

Abstract
Activation of HIF-1α and Nrf2 is a primary component of cellular response to oxidative stress, and activation of HIF-1α and Nrf2 provides neuroprotection in models of neurodegenerative disorders, including ischemic stroke, Alzheimer’s and Parkinson’s diseases. Screening a library of CNS-targeted drugs using novel reporters for HIF-1α and Nrf2 elevation in neuronal cells revealed histone deacetylase (HDAC) inhibitors as potential activators of these pathways. We report the identification of phenylhydroxamates as single agents exhibiting tripartite inhibition of HDAC6, inhibition of HIF-1 prolyl hydroxylase (PHD), and activation of Nrf2. Two superior tripartite agents, ING-6 and ING-66, showed neuroprotection against various cellular insults, associated with stabilization of both Nrf2 and HIF-1, and expression of their respective target genes in vitro and in vivo. Discovery of the innate ability of phenylhydroxamate HDAC inhibitors to activate Nrf2 and HIF provides a novel route to multifunctional neuroprotective agents and cautions against HDAC6 selective inhibitors as chemical probes of specific HDAC isoform function.
Keywords: Phenylhydroxamates, histone deacetylase inhibitors, Nrf2 activators, HIF-1 activators, neuroprotection, oxidative stress
Graphical Abstract
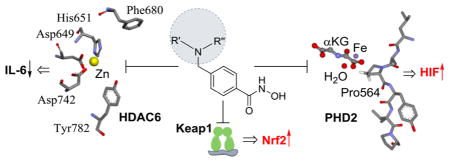
It is widely recognized that the transcription factor, Nrf2 (nuclear factor erythroid 2-related factor), is a master regulator of cellular stress response, providing cytoprotection against cellular insults, notably oxidative stress and disrupted redox balance.1 Oxidants and electrophiles interact with Keap1 (Kelch-like ECH-associated protein 1) to cause accumulation of Nrf2 and translocation to the nucleus,2 where binding to the antioxidant response element (ARE) induces transcription and upregulation of cytoprotective and antioxidant enzymes, including heme oxygenase-1 (HO-1), NAD(P)H quinone oxidoreductase-1 (NQO1), and thioredoxin (TRX).3–7 Pharmacological activation of Nrf2 represents a promising approach to neuroprotection in the treatment of neurodegenerative diseases including Parkinson’s and Alzheimer’s diseases, amyotrophic lateral sclerosis, and acute brain insults such as ischemic stroke and trauma.8,9
It is understood that the reported neuroprotective activity of many natural products results from the presence of Nrf2-activating electrophilic (commonly Michael acceptor) or phenolic moieties.10,11 In contrast, the contribution of Nrf2 to the neuroprotective activity of synthetic chemical probes is less frequently explored, but can contribute to beneficial activity in neurodegenerative diseases that are generally multifactorial in etiology. Recently, we screened, the IRSF SMART library of United States Food and Drug Administration approved and preclinical drugs (https://www.rettsyndrome.org/research/smart-library),12 in the Neh2-luciferase reporter assay, in search of novel Nrf2 activators that function via direct stabilization of the Nrf2 transcription factor. Benzamide-based HDAC inhibitors, tacedinaline and entinostat, were inactive; however, among Nrf2 hits in the IRSF library, were several hydroxamate-based HDAC inhibitors (HDACi), namely, pan-inhibitors SAHA, TSA, and oxamflatin, and a phenylhydroxamate-based HDAC6-selective inhibitor tubastatin13 (Figure 1). Whereas TSA and oxamflatin contain Michael acceptor functionalities that might be expected to activate Keap1/Nrf2, these functionalities are not found in SAHA and tubastatin.
Figure 1.

Structures and activity summary of HDACi in Nrf2 (Neh2-luc) and HIF-1α (ODD-luc) reporter assays in SH-SY5Y cells. (A) Representative HDAC inhibitors from the IRSF SMART library used in the initial screening of Nrf2 and HIF-1 activity. (B) Novel phenylhydroxamate HDAC6 inhibitors (series A), more closely resembling tubastatin, strongly activate Nrf2 and HIF-1, whereas HDACi with acyclic-cap groups (series B) are weak activators.
A second stress response pathway that can protect neurons from hypoxic and other insults is mediated by hypoxia-inducible factor (HIF). Small molecules and natural products that activate HIF-1α signaling have neuroprotective activity.14 Just as Nrf2 is stabilized by small molecules interacting with Keap1, HIF-1α is stabilized by small molecules that interact with and inhibit HIF prolyl hydroxylases (PHDs). Although PHD inhibitors often directly interact with the nonheme iron at the active site,15 PHD inhibitors may also elicit HIF-independent, neuroprotective mechanisms.16 The HIF ODD-luciferase reporter17 was used to assay HDAC inhibitors for their ability to stabilize HIF, and again tubastatin was effective, establishing phenylhydroxamate HDAC inhibitors, exemplified by tubastatin, as potential, tripartite neuroprotective agents, activating Nrf2 and HIF-1α in addition to providing selective inhibition of HDAC6 over other HDAC isoforms.
RESULTS AND DISCUSSION
The syntheses of a series of HDAC inhibitors, with structural similarities to tubastatin, including bicyclic and “cap-less” phenylhydroxamic acids, has previously been reported.18–20 These inhibitors retained potency and selectivity for class-II HDAC6, and were effective in increasing tubulin acetylation.19 To explore structure–activity relationships (SAR), for Nrf2 and HIF-1α stabilization, influenced by the bicyclic cap group, a focused set of compounds was created: Series A (I–IV) consisting of indoline, oxindole, and tetrahydroquinoline derivatives; and Series B (V–VII) represented by substituted 2-aminobenzimidazoles and 4-aminotetrahydroquinoline (Figure 1, Supporting Information Table 1S). All novel compounds (group I–VII) demonstrated (a) excellent potency toward HDAC6 inhibition (1.0 nM < IC50 < 50.0 nM) and (b) high selectivity versus the class I isoform HDAC1, comparable to tubastatin.13 As a representative potent HDAC6 inhibitor (IC50 ~ 9 nM), ING-6 was evaluated across the full panel of Zn-dependent isoforms and found to possess excellent selectivity (Table 2S).
The Neh2-luc SH-SY5Y cell-based reporter system provides a neuronal cell line that expresses the minimum portion of the Nrf2 transcription factor (sufficient for recognition by Keap1 and consequent ubiquitination) fused to firefly luciferase.12 Luciferase response corresponds to stabilization and accumulation of Nrf2 and is elevated by known Nrf2-activators that interact with Keap1, causing Keap1 displacement from Nrf2.12 This assay identifies the first step in Nrf-2 activation, stabilization of the Nrf2 protein via its release from Keap1. The ODD-luc SH-SY5Y cell reporter assay provides a comparable readout for HIF-1α stabilization. PHD marks HIF-1α for ubiquitination and proteasomal degradation and controls the rate-limiting step in the HIF-1α ODD-luc reporter response.17 Under hypoxia, or pharmacological inhibition of PHD, the level of HIF-1α protein increases, reflected by increased HIF-1α ODD-luc response.
Using the Nrf2 and HIF-1α reporter assays to screen the novel analogues of tubastatin, we identified several HDAC6 inhibitors that strongly activate both Nrf2 and HIF-1α (Figure 1). The activation of Nrf2 by tubastatin was matched or exceeded by Series A analogues with an electron-withdrawing substituent in the 5- or 6-position of the aromatic ring in the region commonly referred to as the “cap group” (Figure 1). The introduction of a single chlorine in the aromatic ring of the CAP group, as in compound ING-6 (Figure 2), led to increased potency (5.6 fold activation). Electron-donating groups were not associated with high levels of Nrf2. Interestingly, the 3,3-dimethylindolin-2-one (IV, R′ and R″ = Me) was twice as efficacious as the 3,3-dimethylindoline (III): 3.4- vs 1.8-fold. Furthemore, replacement of the gem-dimethyl group with two fluorines in the 3-position of the oxindole ring (IV, R′ and R″ = F) yielded the most potent Nrf2 activator among all the HDAC inhibitors studied, namely compound ING-66 (9.9 fold activation, Figure 2). The different levels of activation of Nrf2 and HIF-1α by each of these phenylhydroxamates (Figure 1, Table 1S) contrasted with similar potency for HDAC6 inhibition (1.0 nM < IC50 < 50.0 nM). Moreover, these compounds do not contain a classical Keap 1 interacting group, such as a thiophilic Michael acceptor: in contrast to the Keap1-modifying Nrf2-activators dimethyl fumarate (DMF)21 and tert-butylhydroquinone (TBHQ), our HDAC inhibitors did not cause GSH depletion (Figure 1S).21 DMF and TBHQ were used as controls in the Neh-Luc cell-based assay.
Figure 2.

Comparison of concentration dependence for ING-6, ING-66, and tubastatin in SH-SY5Y cells: (A) Nrf2 Neh2-luc activation; (B) HIF-1α ODD-luc reporter activation. TBHQ and the HIF-activating PHD inhibitor, FG-4592, were used as controls. Data show mean and SEM for fold increase in luminescence relative to vehicle control.
Tubastatin activated HIF-1α 2.4-fold in SH-SY5Y cells, which was matched or exceeded by a dozen members of the new phenylhydroxamate HDACi library. SAR analysis revealed lipophilic substituents in the aromatic ring to be most favorable, as demonstrated by chloro- and benzyloxy-substituted indolines (series A in Figure 1). In reverse of observations on Nrf2 activation, of the indoline/indolinone pair (III vs IV), the 3,3-dimethyl-indoline was most active in activation of HIF-1α (Figure 1, Table 1S), and the 6-chloro-substituted indoline ING-6 was found to be the most potent HIF-1 activator (Figure 2). Whereas no functionality commonly associated with Nrf2 activation is present in the Nrf2-activating HDAC inhibitors, PHD is an iron-dependent dioxygenase, and the metal-coordinating hydroxamate functionality of HDACi does provide a potential mechanism for PHD inhibition and HIF-1α stabilization. To test the potential for PHD inhibition by metal coordination, we docked the best HIF-1 stabilizing phenyl-hydroxamates into the binding site of PHD2 (4JZR)22 as exemplified by ING-6 in Figure 3. The active site Fe(II) is tethered by His313, His374, and Asp315, and is capable of coordinating the hydroxamate carbonyl oxygen as a fourth ligand (Fe–O distance 2.15 Å), while the NH group of ING-6 forms a hydrogen bond with the nearby Asp254 residue through water molecule 538. Additional interactions with the enzyme include a potential cation–π interaction with Arg322 and a π-stacking interaction with Trp258. Recent studies highlight the relevance of disruption of the hydrogen bonds between Pro564 in the hydroxylation site of HIF-1α and Arg322 of PHD2;22 and it is possible that the bulky and lipophilic substituents in the CAP region of the best HIF activators contribute to inhibition of HIF hydroxylation by PHD. Thus, this subset of phenylhydroxamates was found to stabilize Nrf2 and HIF-1α and are therefore likely to activate signaling via these pathways contributing to neuroprotection.
Figure 3.

Compound ING-6 docked into the binding site of PHD2 (4JZR).22
Cell-based reporter assays demonstrated that similarly to tubastatin, the potent, selective HDAC6 inhibitors ING-6 and ING-66 stabilized Nrf2 and HIF-1α, causing concentration-dependent accumulation of these transcription factors, with significant stabilization at 10 μM concentration. Activation requires protein stabilization and translocation to the nucleus; therefore, we tested ING-6 and ING-66 in cells, imaging Nrf2 with respect to the Hoechst nuclear stain (Figure 4A). Both ING-6 and ING-66 caused nuclear translocation, consistent with activation.
Figure 4.

(A) Stabilization and accumulation of Nrf2 on treatment with ING-6 and ING-66 visualized by immunofluorescence (IF). HeLa cells were treated with test compounds (10 μM) for 3 h: Nrf2 expression was quantified with anti-Nrf2 antibody (green) and colocalized with Hoechst nuclear stain (blue). (B) Immunoblot showing total, nuclear levels of HO1 and NQO1 after cells were treated with test compounds (10 μM) or DMSO (Cntr) for 4h (HO1) or 16h (NQO1) in N2a cells, using GAPDH as loading control. Band intensity was normalized to GAPDH: n = 3, *p < 0.05 as measured by Student’s t test.
To extend these observations to primary cells, the induction of downstream Nrf2 target genes was tested in mouse embryonic fibroblasts from wild type (WT) and Nrf2 knockout mice, using ING-6, ING-66, and the canonical Nrf2 activator TBHQ. Of the two HDACi tested in this cell line, ING-66 was superior as an activator of Nrf2 and downstream signaling, causing significant increases in Hmox-1 and NQO1 in WT cells, which were largely dependent on Nrf2 (Figure 2S). Consistent with the upregulation of mRNA levels of Nrf2 target genes, the protein levels of HO1 and NQO1 were also significantly increased between 4 and 16 h after the treatments as shown by Western blot (Figure 4B).
Activation of Nrf221 and HIF-1α23 signaling have been reported to provide neuroprotective activity, therefore, to extend our observations on ING-6 and ING-66, it was important to explore any neuroprotective phenotype. ING-6 and ING-66 were tested in neuronal cell lines subjected to a variety of insults that have been reported previously in the literature: H2O2, homocysteic acid (HCA), 1-methyl-4-phenyl-pyridinium (MPP+), and oxygen glucose deprivation (OGD) (Figures 5 and 6).
Figure 5.

Characterization of ING-6 and ING-66 in various neurotoxicity models in cell cultures. (A) Concentration-dependent neuroprotection of SH-SY5Y cells from H2O2-induced toxicity. Cell viability was measured using the MTT assay at 24 h. Data show mean and SEM normalized to control (n = 6): *p < 0.05, ***p < 0.001 versus control insult (n = 6/group), by 1-way ANOVA Dunnett’s test. (B) Test compounds at 5 μM protected N2a cells against MPP+ (100 μM) insult. Cell viability was measured at 24 h using Presto-Blue assay. Data show mean and SEM (n = 3): *p < 0.05 versus control insult. (C) Concentration-dependent protection by ING-66 of HCA-treated immature primary cortical neurons. Cell viability was assessed at 24 h using MTT assay. Data show mean and SEM (n = 3): *p < 0.05 and ***p < 0.001 versus control insult.
Figure 6.

Concentration dependence of ING-6 and ING-66 neuroprotection in SH-SY5Y cells subject to OGD: 24 h pretreatment (A) and treatment post OGD (B). Cell viability was measured using the MTT assay 24 h post insult. Data show mean and SEM normalized to control (n = 6): *p < 0.05, ***p < 0.001 versus insult by1-way ANOVA with Dunnett’s test.
Exposure to the endogenous ROS, H2O2, is frequently used to mimic oxidative stress leading to neuronal loss. Pretreatment of SH-SY5Y cell cultures with ING-6 or ING-66, 24 h prior to H2O2 insult, yielded significant neuroprotection (Figure 5A). The greater efficacy of ING-66 may be attributed to superior Nrf2 stabilization and HO-1 elevation, relative to ING-6 (Figures 2 and 4A; Table 1S). MPP+ is a mitochondrial toxin routinely used in vivo and in vitro to mimic dopaminergic neuronal loss associated with Parkinson’s disease. Both ING-6 and ING-66 at 5 μM produced significant protection against MPP+ neurotoxicity in mouse neuronal N2a cells (Figure 5B). HCA is an NMDA receptor agonist that is used to induce oxidative stress associated with glutamate excitotoxicity in neurons: neuroprotective activity of tubastatin in this model has been reported.13 ING-66 produced concentration-dependent and significant neuroprotection in HCA-treated primary cortical neurons, comparable to tubastatin (Figure 4C). No drug toxicity was observed in these primary cells. Furthermore, ING-6 and ING-66 showed no hepatotoxicity in the “liver-on-chip” assay, routinely used in drug development, testing viability of 3D cultures of HepaRG cells that mimic hepatocytes:24,25 LC50 > 500 μM (Figure 3S).
OGD provides a composite model of ischemia and reperfusion injury, which is responsive to neuroprotection mediated by HIF-1α and Nrf2.26 Pretreatment of cells 24 h prior to the OGD insult, with ING-6 or ING-66, provided significant concentration-dependent neuroprotection (Figure 6A). Ischemia is caused by withdrawal of both O2 and glucose; however, when both O2 and glucose are restored, there is a wave of reperfusion injury initiated by release of ROS. Post-treatment of cells with ING-6 or ING-66 was less effective; although, ING-6 was relatively better, which may be attributed to superior HIF stabilization (Figures 2 and 6B; Table 1S).
Having established the efficacy of ING-6 and ING-66 in four different in vitro neurotoxicity models, relevant to human neurodegenerative diseases, we investigated the expression of Nrf2 target genes in 3 month old male C57BL/6 mice administered ING-66 (25 mg/kg i.p.). Analysis by RT-PCR showed significant increases in Hmox-1 and NQO1 in the ventral midbrain and striatum (Figure 7A–B). Hmox-1 expression was also significantly upregulated in the liver (Figure 4S). These in vivo data support the in vitro observations of HIF-1α and Nrf2 activation by phenylhydroxamate HDAC inhibitors.
Figure 7.

In vivo Nrf2 target gene activation by ING-66. Quantitative RT-PCR analysis showing relative mRNA levels of ARE target genes in the brain tissues of C57BL/6 mice treated with ING-66 or vehicle control. mRNA expression of target genes (A) in striatum and (B) ventral midbrain. Data show mean ± SEM mRNA levels (relative to GAPDH): *p < 0.05, ***p < 0.001 compared to controls, (n = 5).
CONCLUSION
In summary, we established that novel phenylhydroxamates, like tubastatin, stabilized and activated two master regulators of cellular stress response, Nrf2 and HIF-1α, in addition to providing selective HDAC6 inhibition. Activation of Nrf2 and HIF-1α showed some dependence on cap-group structure, but was not consistent with HDAC6 inhibition potency. Further study of the best tripartite activators, ING-6 and ING-66, showed induction of Nrf2-dependent genes in vitro and in vivo. It has been proposed that Class-I HDAC inhibitors, but not HDAC6 inhibitors, may activate the Nrf2-ARE response by promoting the acetylation of Nrf2 or the acetylation of histones that facilitate transcription mediated by ARE.27–29 However, we found the benzamide-based Class-I HDAC inhibitors, tacedina-line and entinostat, to be completely inactive with respect to Nrf2-mediated transcription, in contrast to tubastatin, ING-6, and ING-66, which have little effect on Class-I HDACs,13,18 but stabilize Nrf2. Although the pan-HDAC inhibitors, TSA and SAHA, have been reported to cause acetylation and activation of Nrf2 via inhibition of Class-I HDACs,27–29 the Neh2-luciferase reporter is insensitive to this mechanism. Taken together, these observations demonstrate that HIF-1α and Nrf2 activation are activities of a subset of phenylhydroxamates that also potently inhibit HDAC6.
The selective HDAC6 inhibitors, ING-6 and ING-66, were neuroprotective in four independent in vitro models of neurodegeneration, modeling aspects of oxidative stress, excitotoxicity, Parkinson’s disease, and ischemic stroke. The micromolar, neuroprotective concentrations of ING-6 and ING-66 mirror those at which significant activation of HIF-1α and Nrf2 are observed. Although these concentrations are higher than those anticipated to inhibit HDAC6, the same concentrations of tubastatin were needed to elicit neuroprotection in previous literature reports.13,18 It has also previously been reported that hydroxamate family HDAC inhibitors, acting as metal chelating antioxidants, exert neuroprotective effects via HDAC independent mechanisms.30 Tripartite neuroprotective agents such as ING-6 and ING-66 hold promise as therapeutic candidates for neurodegenerative disorders (see Figure 5S for predicted druglikeness), but care must be taken in use as specific chemical probes for HDAC6 function.
Supplementary Material
Acknowledgments
Funding
This work was supported in part from the National Institute of Health Grants R01 NS060885, NS101967, and NS079183, the International Rett Syndrome Foundation Grant Contract 2769, Michael J. Fox Foundation for Parkinson’s disease, National Parkinson Foundation CSRA Chapter, Par Fore Parkinson Inc., Winifred Masterson Burke Foundation, Thomas Hartman Foundation for Parkinson’s disease, and Russian Science Foundation Grant 16-14-10226. We thank Dr. Alan P. Kozikowski (UIC) for partial financial support.
We thank the International Rett Syndrome Foundation for providing their SMART Library for screening.
Footnotes
Author Contributions
The manuscript was prepared through contributions of all authors. I.N.G., N.A.S., S.H.L., A.G., I.G.G., G.R.J.T., and B.T. conceived and designed the experiments. I.N.G. synthesized all compounds. N.A.K., S.W., and M.W.B. performed cell viability and gene expression studies in vitro. N.A.S. and I.G.G. performed library screening followed by extended SAR analysis. A.G. performed GSH reactivity assay. M.A. performed administration of ING-66 in mice and gene expression analysis in vivo by RT-PCR. M.W.B., S.H.L., and M.B.A. performed cytotoxicity experiments. S.V.N. performed liver-on-chip assay. I.N.G., N.A.S., A.G., A.A.P., I.G.G., and B.T. performed data analysis. B.T., G.R.J.T., and R.R.R. provided resources and supervised the experiments. I.N.G., I.G.G., G.R.J.T., and B.T. wrote the manuscript.
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschemneur-o.7b00435.
Synthesis, characterization, and biological assays (PDF)
References
- 1.Wakabayashi N, Slocum SL, Skoko JJ, Shin S, Kensler TW. When NRF2 talks, who’s listening? Antioxid Redox Signaling. 2010;13:1649–1663. doi: 10.1089/ars.2010.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol. 2004;24:10941–10953. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reiter RJ. Oxidative processes and antioxidative defense mechanisms in the aging brain. FASEB J. 1995;9:526–533. [PubMed] [Google Scholar]
- 4.Gazaryan IG, Ratan RR. Oxidative Damage in Neurodegeneration and Injury. Reference Module in Neuroscience and Biobehavioral Psychology. 2017:1–10. [Google Scholar]
- 5.Elbirt KK, Bonkovsky HL. Heme oxygenase: Recent advances in understanding its regulation and role. Proc Assoc Am Physician. 1999;111:438–447. [PubMed] [Google Scholar]
- 6.Alfieri A, Srivastava S, Siow RC, Cash D, Modo M, Duchen MR, Fraser PA, Williams SC, Mann GE. Sulforaphane preconditioning of the Nrf2/HO-1 defense pathway protects the cerebral vasculature against blood-brain barrier disruption and neurological deficits in stroke. Free Radical Biol Med. 2013;65:1012–1022. doi: 10.1016/j.freeradbiomed.2013.08.190. [DOI] [PubMed] [Google Scholar]
- 7.Kobayashi M, Yamamoto M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid Redox Signaling. 2005;7:385–394. doi: 10.1089/ars.2005.7.385. [DOI] [PubMed] [Google Scholar]
- 8.Calkins MJ, Johnson DA, Townsend JA, Vargas MR, Dowell JA, Williamson TP, Kraft AD, Lee JM, Li J, Johnson JA. The Nrf2/ARE pathway as a potential therapeutic target in neurodegenerative disease. Antioxid Redox Signaling. 2009;11:497–508. doi: 10.1089/ars.2008.2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gazaryan IG, Thomas B. The status of Nrf2-based therapeutics: current perspectives and future prospects. Neural Regener Res. 2016;11:1708–1711. doi: 10.4103/1673-5374.194706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li L, Zhang X, Cui L, Wang L, Liu H, Ji H, Du Y. Ursolic acid promotes the neuroprotection by activating Nrf2 pathway after cerebral ischemia in mice. Brain Res. 2013;1497:32–39. doi: 10.1016/j.brainres.2012.12.032. [DOI] [PubMed] [Google Scholar]
- 11.Kim H, Kim W, Yum S, Hong S, Oh JE, Lee JW, Kwak MK, Park EJ, Na DH, Jung Y. Caffeic acid phenethyl ester activation of Nrf2 pathway is enhanced under oxidative state: structural analysis and potential as a pathologically targeted therapeutic agent in treatment of colonic inflammation. Free Radical Biol Med. 2013;65:552–562. doi: 10.1016/j.freeradbiomed.2013.07.015. [DOI] [PubMed] [Google Scholar]
- 12.Smirnova NA, Haskew-Layton RE, Basso M, Hushpulian DM, Payappilly JB, Speer RE, Ahn YH, Rakhman I, Cole PA, Pinto JT, Ratan RR, Gazaryan IG. Development of Neh2-luciferase reporter and its application for high throughput screening and real-time monitoring of Nrf2 activators. Chem Biol. 2011;18:752–765. doi: 10.1016/j.chembiol.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Butler KV, Kalin J, Brochier C, Vistoli G, Langley B, Kozikowski AP. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J Am Chem Soc. 2010;132:10842–10846. doi: 10.1021/ja102758v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma TC, Langley B, Ko B, Wei N, Gazaryan IG, Zareen N, Yamashiro DJ, Willis DE, Ratan RR. A screen for inducers of p21(waf1/cip1) identifies HIF prolyl hydroxylase inhibitors as neuroprotective agents with antitumor properties. Neurobiol Dis. 2013;49:13–21. doi: 10.1016/j.nbd.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hales NJ, Beattie JF. Novel inhibitors of prolyl 4-hydroxylase. 5 The intriguing structure-activity relationships seen with 2,2′-bipyridine and its 5,5′-dicarboxylic acid derivatives. J Med Chem. 1993;36:3853–3858. doi: 10.1021/jm00076a014. [DOI] [PubMed] [Google Scholar]
- 16.Siddiq A, Aminova LR, Troy CM, Suh K, Messer Z, Semenza GL, Ratan RR. Selective inhibition of hypoxia-inducible factor (HIF) prolyl-hydroxylase 1 mediates neuroprotection against normoxic oxidative death via HIF- and CREB-independent pathways. J Neurosci. 2009;29:8828–8838. doi: 10.1523/JNEUROSCI.1779-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smirnova NA, Rakhman I, Moroz N, Basso M, Payappilly J, Kazakov S, Hernandez-Guzman F, Gaisina IN, Kozikowski AP, Ratan RR, Gazaryan IG. Utilization of an in vivo reporter for high throughput identification of branched small molecule regulators of hypoxic adaptation. Chem Biol. 2010;17:380–391. doi: 10.1016/j.chembiol.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen S, Benoy V, Bergman JA, Kalin JH, Frojuello M, Vistoli G, Haeck W, Van Den Bosch L, Kozikowski AP. Bicyclic-Capped Histone Deacetylase 6 Inhibitors with Improved Activity in a Model of Axonal Charcot-Marie-Tooth Disease. ACS Chem Neurosci. 2016;7:240–258. doi: 10.1021/acschemneuro.5b00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wagner FF, Olson DE, Gale JP, Kaya T, Weiwer M, Aidoud N, Thomas M, Davoine EL, Lemercier BC, Zhang YL, Holson EB. Potent and selective inhibition of histone deacetylase 6 (HDAC6) does not require a surface-binding motif. J Med Chem. 2013;56:1772–1776. doi: 10.1021/jm301355j. [DOI] [PubMed] [Google Scholar]
- 20.Gaisina IN, Tueckmantel W, Ugolkov A, Shen S, Hoffen J, Dubrovskyi O, Mazar A, Schoon RA, Billadeau D, Kozikowski AP. Identification of HDAC6-Selective Inhibitors of Low Cancer Cell Cytotoxicity. ChemMedChem. 2016;11:81–92. doi: 10.1002/cmdc.201500456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahuja M, Kaidery NA, Yang LC, Calingasan N, Smirnova N, Gaisin A, Gaisina IN, Gazaryan I, Hushpulian DM, Kaddour-Djebbar I, Bollag WB, Morgan JC, Ratan RR, Starkov AA, Beal MF, Thomas B. Distinct Nrf2 Signaling Mechanisms of Fumaric Acid Esters and Their Role in Neuroprotection against 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Induced Experimental Parkinson’s-Like Disease. J Neurosci. 2016;36:6332–6351. doi: 10.1523/JNEUROSCI.0426-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deng G, Zhao B, Ma Y, Xu Q, Wang H, Yang L, Zhang Q, Guo TB, Zhang W, Jiao Y, Cai X, Zhang J, Liu H, Guan X, Lu H, Xiang J, Elliott JD, Lin X, Ren F. Novel complex crystal structure of prolyl hydroxylase domain-containing protein 2 (PHD2): 2,8-Diazaspiro[4.5]decan-1-ones as potent, orally bioavailable PHD2 inhibitors. Bioorg Med Chem. 2013;21:6349–6358. doi: 10.1016/j.bmc.2013.08.046. [DOI] [PubMed] [Google Scholar]
- 23.Speer R, Ratan RR. Hypoxic Adaptation in the Nervous System: Promise for Novel Therapeutics for Acute and Chronic Neurodegeneration. Adv Exp Med Biol. 2016;903:221–243. doi: 10.1007/978-1-4899-7678-9_16. [DOI] [PubMed] [Google Scholar]
- 24.Liddle C, Stedman C. Hepatic Metabolism of Drugs. In: Rodes J, Benhamou J, Blei A, Reichen J, Rizzetto M, editors. Textbook of Hepatology: From Basic Science to Clinical Practice. 3. Wiley-Blackwell; 2007. pp. 241–249. [Google Scholar]
- 25.Poloznikov AA, Zakhariants AA, Nikulin SV, Smirnova NA, Hushpulian DM, Gaisina IN, Tonevitsky AG, Tishkov VI, Gazaryan IG. Structure-activity relationship for branched oxyquinoline HIF activators: Effect of modifications to phenylacetamide “tail”. Biochimie. 2017;133:74–79. doi: 10.1016/j.biochi.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 26.Guo SH, Miyake M, Liu KJ, Shi HL. Specific inhibition of hypoxia inducible factor 1 exaggerates cell injury induced by in vitro ischemia through deteriorating cellular redox environment. J Neurochem. 2009;108:1309–1321. doi: 10.1111/j.1471-4159.2009.05877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun Z, Chin YE, Zhang DD. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Molecular and cellular biology. 2009;29:2658–2672. doi: 10.1128/MCB.01639-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang B, Zhu X, Kim Y, Li J, Huang S, Saleem S, Li RC, Xu Y, Dore S, Cao W. Histone deacetylase inhibition activates transcription factor Nrf2 and protects against cerebral ischemic damage. Free Radical Biol Med. 2012;52:928–936. doi: 10.1016/j.freeradbiomed.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Correa F, Mallard C, Nilsson M, Sandberg M. Activated microglia decrease histone acetylation and Nrf2-inducible anti-oxidant defence in astrocytes: restoring effects of inhibitors of HDACs, p38 MAPK and GSK3beta. Neurobiol Dis. 2011;44:142–151. doi: 10.1016/j.nbd.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olson DE, Sleiman SF, Bourassa MW, Wagner FF, Gale JP, Zhang YL, Ratan RR, Holson EB. Hydroxamate-based histone deacetylase inhibitors can protect neurons from oxidative stress via a histone deacetylase-independent catalase-like mechanism. Chem Biol. 2015;22:439–445. doi: 10.1016/j.chembiol.2015.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.