands of genetic markers spanning multiple evolutionary timescales. Syst. Biol. 61, 717–726 (2012). [DOI] [PubMed] [Google Scholar]
- 43.M. MacManes, MacManes salt extraction protocol (2013); http://dx.doi.org/10.6084/m9.figshare.658946.
- 44.Giarla T. C., Esselstyn J. A., The challenges of resolving a rapid, recent radiation: Empirical and simulated phylogenomics of Philippine shrews. Syst. Biol. 64, 727–740 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Streicher J. W., Schulte J. A. II, Wiens J. J., How should genes and taxa be sampled for phylogenomic analyses with missing data? An empirical study in iguanian lizards. Syst. Biol. 65, 128–145 (2016). [DOI] [PubMed] [Google Scholar]
- 46.Faircloth B. C., PHYLUCE is a software package for the analysis of conserved genomic loci. Bioinformatics 32, 786–788 (2016). [DOI] [PubMed] [Google Scholar]
- 47.B. C. Faircloth, Illumiprocessor: A trimmomatic wrapper for parallel adapter and quality trimming (2013); http://dx.doi.org/10.6079/J9ILL.
- 48.Bolger A. M., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grabherr M. G., Haas B. J., Yassour M., Levin J. Z., Thompson D. A., Amit I., Adiconis X., Fan L., Raychowdhury R., Zeng Q., Chen Z., Mauceli E., Hacohen N., Gnirke A., Rhind N., di Palma F., Birren B. W., Nusbaum C., Lindblad-Toh K., Friedman N., Regev A., Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Katoh K., Standley D. M., MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Castresana J., Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552 (2000). [DOI] [PubMed] [Google Scholar]
- 52.Stamatakis A., RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leaché A. D., Rannala B., The accuracy of species tree estimation under simulation: A comparison of methods. Syst. Biol. 60, 126–137 (2011). [DOI] [PubMed] [Google Scholar]
- 54.Seo T.-K., Calculating bootstrap probabilities of phylogeny using multilocus sequence data. Mol. Biol. Evol. 25, 960–971 (2008). [DOI] [PubMed] [Google Scholar]
- 55.Mirarab S., Warnow T., ASTRAL-II: Coalescent-based species tree estimation with many hundreds of taxa and thousands of genes. Bioinformatics 31, i44–i52 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meiklejohn K. A., Faircloth B. C., Glenn T. C., Kimball R. T., Braun E. L., Analysis of a rapid evolutionary radiation using ultraconserved elements (UCEs): Evidence for a bias in some multi-species coalescent methods. Syst. Biol. 65, 612–627 (2016). [DOI] [PubMed] [Google Scholar]
- 57.Sukumaran J., Holder M. T., DendroPy: A Python library for phylogenetic computing. Bioinformatics 26, 1569–1571 (2010). [DOI] [PubMed] [Google Scholar]
- 58.Popescu A.-A., Huber K. T., Paradis E., ape 3.0: New tools for distance-based phylogenetics and evolutionary analysis in R. Bioinformatics 28, 1536–1537 (2012). [DOI] [PubMed] [Google Scholar]
- 59.Harmon L. J., Weir J. T., Brock C. D., Glor R. E., Challenger W., GEIGER: Investigating evolutionary radiations. Bioinformatics 24, 129–131 (2008). [DOI] [PubMed] [Google Scholar]
- 60.Huelsenbeck J. P., Nielsen R., Bollback J. P., Stochastic mapping of morphological characters. Syst. Biol. 52, 131–158 (2003). [DOI] [PubMed] [Google Scholar]
- 61.Revell L. J., phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 3, 217–223 (2012). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/5/eaao5017/DC1
fig. S1. ML tree for ONE-i100.
fig. S2. Species tree for ONE-i100.
fig. S3. ML tree for ONE-p70.
fig. S4. Species tree for ONE-p70.
fig. S5. ML tree for TWO-i100.
fig. S6. Species tree for TWO-i100.
fig. S7. Species tree for TWO-p70 data set.
table S1. Sampling information and data processing results.
table S2. SH test results from the SH tests for monophyly of Prasinohaema.
table S3. Partial and full monophyly of Prasinohaema.
table S4. Model test results for the ONE-p70 and TWO-p70 data sets.
table S5. Results from marginal ancestral state reconstructions and stochastic character mapping using the ONE and TWO data set as input.
table S6. Results from stochastic mapping across gene trees and posterior trees.