

Abstract
An increase in cytosolic Ca2+ concentration ([Ca2+]cyt) in pulmonary artery smooth muscle cells (PASMCs) triggers pulmonary vasoconstriction and stimulates PASMC proliferation leading to vascular wall thickening. Here, we report that STIM2, a Ca2+ sensor in the sarcoplasmic reticulum (SR) membrane, is required for raising the resting [Ca2+]cyt in PASMCs from patients with pulmonary arterial hypertension (PAH) and activating signaling cascades that stimulate PASMC proliferation and inhibit PASMC apoptosis. Downregulation of STIM2 in PAH-PASMCs reduces the resting [Ca2+]cyt, while overexpression of STIM2 in normal PASMCs increases the resting [Ca2+]cyt. The increased resting [Ca2+]cyt in PAH-PASMCs is associated with enhanced phosphorylation (p) of CREB, STAT3 and AKT, increased NFAT nuclear translocation, and elevated level of Ki67 (a marker of cell proliferation). Furthermore, the STIM2-associated increase in the resting [Ca2+]cyt also upregulates the anti-apoptotic protein Bcl-2 in PAH-PASMCs. Downregulation of STIM2 in PAH-PASMCs with siRNA a) decreases the level of pCREB, pSTAT3 and pAKT and inhibits NFAT nuclear translocation, thereby attenuating proliferation, and b) decreases Bcl-2, which leads to an increase of apoptosis. In summary, these data indicate that upregulated STIM2 in PAH-PASMCs, by raising the resting [Ca2+]cyt, contributes to enhancing PASMC proliferation by activating the CREB, STAT3, AKT and NFAT signaling pathways and stimulating PASMC proliferation. The STIM2-associated increase in the resting [Ca2+]cyt is also involved in upregulating Bcl-2 that makes PAH-PASMCs resistant to apoptosis, and thus plays an important role in sustained pulmonary vasoconstriction and excessive pulmonary vascular remodeling in patients with PAH.
Keywords: pulmonary arterial hypertension, store-operated Ca2+ entry, Ca2+ signaling, cell proliferation, cell apoptosis
Introduction
Idiopathic pulmonary arterial hypertension (IPAH) is a fatal and progressive disease. Sustained pulmonary vasoconstriction and concentric pulmonary artery wall thickening are the primary causes of the elevated pulmonary vascular resistance (PVR) and pulmonary arterial pressure (PAP) in patients with IPAH. An increase in cytosolic free Ca2+ concentration ([Ca2+]cyt) in pulmonary artery smooth muscle cells (PASMCs) is a major trigger for pulmonary vasoconstriction and an important cause for pulmonary vascular remodeling via stimulation of PASMC proliferation and inhibition of PASMC apoptosis. The current treatment for pulmonary arterial hypertension (PAH) is mainly based on the correction of increased ratio of circulating vasoconstrictors (e.g., endothelin) to vasodilators (e.g., prostacyclin and nitric oxide) by blocking endothelin receptors, increasing circulating prostacyclin (PGI2) or orally intaking PGI2 analogues, inhaling nitric oxide (NO), stimulating soluble guanylate cyclase or inhibiting phosphodiesterase.1 The mortality, however, remains high in patients with idiopathic PAH. It is thus important to define precise mechanisms involved in PASMC contraction, proliferation, and apoptosis, identify novel therapeutic targets and develop new and effective therapeutic interventions for PAH. One of the potential pathogenic mechanisms for PAH for developing novel therapies is the enhanced intracellular Ca2+ signaling in PAH-PASMCs, which not only induces PASMC contraction and migration but also stimulates PASMC proliferation and inhibits PASMC apoptosis.
In addition to causing PASMC contraction and pulmonary vasoconstriction, a rise in [Ca2+]cyt in PASMC can activate transcription factors and signal transduction proteins essential for cell proliferation and migration.2–6 Removal or chelation of extracellular free Ca2+ inhibits PASMC proliferation and migration, indicating that an increase in [Ca2+]cyt due to Ca2+ influx is a requisite for PASMCs to contract, proliferate and migrate.7, 8 We have previously reported that the resting [Ca2+]cyt in PAH-PASMCs is significantly higher than in normal PASMCs, while receptor-operated Ca2+ entry (ROCE) and store-operated Ca2+ entry (SOCE) are both enhanced in PAH-PASMCs compared to normal PASMCs.6, 9–11 The enhanced ROCE and SOCE have been demonstrated to result, at least partially, from upregulated protein expression and enhanced function of the transient receptor potential (TRP) channels (e.g., TRPC6/C3/C1) and Orai channels (Orai1/2).12–16 Tight control of the resting [Ca2+]cyt is vital in maintaining PASMC function and regulating PASMC contraction, proliferation and apoptosis.10, 17 Sustained and prolonged increase of the resting [Ca2+]cyt to an abnormal level has been implicated in many neurological and cardiovascular diseases.18, 19
Stromal interaction molecule 1 (STIM1) protein is a Ca2+ sensor expressed in the endoplasmic (ER) or sarcoplasmic (SR) reticulum membrane in a variety of cell types. Depletion or reduction of Ca2+ in the ER/SR results in dissociation of Ca2+ from the EF domain of STIM1, leading to STIM1 dimerization and translocation to the puncta area to recruit Orai1 channels in the plasma membrane to form store-operated Ca2+ channels (SOC) and inducing SOCE.20 STIM2 structurally shares significant homology with STIM1, the major differences between STIM2 and STIM1 are in the EF domain,21 a Ca2+-binding domain in the N-terminal region located in the luminal area of the ER/SR. When compared with STIM1, the relatively lower Ca2+ affinity of EF domain, together with the kinetics of Ca2+ dissociation, enables STIM2 to cluster at ER-PM junctions in response to small changes or minimal depletion of Ca2+ in the ER/SR stores. Thus, STIM2 has been proposed to regulate Ca2+ influx in unstimulated cells for maintaining [Ca2+]cyt or recruiting or gating Orai1 (and other SOC) at low concentrations of agonist stimulation.22, 23
Through siRNA screening of human signaling proteome, STIM2 was identified as the most potent positive regulator of the resting [Ca2+]cyt.22 Thus, it has been demonstrated that STIM2 is more involved in regulating the resting [Ca2+]cyt, while STIM1 is more involved in the regulation of SOCE.22 The reason that STIM2 acts as a predominant regulator of the resting [Ca2+]cyt is, as mentioned earlier, due to its low affinity to Ca2+ and/or its high sensitivity to changes of [Ca2+] in the ER/SR. A small decrease in ER/SR [Ca2+] would cause STIM2 to translocate to the ER/SR-plasma membrane (ER-PM) junctions to induce Ca2+ influx by recruiting Orai and TRPC to form store-operated Ca2+ channels. In addition to the store-dependent regulation of the resting [Ca2+]cyt, studies also showed store-independent controls of STIM2/Orai1 complex and resting [Ca2+]cyt. First, a cytosolic STIM2 preprotein created by signal peptide inefficiency can activate Orai1 in a store-independent manner.24 Second, the STIM2/Orai1 complex formation is under control of cytoplasmic Ca2+/calmodulin (CaM); low [Ca2+]cyt or low activity of CaM promotes STIM2/Orai1 formation independent of [Ca2+] in the ER/SR.25
In this study, we show that a) STIM2 is involved in the elevated resting [Ca2+]cyt in PAH-PASMCs and b) the STIM2-associated increase in the resting [Ca2+]cyt is responsible for stimulating PASMC proliferation and inhibiting PASMC apoptosis in PAH. The critical role of STIM2 in the pathogenesis of PAH suggests that targeting STIM2 may potentially be a good strategy to develop novel therapeutic approaches for PAH.
Materials and Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Subjects
The diagnosis of PAH was established clinically in five patients by NIH PAH Registry with an average mean PAP of 56±5 mmHg. Use of human pulmonary tissue and cell for these experiments was approved by the University of Arizona Institutional Review Board.
[Ca2+]cyt Measurement
Fura-2 fluorescence was measured in PASMCs from normal subjects and PAH patients as described previously,26 with an NIS Elements 3.2 software on a Nikon inverted fluorescent microscope (Eclipse Ti-E; Nikon, Tokyo, Japan).
Drugs and Chemicals
Cyclopiazonic acid (CPA) (Sigma-Aldrich) was prepared and aliquoted as stock solutions in dimethyl sulfoxide (DMSO) and stored at −20°C until use.
Statistical Analysis
Data are expressed as the mean±standard error (SE). Statistical significance between two groups or among multiple groups was examined using Student’s t-test or Scheffé’s test after One Way ANOVA, respectively. Significant difference is expressed in the figure legends as *P<0.05, **P<0.01 and ***P<0.001.
An expanded materials and methods section is available in the online-only Data Supplement.
Results
Enhanced Resting [Ca2+]cyt and Upregulated STIM2 in PAH-PASMC
We first compared the resting [Ca2+]cyt in PASMCs isolated from normal subjects (n=3) and PAH patients (n=5). The results showed a significant increase of resting [Ca2+]cyt in PAH-PASMCs (111.7±2.00 nM, n=256 cells, 3-5 repeats for each patient from 5 patients) in comparison to normal PASMCs (82.7±2.06 nM, n=140 cells, 3-5 repeats for each subject from 3 normal subjects) (Fig. 1A and B). The relative 30 nM difference in the resting [Ca2+]cyt in PAH-PASMCs and normal PASMCs (approximately 35% increase in the resting [Ca2+]cyt for PAH-PASMCs compared to normal PASMCs), is critical for activating downstream Ca2+-sensitive cell signaling cascade responsible for cell contraction, migration, and proliferation.
Figure 1.
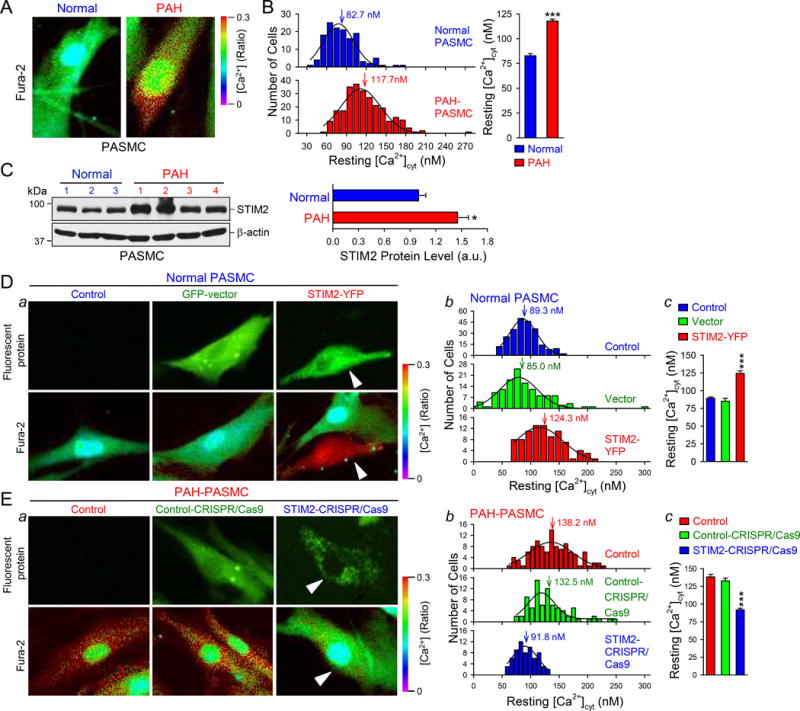
STIM2 protein expression is upregulated in PASMC from patients with PAH compared with PASMC from normal subjects and is sufficient and necessary for increasing the resting [Ca2+]cyt in PASMCs. A Representative fluorescent images showing the resting [Ca2+]cyt in a normal PASMC and a PAH PASMC. B, Histogram (left) showing the distribution of the resting [Ca2+]cyt in normal (n=3 normal subjects) and PAH (n=5 patients) PASMCs. Summarized data (right, means±SE) showing the averaged resting [Ca2+]cyt in normal and PAH PASMCs. ***P<0.001 vs. Normal. C, Western blot analyses on STIM2 (left) in normal and PAH PASMCs. Summarized data (right, means±SE, n=3 normal subjects, and 4 PAH patients) showing the protein level of STIM2 in normal and PAH PASMCs. *P<0.05 vs. Normal. D, Representative fluorescent images (a) showing the resting [Ca2+]cyt in normal PASMCs and normal PASMCs transfected with GFP-vector and STIM2 gene (STIM2-YFP). Arrows indicate the normal PASMCs that are positively transfected with STIM2-YFP. Histogram (b) showing the distribution of the resting [Ca2+]cyt in Control, GFP-vector-transfected and STIM2-YFP-transfected normal PASMCs. Vertical arrows indicate the peak values of the resting [Ca2+]cyt. Summarized data (c, means±SE, n=3 normal subjects) showing the averaged resting [Ca2+]cyt in Control, Vector-transfected and STIM2-transfected normal PASMCs. ***P<0.001 vs. Control and GFP-vector. E, Representative fluorescent images (a) showing the resting [Ca2+]cyt in PAH-PASMCs (Control) and PAH-PASMCs transfected with a Control-CRISPR/Cas9 plasmid (Control-CRISPR/Cas9) and the STIM2-CRISPR/Cas9 plasmid (STIM2-CRISPR/Cas9). Arrows indicate the PAH-PASMCs that are positively transfected with STIM2-CRISPR/Cas9 plasmid. Histogram (b) showing the distribution of the resting [Ca2+]cyt in Control, Control-CRISPR/Cas9-transfected and STIM2-CRISPR/Cas9-transfected PAH-PASMCs, with the averaged values of the resting [Ca2+]cyt at 138.2, 132.5 and 91.8 nM, respectively. Summarized data (c, means±SE, n=5 PAH patients) showing the level of the resting [Ca2+]cyt in Control, Control-CRISPR/Cas9-transfected and STIM2-CRISPR/Cas9-transfected PAH-PASMC. ***P<0.001 vs. Control and Control-CRISPR/Cas9.
STIM proteins, especially STIM2, are involved in both store-dependent and store-independent regulation of [Ca2+]cyt.22 In small pulmonary artery (PA) from normal subjects, we found that the protein expression of STIM2 was low in smooth muscle. In contrast, in PAs from PAH patients, the protein expression level of STIM2 was high in smooth muscle layer (Fig. S1A). In isolated PASMCs, STIM2 exhibited significantly higher expression level in PAH patients than in normal subjects (Fig. 1C). Similarly, STIM2 levels in PAs isolated from rats with MCT-induced pulmonary hypertension (PH) and mice with hypoxia-induced PH, were both greater than in PAs from normal controls (data not shown). These results indicate that the enhanced resting [Ca2+]cyt in PASMCs from PAH patients (or PH animals) is associated with upregulated expression of STIM2.
These results directed us to speculate that upregulation of STIM2 is important for initiating and/or maintaining the highly proliferative phenotype of PAH-PASMCs via enhancing the resting [Ca2+]cyt.
STIM2 is Sufficient and Necessary to Increase the Resting [Ca2+]cyt in PASMC
Downregulation of STIM2 in PAH-PASMCs (where STIM2 is upregulated) by CRISPR/Cas9 reduced the resting [Ca2+]cyt from 138.3±3.55 nM (n=111 cells, 3-5 repeats for each patient from 5 patients) to 91.8±2.22 nM (n=68 cells, 3-5 repeats for each patient from 5 patients), while overexpression of STIM2 in normal PASMCs (where STIM2 level is low) increased the resting [Ca2+]cyt from 89.3±1.59 nM (n=234 cells, 3-5 repeats for each subject from 3 normal subjects) to 124.3±3.72 nM (n=90 cells, 3-5 repeats for each subject from 3 normal subjects) (Fig. 1D and E). The approximate 35 nM enhancement in the resting [Ca2+]cyt in STIM2-transfected PASMCs compared to control PASMCs, is similar to the difference (~46.5 nM) between normal PASMCs and PAH-PASMCs (see Fig. 1A and B). We also found that overexpression of STIM2 in HEK293 cells significantly enhanced the resting [Ca2+]cyt (Fig. S1B and C). These data indicate that STIM2 is sufficient to increase the resting [Ca2+]cyt in normal PASMCs and necessary for the enhanced resting [Ca2+]cyt in PAH-PASMCs.
Upregulated STIM2 Activates Ca2+-dependent Signaling Cascades in PASMC
Upregulation of STIM2 correlates with enhanced resting [Ca2+]cyt in PASMCs. The next set of experiments was designed to examine whether the STIM2-mediated increase in the resting [Ca2+]cyt enhances phosphorylation of the signaling proteins and transcription factors directly or indirectly involved in stimulating PASMC proliferation (e.g., CREB, STAT3 and NFATc2) and inhibiting PASMC apoptosis (e.g., Bcl-2).3, 4, 27 In addition, we also examined the effect of overexpressing STIM2 and enhancing [Ca2+]cyt on the AKT signaling pathway, which has been shown to be Ca2+/CaM dependent.28 The level of phosphorylated (p) CREB, STAT3, and AKT in PAH-PASMCs was significantly higher than in normal PASMCs under the resting condition (Fig. 2A). The nuclear translocation of NFATc2 was detected (i.e., activated NFATc2) in ~40% PAH-PASMCs, while NFATc2 was mostly located in the cytoplasm (i.e., inactive NFATc2) in >90% normal PASMCs (Fig. 2C). Downregulation of STIM2 in PAH-PASMC with siRNA significantly reduced the level of pCREB, pSTAT3 and pAKT (Fig. 2B) and decreased the nuclear translocation of NFATc2 (Fig. 2C). We then overexpressed STIM2 in normal PASMCs to determine whether upregulation of STIM2 and increased resting [Ca2+]cyt were sufficient to activate Ca2+-dependent signaling pathways. As shown in Figure 3A, overexpression of STIM2 in normal PASMCs increased pCREB, pSTAT3 and pAKT (Fig. 3A) and enhanced NFATc2 nuclear translocation (Fig. 3C). These results indicate that STIM2 is sufficient to increase phosphorylation of CREB, STAT3, and AKT, and activated NFATc2 in normal PASMC.
Figure 2.
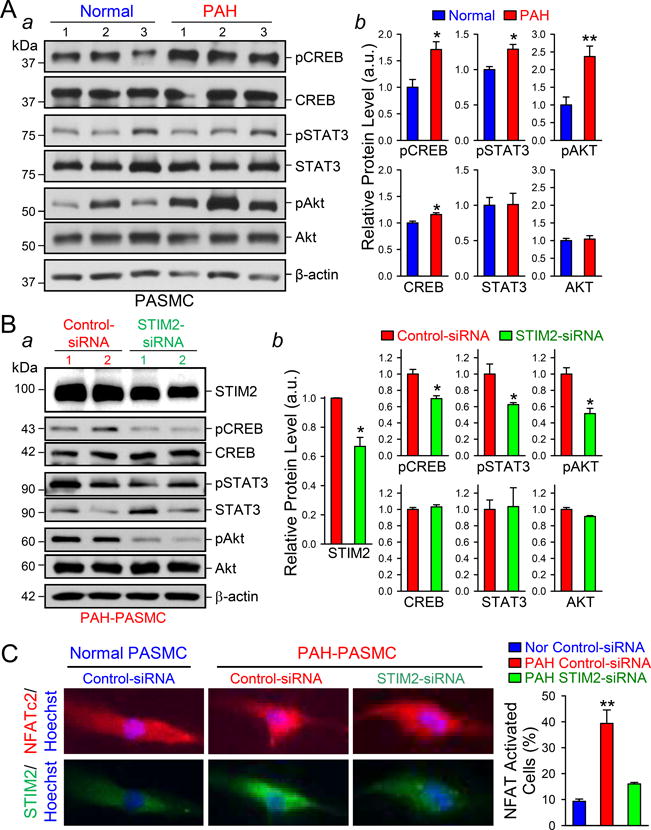
STIM2 is necessary for increasing the level of phosphorylated (p) CREB, STAT3, and AKT and enhancing the nuclear translocation of NFATC2 in PASMCs. A Western blot analyses (a) on pCREB, CREB, pSTAT3, STAT3, pAKT, and AKT in normal and PAH PASMCs. Summarized data (b, means±SE, n=3 normal subjects and 5 PAH patients) showing the protein level of pCREB, CREB, pSTAT3, STAT3, pAKT and AKT in normal and PAH PASMC. *P<0.05 and **P<0.01 vs. Normal. B, Western blot analyses (a) on pCREB, CREB, pSTAT3, STAT3, pAKT, and AKT in PAH-PASMCs transfected with control-siRNA and STIM2-siRNA. Summarized data (b, means±SE, n=5 PAH patients) showing the protein level of pCREB, CREB, pSTAT3, STAT3, pAKT and AKT in control-siRNA- and STIM2-siRNA-transfected PAH-PASMCs. *P<0.05 vs. Control-siRNA. C, Representative fluorescent images showing NFATC2 (red), STIM2 (green) and nuclei (blue) in control-siRNA-transfected normal and PAH-PASMCs and STIM2-siRNA-transfected PAH-PASMCs. Summarized data (right, means±SE, n=3 normal subjects, and 5 PAH patients) showing the percentage of NFATC2-activated cells in control-siRNA-transfected normal and PAH PASMCs and STIM2-siRNA-transfected PAH-PASMCs. **P<0.01 vs. Nor control-siRNA and PAH STIM2-siRNA.
Figure 3.
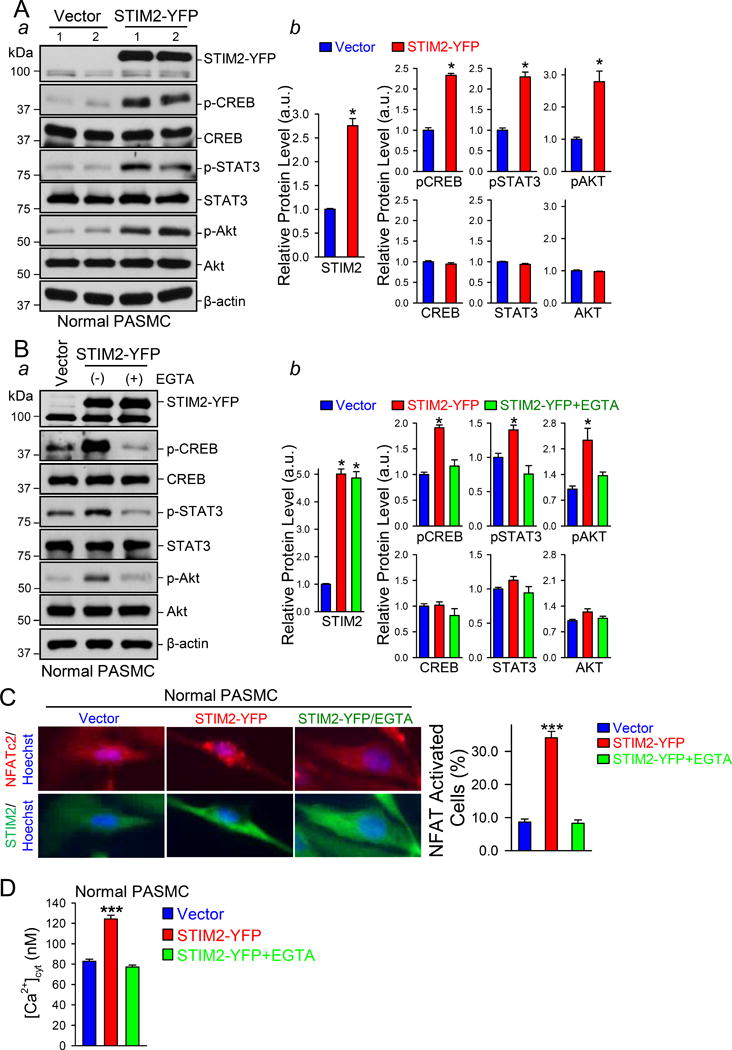
STIM2 is sufficient for increasing the level of phosphorylated (p) CREB, STAT3, and AKT and enhancing the nuclear translocation of NFATC2 in PASMCs. A Western blot analyses (a) on pCREB, CREB, pSTAT3, STAT3, pAKT and AKT in vector- and STIM2-YFP-transfected normal PASMCs. Summarized data (b, means±SE, n=3 normal subjects) showing the protein level of pCREB, CREB, pSTAT3, STAT3, pAKT and AKT in vector- and STIM2-YFP-transfected normal PASMC. *P<0.05 vs. Vector. B, Western blot analyses (a) on pCREB, CREB, pSTAT3, STAT3, pAKT and AKT in vector- and STIM2-YFP-transfected normal PASMCs treated without and with EGTA. Summarized data (b, means±SE, n=3 normal subjects) showing the protein level of pCREB, CREB, pSTAT3, STAT3, pAKT and AKT in vector- and STIM2-YFP-transfected normal PASMCs treated without and with EGTA. *P<0.05 vs. Vector and STIM2-YFP+EGTA. C, Representative fluorescent images showing NFATC2 (red), STIM2 (green) and nuclei (blue) in vector- and STIM2-YFP-transfected normal PASMCs treated without and with EGTA. Summarized data (right, means±SE, n=3 normal subjects) showing the percentage of NFATC2-activated cells in vector- and STIM2-YFP-transfected normal PASMCs treated without and with EGTA. ***P<0.001 vs. Vector and STIM2-YFP+EGTA. D, Summarized data (means±SE, n=3 normal subjects) showing the resting [Ca2+]cyt in vector- and STIM2-YFP-transfected normal PASMCs treated without and with EGTA. ***P<0.001 vs. Vector and STIM2-YFP+EGTA.
To examine whether the STIM2-mediated activation of the signaling cascades results from the enhanced resting [Ca2+]cyt, we repeated the experiments in the presence of EGTA (2 mM) which reduces extracellular free [Ca2+] to 500 nM.14 Reduction of extracellular Ca2+ with EGTA (2 mM for 24 hrs) abolished the increase in the resting [Ca2+]cyt induced by overexpression of STIM2 (Fig. 3D), but didn’t affect the transfection efficiency of STIM2 in normal PASMCs (Fig. 3B). The level of pCREB, pSTAT3, and pAKT, and the nuclear translocation of NFATc2 in STIM2-transfected cells were significantly decreased when the cells were cultured in EGTA-containing media compared with the cells cultured in control (or EGTA-free) media (Fig. 3B). These results suggest that STIM2 enhances phosphorylation of transcription factors CREB and STAT3, activates the AKT signaling cascade, and induces NFAT nuclear translocation by increasing the resting [Ca2+]cyt in human PASMCs.
Upregulation of STIM2 Enhances PASMC Proliferation
It has been demonstrated that activated CREB, STAT3, AKT, and NFATc2 are involved in stimulating PASMC proliferation.2–4 Upregulation of STIM2 in normal PASMCs significantly increased cell proliferation, determined by measuring 5-ethynyl-2-deoxyuridine (EdU) incorporation or DNA synthesis, in comparison to cells transfected with an empty vector (Fig. 4A); while downregulation of STIM2 in PAH-PASMCs with siRNA significantly decreased EdU incorporation (Fig. 4B). Furthermore, overexpression of STIM2 in normal PASMCs considerably increased the cell proliferation marker Ki67; whereas downregulation of STIM2 with siRNA in PAH-PASMCs decreased Ki67 (Fig. 4C). These results suggest that upregulation of STIM2 is sufficient to enhance cell proliferation in normal PASMCs and necessary for the enhanced cell proliferation in PAH-PASMCs.
Figure 4.
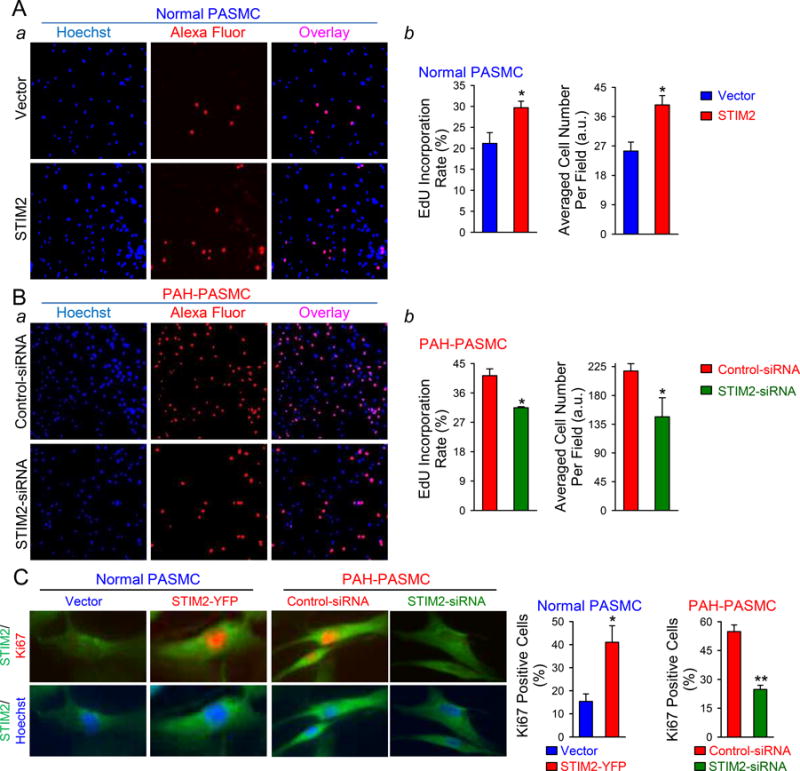
STIM2 is sufficient and necessary for increasing cell proliferation in PASMC. A Representative fluorescent images (a) showing nuclei (blue) and EdU (red), along with the overlay images (purple), in vector- and STIM2-transfected normal PASMCs. Summarized data (b, means±SE, n=3) showing the percentage of cells with EdU incorporation (middle panel) and averaged cell number per field (right panel) in vector- and STIM2-transfected normal PASMCs. *P<0.05 vs. Vector. B, Representative fluorescent images (a) showing nuclear (blue) and EdU staining (red), as well as the overlay images (purple), in control-siRNA- and STIM2-siRNA-transfected PAH-PASMCs. Summarized data (b, means±SE, n=5) showing the percentage of cells with EdU incorporation (middle panel) and averaged cell number per field (right panel) in control-siRNA- and STIM2-siRNA-transfected PAH-PASMCs. *P<0.05 vs. Control-siRNA. C, Representative fluorescent images showing STIM2 (green), Ki67 (red) and nuclei (blue) in normal PASMCs transfected with vector and STIM2-YFP, and PAH-PASMCs transfected with control-siRNA and STIM2-siRNA. Summarized data (left, means±SE, n=3 normal subjects and 5 PAH patients) showing the percentage of Ki67-positive normal PASMCs (left) transfected with vector and STIM2-YFP, and Ki67-positive PAH-PASMCs (right) transfected with control-siRNA and STIM2-siRNA, respectively. *P<0.05 vs Vector and **P<0.01 vs Control-siRNA.
Upregulation of STIM2 Inhibits PASMC Apoptosis by Increasing Bcl-2/Bax Ratio
Bcl-2, an anti-apoptotic protein, and Bax, a pro-apoptotic protein, both play an important role in regulating apoptosis in PASMCs. NFATc2 activation has been shown to contribute to Bcl-2 upregulation in PASMCs from idiopathic PAH patients.27 Our study showed that NFATc2 was more activated in PAH-PASMCs than in normal PASMCs (Figs. 2C and 3C). Downregulation of STIM2 in PAH-PASMCs decreased the resting [Ca2+]cyt and inhibited NFATc2 activation; while upregulation of STIM2 in normal PAMSCs increased [Ca2+]cyt and enhanced NFATc2 activation. To examine whether STIM2 or STIM2-mediated increase in the resting [Ca2+]cyt affects PASMC apoptosis via NFAT-regulated Bcl-2 protein expression, we first compared Bcl-2 (and Bax) expression level and the Bcl-2/Bax ratio in normal and PAH PASMCs. The protein expression of Bcl-2 in PAH-PASMCs was significantly higher than in normal PASMCs, while the protein level of Bax was markedly lower in PAH-PASMCs than in normal PASMCs. Thus, the Bcl-2/Bax ratio was substantially higher in PAH-PASMCs than in normal PASMCs (Fig. 5A). Furthermore, knockdown of STIM2 in PAH-PASMCs increased the percentage of TUNEL-positive cells compared with PAH-PASMCs treated with control-siRNA (Fig. 5B), which was due apparently to the decreased Bcl-2/Bax ratio (Fig. 5C). Reduction of extracellular Ca2+ with EGTA also significantly decreased the protein level of Bcl-2 in PAH-PASMCs, resulting in the decrease of the Bcl-2/Bax ratio (Fig. 5D). These results further support the hypothesis that the STIM2/Ca2+/NFATc2/Bal-2 pathway plays an essential role in the regulation of apoptosis in PASMC and upregulation of STIM2 is one of the important contributors to inhibit PAH-PASMC apoptosis.
Figure 5.
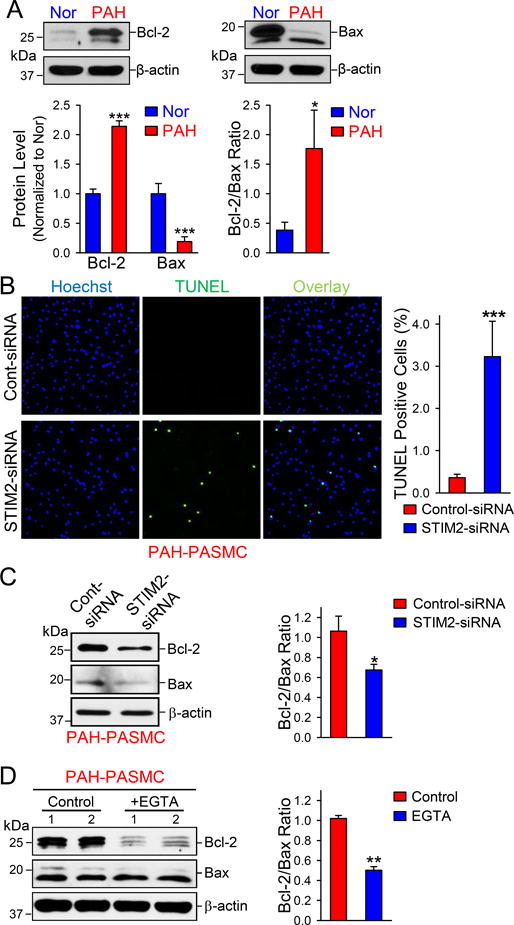
STIM2 contributes to the decreased cell apoptosis in PAH-PASMCs. A Western blot analyses (upper panels) on Bcl-2 and Bax in normal and PAH PAMSCs. Summarized data (lower panels, means±SE, n=3-5) showing the protein level of Bcl-2 and Bax, and the ratio of Bcl-2 to Bax, in normal and PAH PASMCs. *P<0.05 and ***P<0.001 vs. Normal. B, Representative fluorescent images (left) showing nuclear (blue) and TUNEL (green) staining, along with the overlay images (cyan) of nuclei and TUNEL, in control-siRNA- and STIM2-siRNA-transfected PAH-PASMCs. Summarized data (right, means±SE, n=5) showing the percentage of TUNEL positive PAH-PASMCs transfected with control-siRNA and STIM2-siRNA. ***P<0.001 vs Control-siRNA. C, Western blot analyses (left) on Bcl-2 and Bax in control-siRNA- and STIM2-siRNA-transfected PAH-PASMCs. Summarized data (right, means±SE, n=5) showing the protein level of Bcl-2 and Bax in control-siRNA- and STIM2-siRNA-transfected PAH-PASMCs. *P<0.05 vs. Control-siRNA. D, Western blot (left) analyses on Bcl-2 and Bax in PAH-PASMCs in the absence (Control) and presence of EGTA. Summarized data (right, means±SE, n=5) showing the protein level of Bcl-2 and Bax in control and EGTA-treated PAH-PASMCs. **P<0.01 vs. control.
STIM2 Clusters are Increased in PAH-PASMCs
Translocation of polymerized STIM2 to the ER/SR-plasma membrane (PM) junctions develops STIM2 clusters that recruit Orai to form store-operated Ca2+ channels inducing SOCE.23 Under the resting condition, there were no or very few STIM2 clusters and STIM2-Orai1 co-localization in normal PASMCs; however, there were more STIM2 clusters and STIM2-Orai1 co-localization in PAH-PASMCs where STIM2 expression was upregulated (Fig. 6B). Passive depletion of Ca2+ from intracellular Ca2+ stores (or SR) using cyclopiazonic acid (CPA), an SR Ca2+-pump inhibitor, further induced STIM2 clusters and enhanced STIM2-Orai1 co-localization in both normal and PAH PASMCs (Fig. 6A). These results indicate that upregulated STIM2 in PAH-PASMCs can a) translocate to the ER/SR-PM junctions to recruit and interact with Orai1 channels to mediate Ca2+ influx without store depletion, and b) further translocate to the puncta area to enhance SOCE when intracellular stores are depleted.
Figure 6.
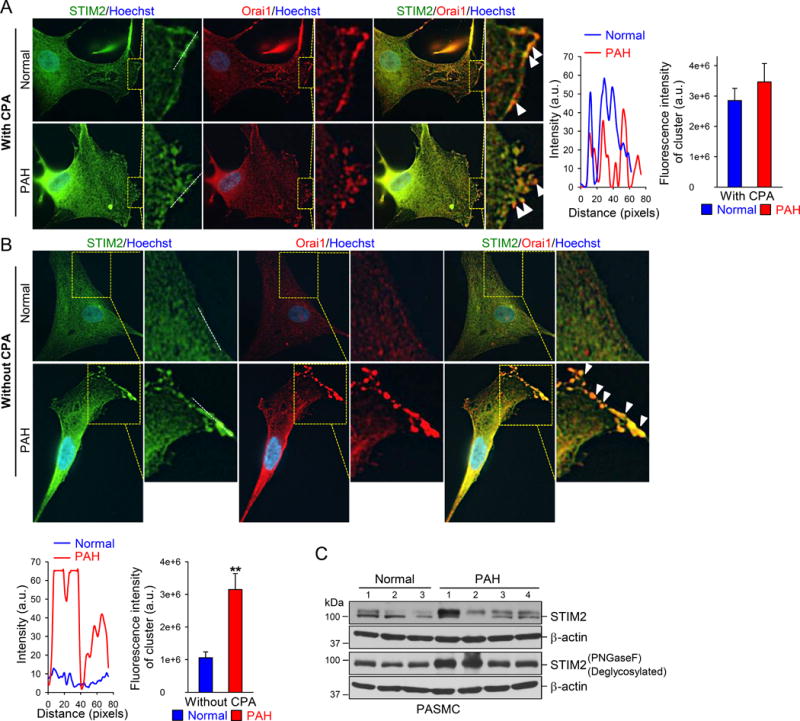
STIM2 clustering is increased in PAH-PASMCs. A Representative fluorescent images (left) showing the nuclei (blue), STIM2 (green), and Orai1 (red), along with the overlay images (orange), in normal and PAH PASMCs treated with CPA (10 μM). PASMCs treated with CPA exhibit a STIM2 distribution in the plasma membrane. Overlay images showing the co-localization of STIM2 and Orai1 in the plasma membrane in PASMCs after CPA treatment (indicated by arrows). The magnified images (highlighted in yellow) showing the STIM2 clusters (red) and STIM2/Orai1 interactions in the SR-plasma membrane junction area. Line scan measurement (middle) showing different fluorescence intensity (representing STIM2 and STIM2/Orai1 levels) along the line of the region indicated in the left images. Summarized data (right, means±SE, n=3 normal subjects, and 5 patients) showing the fluorescent intensity of the clusters in normal and PAH PASMCs with CPA treatment. B, Representative fluorescent images (upper panels) showing the nuclei (blue), STIM2 (green) and Orai1 (red), along with the overlay images (orange), in normal and PAH PASMCs without CPA treatment. PAH-PASMCs exhibit STIM2 clustering distributions in the plasma membrane. Overlay images showing the co-localization of STIM2 and Orai1 in the plasma membrane (arrows). The magnified images (highlighted in yellow) showing the STIM2 clusters (red) and STIM2/Orai1 interactions in the SR-plasma membrane junction area. Line scan measurement (middle) showing the different fluorescence intensity (representing STIM2 and STIM2/Orai1 levels) along the line of the region indicated in the left images. Summarized data (lower panel, right, means±SE, n=3 normal subjects and 5 patients) showing the cluster intensity in normal and PAH PASMCs without CPA treatment. C, Western blot analyses on STIM2 in normal and PAH PASMCs before and after deglycosylation using PNGaseF.
It has been reported that a STIM2 pre-protein is involved in STIM2-mediated and store-independent Ca2+ influx.24 Our Western blot data showed that there were two closely located bands of STIM2 in both normal and PAH-PASMCs; the upper band could be the STIM2 pre-protein or the glycosylated STIM2. Using deglycosylation enzyme PNGaseF, we were able to eliminate the upper band indicating that the observed two bands of STIM2 were STIM2 and the glycosylated STIM2 rather than STIM2 and the STIM2 pre-protein (Fig. 6C).
Discussion
The results of this study indicate that i) the resting [Ca2+]cyt and the protein expression level of STIM2 are significantly greater in PASMCs from PAH patients than in PASMCs from normal subjects; ii) upregulation of STIM2 in normal PASMCs (where STIM2 is low) significantly enhances the resting [Ca2+]cyt, while downregulation of STIM2 in PAH-PASMCs (where STIM2 is high or upregulated) significantly attenuates the resting [Ca2+]cyt; v) the STIM2-mediated increase in the resting [Ca2+]cyt activates multiple intracellular signaling cascades (e.g., AKT, STAT3, CREB, and NFAT) to stimulate PASMC proliferation; and v) the STIM2-mediated increase in the resting [Ca2+]cyt also contributes to inhibiting PASMC apoptosis by increasing the ratio of Bcl-2/Bax. These observations imply that STIM2 functions as a positive regulator of the resting [Ca2+]cyt in PASMCs. Upregulation of STIM2 in PASMCs, such as that in PAH-PASMCs, is likely an important cause for the sustained pulmonary vasoconstriction and excessive pulmonary vascular remodeling in patients with PAH and animals with severe experimental pulmonary hypertension (PH).
STIM2-mediated Regulation of Intracellular Ca2+ Homeostasis
As an ER/SR Ca2+ sensor, the commonly reported function of STIM2 is its contribution to the initiation and regulation of store-operated Ca2+ entry (SOCE). Enhanced SOCE due partially to upregulation of STIM proteins as well as Orai and TRP channels has been implicated in stimulating PASMC proliferation, phenotypical transition of contractile PASMC to proliferative PASMC, and the development of PAH.13, 29
There are at least two STIM2 splice variants, STIM2.1 and STIM2.2, which are both expressed in PASMCs. Functional studies have revealed that STIM2.2 promotes SOCE, whereas STIM2.1 inhibits SOCE.30, 31 The effect of STIM2 overexpression on SOCE varies significantly among different cell types; for example, overexpression of STIM2 in T cells and fibroblasts enhances SOCE,32 while overexpression of STIM2 in HEK293 cells and PC12 cells actually inhibits SOCE.33 Nonetheless, overexpression of STIM2 has consistently produced a significant enhancement in the resting [Ca2+]cyt in many different types of cells.30, 31 Our study confirmed the critical role of STIM2, as a potent positive regulator, in the regulation of the resting [Ca2+]cyt in human PASMCs. Overexpression of STIM2 in normal human PASMCs, or upregulation of STIM2 in PAH-PASMCs, both lead to significantly increased resting [Ca2+]cyt and activation of Ca2+-sensitive signaling proteins and Ca2+-responsive transcription factors (e.g., CREB, STAT3, and NFAT) (Fig. 1–3). An interesting finding from this study is that STIM2-associated increase in the resting [Ca2+]cyt in human PASMCs also results in the rise of Bcl-2/Bax ratio, which is potentially and partially responsible for the apoptosis resistance of PAH-PASMCs.27, 34 The observations from this study imply that downregulation or inhibition of STIM2, more specifically, STIM2.2, may potentially be a critical target to develop novel drugs to inhibit the development of pulmonary arterial remodeling and/or regress the remodeled pulmonary arteries responsible for the elevated pulmonary vascular resistance in patients with PAH.
STIM2-associated Increase in the Resting [Ca2+]cyt and PASMC Proliferation
There are at least four steps in the cycle that are sensitive to Ca2+ and its downstream signaling cascades: the transition from the G0 to G1 phase, the transition from the G1 to S phase, the transition from the G2 to M phase, and the whole M phase (or mitosis).35 Removal or chelation of extracellular Ca2+, blockade of Ca2+ channels and chelation of intracellularly stored Ca2+ all attenuate PASMC proliferation7. These data indicate that an increase in [Ca2+]cyt due to the Ca2+ influx and Ca2+ release is required for PASMC proliferation. An increase in [Ca2+]cyt induces PASMC proliferation by initiating or promoting the cell cycle via activation of pro-proliferative signaling cascades and transfection factors. In this study, we provide substantial evidence that STIM2-associated increase in the resting [Ca2+]cyt in PASMCs enhances phosphorylation of AKT, STAT3, and CREB, promotes the nuclear translocation of NFAT, and stimulates cell proliferation (determined by EdU incorporation and expression of Ki67). STIM2 is sufficient to increase the resting [Ca2+]cyt and promote cell proliferation in normal PASMCs, while upregulated STIM2 is necessary for the enhanced resting [Ca2+]cyt and augmented cell proliferation in PAH-PASMCs. Specific downregulation and/or inhibition of STIM2 or STIM2.2 would potentially be an excellent strategy to develop novel therapies for the treatment of PAH and other forms of pulmonary hypertension with severe pulmonary vascular remodeling.
The PI3K-AKT-mTOR signaling cascade is a critical intracellular pathway that regulates the cell cycle, protein expression and cell proliferation.36 Growth factor-mediated activation of G protein-coupled receptors (GPCR, e.g., CaSR and ETA receptors) and tyrosine kinase receptors (TKR, e.g., PDGF receptors), along with an increase in [Ca2+]cyt, can promote the production of PI3K37 that subsequently activates or phosphorylates AKT and promotes the AKT/mTOR signaling and other AKT-dependent signaling pathways. In this study, we showed that STIM2-mediated increase in the resting [Ca2+]cyt in PAH-PASMCs (or in normal PASMCs transiently transfected with STIM2) was associated with significant phosphorylation of AKT. Phosphorylated AKT can further activate or phosphorylate CREB and mTOR, leading to cell proliferation and gene expression. It has been demonstrated that Ca2+/calmodulin (CaM)-dependent kinase 4 (CaMK4) is also involved in activating AKT and thus promoting the AKT/mTOR signaling pathways to stimulate cell proliferation.28 In addition to AKT, our data also indicated significant phosphorylation of STAT3 and nuclear translocation of NFAT in PAH-PASMCs in which STIM2 was upregulated in comparison to normal PASMCs. The JAK/STAT3 signaling cascade is another important intracellular signaling pathway that mediates cytokine- and mitogen-induced cell proliferation.3 The STIM2-mediated STAT3 phosphorylation via increased resting [Ca2+]cyt in PAH-PASMCs would enhance inflammation-induced PASMC proliferation and thus concentric pulmonary vascular remodeling. It is, however, unclear whether the STIM2-associated increase in the resting [Ca2+]cyt activates STAT3 directly by Ca2+/CaM and Ca2+/CaMK or indirectly by the AKT/mTOR cascade. The data from this study indicate that the STIM2-mediated increases in the resting [Ca2+]cyt in PASMC from PAH patients activate multiple intracellular signaling pathways that promote PASMC proliferation contributing to pulmonary vascular remodeling.
STIM2-associated Increase in the Resting [Ca2+]cyt and PASMC Apoptosis
Bcl-2, an anti-apoptosis protein that mediates anti-apoptotic effect on a variety of cells, has been reported to be highly expressed in PASMCs isolated from patients with IPAH.27 The data from this study not only confirmed the upregulation of Bcl-2 or increase of the Bcl-2/Bax ratio in PAH-PASMCs but also revealed that the STIM2-mediated increase of the resting [Ca2+]cyt is one of the causes for the upregulated Bcl-2 or increased Bcl-2/Bax ratio. The STIM2-associated increase in the Bcl-2/Bax ratio would ultimately contribute to the resistance of PAH-PASMCs to apoptosis.34 Excessive activation of NFAT or enhanced nuclear translocation of NFAT due to elevated resting [Ca2+]cyt is considered as the driving force behind in regulating Bcl-2 expression.27 In lung cancer cells, ATP-induced increase in [Ca2+]cyt has been shown to upregulate Bcl-2 expression as well.38 These observations indicate that intracellular Ca2+ is an important regulator of Bcl-2 expression; an increase in [Ca2+]cyt would upregulate Bcl-2 expression via activation of NAFT and other Ca2+/CaM-sensitive signaling cascades, which result in an anti-apoptotic effect on PASMCs and cancer cells.
Increased PASMC proliferation and decreased PASMC apoptosis in the pulmonary arterial wall both contribute to the development and progression of pulmonary vascular wall thickening, a major cause for the elevated pulmonary vascular resistance and pulmonary arterial pressure in patients with PAH and animals with several PH. The indirect anti-apoptotic effect of STIM2, by increasing the resting [Ca2+]cyt, activation of NFAT and upregulation of Bcl-2 in PASMCs, suggests a new pathogenic role of the STIM2-mediated Ca2+ signaling in the development of pulmonary vascular remodeling in patients with PAH. Given the previous observation that only STIM2 protein level was increased in PASMCs from PAH patients (but not STIM1),12 we believe that upregulated STIM2 may play an important pathogenic role in PAH by stimulating PASMC proliferation and inhibiting PASMC apoptosis.
STIM2 Clusters and Activates Orai1 Channels under Resting Conditions in PAH-PASMCs
We observed that, under resting conditions or when intracellular stores were not passively or actively depleted or reduced, more STIM2 clusters co-localized with Orai1 near the plasma membrane area in PAH-PASMCs than in normal PAMSCs (Fig. 6B). These data indicate that upregulation of STIM2, such as in PASMCs from PAH patients, results in STIM2 clustering with Orai1 in the plasma membrane, which may tentatively induce a “basal” or gradual Ca2+ influx through Orai1 channels to increase the resting [Ca2+]cyt. The increased STIM2 clustering with Orai1, due apparently to STIM2 overexpression or upregulation, seems not to require significant reduction or depletion of Ca2+ in the SR. These data imply that STIM2 can recruit and activate Orai1 channels to increase the resting [Ca2+]cyt in PASMCs where STIM2 expression level is high.
A cytoplasmic STIM2 pre-protein that cannot target ER/SR has been discovered recently to play an important role in recruiting and activating Orai1 channels in a store-independent manner.24 In our Western blot experiments, we often detected an upper band in both normal and PAH PASMCs (Fig. 6C), which is similar to the STIM2 pre-protein band as indicated by other investigators.24 However, treatment of with PNGaseF, an enzyme that catalyzes the cleavage of an internal glycoside bond in an oligosaccharide, abolished the upper STIM2 band, indicating that the upper STIM2 band is the glycosylated STIM2 band rather than the STIM2 pre-protein band. It is thus possible that STIM2 pre-protein may express in human PASMCs at a very low level and its functional role is unclear. Furthermore, it also remains unclear whether glycosylation of STIM2 contributes to enhancing its function to cluster and recruit Orai1 channels in the plasma membrane.
In summary, this study provides compelling evidence that STIM2, a Ca2+ sensor expressed in the ER/SR membrane, plays an important role in regulating the resting [Ca2+]cyt in human PASMC. STIM2 is sufficient to increase the resting [Ca2+]cyt in normal PASMCs and necessary for the elevated resting [Ca2+]cyt in PAH-PASMCs. Upregulation of STIM2 in PASMC from patients with PAH, via elevated resting [Ca2+]cyt and activated Ca2+-sensitive signaling pathways (e.g., AKT/mTOR, AKT/CREB, STAT3, and NFAT), contributes to enhancing PASMC proliferation and inhibiting PASMC apoptosis and ultimately lead to the development and progression of pulmonary vascular remodeling in PAH. Pulmonary vascular smooth muscle STIM2 is potentially a promising drug target for developing novel therapies for PAH and other forms of pulmonary hypertension.
Limitations
For the in vitro experiments, we used PASMC isolated from a limited number of donors (n=3 normal subjects) and patients (n=5 PAH patients). The significance of our findings thus needs to be further confirmed by examining PASMCs from more patients with PAH and patients with other forms of pulmonary arterial hypertension. Also, we used primary cultured PAMSCs, which might undergo phenotypical changes to unmask certain pathogenic or pathological characteristics. We will conduct in vivo experiments using animal models to confirm the findings shown in this study and will investigate whether smooth muscle-specific knockout of STIM2 ameliorates the development and progression of experimental pulmonary hypertension in animals. Sustained pulmonary vasoconstriction and concentric pulmonary vascular wall thickening both directly contribute to the elevated pulmonary vascular resistance and, subsequently, increased pulmonary arterial pressure and right heart failure in PAH patients. Medial hypertrophy caused by excessive PASMC proliferation and attenuated PASMC apoptosis in distal vessels account for excessive pulmonary vascular remodeling in patients with PAH. Identifying the pathogenic mechanisms involved in enhanced PASMC proliferation, and impaired PASMC apoptosis is important to develop effective therapies for PAH. This study suggests that upregulated STIM2 in PASMCs is potentially a critical drug target for developing novel therapies to treat PAH or pulmonary hypertension in general.
Supplementary Material
Perspectives.
The current work identifies STIM2 as one of the major regulators that increases the resting [Ca2+]cyt in PASMC from patients with pulmonary arterial hypertension (PAH). The STIM2-associated rise in the resting [Ca2+]cyt stimulates PASMC proliferation by activating AKT, STAT3 and NF-AT signaling pathways and inhibits PASMC apoptosis by upregulating Bcl-2. Inhibition or downregulation of STIM2 may restore the pathogenic rise in the resting [Ca2+]cyt in PASMC, and attenuate or reverse pulmonary vascular remodeling in PAH. The findings of this study, therefore, warrant further translational studies to exploit STIM2 as a drug target for developing novel therapies for PAH.
Novelty and Significance.
What Is new?
This is the first study to identify STIM2 as a critical regulator of the resting [Ca2+]cyt in human pulmonary arterial smooth muscle cell (PASMC) and upregulated STIM2 as a new cause for the increased resting [Ca2+]cyt in PASMC from patients with pulmonary arterial hypertension (PAH).
STIM2-mediated Ca2+ entry under the resting conditions is not dependent of complete store depletion; a small decrease in stored [Ca2+] is sufficient to activate STIM2-associated Ca2+ entry.
This study is also among the first to link a STIM2-mediated increase in the resting [Ca2+]cyt to the activation of different signaling pathways that ultimately leads to stimulation of PASMC proliferation and pulmonary vascular remodeling.
What Is Relevant?
Our observations indicate that upregulated expression and increased function of STIM2 in PASMCs, which is more sensitive to the changes of [Ca2+] in the SR/ER than STIM1, play a pathogenic role in the development and progression of PAH. Selective blockade of STIM2 is potentially a novel therapeutic approach for PAH.
Summary.
This study provides original findings to demonstrate that STIM2 plays an important role in regulating the resting [Ca2+]cyt in PASMC. Upregulation of STIM2 in PASMC from patients with PAH, via elevating the resting [Ca2+]cyt and activating Ca2+-sensitive signaling pathways (e.g., AKT/mTOR, AKT/CREB, STAT3, and NFAT), contributes to enhancing PASMC proliferation and inhibiting PASMC apoptosis, and ultimately lead to the development and progression of pulmonary vascular remodeling in PAH.
Acknowledgments
Sources of Funding
This work was supported in part by the grants from the National Heart, Lung and Blood Institute of the National Institutes of Health (HL135807 and HL125208).
Footnotes
Disclosures
None.
References
- 1.Humbert M, Ghofrani HA. The molecular targets of approved treatments for pulmonary arterial hypertension. Thorax. 2016;71:73–83. doi: 10.1136/thoraxjnl-2015-207170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hou X, Chen J, Luo Y, Liu F, Xu G, Gao Y. Silencing of STIM1 attenuates hypoxia-induced PASMCs proliferation via inhibition of the SOC/Ca2+/NFAT pathway. Respir Res. 2013;14:2. doi: 10.1186/1465-9921-14-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paulin R, Meloche J, Bonnet S. STAT3 signaling in pulmonary arterial hypertension. JAKSTAT. 2012;1:223–233. doi: 10.4161/jkst.22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pulver RA, Rose-Curtis P, Roe MW, Wellman GC, Lounsbury KM. Store-operated Ca2+ entry activates the CREB transcription factor in vascular smooth muscle. Circ Res. 2004;94:1351–1358. doi: 10.1161/01.RES.0000127618.34500.FD. [DOI] [PubMed] [Google Scholar]
- 5.Berridge MJ. Calcium signalling and cell proliferation. Bioessays. 1995;17:491–500. doi: 10.1002/bies.950170605. [DOI] [PubMed] [Google Scholar]
- 6.Golovina VA, Platoshyn O, Bailey CL, Wang J, Limsuwan A, Sweeney M, Rubin LJ, Yuan JX. Upregulated TRP and enhanced capacitative Ca2+ entry in human pulmonary artery myocytes during proliferation. Am J Physiol Heart Circ Physiol. 2001;280:H746–755. doi: 10.1152/ajpheart.2001.280.2.H746. [DOI] [PubMed] [Google Scholar]
- 7.Sweeney M, McDaniel SS, Platoshyn O, Zhang S, Yu Y, Lapp BR, Zhao Y, Thistlethwaite PA, Yuan JX. Role of capacitative Ca2+ entry in bronchial contraction and remodeling. J Appl Physiol (1985) 2002;92:1594–1602. doi: 10.1152/japplphysiol.00722.2001. [DOI] [PubMed] [Google Scholar]
- 8.Leggett K, Maylor J, Undem C, Lai N, Lu W, Schweitzer K, King LS, Myers AC, Sylvester JT, Sidhaye V, Shimoda LA. Hypoxia-induced migration in pulmonary arterial smooth muscle cells requires calcium-dependent upregulation of aquaporin 1. Am J Physiol Lung Cell Mol Physiol. 2012;303:L343–353. doi: 10.1152/ajplung.00130.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan JX, Aldinger AM, Juhaszova M, Wang J, Conte JV, Jr, Gaine SP, Orens JB, Rubin LJ. Dysfunctional voltage-gated K+ channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension. Circulation. 1998;98:1400–1406. doi: 10.1161/01.cir.98.14.1400. [DOI] [PubMed] [Google Scholar]
- 10.Undem C, Luke T, Shimoda LA. Contribution of elevated intracellular calcium to pulmonary arterial myocyte alkalinization during chronic hypoxia. Pulm Circ. 2016;6:93–102. doi: 10.1086/685053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamamura A, Yamamura H, Zeifman A, Yuan JX. Activity of Ca2+-activated Cl− channels contributes to regulating receptor- and store-operated Ca2+ entry in human pulmonary artery smooth muscle cells. Pulm Circ. 2011;1:269–279. doi: 10.4103/2045-8932.83447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song MY, Makino A, Yuan JX. STIM2 Contributes to Enhanced Store-operated Ca2+ Entry in Pulmonary Artery Smooth Muscle Cells from Patients with Idiopathic Pulmonary Arterial Hypertension. Pulm Circ. 2011;1:84–94. doi: 10.4103/2045-8932.78106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ogawa A, Firth AL, Smith KA, Maliakal MV, Yuan JX. PDGF enhances store-operated Ca2+ entry by upregulating STIM1/Orai1 via activation of Akt/mTOR in human pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol. 2012;302:C405–411. doi: 10.1152/ajpcell.00337.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sweeney M, Yu Y, Platoshyn O, Zhang S, McDaniel SS, Yuan JX. Inhibition of endogenous TRP1 decreases capacitative Ca2+ entry and attenuates pulmonary artery smooth muscle cell proliferation. Am J Physiol Lung Cell Mol Physiol. 2002;283:L144–155. doi: 10.1152/ajplung.00412.2001. [DOI] [PubMed] [Google Scholar]
- 15.Yu Y, Fantozzi I, Remillard CV, Landsberg JW, Kunichika N, Platoshyn O, Tigno DD, Thistlethwaite PA, Rubin LJ, Yuan JX. Enhanced expression of transient receptor potential channels in idiopathic pulmonary arterial hypertension. Proc Natl Acad Sci U S A. 2004;101:13861–13866. doi: 10.1073/pnas.0405908101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu Y, Sweeney M, Zhang S, Platoshyn O, Landsberg J, Rothman A, Yuan JX. PDGF stimulates pulmonary vascular smooth muscle cell proliferation by upregulating TRPC6 expression. Am J Physiol Cell Physiol. 2003;284:C316–330. doi: 10.1152/ajpcell.00125.2002. [DOI] [PubMed] [Google Scholar]
- 17.Platoshyn O, Yu Y, Ko EA, Remillard CV, Yuan JX. Heterogeneity of hypoxia-mediated decrease in IK(V) and increase in [Ca2+]cyt in pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2007;293:L402–416. doi: 10.1152/ajplung.00391.2006. [DOI] [PubMed] [Google Scholar]
- 18.Ter Keurs HE, Boyden PA. Calcium and arrhythmogenesis. Physiol Rev. 2007;87:457–506. doi: 10.1152/physrev.00011.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell. 2006;126:981–993. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stathopulos PB, Zheng L, Ikura M. Stromal interaction molecule (STIM) 1 and STIM2 calcium sensing regions exhibit distinct unfolding and oligomerization kinetics. J Biol Chem. 2009;284:728–732. doi: 10.1074/jbc.C800178200. [DOI] [PubMed] [Google Scholar]
- 22.Brandman O, Liou J, Park WS, Meyer T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell. 2007;131:1327–1339. doi: 10.1016/j.cell.2007.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kar P, Bakowski D, Di Capite J, Nelson C, Parekh AB. Different agonists recruit different stromal interaction molecule proteins to support cytoplasmic Ca2+ oscillations and gene expression. Proc Natl Acad Sci U S A. 2012;109:6969–6974. doi: 10.1073/pnas.1201204109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Graham SJ, Dziadek MA, Johnstone LS. A cytosolic STIM2 preprotein created by signal peptide inefficiency activates ORAI1 in a store-independent manner. J Biol Chem. 2011;286:16174–16185. doi: 10.1074/jbc.M110.206946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parvez S, Beck A, Peinelt C, Soboloff J, Lis A, Monteilh-Zoller M, Gill DL, Fleig A, Penner R. STIM2 protein mediates distinct store-dependent and store-independent modes of CRAC channel activation. FASEB J. 2008;22:752–761. doi: 10.1096/fj.07-9449com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song S, Yamamura A, Yamamura H, Ayon RJ, Smith KA, Tang H, Makino A, Yuan JX. Flow shear stress enhances intracellular Ca2+ signaling in pulmonary artery smooth muscle cells from patients with pulmonary arterial hypertension. Am J Physiol Cell Physiol. 2014;307:C373–383. doi: 10.1152/ajpcell.00115.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bonnet S, Rochefort G, Sutendra G, Archer SL, Haromy A, Webster L, Hashimoto K, Bonnet SN, Michelakis ED. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc Natl Acad Sci U S A. 2007;104:11418–11423. doi: 10.1073/pnas.0610467104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koga T, Hedrich CM, Mizui M, Yoshida N, Otomo K, Lieberman LA, Rauen T, Crispin JC, Tsokos GC. CaMK4-dependent activation of AKT/mTOR and CREM-α underlies autoimmunity-associated Th17 imbalance. J Clin Invest. 2014;124:2234–2245. doi: 10.1172/JCI73411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fernandez RA, Wan J, Song S, Smith KA, Gu Y, Tauseef M, Tang H, Makino A, Mehta D, Yuan JX. Upregulated expression of STIM2, TRPC6, and Orai2 contributes to the transition of pulmonary arterial smooth muscle cells from a contractile to proliferative phenotype. Am J Physiol Cell Physiol. 2015;308:C581–593. doi: 10.1152/ajpcell.00202.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miederer AM, Alansary D, Schwar G, Lee PH, Jung M, Helms V, Niemeyer BA. A STIM2 splice variant negatively regulates store-operated calcium entry. Nat Commun. 2015;6:6899. doi: 10.1038/ncomms7899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rana A, Yen M, Sadaghiani AM, Malmersjo S, Park CY, Dolmetsch RE, Lewis RS. Alternative splicing converts STIM2 from an activator to an inhibitor of store-operated calcium channels. J Cell Biol. 2015;209:653–669. doi: 10.1083/jcb.201412060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oh-Hora M, Yamashita M, Hogan PG, Sharma S, Lamperti E, Chung W, Prakriya M, Feske S, Rao A. Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat Immunol. 2008;9:432–443. doi: 10.1038/ni1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Soboloff J, Spassova MA, Hewavitharana T, He LP, Xu W, Johnstone LS, Dziadek MA, Gill DL. STIM2 is an inhibitor of STIM1-mediated store-operated Ca2+ Entry. Curr Biol. 2006;16:1465–1470. doi: 10.1016/j.cub.2006.05.051. [DOI] [PubMed] [Google Scholar]
- 34.Zhang S, Fantozzi I, Tigno DD, Yi ES, Platoshyn O, Thistlethwaite PA, Kriett JM, Yung G, Rubin LJ, Yuan JX. Bone morphogenetic proteins induce apoptosis in human pulmonary vascular smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2003;285:L740–754. doi: 10.1152/ajplung.00284.2002. [DOI] [PubMed] [Google Scholar]
- 35.Choi J, Husain M. Calmodulin-mediated cell cycle regulation: new mechanisms for old observations. Cell Cycle. 2006;5:2183–2186. doi: 10.4161/cc.5.19.3265. [DOI] [PubMed] [Google Scholar]
- 36.Houssaini A, Abid S, Mouraret N, Wan F, Rideau D, Saker M, Marcos E, Tissot CM, Dubois-Rande JL, Amsellem V, Adnot S. Rapamycin reverses pulmonary artery smooth muscle cell proliferation in pulmonary hypertension. Am J Respir Cell Mol Biol. 2013;48:568–577. doi: 10.1165/rcmb.2012-0429OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Danciu TE, Adam RM, Naruse K, Freeman MR, Hauschka PV. Calcium regulates the PI3K-Akt pathway in stretched osteoblasts. FEBS Lett. 2003;536:193–197. doi: 10.1016/s0014-5793(03)00055-3. [DOI] [PubMed] [Google Scholar]
- 38.Song S, Jacobson KN, McDermott KM, Reddy SP, Cress AE, Tang H, Dudek SM, Black SM, Garcia JG, Makino A, Yuan JX. ATP promotes cell survival via regulation of cytosolic [Ca2+] and Bcl-2/Bax ratio in lung cancer cells. Am J Physiol Cell Physiol. 2016;310:C99–114. doi: 10.1152/ajpcell.00092.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.