

Abstract
Organophosphate compounds (OPCs) are commonly used as pesticides and were developed as nerve agents for chemical warfare. Exposure to OPCs results in toxicity due to their covalent binding and inhibition of acetylcholinesterase (AChE). Treatment for toxicity due to OPC exposure has been largely focused on the reactivation of AChE by oxime-based compounds via direct nucleophilic attack on the phosphorous center. However, due to the disadvantages to existing oxime-based reactivators for treatment of OPC poisoning, we considered non-oxime mechanisms of reactivation. A high throughput screen of compound libraries was performed to discover previously unidentified reactivation compounds, followed by studies on their analogs. In the process, we discovered multiple non-oxime classes of compounds, the most robust of which we have already reported.(1) Herein, we report other classes of compounds we identified in our screen that are efficient at reactivation. During biochemical characterization, we also found some compounds with other activities that may inspire novel therapeutic approaches to OPC toxicity. Specifically, we found compounds that (1) increase the rate of substrate hydrolysis by AChE and, (2) protect the enzyme from inhibition by OPC. Further, we discovered that a subset of reactivator compounds recover activity from both AChE and the related enzyme butyrylcholinesterase (BuChE). We now report these compounds, their activities and discuss how each relates to therapeutic approaches that would provide alternatives to traditional oxime-based reactivation.
Keywords: Acetylcholinesterase (AChE), Butyrylcholinesterase (BuChE), Organophosphate compounds (OPCs), Non-oxime reactivators, Structure activity relationship (SAR) study
Graphical Abstract
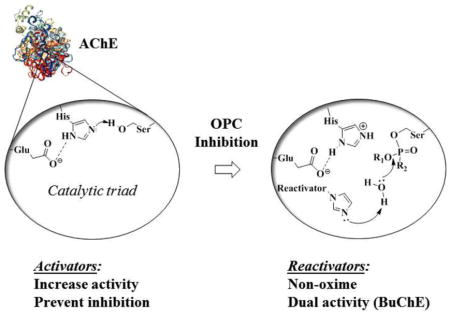
1. Introduction
Recent use of organophosphorus compounds (OPCs) for poisoning, either in terrorist attacks (Figure 1, sarin 1, soman 2, etc.) (2, 3) or in self-poisonings (both suicides and accidental exposure) using readily available pesticides (Figure 1, DFP 3, paraoxon 4, etc.) (4), has intensified the interest in research on reversing the effects of OPC toxicity. The mechanism of most acute toxicities originates with OPC-inhibition of acetylcholinesterase (AChE, EC 3.1.1.7), the enzyme that is responsible for the hydrolysis of the neurotransmitter acetylcholine. OPCs inhibit AChE via covalent binding to the serine hydroxyl group in the enzyme active site (Scheme 1A). This inhibition results in the accumulation of the neurotransmitter acetylcholine in the synaptic cleft, which in turn leads to saturation of cholinergic receptors and an inability to control the muscles involved in breathing, leading to asphyxiation, paralysis, and eventually death.(5)
Figure 1.
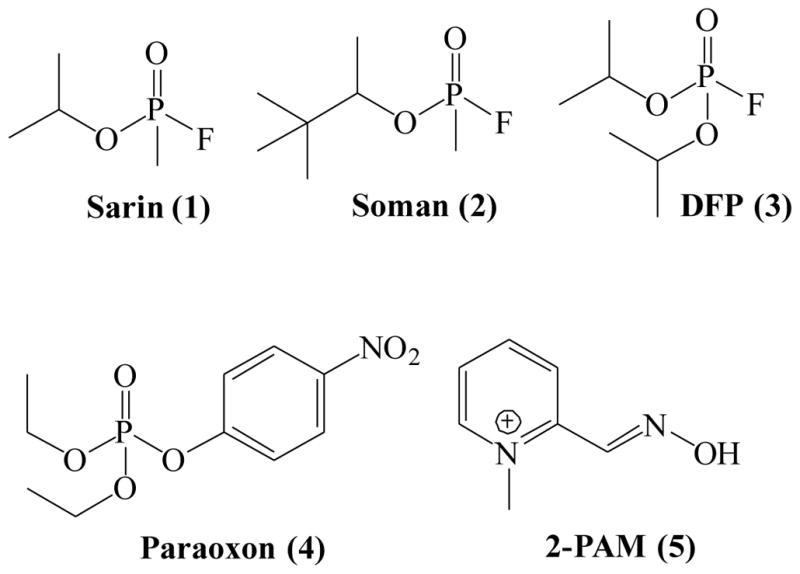
Chemical structures of common OPCs and oxime reactivator 2-PAM
Scheme 1.

The active site gorge of AChE contains a catalytic triad, consisting of the amino acids Ser200, His440 and Glu327 (numbering for the human AChE enzyme). Once OPCs bind to the active site, the nucleophilic serine attacks the phosphorus, forming a phosphyl adduct.(6) This first step is similar to the formation of the transition state when AChE binds acetylcholine. However, any hydrolysis of OPCs is very slow, requiring several days for some nerve agents.(7) Instead, with most nerve agents a dealkylation (often a unimolecular SN1 reaction involving a carbocationic intermediate) occurs, yielding a negatively charged “aged” adduct, which is stable and does not undergo further hydrolysis to free enzyme.(8)
The current approved medical guidelines for treatment of organophosphate toxicity, e.g., both nerve agent (NA) exposure and accidental pesticide poisonings, are administration of atropine and pralidoxime (2-PAM 5, Figure 1).(9) The discovery of nucleophilic reactivation by 2-PAM, and oximes in general (Scheme 1B), was first described by Wilson in the early 1950’s.(10, 11) Since then, it has been shown that the currently available oximes suffer from several drawbacks making them potentially impractical for actual therapeutic use. First, many of the reported oximes have difficulty crossing the blood-brain barrier (BBB).(12) Increasing lipophilicity improves the penetration of the BBB, but also increases toxicity.(13) It has been shown that in some cases the resulting oxime adduct can be toxic itself, perhaps also enabling transfer of OPCs across the BBB.(14) The use of pro-drugs has been reported as an alternative approach to address the problem of BBB penetration. The reported pro-drugs undergo oxidation after crossing the BBB, producing an active quaternary oxime in vivo in the CNS.(15) However, the pro-drugs are reportedly difficult to synthesize and are unstable due to auto-oxidation in moisture.(16, 17) Additionally, oximes are not equally effective at reactivating different OPCs and further studies in human tissue models revealed optimal efficacy only at concentrations that objectively could not be achieved in vivo.(18) The effectiveness of oximes is further reduced in the case of nerve agents that rapidly age (e.g., soman).(19)
While significant insight into the mechanism of action of oximes has been gained from the extensive structure-activity relationship studies on them (20–25), there is still an unmet need to find adequate therapeutics to treat OPC poisoning. We explored the possibility of other mechanisms for reactivation of OPC-inhibited AChE that may be more efficient and would be the starting point for improved treatment strategies. We established a solid-state screen for reactivation of OPC-inhibited AChE and identified several new non-oxime classes of compounds, some of which we have already reported.(1) We propose that these compounds reactivate AChE using a general acid-base catalysis mechanism, whereas oximes use a direct nucleophilic mechanism (Scheme 1B). Here we report other compounds from our screening efforts and from the testing of readily available analogs of our initial hits, that had additional activities which we now report: i) analogs of our initial hits eliminating the potential for toxicity had different reactivation profiles; ii) completely new classes of compounds that reactivate AChE that up until now remained unreported; iii) compounds that stimulate AChE activity; iv) compounds that protect AChE from inhibition by OPCs; and v) compounds that are efficient at reactivating both AChE and the related enzyme, butyrylcholinesterase (BuChE). We discuss each of these in the context of their utility in novel therapeutic approaches, presenting them as mechanistic leads for further studies. We present these compounds for their consideration as different pathways for treatment, while recognizing that further investigation and characterization is needed to understand their mechanism of action and potential for efficacy.
2. Materials and Methods
2.1 Chemicals
All solvents and reagents were obtained from Aldrich and used without further purification. Analytical thin-layer chromatography (TLC) was performed on aluminum plates pre-coated with silica gel also obtained from Aldrich. Column chromatography was carried out on Merck 938S silica gel. Proton NMR spectra were recorded using Varian 400 MHz NMR spectrometer. Spectra were referenced to the residual solvent peak and chemical shifts expressed in ppm from the internal reference peak. Data are reported as follows: chemical shifts, multiplicity (s=singlet, d=doublet, t=triplet, q=quartet, quin=quintet, br=broad, m=multiplet), coupling constants (Hz), and number of protons. All compounds described were of >95% purity. Purity was confirmed by analytical LC/MS recorded on a Shimadzu system. Elution started with 95% water (+0.1% formic acid) and 5% acetonitrile (+0.1% formic acid) and ended with 95% acetonitrile (0.1% formic acid) and 5% water (0.1%formic acid) and used a linear gradient at a flow rate of 0.2 mL/min. The molecular ion M+, with intensities in parentheses, is given followed by peaks corresponding to major fragment losses. Melting points were performed using a MEL-TEMP II melting point apparatus and are reported uncorrected.
2.2. Chemical structures of compounds used in this report
2.2.1 Original hit with less toxic analog
We synthesized an analog of amodiaquine (ADQ), isoquine (ISQ) (Figure 2A), that has less potential for toxicity.
Figure 2.




Chemical structures of compounds used in this report
2.2.2 Non-oxime reactivation compounds
During our screening efforts, we identified several compounds that did not have oxime functionality but had significant reactivation activity (Figure 2B).
2.2.3 Analogs of SP138
We synthesized several compounds in order to perform SAR studies on one of our most active hits, SP138 (Figure 2C).
2.2.4 SAR studies on pyridine-containing reactivation compounds
From our initial screening, we identified compounds containing pyridine that reactivated OPC-inhibited AChE. We tested the effect of the length of the linker on reactivation activity with these compounds (Figure 2D).
2.2.5 Structural analogs of donepezil
One of the hits we identified in our initial screen was structurally similar to donepezil. We synthesized a series of analogs and tested them for reactivation activity (Figure 2E)
2.2.6 Compounds that activate AChE
Compounds shown to activate the rate of AChE activity are shown in Figure 2F. Pre-incubation with a subset of these were shown to prevent inhibition by OPC.
2.2.7 Compounds that reactivate BuChE
Addition of compounds shown in Figure 2G to paraoxon-inhibited BuChE resulted in significant reactivation of the enzyme. A subset of these compounds was also efficient at reactivating AChE.
2.3 Activity assays
2.3.1 Reactivation assays
Herein, we describe characterization of compounds that we identified in our screening for compounds that reactivate AChE. A more complete description of our screening conditions can be found in our report on our initial findings.(1) From our initial screen, we identified several classes of compounds that reactivated the immobilized muAChE. We then confirmed these hits using a solution phase assay. We used muAChE for our screen because it most readily available and our initial solution phase confirmations were also done on the mouse form of the enzyme. Further analysis was pursued with huAChE and huBuChE to determine if activity would be translatable. In addition, our model OPC for screening was paraoxon, while we used DFP for further characterization, as model for more sterically-hindered OPCs.
For all activity assays, enough enzyme was used to reach a rate of hydrolysis for acetylthiocholine (ATCh) (or for BuChE, butyrylthiocholine, BTCh), resulting in the breakdown of DTNB, that gave an OD412 of around 1 within 7–10min. The amount of enzyme used was adjusted for differences in the protein stock used. A minimum of 4-fold molar excess of OPC (either paraoxon or DFP) was used to inhibit the enzyme. The enzyme was allowed to incubate with OPC for 10–15 min. to achieve greater than 95% inhibition, followed by removal of unbound OPC by passage of the inhibited protein through a PD10 column. This was done in parallel with uninhibited AChE as a standard for full activity. Compounds were added to OPC-inhibited AChE and allowed to incubate 3 hrs for paraoxon reactions and for muAChE with DFP; a more extended (overnight) incubation was allowed for huAChE inhibited by DFP and huBuChE. In order to eliminate effects of high concentrations of compounds on enzyme activity, the reaction was diluted 1:100 before adding ATCh (or BTCh) and DTNB (Ellman’s assay).(26)
2.3.2 Activation assays
To determine the direct effect of compound on enzyme activity, we added compounds at the indicated final concentrations to ~0.8nM muAChE (final concentration of monomer) and allowed incubation for 15 minutes prior to addition of ATCh and DTNB (2mM each). AChE activity was monitored at 412nm.
2.3.3. Protection assays
Compounds at the indicated concentrations were incubated for 15 minutes with ~0.8nM muAChE (final concentration of monomer) prior to incubation for an additional 15 minutes with 1μM DFP. Then, ATCh and DTNB (2mM each) were added and enzyme activity was monitored at 412nm.
3 Results
3.1. Modification of original hits to reduce potential for toxicity
Using the solid-state assay platform that we developed(1), we identified the antimalarial drug amodiaquine (ADQ) (Figure 2A) in our initial screen of FDA-approved drugs, but we hesitated to further pursue it because of reports that active doses of ADQ cause hepatotoxicity resulting from the formation of a p-aminoquinone during metabolism in the liver.(27) To reduce potential for toxicity, we synthesized (Scheme 2, Supplementary data) and evaluated a known analog of ADQ, isoquine (ISQ, Figure 2A), which eliminates the reactive electrophilic quinoneimine intermediate associated with ADQ toxicity by changing the position of its amino group (change in Mannich base: 4-amino-alpha-diethylamino-o-cresol (ADOC)(1) structure from 4- to 3-amino group). ISQ cannot form the quinoneimine intermediate and, thus, has been reported to have greatly reduced toxicity. This structural change was shown not to affect antimalarial activity, with reports showing that ISQ is equally effective against malaria as ADQ.(28) We found that reactivation of paraoxon-inhibited AChE by ISQ was similar to reactivation by ADQ (Figure 3A); however, the reactivation of DFP-inhibited AChE was somewhat less efficient (see Figure 3B), suggesting that the proper orientation and positions of functional groups in ADQ are important for binding to OPC-inhibited enzyme. Isoquine, however, remains a valid lead for further optimization, as it indicates that the attachment at 4- position is not needed for activity. In this respect, the Mannich base ADOC (as we reported previously(1)) is similar to oxime, which can be attached at various linkers to other anchoring groups.
Scheme 2.

Reagents and condition: EtOH, reflux, 12h
Figure 3. Concentration dependence of reactivation of huAChE by ISQ.

HuAChE was incubated with either A. paraoxon for 15 minutes or B. DFP for 10 minutes before addition of the indicated concentrations of ADQ (red-brown) or a less toxic analog, ISQ (teal). Reactivation for paraoxon was allowed for 3 hours, and overnight for DFP. Reactions were diluted 1:100 to reduce background effects from the reactivation compound prior to measurement of enzyme activity. Activities are given relative to activity in the absence of inhibitor or reactivation compound.
3.2. Classes of non-oxime reactivation compounds
Several non-oxime reactivation compounds emerged from our screen and can be considered as new starting points for reactivation-based therapeutics. We classified these compounds into three groups based on the similarities in their scaffolds: Representative compounds from each group are shown in Figure 2B and their activities are shown in Figure 4. Group 1 – Imidazole reactivators; Group 2 – Pyridine reactivators and Group 3 – Piperazine reactivators.
Figure 4. Non-oxime reactivators emerged from screening.

Three groups of structural families were identified from our screen for novel reactivators. The newly identified reactivation compounds (25μM) were allowed to incubate with paraoxon-inhibited muAChE for 3 hours prior to measurement of enzyme activity. The bar graph shows activity relative to uninhibited, untreated enzyme under the same conditions.
3.2.1. Group 1 – Imidazole reactivators
Some of the compounds in Group 1 had the highest activity and we decided to explore several of their structural domains that were similar (see Figure 5). Domain A was designated as the potential functional domain; Domain B is a linker of several methylene groups between the domains; and Domain C was assigned the putative binding domain. Our initial structure-activity relationship (SAR) study started with synthesis of several SP134 and SP138 analogs (see Figure 2C) designed to test the importance of Domain B, i.e., the length of the linker between the aromatic moiety and the imidazole group. The results with these analogs are reported in our previous publication (1).
Figure 5. Common structural domains of our top hits.

The domains of the molecule have been outlined for initial SAR studies.
After identifying the optimal linker length, we continued our preliminary SAR evaluation of Domain C in SP138. Domain C was unique to SP138, so we were interested in whether or not it was essential, and if any changes in this domain would enhance activity. First, we found that removal of the phenyl ring (SP163) resulted in a loss of reactivation activity (reactivation reached only ~25%, see Figure 6). We next turned our attention to the bromine in the para position of the phenyl moiety. We decided to prepare a set of analogs containing various halogens in place of the bromine in SP138 because halogens can enhance polarity and decrease the rate of metabolic degradation due to their electron withdrawing effect on the aromatic ring. Replacement of bromine in the para position with chlorine, SP168, and fluorine, SP170, did not significantly change reactivation activity (Figure 6). Inclusion of an additional non-polar phenyl ring in the para position, SP171 did not show significant improvement in activity either. Placement of a polar hydroxyl group in the para position, SP172 also had little effect on reactivation activity. These results indicate that, unlike the phenyl group itself, the para position is tolerant to many different moieties and has little effect on reactivation, therefore presenting an opportunity to improve physical properties and ease of formulation. Analogs SP138, SP159, SP163, SP168, SP170, SP171 and SP172 were synthesized according to Scheme 3 in Supplementary data.
Figure 6. Initial SAR to determine role of phenyl moiety of Domain C.

Analogs of the initial hit SP138 were synthesized and tested for the concentration dependence of reactivation of huAChE inhibited by DFP. Enzyme was treated with DFP for 10 min. prior to removal of excess DFP by PD10 column. After compounds were added at the indicated concentrations, reactivation was allowed to proceed overnight. Prior to measuring activity, the reactions were diluted 1:100. Each graph shows the controls of SP138 and 2-PAM.
3.2.2 Group 2 – Pyridine reactivators
In order to explore Group 2 – pyridine-based reactivators, we initially decided to keep the pyridine in the Domain A position and combine it with the 2,4-dichlorophenyl moiety from Group 1 in the putative binding position, Domain C, because this structural feature is expected to fit in the proximity of the OPC-inhibited serine in the active site of AChE, based on our previously published SAR and crystallography data(1). We also tested the effect of the length of the linker on reactivation activity with these compounds (SP94, SP96 and SP98, Figures 2D). These 3 analogs were obtained in good yields (75–85%) by condensing 2,4-dichlorobenzoic acid and pyridine-amines (varying in linker length between the two moieties), using standard EDC coupling conditions. These compounds were not soluble at high concentrations and we were not able to test them at concentrations above 75μM. They resulted in significant reactivation above background; however, they did not reach over ~50% activity and were not as efficient as other compounds we have identified (Figure 7). Specifically, we compared these compounds to SP134 which has the same 2,4-dichlorophenyl moiety in Domain C, so that we could directly assess the effects of the pyridine in Domain A.
Figure 7. Pyridine analogs SP94, SP96, SP98.
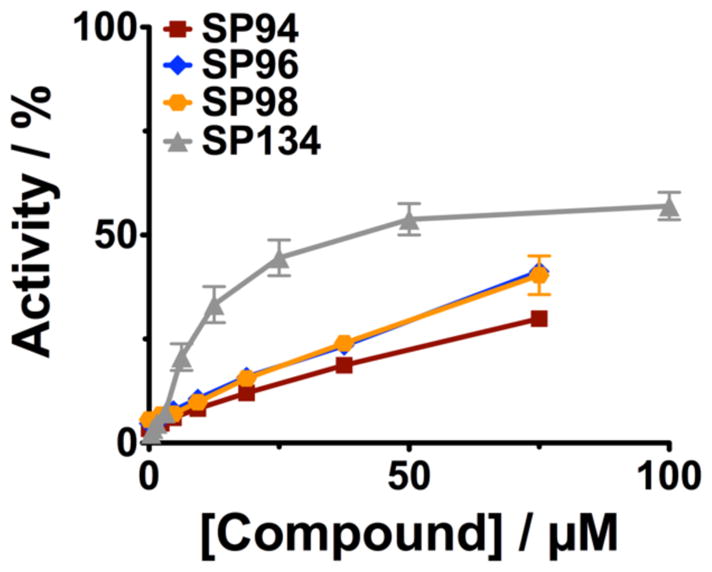
Analogs combining features of Group 1 and Group 2 were synthesized and tested for reactivation activity on paraoxon- inhibited muAChE. Paraoxon was incubated with muAChE for 15 minutes prior to removal of excess OPC using a PD10 column. Inhibited enzyme was incubated with the indicated concentrations of compounds for 3 hours. Reactions were diluted 1:100 before measuring enzyme activity.
3.2.3. Group 3 – Piperazine reactivators
Group 3 reactivators contain a piperazine ring as the middle part of the structure (Figure 2B), indicating that it is possible to use this motif as a general base to enhance hydrolysis of OPC adducts. We classified representative compound C47 into both Group 2 and Group 3, because it contains a pyridine moiety (Group 2 feature) and is also linear as the Group 3 compounds, differing only in that its middle group is a piperidine instead of a piperazine. Interestingly, C47 has some structural resemblance to donepezil, a known potent inhibitor of AChE that is used for treatment of Alzheimer’s disease. We designed and synthesized three donepezil analogs, SP111, SP112 and SP117 (Figure 2E), containing either piperidine or piperazine groups while also containing the 1-indanone moiety of donepezil, which was shown to bind to the peripheral active site (PAS) of AChE(29), with the expectation that this binding would help bring the general base activity in proximity of the OPC-inhibited site in AChE and increase reactivation. Analogs SP111 and SP112 were synthesized in a one-step reaction using reductive amination conditions with triacetoxyborohydride, and synthesis of analog SP117 was completed in a two-step reaction, first introducing the aldehyde group to commercially available 5,6-dimethoxy-1-indanone(30) followed by reductive amination. Unfortunately, these analogs exhibited very strong inhibition of AChE activity, retaining the potent inhibition profile seen with donepezil, and even upon several dilutions, we were not able to reduce this inhibition (data not shown). Thus, these compounds could not be evaluated for their reactivation activity, and their strong inhibitory properties make them unsuitable to treat scenarios in which the AChE is already compromised; however, they may be of interest for other indications.
3.3. Activation of residual AChE to compensate for OPC-inhibition
As a control for our screening conditions, we added library compounds to uninhibited AChE, and to our surprise, we discovered a family of compounds that stimulated the rate of acetylthiocholine (ATCh) hydrolysis by AChE (Figure 2F). We identified trifluoperazine 6 and prochlorperazine 7 from our initial screening, using our solid-state assay(1) and we confirmed these results in solution phase, with prochlorperazine 7 giving the most significant stimulation (Figure 8). We then expanded these studies, first to structurally related thioperazines such as chlorpromazine 8, and acepromazine 9, and further to other tricyclics, including four dibenzazepines: imipramine 10, desipramine 11, amitriptyline 12, and doxepin 13 (Figure 8). We determined the effect of increasing concentrations of compound on the rate of hydrolysis at a saturating concentration of ATCh (2mM) and we plotted the dependence of concentration of compound on the fold increase in rate relative to the rate in the absence of compound in Figure 8. Previous studies indicated that some tricyclic compounds are inhibitors of AChE, but they were shown to have inhibition constants(31–33) that were much higher than the concentrations needed for activation; as a result, activation activity can be separated from other effects, and these compounds now present interesting leads.
Figure 8. Compounds that activate hydrolysis activity of AChE.

The concentration dependence of hydrolysis of acetylthiocholine by AChE was measured by a standard Ellman’s assay at saturating substrate concentration. The indicated concentrations of compound were incubated with muAChE for 15 minutes prior to addition measuring activity. Activity is shown relative to untreated enzyme.
From our results with this set of compounds, we observed several SARs; (i) the methylpiperazinyl group is important for activation (Figure 8, analogs 7–9), while (ii) removal of a single methyl group on nitrogen decreases activation rate (analog 10 vs 11), both hinting at potential for further improvement by adding alkyl groups or longer chains. These results suggest that the family of compounds that activate AChE is characterized by a three-membered ring moiety separated from a piperazine ring via a linker. We propose, based on the structural similarity with tacrine, that these compounds are likely binding to a peripheral anionic site of AChE, but in orientation that does not block access of the substrate; further mechanistic studies to test this hypothesis are necessary in the future.
The identification of compounds that can stimulate AChE activity made us consider the utility of stimulating residual AChE in event of OPC exposure. It has been demonstrated by us(1) and others(34, 35) that only a small percentage of AChE needs to be active in vivo in order to retain viability after exposure to OPC. Thus, it is possible that administration of an activation compound would stimulate enough AChE activity to promote survival in the event of OPC poisoning. Whether such activity is feasible in vivo, where AChE has been proposed to operate at diffusion limited rates, and whether our results are due to using a thio-analog of the native substrate(36), cannot be answered at this time.
3.4. Prevention of OPC-inhibition as a potential pre-treatment
The ability of these compounds to activate AChE made us consider the possibility of using them as pre-treatments to keep AChE activity levels high enough to prevent the effects of OPC poisoning. Activation activity may be a result of the compound binding in the active site gorge; thus, we wanted to explore the effect of the presence of these compounds on inhibition by OPC if they were bound to AChE prior to exposure. We initially studied the effect of trifluoperazine 6 or prochlorperazine 7 on the protection of AChE from inhibition by DFP in solution. For these experiments, we added compound at the indicated concentrations to AChE, followed by addition of 1μM DFP and allowed the DFP to incubate for 15 minutes before adding ATCh and DTNB to measure AChE activity. Initial rates were calculated and were compared relative to the activity of AChE that had been exposed to neither compound nor DFP (the latter was set as 100% activity). The results from these studies (see Figure 9) show that if either trifluoperazine 6 or prochlorperazine 7 are present (at concentrations above ~5μM) before AChE is challenged with DFP, the AChE retains most of its initial activity. We also demonstrated that tricyclic compounds, such as amitriptyline 12, and doxepin 13 protected AChE against inhibition by DFP. The ability of these compounds to protect against OPC inhibition and activate residual AChE activity make them an intriguing starting point for future development for novel treatments for OPC poisoning.
Figure 9. Protection of AChE from OPC inhibition.

The indicated concentrations of compound was incubated for 15 minutes with muAChE prior to addition of 1μM DFP. This was then incubated for an additional 15 minutes prior to measuring enzyme activity. In the absence of compound, addition of OPC resulted in nearly complete inhibition. The presence of compound prior to exposure to OPC allowed significant to nearly full activity levels.
3.5. Reactivation of BuChE
The active sites of BuChE and AChE have similarities (37–39) and BuChE is efficient at catalyzing the breakdown of acetylcholine. BuChE is inhibited by OPCs itself, but loss of BuChE activity is not thought to contribute to toxicity, since it is not known to perform an essential function in vivo(40). Thus, the use of BuChE as a bioscavanger to sequester OPCs as a treatment for toxicity has been investigated extensively (38, 41–44). One important drawback is that it acts stoichiometrically and thus needs to be administered in very high doses. A compound that could reactivate BuChE would essentially turn it into a catalytic scavenger (38, 45–47). While the active sites of AChE and BuChE are similar, there are some well-characterized differences that have made it difficult to find compounds that interact efficiently with both active sites(48–50). We thought that some of the compounds we identified could potentially reactivate BuChE or even both AChE and BuChE.
We tested a panel of our hits from our AChE screen and found a subset of those compounds also reactivated BuChE (Figures 2G and 10). While #17 did not efficiently reactivate AChE, it showed significant reactivation of BuChE. Interestingly, ADQ was efficient at reactivating both AChE and BuChE, however, ADOC, the minimal Mannich phenol of Domain A in ADQ, which has robust reactivation activity of AChE(1) was very poor at reactivating BuChE. Of the dual reactivators, ICQ, which retains the Domain C of ADQ but has the imidazole of our Group 1 analogs in the functional group (Domain A), gave significant reactivation of both BuChE and AChE (nearly complete reactivation for human AChE(1)). Further, while SP138 was only efficient at reactivating AChE, the related compound SP134 gave significant reactivation of both AChE and BuChE. In addition, we explored the effect of the length of linker of Domain B on reactivation of BuChE by SP134 and we found that the 5-carbon linker of SP134 was the optimal length. These represent new leads for dual reactivators that can both help with initial toxicity and lead to recovery of scavenging agents.
Figure 10. Solution phase reactivation of human BuChE.
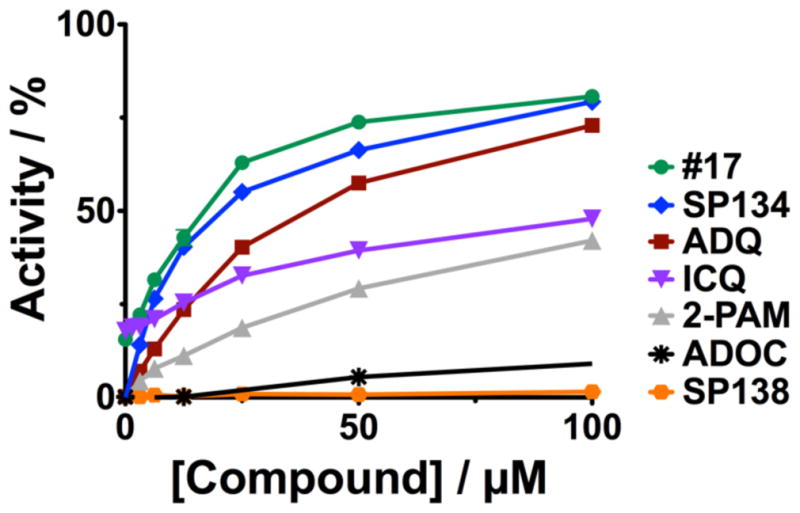
The indicated concentrations of compounds were added to paraoxon-inhibited huBuChE. Compounds were allowed to incubate with enzyme overnight and then diluted to reduce inhibition by compound before adding butyrylthioholine and DTNB to measure activity. Percent activity is reported relative to activity in the absence of compound or inhibitor.
4. Discussion
A way to improve treatments for OPC toxicity has been sought for decades. The main focus has been on increasing the efficacy of oximes; however, we took the approach of finding new mechanisms of action by performing a screen for compounds that reactivate OPC-inhibited AChE. Through this screening effort, not only did we find previously unreported non-oxime chemical groups that are capable of reactivating AChE, but also the following new starting points for potentially novel therapeutic approaches:
Further classes of non-oxime reactivators: containing imidazoles, pyridine, or piperazine moieties
Compensation for OPC-inhibited AChE, by stimulation of residual AChE using compounds that increase the rate of AChE activity, thus keeping baseline AChE above the threshold for toxic effects
Prevention of OPC inhibition, by compounds that bind in the active site of AChE and delay OPC inhibition, as a potential pre-treatment for warfighters or agricultural workers or civilians at risk of exposure. Any compound that binds in the active site has the potential to be inhibitory itself; however, we found compounds that stimulate AChE activity and also protect against OPC inhibition. This should eliminate potential for side effects due to AChE inhibition.
Dual reactivation of AChE and BuChE, using compounds that are efficient at reactivating both enzymes. This type of treatment would not only increase the efficacy of BuChE bioscavenger-based treatments, the compounds would also act regenerate active AChE enzyme, making this a highly attractive new line of treatment.
Of note, complete reactivation of the enzyme was not achieved in many cases we present here. It remains unclear if this is due to a slow rate of reactivation, promotion of aging during the reaction, or some other inhibitory effect of the compounds. We limited our characterization of these compounds to a survey of the concentration dependence of their effect on enzyme and we present them as important starting points, while a more complete understanding of the mechanism of their action is needed to determine their full potential.
Supplementary Material
Scheme 3. Reagents and condition: CDI, THF, rt, 12 h, reflux, 5h
Highlights.
Previously unreported non-oxime reactivators have been discovered
These compounds have been optimized through SAR studies
Compounds have been identified that increase AChE activity and stimulate the enzyme
Inhibition of AChE by OPC can be prevented by pre-incubation with compounds
Dual reactivators were active on both AChE and BuChE
Acknowledgments
This research was supported by HDTRA1-11-C-0006, HDTRA1-16-1-0053, and by NIH CounterACT grant number 5R21NS080790-02. Equipment used to characterize synthetic compounds was supplied through ONR grant N00014-11-1-0900 and NIH 1S10OD018121-01
Appendix: Supplementary data
Synthesis of Isoquine (ISQ)
Isoquine (ISQ) was prepared following the two-step procedure previously reported by O’Neill et al (Scheme 2).(28) The first step involves synthesis of Mannich 3-aminophenol product, and we reported it in our previous work (1) and second step of the synthetic sequence involves coupling with 4,7-dichloroquinoline to provide target molecule ISQ. After purification with flash chromatography using 10% MeOH/dichloromethane solvent system as eluent, ISQ was obtained as a hydrochloride salt. This salt was free based by dissolving in distilled water and adding saturated sodium bicarbonate solution. The precipitated free-based ISQ was collected after filtration as an off-white solid.
5-((7-chloroquinolin-4-yl)amino)-2-((diethylamino)methyl)phenol, ISQ
Off-white solid (64%). 1H NMR (CDCl3, 400 MHz): δ 8.69 (d, J = 5.2 Hz, 2H), 8.12 (d, J = 8.4 Hz, 2H), 7.99 (s, 2H), 7.40 (dd, J = 6.8, 2.0 Hz, 2H), 6.66 (d, J = 5.2 Hz, 2H), 4.22 (q, J = 7.2 Hz, 4H), 1.54 (t, J = 6.4 Hz, 8H). 13C NMR (CDCl3, 100 MHz): δ 161.7, 152.6, 149.8, 135.7, 127.9, 126.4, 123.6, 119.9, 100.9, 64.4, 14.5. ESI-MS: [m/z+H]: 357.
General procedure for synthesis of amide quinolines
General route for synthesis of amide quinoline derivatives is shown in Scheme 3 below. The appropriate commercially available quinoline-4-carboxylic acids were activated by 1,1-carbonyldiimidazole and N-(3-aminopropyl) imidazole was added to obtain the target structures. These conversions provided the desired derivatives in good yields and all target compounds were isolated either as crystalline solids, or we converted them into suitable salts.
General procedure for the preparation of SP138 analogs: A mixture of 2.75 mmol of the appropriate carboxylic acid and 3.025 mmol of 1,1′-carbonyldiimidazole in 15 mL of THF was stirred at room temperature for 1.5 h. The reaction mixture was cooled to 0°C and 2.75 mmol of the N-(3-aminopropyl) imidazole was added. The reaction mixture was then stirred at room temperature for 18–24 h, and refluxed for 5 h. The solvent was removed in vacuo and the residue was dissolved in 20 mL of diethyl ether and treated with 1N HCl. Acid layer was separated, basified with saturated NaHCO3, and extracted with ethyl acetate (3×25 mL). The organic layers were combined, dried over Na2SO4, and concentrated to give the desired crude product.
N-(3-(1H-imidazol-1-yl)propyl)-2-(4-bromophenyl)quinoline-4-carboxamide, SP138
The crude compound was purified by column chromatography in 5% methanol/dichloromethane solvent system to give SP138 as a yellow solid (0.84 g, 69%). 1H NMR (CDCl3, 400 MHz): δ 8.06 (s, 1H), 7.95 (d, J = 8.8 Hz, 1H), 7.79 (d, J = 7.6 Hz, 2H), 7.57 (d, J = 8.8 Hz, 2H), 7.45 (d, J = 8 Hz, 2H), 7.37 (t, J = 7.6 Hz, 1H), 7.13 (s, 1H), 6.75 (t, J = 15 Hz, 2H), 3.83 (t, J = 6.4 Hz, 2H), 3.26 (d, J = 8 Hz, 2H), 1.92 (t, J = 6.8 Hz, 2H). 13C NMR (CDCl3, 100 MHz): δ 167.9, 155.3, 148.6, 142.6, 137.5, 136.9, 132.0, 130.4, 130.0, 129.3, 128.9, 127.5, 124.9, 124.4, 123.4, 118.9, 116.1, 44.7, 37.3, 30.9. ESI-MS: [m/z+H]: 436.
N-(3-(1H-imidazol-1-yl)propyl)-2-phenylquinoline-4-carboxamide, SP159
The crude compound was purified by column chromatography in 5–10% methanol/dichloromethane solvent system to give SP159 as a pale yellow solid (0.65 g, 66%). 1H NMR (CDCl3, 400 MHz): δ 8.10 (d, J = 8.8 Hz, 2H), 8.07 (d, J = 6.4 Hz, 2H), 7.78 (s, 1H), 7.70 (t, J = 7.2 Hz, 1H), 7.57-7.35 (m, 6H), 6.89 (t, J = 18 Hz, 2H), 3.98 (t, J = 6.8 Hz, 2H), 3.41 (d, J = 6 Hz, 2H), 2.67 (bs, 1H), 2.14-2.03 (m, 2H). 13C NMR (CDCl3, 100 MHz): δ 168.2, 156.7, 148.7, 142.6, 138.8, 137.0, 130.3, 130.1, 129.8, 129.2, 129.0, 127.5, 127.4, 125.0, 123.4, 119.0, 116.6, 44.7, 37.3, 31.0. ESI-MS: [m/z+H]: 357.
N-(3-(1H-imidazol-1-yl)propyl)quinoline-4-carboxamide, SP163
The crude compound was purified by column chromatography in 10% methanol/dichloromethane solvent system to give SP163 as a clear oil (0.30 g, 39%). 1H NMR (CDCl3, 400 MHz): δ 8.71 (d, J = 4.4 Hz, 1H), 8.47 (t, J = 6 Hz, 1H), 8.10 (d, J = 8.4 Hz, 1H), 8.01 (d, J = 8.4 Hz, 1H), 7.67 (t, J = 8 Hz, 1H), 7.52 (t, J = 8 Hz, 1H), 7.27 (s, 1H), 6.88 (s, 1H), 6.81 (s, 1H), 3.96 (t, J = 6.8 Hz, 2H), 3.38 (d, J = 6 Hz, 2H), 2.04 (t, J = 6.8 Hz, 2H). 13C NMR (CDCl3, 100 MHz): δ 167.9, 149.4, 148.2, 141.8, 136.8, 129.9, 129.4, 128.9, 127.5, 125.1, 124.4, 118.9, 118.5, 44.4, 36.9, 30.7. ESI-MS: [m/z+H]: 281.
N-(3-(1H-imidazol-1-yl)propyl)-2-(4-chlorophenyl)quinoline-4-carboxamide, SP168
The crude compound was purified by column chromatography in 5–10% methanol/dichloromethane solvent system to give SP168 as an yellow oil (0.68 g, 73%). 1H NMR (CDCl3, 400 MHz): δ 8.08 (d, J = 9.2 Hz, 2H), 8.00 (d, J = 8.8 Hz, 2H), 7.73 (s, 1H), 7.70 (t, J = 8.4 Hz, 1H), 7.62 (t, J = 5.6 Hz, 1H), 7.50 (t, J = 8 Hz, 1H), 7.43-7.40 (m, 2H), 6.90 (s, 2H), 4.00 (t, J = 6.8 Hz, 2H), 3.42 (dd, J = 6.4 Hz, 3H), 2.08 (d, J = 6.4 Hz, 2H). 13C NMR (CDCl3, 100 MHz): δ 167.9, 155.1, 148.5, 142.5, 137.0, 135.9, 130.2, 129.9, 129.0, 128.8, 128.6, 127.4, 124.8, 123.3, 116.1, 44.7, 37.2, 30.8. ESI-MS: [m/z+H]: 391.
N-(3-(1H-imidazol-1-yl)propyl)-2-(4-fluorophenyl)quinoline-4-carboxamide, SP170
The crude compound was purified by column chromatography in 5–10% methanol/dichloromethane solvent system to give SP170 as a clear oil (0.88 g, 86%). 1H NMR (CDCl3, 400 MHz): δ 8.05-7.99 (m, 4H), 7.66 (t, J = 7.2 Hz, 2H), 7.45 (t, J = 6.8 Hz, 1H), 7.22 (s, 1H), 7.10 (t, J = 8.4 Hz, 2H), 6.83 (s, 1H), 6.79 (s, 1H), 3.89 (t, J = 6.8 Hz, 2H), 3.31 (dd, J = 6.8 Hz, 2H), 2.01-1.95 (m, 2H). 13C NMR (CDCl3, 100 MHz): δ 168.0, 165.1, 162.6, 155.3, 148.4, 142.5, 136.8, 134.7, 130.1, 129.8, 129.2, 129.0, 127.2, 124.8, 123.1, 118.9, 116.2, 115.8, 115.6, 44.5, 37.0, 30.7. ESI-MS: [m/z+H]: 375.
N-(3-(1H-imidazol-1-yl)propyl)-2-([1,1′-biphenyl]-4-yl)quinoline-4-carboxamide, SP171
The crude compound was purified by column chromatography in 0–5% methanol/dichloromethane solvent system to give SP171 as a clear oil (0.80 g, 67%). 1H NMR (CDCl3, 400 MHz): δ 8.14 (d, J = 8.4 Hz, 2H), 8.11-8.06 (m, 2H), 7.81 (s, 1H), 7.71-7.63 (m, 4H), 7.62 (d, J = 7.2 Hz, 2H), 7.50-7.43 (m, 3H), 7.37 (d, J = 9.6 Hz, 2H), 6.87 (s, 2H), 3.96 (t, J = 6.8 Hz, 2H), 3.39 (dd, J = 6.4 Hz, 2H), 2.06 (t, J = 6.8 Hz, 2H). 13C NMR (CDCl3, 100 MHz): δ 168.1, 156.0, 148.6, 142.5, 142.4, 140.1, 137.4, 136.9, 130.1, 129.9, 128.9, 128.9, 127.8, 127.5, 127.2, 127.0, 124.9, 123.3, 118.9, 116.5, 44.7, 37.2, 30.8. ESI-MS: [m/z+H]: 433.
N-(3-(1H-imidazol-1-yl)propyl)-2-(4-hydroxyphenyl)quinoline-4-carboxamide, SP172
The crude compound was purified by column chromatography in 10% methanol/dichloromethane solvent system to give SP172 as a pale yellow oil (0.68 g, 66%). 1H NMR (MeOD, 400 MHz): δ 8.08 (t, J = 9.2 Hz, 2H), 8.03 (d, J = 9.2 Hz, 2H), 7.91 (s, 1H), 7.76-7.71 (m, 2H), 7.68 (s, 1H), 7.55 (t, J = 6.8 Hz, 1H), 7.19 (s, 1H), 7.04 (s, 2H), 6.98 (s, 1H), 6.93 (d, J = 7.2 Hz, 2H), 4.16 (t, J = 6.8 Hz, 2H), 3.47 (t, J = 6.8 Hz, 2H), 2.15 (t, J = 6.8 Hz, 2H). 13C NMR (MeOD, 100 MHz): δ 168.9, 159.3, 157.1, 148.2, 142.8, 137.1, 134.8, 129.9, 129.8, 128.8, 128.6, 127.6, 126.4, 124.7, 122.9, 121.1, 119.2, 116.4, 115.3, 44.3, 36.7, 30.4. ESI-MS: [m/z+H]: 373.
Footnotes
Conflict of interest
The authors declare the existence of a patent filed by Columbia University on non-oxime reactivators and activators.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Katz FS, Pecic S, Tran TH, Trakht I, Schneider L, Zhu Z, et al. Discovery of New Classes of Compounds that Reactivate Acetylcholinesterase Inhibited by Organophosphates. Chembiochem. 2015;16(15):2205–15. doi: 10.1002/cbic.201500348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suzuki T, Morita H, Ono K, Maekawa K, Nagai R, Yazaki Y. Sarin poisoning in Tokyo subway. Lancet. 1995;345(8955):980. [PubMed] [Google Scholar]
- 3.Vogel L. WHO releases guidelines for treating chemical warfare victims after possible Syria attacks. Cmaj. 2013;185(14):E665. doi: 10.1503/cmaj.109-4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eddleston M, Buckley NA, Eyer P, Dawson AH. Management of acute organophosphorus pesticide poisoning. Lancet. 2008;371(9612):597–607. doi: 10.1016/S0140-6736(07)61202-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mercey G, Verdelet T, Renou J, Kliachyna M, Baati R, Nachon F, et al. Reactivators of acetylcholinesterase inhibited by organophosphorus nerve agents. Acc Chem Res. 2012;45(5):756–66. doi: 10.1021/ar2002864. [DOI] [PubMed] [Google Scholar]
- 6.Carletti E, Colletier JP, Dupeux F, Trovaslet M, Masson P, Nachon F. Structural evidence that human acetylcholinesterase inhibited by tabun ages through O-dealkylation. J Med Chem. 2010;53(10):4002–8. doi: 10.1021/jm901853b. [DOI] [PubMed] [Google Scholar]
- 7.Trovaslet-Leroy M, Musilova L, Renault F, Brazzolotto X, Misik J, Novotny L, et al. Organophosphate hydrolases as catalytic bioscavengers of organophosphorus nerve agents. Toxicol Lett. 2011;206(1):14–23. doi: 10.1016/j.toxlet.2011.05.1041. [DOI] [PubMed] [Google Scholar]
- 8.Sirin GS, Zhou Y, Lior-Hoffmann L, Wang S, Zhang Y. Aging mechanism of soman inhibited acetylcholinesterase. J Phys Chem B. 2012;116(40):12199–207. doi: 10.1021/jp307790v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peter JV, Moran JL, Graham PL. Advances in the management of organophosphate poisoning. Expert Opin Pharmacother. 2007;8(10):1451–64. doi: 10.1517/14656566.8.10.1451. [DOI] [PubMed] [Google Scholar]
- 10.Wilson IB. Acetylcholinesterase. XI. Reversibility of tetraethyl pyrophosphate. J Biol Chem. 1951;190(1):111–7. [PubMed] [Google Scholar]
- 11.Wilson IB, Ginsburg B. A powerful reactivator of alkylphosphate-inhibited acetylcholinesterase. Biochim Biophys Acta. 1955;18(1):168–70. doi: 10.1016/0006-3002(55)90040-8. [DOI] [PubMed] [Google Scholar]
- 12.Lorke DE, Kalasz H, Petroianu GA, Tekes K. Entry of oximes into the brain: a review. Curr Med Chem. 2008;15(8):743–53. doi: 10.2174/092986708783955563. [DOI] [PubMed] [Google Scholar]
- 13.Okuno S, Sakurada K, Ohta H, Ikegaya H, Kazui Y, Akutsu T, et al. Blood-brain barrier penetration of novel pyridinealdoxime methiodide (PAM)-type oximes examined by brain microdialysis with LC-MS/MS. Toxicol Appl Pharmacol. 2008;227(1):8–15. doi: 10.1016/j.taap.2007.09.021. [DOI] [PubMed] [Google Scholar]
- 14.Read RW, Riches JR, Stevens JA, Stubbs SJ, Black RM. Biomarkers of organophosphorus nerve agent exposure: comparison of phosphylated butyrylcholinesterase and phosphylated albumin after oxime therapy. Arch Toxicol. 2010;84(1):25–36. doi: 10.1007/s00204-009-0473-4. [DOI] [PubMed] [Google Scholar]
- 15.Demar JC, Clarkson ED, Ratcliffe RH, Campbell AJ, Thangavelu SG, Herdman CA, et al. Pro-2-PAM therapy for central and peripheral cholinesterases. Chem Biol Interact. 2010;187(1–3):191–8. doi: 10.1016/j.cbi.2010.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shek E, Higuchi T, Bodor N. Improved delivery through biological membranes. 3. Delivery of N-methylpyridinium-2-carbaldoxime chloride through the blood-brain barrier in its dihydropyridine pro-drug form. J Med Chem. 1976;19(1):113–7. doi: 10.1021/jm00223a019. [DOI] [PubMed] [Google Scholar]
- 17.Bodor N, Shek E, Higuchi T. Improved delivery through biological membranes. 1. Synthesis and properties of 1-methyl-1,6-dihydropyridine-2-carbaldoxime, a pro-drug of N-methylpyridinium-2-carbaldoxime chloride. J Med Chem. 1976;19(1):102–7. doi: 10.1021/jm00223a017. [DOI] [PubMed] [Google Scholar]
- 18.Antonijevic B, Stojiljkovic MP. Unequal efficacy of pyridinium oximes in acute organophosphate poisoning. Clin Med Res. 2007;5(1):71–82. doi: 10.3121/cmr.2007.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanson B, Nachon F, Colletier JP, Froment MT, Toker L, Greenblatt HM, et al. Crystallographic snapshots of nonaged and aged conjugates of soman with acetylcholinesterase, and of a ternary complex of the aged conjugate with pralidoxime. J Med Chem. 2009;52(23):7593–603. doi: 10.1021/jm900433t. [DOI] [PubMed] [Google Scholar]
- 20.Musilek K, Dolezal M, Gunn-Moore F, Kuca K. Design, evaluation and structure-activity relationship studies of the AChE reactivators against organophosphorus pesticides. Med Res Rev. 2011;31(4):548–75. doi: 10.1002/med.20192. [DOI] [PubMed] [Google Scholar]
- 21.Jokanovic M. Structure-activity relationship and efficacy of pyridinium oximes in the treatment of poisoning with organophosphorus compounds: a review of recent data. Curr Top Med Chem. 2012;12(16):1775–89. [PubMed] [Google Scholar]
- 22.Worek F, Wille T, Koller M, Thiermann H. Reactivation kinetics of a series of related bispyridinium oximes with organophosphate-inhibited human acetylcholinesterase--Structure-activity relationships. Biochem Pharmacol. 2012;83(12):1700–6. doi: 10.1016/j.bcp.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 23.Kalisiak J, Ralph EC, Cashman JR. Nonquaternary reactivators for organophosphate-inhibited cholinesterases. J Med Chem. 2012;55(1):465–74. doi: 10.1021/jm201364d. [DOI] [PubMed] [Google Scholar]
- 24.Milan J. Structure-Activity Relationship and Efficacy of Pyridinium Oximes in the Treatment of Poisoning with Organophosphorus Compounds: A Review of Recent Data. Current Topics in Medicinal Chemistry. 2012;12(16):1775–89. [PubMed] [Google Scholar]
- 25.Choudhary MI, Reitz AB. Frontiers in Medicinal Chemistry. Vol. 8. Bentham Science Publishers; 2016. [Google Scholar]
- 26.Ellman GL, Courtney KD, Andres V, Jr, Feather-Stone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 27.Tingle MD, Jewell H, Maggs JL, O’Neill PM, Park BK. The bioactivation of amodiaquine by human polymorphonuclear leucocytes in vitro: chemical mechanisms and the effects of fluorine substitution. Biochem Pharmacol. 1995;50(7):1113–9. doi: 10.1016/0006-2952(95)00236-s. [DOI] [PubMed] [Google Scholar]
- 28.O’Neill PM, Mukhtar A, Stocks PA, Randle LE, Hindley S, Ward SA, et al. Isoquine and related amodiaquine analogues: a new generation of improved 4-aminoquinoline antimalarials. J Med Chem. 2003;46(23):4933–45. doi: 10.1021/jm030796n. [DOI] [PubMed] [Google Scholar]
- 29.Pecic S, McAnuff MA, Harding WW. Nantenine as an acetylcholinesterase inhibitor: SAR, enzyme kinetics and molecular modeling investigations. J Enzyme Inhib Med Chem. 2011;26(1):46–55. doi: 10.3109/14756361003671078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sugimoto H, Iimura Y, Yamanishi Y, Yamatsu K. Synthesis and structure-activity relationships of acetylcholinesterase inhibitors: 1-benzyl-4-[(5,6-dimethoxy-1-oxoindan-2-yl)methyl]piperidine hydrochloride and related compounds. J Med Chem. 1995;38(24):4821–9. doi: 10.1021/jm00024a009. [DOI] [PubMed] [Google Scholar]
- 31.Muller TC, Rocha JB, Morsch VM, Neis RT, Schetinger MR. Antidepressants inhibit human acetylcholinesterase and butyrylcholinesterase activity. Biochim Biophys Acta. 2002;1587(1):92–8. doi: 10.1016/s0925-4439(02)00071-6. [DOI] [PubMed] [Google Scholar]
- 32.Ahmed M, Batista J, Rocha T, Mazzanti CM, Hassan W, Morsch VM, et al. Comparative study of the inhibitory effect of antidepressants on cholinesterase activity in Bungarus sindanus (krait) venom, human serum and rat striatum. J Enzyme Inhib Med Chem. 2008;23(6):912–7. doi: 10.1080/14756360701809977. [DOI] [PubMed] [Google Scholar]
- 33.Nunes-Tavares N, Nery da Matta A, Batista e Silva CM, Araujo GM, Louro SR, Hasson-Voloch A. Inhibition of acetylcholinesterase from Electrophorus electricus (L.) by tricyclic antidepressants. Int J Biochem Cell Biol. 2002;34(9):1071–9. doi: 10.1016/s1357-2725(02)00027-4. [DOI] [PubMed] [Google Scholar]
- 34.Chaudhary SC, Singh K, Sawlani KK, Jain N, Vaish AK, Atam V, et al. Prognostic significance of estimation of pseudocholinesterase activity and role of pralidoxime therapy in organophosphorous poisoning. Toxicol Int. 2013;20(3):214–7. doi: 10.4103/0971-6580.121669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sumathi ME, Kumar SH, Shashidhar KN, Takkalaki N. Prognostic significance of various biochemical parameters in acute organophosphorus poisoning. Toxicol Int. 2014;21(2):167–71. doi: 10.4103/0971-6580.139800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neves PAAdA, Silva EN, Beirão PSL. Microcalorimetric Study of Acetylcholine and Acetylthiocholine Hydrolysis by Acetylcholinesterase. Advances in Enzyme Research. 2017;05(01):12. [Google Scholar]
- 37.Reiner E, Sinko G, Skrinjaric-Spoljar M, Simeon-Rudolf V. Comparison of protocols for measuring activities of human blood cholinesterases by the Ellman method. Arh Hig Rada Toksikol. 2000;51(1):13–8. [PubMed] [Google Scholar]
- 38.Masson P, Lockridge O. Butyrylcholinesterase for protection from organophosphorus poisons: catalytic complexities and hysteretic behavior. Arch Biochem Biophys. 2010;494(2):107–20. doi: 10.1016/j.abb.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee JC, Harpst JA. Purification and properties of butyrylcholinesterase from horse serum. Arch Biochem Biophys. 1971;145(1):55–63. doi: 10.1016/0003-9861(71)90009-9. [DOI] [PubMed] [Google Scholar]
- 40.Li B, Duysen EG, Saunders TL, Lockridge O. Production of the butyrylcholinesterase knockout mouse. J Mol Neurosci. 2006;30(1–2):193–5. doi: 10.1385/JMN:30:1:193. [DOI] [PubMed] [Google Scholar]
- 41.Broomfield CA, Maxwell DM, Solana RP, Castro CA, Finger AV, Lenz DE. Protection by butyrylcholinesterase against organophosphorus poisoning in nonhuman primates. J Pharmacol Exp Ther. 1991;259(2):633–8. [PubMed] [Google Scholar]
- 42.Lenz DE, Maxwell DM, Koplovitz I, Clark CR, Capacio BR, Cerasoli DM, et al. Protection against soman or VX poisoning by human butyrylcholinesterase in guinea pigs and cynomolgus monkeys. Chem Biol Interact. 2005;157–158:205–10. doi: 10.1016/j.cbi.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 43.Lenz DE, Clarkson ED, Schulz SM, Cerasoli DM. Butyrylcholinesterase as a therapeutic drug for protection against percutaneous VX. Chem Biol Interact. 2010;187(1–3):249–52. doi: 10.1016/j.cbi.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 44.Mumford H, Docx CJ, Price ME, Green AC, Tattersall JE, Armstrong SJ. Human plasma-derived BuChE as a stoichiometric bioscavenger for treatment of nerve agent poisoning. Chem Biol Interact. 2013;203(1):160–6. doi: 10.1016/j.cbi.2012.08.018. [DOI] [PubMed] [Google Scholar]
- 45.Lockridge O, Blong RM, Masson P, Froment MT, Millard CB, Broomfield CA. A single amino acid substitution, Gly117His, confers phosphotriesterase (organophosphorus acid anhydride hydrolase) activity on human butyrylcholinesterase. Biochemistry. 1997;36(4):786–95. doi: 10.1021/bi961412g. [DOI] [PubMed] [Google Scholar]
- 46.Millard CB, Lockridge O, Broomfield CA. Design and expression of organophosphorus acid anhydride hydrolase activity in human butyrylcholinesterase. Biochemistry. 1995;34(49):15925–33. doi: 10.1021/bi00049a007. [DOI] [PubMed] [Google Scholar]
- 47.Millard CB, Lockridge O, Broomfield CA. Organophosphorus acid anhydride hydrolase activity in human butyrylcholinesterase: synergy results in a somanase. Biochemistry. 1998;37(1):237–47. doi: 10.1021/bi972057c. [DOI] [PubMed] [Google Scholar]
- 48.Bhattacharjee AK, Pomponio JW, Evans SA, Pervitsky D, Gordon RK. Discovery of subtype selective muscarinic receptor antagonists as alternatives to atropine using in silico pharmacophore modeling and virtual screening methods. Bioorg Med Chem. 2013;21(9):2651–62. doi: 10.1016/j.bmc.2013.01.072. [DOI] [PubMed] [Google Scholar]
- 49.Duysen EG, Cashman JR, Schopfer LM, Nachon F, Masson P, Lockridge O. Differential sensitivity of plasma carboxylesterase-null mice to parathion, chlorpyrifos and chlorpyrifos oxon, but not to diazinon, dichlorvos, diisopropylfluorophosphate, cresyl saligenin phosphate, cyclosarin thiocholine, tabun thiocholine, and carbofuran. Chem Biol Interact. 2012;195(3):189–98. doi: 10.1016/j.cbi.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Darvesh S, McDonald RS, Penwell A, Conrad S, Darvesh KV, Mataija D, et al. Structure-activity relationships for inhibition of human cholinesterases by alkyl amide phenothiazine derivatives. Bioorg Med Chem. 2005;13(1):211–22. doi: 10.1016/j.bmc.2004.09.059. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Scheme 3. Reagents and condition: CDI, THF, rt, 12 h, reflux, 5h