

The airway epithelium, from the nasal cavity to the alveoli, persistently encounters toxins and pathogens. If the invader clears the mucus and breaches the epithelium, it must be contained by an innate immune system while awaiting arrival of the adaptive immune system. Hence, we need a biological process to immediately recognize, engage, and destroy a pathogen. The complement cascade is the prototype of such an innate process. It quickly amplifies based on cleavage of its most abundant complement protein: C3 to C3a and C3b (Fig 1, A). C3a is a vasodilator and chemoattractant for neutrophils and monocytes, whereas C3b is an opsonin for pathogens and debris. This cleavage (and thus, activation) is triggered primarily in 3 ways: antibody binding to antigen (classical pathway), mannose-binding lectin attaching to oligosaccharides/sugars (lectin pathway), or engagement of a feedback loop to deposit C3b onto a target (alternative pathway).1
FIG 1.
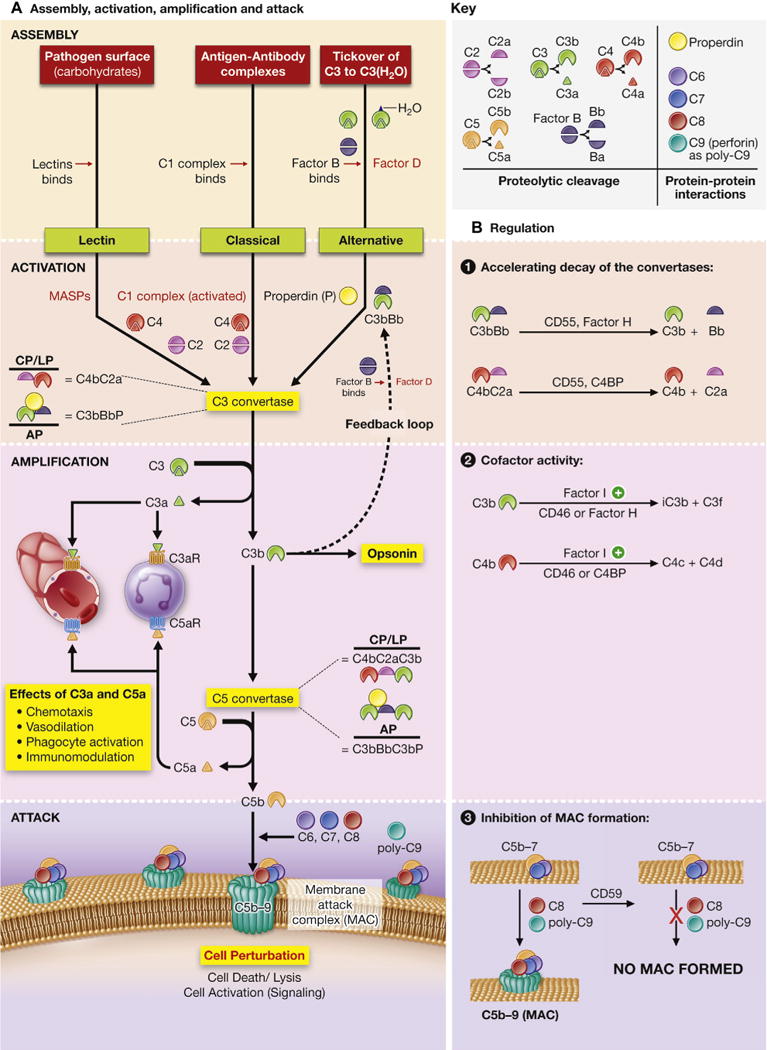
A, Assembly, activation, amplification, and attack. The complement system is commonly activated by one of 3 triggers. The lectin pathway (LP) is triggered when lectins bind carbohydrates (oligosaccharides) on the pathogen surface. Next, mannose-associated serine proteases (MASPs) are activated, and they cleave C4 and C2. The classical pathway (CP) is triggered by antigen-antibody complexes interacting with the C1 complex (C1[qr2s2]). This activates C1s to cleave C4 and C2. The alternative pathway (AP) is constitutively active at low levels because C3 metabolizes to C3(H2O) at approximately 1% per hour. It is amplified when C3(H2O) binds to Factor B and enhanced with binding of the positive regulator properdin (P). Factor B is cleaved by Factor D to Bb, forming C3bBbP. This forms an autocrine feedback loop that can be amplified. All 3 proteases are denoted in red. The 3 pathways converge to form the C3 convertases (formed by enzyme subunits: either C4bC2a for the classical and lectin pathways or C3bBbP for the alternative pathway). The C3 convertases cleave C3 to C3a and C3b. Attachment of C3b to C3 convertases changes their substrate specificity, allowing them to become C5 convertases (C4bC2aC3b or C3bBbC3bP). The C5 convertases cleave C5 to C5a and C5b. C5b then forms the membrane-attack complex with C6, C7, C8, and C9, resulting in cell perturbation. C3a and C5a bind to the G protein–coupled receptors C3aR and C5aR1, respectively, leading to chemotactic, vasodilatory, phagocyte-activating, and immunomodulatory effects. B, Complement regulation. The complement cascade is tightly regulated by membrane-associated and plasma proteins to prevent excessive inflammation. CD46 (known as membrane cofactor protein) and CD55 (known as decayaccelerating factor) are membrane regulators, whereas Factor H, Factor I, and C4BP (C4-binding protein) are fluid-phase regulators. CD55 irreversibly decays (dissociates) C2a from C4b, as well as Bb from C3b, to inactivate C3 and C5 convertases. Not shown in the figure is the ability of CD55 to competitively inhibit binding of Bb to C3b and C2a to C4b. Factor H also decays the C3b-containing convertases, whereas C4BP decays the C4b-containing convertases. Factor I proteolytically degrades C3b or C4b but only in the presence of the appropriate cofactors. Complement receptor 1 (CR1; CD35) is another membrane-associated receptor and regulator present almost exclusively on hematopoietic cells, which has both decay-accelerating and cofactor activity. CD59 is a membrane regulator that functions independently from CD46 and CD55. It prevents insertion of C8 and C9 into the membrane, thus inhibiting formation of the membrane attack complex and preventing cell perturbation.
Complement activation is viewed traditionally as occurring in the blood because of abundant hepatic production and secretion of most of its components. However, multiple cell types are also known to synthesize complement proteins.1 Those synthesized by resident and invading immune cells have been summarized recently.2 In contrast, less is known about production and activation of complement in local tissues in contact with the environment, such as the airway. Moreover, whether local complement activation worsens or mitigates injury has been inadequately investigated in most organ systems. In this minireview, we appraise the literature published in the last 3 years on locally synthesized complement proteins in the airway and their potential role in patients with respiratory disease.
ALLERGIC RHINITIS AND ASTHMA
Unbiased transcriptomic approaches have provided insight into complement expression after environmental exposures. Patients with allergic rhinitis who underwent nasal allergen challenge with grass pollen demonstrated upregulated transcripts of the complement protein properdin (CFP) and C5a receptor (C5aR1) in their nasal mucosal specimens, along with the IL-1 family of cytokines (IL-1α and IL-1β), TH2 cytokines (IL-4, IL-5, and IL-13), and interferons.3 Prednisone downregulated CFP and C5aR1 in some but failed in 7 of 19 subjects, which highlights variation in the immune response from patient to patient in those with allergic rhinitis and asthma. These patients also had variable baseline IL33 mRNA levels. Thus, the underlying genetic predisposition, as well as baseline levels of cytokines, influenced complement gene expression and accounted for why patients had a variable response to prednisone.
Expression of properdin, a positive regulator of the alternative pathway (Fig 1, A), is increased in the bronchoalveolar lavage (BAL) fluid of asthmatic patients. The pathophysiologic correlation in a mouse model has been described elsewhere.E44 C5aR1 binds C5a, with the latter generated by means of cleavage of C5 downstream of C3 activation (Fig 1, A). Although there are no clinical trials yet targeting C5aR in asthmatic patients, preclinical studies showed that C5aR1 antagonism decreased BAL fluid and parenchymal inflammatory cells, IL-13 levels, and TH2 cytokine gene expression without reducing serum IgE levels in OVA- sensitized and challenged mice.4 Hence, properdin and C5aR1 should be evaluated further as potential therapeutic targets for difficult-to-treat allergic rhinitis and asthma.
CYSTIC FIBROSIS
Complement proteins have also been shown to be produced and secreted by airway epithelial cells. C3 was identified as one of the most abundant proteins, along with mucin 5B, mucin 5AC, and fibronectin, in apical secretions from cultures of bronchial epithelial cell lines derived from patients with cystic fibrosis (CF).5 Factor B was increased, but Factor I was decreased in the CF secretome, as detected by using mass spectrometry–based proteomics. Factor B is a critical enzymatic component of the alternative pathway and promotes inflammation (Fig 1, A), whereas Factor I is a fluid-phase serine protease that cleaves C3b to inactivated C3b (iC3b, relative to hemolytic activity) and thus, mitigates inflammation (Fig 1, B). Hence, low levels of Factor I could lead to poorly controlled C3 convertase formation, excessive cascade activation, and a potentially heightened airway inflammatory response. Supporting this concept, there was significantly more C3 activation in the CF secretome, as detected by using immunoblotting.5 Levels of proteases, such as matrix metalloproteinase 9 and cathepsins B and D, were also increased. Whether these proteases are involved directly in cleaving C3 is unknown; however, it suggests that the CF airway epithelial microenvironment can facilitate C3 cleavage in vivo.
OBLITERATIVE BRONCHIOLITIS
Obliterative bronchiolitis (OB) is a form of chronic airway epithelial cell injury that is a predominant manifestation of chronic rejection in patients after lung transplantation and a major impediment to long-term graft survival. Epithelial injury resulting in OB after lung transplantation has been associated with complement-dependent cytotoxicity; however, its precise mechanism is unclear. C3a and C5a upregulate proinflammatory mediators associated with airway epithelial cell injury (Fig 1, A).E34 Conversely, CD55 (decay-accelerating factor) is downregulated on airway epithelial cells of patients with severe asthma in response to injury (viral infection or ozone).E1 CD55 is a ubiquitously expressed membrane inhibitor that prevents binding of Factor B to the membrane and facilitates decay of the C3 convertase (Fig 1, B). CD46 is also a widely expressed membrane regulator and a cofactor for Factor I to inactivate C3b (Fig 1, B).1 Thus, downregulation of these complement inhibitory proteins (CIPs) could lead to poorly controlled airway inflammation and its downstream consequences, such as fibrosis.
Accordingly, reduced expression of CD55 and CD46 was implicated in driving OB after lung transplantation. Both were decreased on the epithelium of patients with OB.6 These recipients had increased C3a levels in their BAL fluid, indicating C3 activation. In a mouse model in which OB developed at day 21 after lung transplantation, levels of CD55 and Crry (the murine functional homolog of CD46) were decreased in the allograft at day 1, establishing that these changes occurred early. When CD31 T cells were cocultured with allograft-specific antigen-presenting cells, C3a increased IL-17a production, which subsequently downregulated Crry. Additionally, treatment with an anti-C5 antibody mitigated OB in mice. This report proposed cross-talk between IL-17a and C3a generation that downregulated CD55 and CD46 expression, thereby increasing the risk of developing epithelial injury and OB after lung transplantation.
INTERSTITIAL LUNG DISEASE
In line with the findings described above in OB, patients with idiopathic pulmonary fibrosis (IPF) had increased C3a and C5a levels in plasma and lung homogenates, leading to speculation that complement is activated both systemically and locally in patients with this irreversible lung disease.7 In small-airway epithelial cells (SAECs) the fibrogenesis mediator TGF-β induced dose- dependent downregulation of CD55 and CD46 expression. This was associated with increased cleaved poly-ADP ribose polymerase expression, a marker of injury, and a concurrent decrease in E-cadherin, a marker of epithelial-mesenchymal transition (EMT), both of which are hallmarks of IPF. Treatment of SAECs with the anaphylatoxins C3a and C5a at concentrations similar to those identified in the lungs of patients with IPF also resulted in loss of CD55 and CD46, implicating that this downregulation occurred independently with both anaphylatoxins and TGF-β.
The argument for exuberant complement activation in patients with IPF was strengthened by finding increased soluble C5b-9 levels in lung lavage fluid from these patients, suggesting that terminal membrane attack complex formation can mediate cell damage (Fig 1, A).8 In a mouse model of bleomycin-induced lung injury, inhibiting C3aR or C5aR with systemically administered antagonists arrested fibrosis by downregulating TGF-β/bone morphologic protein signaling pathways and reducing local complement activation. Of note, this work focused predominantly on C5aR1. Although TGF-β upregulated C5aR2 in SAECs, its role in patients with IPF is still unclear.
These recent reports renew the importance of local complement activation in the respiratory tract. Complement proteins produced by human airway epithelial cells are listed in Table I. Additionally, the complement system does not act independently in the lung microenvironment. There is a bidirectional interaction between complement proteins and other components of the innate immune system, such as antimicrobial peptides (neutrophil- derived α-defensins and human β-defensin 2), that facilitates host defense at the air-liquid interface.9 An emerging concept for lung disease pathogenesis is that external stress (eg, hypoxia or allergen challenge) or the inherent proinflammatory environment (eg, in those with CF and IPF) increases local synthesis of complement proteins. At the same time, mediators activating the complement cascade also downregulate CIPs, thus putatively propagating ongoing epithelial injury.
TABLE I.
Complement components produced by airway epithelial cells
| Components | Evidence | Primary cell data | Tissue data |
|---|---|---|---|
| Classical pathway | |||
|
| |||
| C1q | Not expressedE1 | ||
|
| |||
| C1r | mRNAE2–E5 | Zhou et alE2 | |
| ProteinE6 | |||
|
| |||
| C1s | mRNAE3,E7 | ||
| ProteinE6 | |||
|
| |||
| C4 | mRNAE8–E10 | Scheetz et alE9 | |
| ProteinE6 | Walters et alE10 | ||
| SupernatantE11,E12 | Peters-Hall et alE11 | ||
| Brass et alE12 | |||
|
| |||
| C2* | mRNAE8 | ||
|
| |||
| Lectin pathway | |||
|
| |||
| MBL | mRNAE5,E13 | Vandermeer et alE13 | |
|
| |||
| H-ficolin | mRNAE14 | Akaiwa et alE14 | |
| ProteinE14,E15 | |||
|
| |||
| Alternative pathway | |||
|
| |||
| C3 | mRNAE7,E8,E16–E18 | Ali et alE20 | Schlosser et alE17 |
| ProteinE6,E18,E19 | Pillai et alE21 | Lane et alE18 | |
| SupernatantE1,E16,E20,E21 | |||
|
| |||
| Factor B | mRNAE8,E13,E16,E17,E22 | Peters-Hall et alE11 | Vandermeer et alE13 |
| ProteinE6 | Brass et alE12 | Schlosser et alE17 | |
| SupernatantE11,E12,E16,E21 | Pillai et alE21 | ||
| Cooper et alE22 | |||
|
| |||
| Factor D | No data and not expected to occur (primary source: adipocytes) | ||
|
| |||
| Properdin* | mRNAE13,E18,E23 | Vandermeer et alE13 | |
| Lane et alE18 | |||
|
| |||
| Terminal complement complex | |||
|
| |||
| C5 | mRNAE7,E8,E17 | SupernatantE24 | |
| ProteinE6 | |||
| Schlosser et alE17 | |||
|
| |||
| C6, C7, C8, C9* | mRNAE6,E17,E25 | Schlosser et alE17 | |
| ProteinE6 | Ying et alE25 | ||
|
| |||
| Regulators, fluid phase | |||
|
| |||
| C1INH | No data | ||
|
| |||
| C4BP* | SupernatantE26,E27 | ||
|
| |||
| Factor H | mRNAE13,E28 | Pillai et alE21 | Vandermeer et alE13 |
| ProteinE6 | |||
| SupernatantE21,E28 | |||
|
| |||
| Factor I | mRNAE13 | ||
| ProteinE6 | |||
| SupernatantE21,E26 | |||
|
| |||
| Clusterin | mRNAE5,E29 | Peters-Hall et alE11 | |
| SupernatantE11 | Hackett et alE29 | ||
|
| |||
| Regulators, membrane bound | |||
|
| |||
| CD46 (MCP) | mRNAE26,E30 | Suzuki et alE32 | |
| ProteinE1,E31–E33 | Varsano et alE33 | ||
|
| |||
| CD55 (DAF) | mRNAE34–E37 | Agrawal et alE34 | Suzuki et alE32 |
| ProteinE1,E32,E33,E35,E37 | Pandya et alE35 | Varsano et alE33 | |
| Supernatant (shedding)E36 | Vainer et alE37 | ||
|
| |||
| CD59 | mRNAE4,E38 | Brass et alE12 | Varsano et alE33 |
| ProteinE1,E33,E39 | Ali et alE20 | ||
| Supernatant (shedding/exosomes)E12,E20,E40 | Castillon et alE39 | ||
| Kesimer et alE40 | |||
|
| |||
| Receptors | |||
|
| |||
| C3aR1 | mRNAE41 | Drouin et alE41 | Drouin et alE41 |
| ProteinE41–E43 | Fregonese et alE42 | ||
|
| |||
| C5aR1 | mRNAE29,E41 | Hackett et alE29 | Gu et alE43 |
| ProteinE41–E43 | Drouin et alE41 | ||
|
| |||
| C5aR2 | No data | ||
|
| |||
| CR1 (CD35) | Not expressedE33 | ||
Components that are not expressed or have no data supporting their production by airway epithelial cells are indicated by italics in the Evidence column. Evidence for protein content was based on immunoblotting, flow cytometry, or mass spectrometry of the cells/cell lysate. Supernatant denotes that the cells secreted the protein, unless otherwise specified (in which case it was either shed or secreted as exosomes).
C1INH, C1-inhibitor; C4BP, C4b-binding protein; DAF, decay-accelerating factor; MBL, mannose-binding lectin; MCP, membrane cofactor protein.
Studies from a single research group or conflicting evidence existing in the literature.
Why CIP downregulation is part of the airway epithelial response is puzzling. We speculate that one reason might be that in an acute insult (eg, pneumonia) the airway requires a proinflammatory status to upregulate the complement cascade and downregulate CIPs for the rapid clearing of pathogen or debris. However, chronic complement activation becomes deleterious if this proinflammatory status is prolonged, resulting in irreversible lung disease. Unfortunately, there is no clear evidence that CH50, AH50, and mannose-binding lectin levels are diagnostic biomarkers in any pulmonary disease. Novel therapies targeting locally active complement proteins need to be designed and tested in these difficult-to-treat respiratory diseases to mitigate dysregulation of the complement system.
Although there are no clinical trials currently targeting locally generated complement proteins in the lung, complement inhibitors are now being evaluated for respiratory diseases. These include use of intravenous C1-inhibitor to reduce house dust mite plus LPS–induced bronchial inflammation (NCT03051698, phase IV, recruiting) and use of IFX-1, an anti-human C5a mAb for treating early severe sepsis or septic shock displaying at least 1 newly developed organ dysfunction and showing clinical evidence of pulmonary or abdominal infection (NCT02246595, phase II, completed). A recent review provides a detailed description of historical and current complement-modulating therapies being evaluated in clinical trials.10
Additionally, locally delivered complement inhibitors (eg, lampalizumab, a specific inhibitor of Factor D in age-related macular degeneration, NCT02288559) are now being tested. Hence, one approach would be to consider locally delivering already approved specific inhibitors, such as purified C1-esterase inhibitor and anti-C5 mAbs to the lungs (through nebulizers).
Additionally, a relatively unexplored area is enhancing complement regulatory proteins to reduce inflammation, especially in diseases such as IPF and OB that have a known imbalance in regulators. Although recombinant and soluble forms of CD46 and CD55 have halted at the preclinical stage in patients with nonpulmonary disease, soluble CR1 was tested in patients with acute lung injury but remains to be evaluated in patients with chronic fibrotic lung injury.E45 This is an exciting but challenging and rapidly expanding field that now more than ever needs to be better understood and tapped for mitigating lung diseases.
Acknowledgments
Supported by the National Institutes of Health (NIH; R01 GM0099111 to J.P.A. and R01HL122585 to S.L.B.) and NIH training grants in the Principles of Pulmonary Research (5T32 HL007317 to H.S.K., and the Immunobiology of Rheumatic Diseases (5T32-AR007279 to H.S.K.). Research reported in this publication is supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases, part of the NIH (P30AR048335); an NIH grant for the Washington University Institute of Clinical and Translational Sciences (3UL1 TR000448); and the Hubert C. and Dorothy R. Moog Professorship (to S.L.B.) supported by the Barnes-Jewish Hospital Foundation.
J. P. Atkinson has received grants from the National Institutes of Health (2 R01 GM099111 and R21 AR069833) and the Rheumatology Research Foundation; has board memberships with Compliment Corporation, Kypha Inc, and Gemini Therapeutics; has consultant arrangements with Achillion Pharmaceuticals, BioMarin Pharmaceuticals, Annexon Biosciences, Celldex Therapeutics, Clinical Pharmacy Services CDMI, Kypha Inc, Omeros Corporation, and True North Therapeutics; and receives stock/stock options from Compliment Corporation, Kypha Inc, and Gemini Therapeutics.
Footnotes
Disclosure of potential conflict of interest: The rest of the authors declare that they have no relevant conflicts of interest.
References
- 1.Barnum SR. Complement: a primer for the coming therapeutic revolution. Pharmacol Ther. 2017;172:63–72. doi: 10.1016/j.pharmthera.2016.11.014. [DOI] [PubMed] [Google Scholar]
- 2.Lubbers R, van Essen MF, van Kooten C, Trouw LA. Production of complement components by cells of the immune system. Clin Exp Immunol. 2017;188:183–94. doi: 10.1111/cei.12952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leaker BR, Malkov VA, Mogg R, Ruddy MK, Nicholson GC, Tan AJ, et al. The nasal mucosal late allergic reaction to grass pollen involves type 2 inflammation (IL-5 and IL-13), the inflammasome (IL-1β), and complement. Mucosal Immunol. 2017;10:408–20. doi: 10.1038/mi.2016.74. [DOI] [PubMed] [Google Scholar]
- 4.Staab EB, Sanderson SD, Wells SM, Poole JA. Treatment with the C5a receptor/CD88 antagonist PMX205 reduces inflammation in a murine model of allergic asthma. Int Immunopharmacol. 2014;21:293–300. doi: 10.1016/j.intimp.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peters-Hall JR, Brown KJ, Pillai DK, Tomney A, Garvin LM, Wu X, et al. Quantitative proteomics reveals an altered cystic fibrosis in vitro bronchial epithelial secretome. Am J Respir Cell Mol Biol. 2015;53:22–32. doi: 10.1165/rcmb.2014-0256RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suzuki H, Lasbury ME, Fan L, Vittal R, Mickler EA, Benson HL, et al. Role of complement activation in obliterative bronchiolitis post-lung transplantation. J Immunol. 2013;191:4431–9. doi: 10.4049/jimmunol.1202242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gu H, Mickler EA, Cummings OW, Sandusky GE, Weber DJ, Gracon A, et al. Crosstalk between TGF-β1 and complement activation augments epithelial injury in pulmonary fibrosis. FASEB J. 2014;28:4223–34. doi: 10.1096/fj.13-247650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gu H, Fisher AJ, Mickler EA, Duerson F, Cummings OW, Peters-Golden M, et al. Contribution of the anaphylatoxin receptors, C3aR and C5aR, to the pathogenesis of pulmonary fibrosis. FASEB J. 2016;30:2336–50. doi: 10.1096/fj.201500044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hiemstra PS. Parallel activities and interactions between antimicrobial peptides and complement in host defense at the airway epithelial surface. Mol Immunol. 2015;68:28–30. doi: 10.1016/j.molimm.2015.07.030. [DOI] [PubMed] [Google Scholar]
- 10.Morgan BP, Harris CL. Complement, a target for therapy in inflammatory and degenerative diseases. Nat Rev Drug Discov. 2015;14:857–77. doi: 10.1038/nrd4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
Supplementary References
- E1.Varsano S, Kaminsky M, Kaiser M, Rashkovsky L. Generation of complement C3 and expression of cell membrane complement inhibitory proteins by human bronchial epithelium cell line. Thorax. 2000;55:364–9. doi: 10.1136/thorax.55.5.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E2.Zhou H, Brekman A, Zuo W-L, Ou X, Shaykhiev R, Agosto-Perez FJ, et al. POU2AF1 Functions in the Human Airway Epithelium To Regulate Expression of Host Defense Genes. J Immunol. 2016;196:3159–67. doi: 10.4049/jimmunol.1502400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E3.Song M, Kim Y-J, Ryu J-C. Identification of genes induced by carbamazepine in human bronchial epithelial BEAS-2B cells. Toxicol Environ Health Sci. 2011;3:106–13. [Google Scholar]
- E4.Chen D, Stueckle TA, Luanpitpong S, Rojanasakul Y, Lu Y, Wang L. Gene expression profile of human lung epithelial cells chronically exposed to single-walled carbon nanotubes. Nanoscale Res Lett. 2015;10:12. doi: 10.1186/s11671-014-0707-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E5.Park E-J, Park K. Induction of pro-inflammatory signals by 1-nitropyrene in cultured BEAS-2B cells. Toxicol Lett. 2009;184:126–33. doi: 10.1016/j.toxlet.2008.10.028. [DOI] [PubMed] [Google Scholar]
- E6.Rothman BL, Merrow M, Bamba M, Kennedy T, Kreutzer DL. Biosynthesis of the third and fifth complement components by isolated human lung cells. Am Rev Respir Dis. 1989;139:212–20. doi: 10.1164/ajrccm/139.1.212. [DOI] [PubMed] [Google Scholar]
- E7.Boesewetter DE, Collier JL, Kim AM, Riley MR. Alterations of A549 lung cell gene expression in response to biochemical toxins. Cell Biol Toxicol. 2006;22:101–18. doi: 10.1007/s10565-006-0150-9. [DOI] [PubMed] [Google Scholar]
- E8.Strunk RC, Eidlen DM, Mason RJ. Pulmonary alveolar type II epithelial cells synthesize and secrete proteins of the classical and alternative complement pathways. J Clin Invest. 1988;81:1419–26. doi: 10.1172/JCI113472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E9.Scheetz TE, Zabner J, Welsh MJ, Coco J, Eyestone M de F, Bonaldo M, et al. Large-scale gene discovery in human airway epithelia reveals novel transcripts. Physiol Genomics. 2004;17:69–77. doi: 10.1152/physiolgenomics.00188.2003. [DOI] [PubMed] [Google Scholar]
- E10.Walters MS, De BP, Salit J, Buro-Auriemma LJ, Wilson T, Rogalski AM, et al. Smoking accelerates aging of the small airway epithelium. Respir Res. 2014;15:94. doi: 10.1186/s12931-014-0094-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E11.Peters-Hall JR, Brown KJ, Pillai DK, Tomney A, Garvin LM, Wu X, et al. Quantitative proteomics reveals an altered cystic fibrosis in vitro bronchial epithelial secretome. Am J Respir Cell Mol Biol. 2015;53:22–32. doi: 10.1165/rcmb.2014-0256RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E12.Brass DM, Gwinn WM, Valente AM, Kelly FL, Brinkley CD, Nagler AE, et al. The Diacetyl-Exposed Human Airway Epithelial Secretome: New Insights into Flavoring-Induced Airways Disease. Am J Respir Cell Mol Biol. 2017;56:784–95. doi: 10.1165/rcmb.2016-0372OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E13.Vandermeer J, Sha Q, Lane AP, Schleimer RP. Innate immunity of the sinonasal cavity: expression of messenger RNA for complement cascade components and toll-like receptors. Arch Otolaryngol Head Neck Surg. 2004;130:1374–80. doi: 10.1001/archotol.130.12.1374. [DOI] [PubMed] [Google Scholar]
- E14.Akaiwa M, Yae Y, Sugimoto R, Suzuki SO, Iwaki T, Izuhara K, et al. Hakata antigen, a new member of the ficolin/opsonin p35 family, is a novel human lectin secreted into bronchus/alveolus and bile. J Histochem Cytochem. 1999;47:777–86. doi: 10.1177/002215549904700607. [DOI] [PubMed] [Google Scholar]
- E15.Bidula S, Sexton DW, Yates M, Abdolrasouli A, Shah A, Wallis R, et al. H-ficolin binds Aspergillus fumigatus leading to activation of the lectin complement pathway and modulation of lung epithelial immune responses. Immunology. 2015;146:281–91. doi: 10.1111/imm.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E16.Zhao YX, Andoh A, Shimada M, Takaya H, Hata K, Fujiyama Y, et al. Secretion of complement components of the alternative pathway (C3 and factor B) by the human alveolar type II epithelial cell line A549. Int J Mol Med. 2000;5:415–9. doi: 10.3892/ijmm.5.4.415. [DOI] [PubMed] [Google Scholar]
- E17.Schlosser RJ, Mulligan RM, Casey SE, Varela JC, Harvey RJ, Atkinson C. Alterations in gene expression of complement components in chronic rhinosinusitis. Am J Rhinol Allergy. 2010;24:21–5. doi: 10.2500/ajra.2010.24.3399. [DOI] [PubMed] [Google Scholar]
- E18.Lane AP, Truong-Tran Q-A, Myers A, Bickel C, Schleimer RP. Serum amyloid A, properdin, complement 3, and toll-like receptors are expressed locally in human sinonasal tissue. Am J Rhinol. 2006;20:117–23. [PMC free article] [PubMed] [Google Scholar]
- E19.Hill LD, Sun L, Leuschen MP, Zach TL. C3 synthesis by A549 alveolar epithelial cells is increased by interferon-gamma and dexamethasone. Immunology. 1993;79:236–40. [PMC free article] [PubMed] [Google Scholar]
- E20.Ali M, Lillehoj EP, Park Y, Kyo Y, Kim KC. Analysis of the proteome of human airway epithelial secretions. Proteome Sci. 2011;9:4. doi: 10.1186/1477-5956-9-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E21.Pillai DK, Sankoorikal B-JV, Johnson E, Seneviratne AN, Zurko J, Brown KJ, et al. Directional secretomes reflect polarity-specific functions in an in vitro model of human bronchial epithelium. Am J Respir Cell Mol Biol. 2014;50:292–300. doi: 10.1165/rcmb.2013-0188OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E22.Cooper P, Potter S, Mueck B, Yousefi S, Jarai G. Identification of genes induced by inflammatory cytokines in airway epithelium. Am J Physiol Lung Cell Mol Physiol. 2001;280:L841–852. doi: 10.1152/ajplung.2001.280.5.L841. [DOI] [PubMed] [Google Scholar]
- E23.Sha Q, Truong-Tran AQ, Plitt JR, Beck LA, Schleimer RP. Activation of airway epithelial cells by toll-like receptor agonists. Am J Respir Cell Mol Biol. 2004;31:358–64. doi: 10.1165/rcmb.2003-0388OC. [DOI] [PubMed] [Google Scholar]
- E24.Rothman BL, Despins AW, Kreutzer DL. Cytokine regulation of C3 and C5 production by the human type II pneumocyte cell line, A549. J Immunol Baltim Md 1950. 1990;145:592–8. [PubMed] [Google Scholar]
- E25.Ying L, Zhang F, Pan X, Chen K, Zhang N, Jin J, et al. Complement component 7 (C7), a potential tumor suppressor, is correlated with tumor progression and prognosis. Oncotarget. 2016;7:86536–46. doi: 10.18632/oncotarget.13294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E26.Okroj M, Corrales L, Stokowska A, Pio R, Blom AM. Hypoxia increases susceptibility of non-small cell lung cancer cells to complement attack. Cancer Immunol Immunother. 2009;58:1771–80. doi: 10.1007/s00262-009-0685-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E27.Luo X, Liu Y, Wang R, Hu H, Zeng R, Chen H. A high-quality secretome of A549 cells aided the discovery of C4b-binding protein as a novel serum biomarker for non-small cell lung cancer. J Proteomics. 2011;74:528–38. doi: 10.1016/j.jprot.2011.01.011. [DOI] [PubMed] [Google Scholar]
- E28.Ajona D, Hsu Y-F, Corrales L, Montuenga LM, Pio R. Down-regulation of human complement factor H sensitizes non-small cell lung cancer cells to complement attack and reduces in vivo tumor growth. J Immunol. 2007;178:5991–8. doi: 10.4049/jimmunol.178.9.5991. [DOI] [PubMed] [Google Scholar]
- E29.Hackett NR, Butler MW, Shaykhiev R, Salit J, Omberg L, Rodriguez-Flores JL, et al. RNA-Seq quantification of the human small airway epithelium transcriptome. BMC Genomics. 2012;13:82. doi: 10.1186/1471-2164-13-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E30.Zell S, Geis N, Rutz R, Schultz S, Giese T, Kirschfink M. Down-regulation of CD55 and CD46 expression by anti-sense phosphorothioate oligonucleotides (S-ODNs) sensitizes tumour cells to complement attack. Clin Exp Immunol. 2007;150:576–84. doi: 10.1111/j.1365-2249.2007.03507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E31.de Astorza B, Cortés G, Crespí C, Saus C, Rojo JM, Albertí S. C3 promotes clearance of Klebsiella pneumoniae by A549 epithelial cells. Infect Immun. 2004;72:1767–74. doi: 10.1128/IAI.72.3.1767-1774.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E32.Suzuki H, Lasbury ME, Fan L, Vittal R, Mickler EA, Benson HL, et al. Role of complement activation in obliterative bronchiolitis post-lung transplantation. J Immunol. 2013;191:4431–9. doi: 10.4049/jimmunol.1202242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E33.Varsano S, Frolkis I, Ophir D. Expression and distribution of cell-membrane complement regulatory glycoproteins along the human respiratory tract. Am J Respir Crit Care Med. 1995;152:1087–93. doi: 10.1164/ajrccm.152.3.7545058. [DOI] [PubMed] [Google Scholar]
- E34.Agrawal A, Sinha A, Ahmad T, Aich J, Singh P, Sharma A, et al. Maladaptation of critical cellular functions in asthma: bioinformatic analysis. Physiol Genomics. 2009;40:1–7. doi: 10.1152/physiolgenomics.00141.2009. [DOI] [PubMed] [Google Scholar]
- E35.Pandya PH, Fisher AJ, Mickler EA, Temm CJ, Lipking KP, Gracon A, et al. Hypoxia-Inducible Factor-1α Regulates CD55 in Airway Epithelium. Am J Respir Cell Mol Biol. 2016;55:889–98. doi: 10.1165/rcmb.2015-0237OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E36.Zhou J, To KK-W, Dong H, Cheng Z-S, Lau CC-Y, Poon VKM, et al. A functional variation in CD55 increases the severity of 2009 pandemic H1N1 influenza A virus infection. J Infect Dis. 2012;206:495–503. doi: 10.1093/infdis/jis378. [DOI] [PubMed] [Google Scholar]
- E37.Vainer ED, Meir K, Furman M, Semenenko I, Konikoff F, Vainer GW. Characterization of novel CD55 isoforms expression in normal and neoplastic tissues. Tissue Antigens. 2013;82:26–34. doi: 10.1111/tan.12138. [DOI] [PubMed] [Google Scholar]
- E38.Goswami MT, Reka AK, Kurapati H, Kaza V, Chen J, Standiford TJ, et al. Regulation of complement-dependent cytotoxicity by TGF-β-induced epithelial-mesenchymal transition. Oncogene. 2016;35:1888–98. doi: 10.1038/onc.2015.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E39.Castillon N, Hinnrasky J, Zahm J-M, Kaplan H, Bonnet N, Corlieu P, et al. Polarized expression of cystic fibrosis transmembrane conductance regulator and associated epithelial proteins during the regeneration of human airway surface epithelium in three-dimensional culture. Lab Invest. 2002;82:989–98. doi: 10.1097/01.lab.0000022221.88025.43. [DOI] [PubMed] [Google Scholar]
- E40.Kesimer M, Scull M, Brighton B, DeMaria G, Burns K, O’Neal W, et al. Characterization of exosome-like vesicles released from human tracheobronchial ciliated epithelium: a possible role in innate defense. FASEB J. 2009;23:1858–68. doi: 10.1096/fj.08-119131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E41.Drouin SM, Kildsgaard J, Haviland J, Zabner J, Jia HP, McCray PB, et al. Expression of the complement anaphylatoxin C3a and C5a receptors on bronchial epithelial and smooth muscle cells in models of sepsis and asthma. J Immunol. 2001;166:2025–32. doi: 10.4049/jimmunol.166.3.2025. [DOI] [PubMed] [Google Scholar]
- E42.Fregonese L, Swan FJ, van Schadewijk A, Dolhnikoff M, Santos MA, Daha MR, et al. Expression of the anaphylatoxin receptors C3aR and C5aR is increased in fatal asthma. J Allergy Clin Immunol. 2005;115:1148–54. doi: 10.1016/j.jaci.2005.01.068. [DOI] [PubMed] [Google Scholar]
- E43.Gu H, Fisher AJ, Mickler EA, Duerson F, Cummings OW, Peters-Golden M, et al. Contribution of the anaphylatoxin receptors, C3aR and C5aR, to the pathogenesis of pulmonary fibrosis. FASEB J. 2016;30:2336–50. doi: 10.1096/fj.201500044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E44.Wang Y, Miwa T, Ducka-Kokalari B, Redai IG, Sato S, Gullipalli D, et al. Properdin Contributes to Allergic Airway Inflammation through Local C3a Generation. J Immunol. 2015;195:1171–81. doi: 10.4049/jimmunol.1401819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E45.Zimmerman JL, Dellinger RP, Straube RC, Levin JL. Phase I trial of the recombinant soluble complement receptor 1 in acute lung injury and acute respiratory distress syndrome. Crit Care Med. 2000;28:3149–54. doi: 10.1097/00003246-200009000-00004. [DOI] [PubMed] [Google Scholar]