

Abstract
Activation of phosphoinositide 3-kinase (PI3K) signaling is frequently observed in triple-negative breast cancer (TNBC), yet PI3K inhibitors have shown limited clinical activity. To investigate intrinsic and adaptive mechanisms of resistance, we analyzed a panel of patient-derived xenograft models of TNBC with varying responsiveness to buparlisib, a pan-PI3K inhibitor. In a subset of patient-derived xenografts, resistance was associated with incomplete inhibition of PI3K signaling and upregulated MAPK/MEK signaling in response to buparlisib. Outlier phosphoproteome and kinome analyses identified novel candidates functionally important to buparlisib resistance, including NEK9 and MAP2K4. Knockdown of NEK9 or MAP2K4 reduced both baseline and feedback MAPK/MEK signaling and showed synthetic lethality with buparlisib in vitro. A complex in/del frameshift in PIK3CA decreased sensitivity to buparlisib via NEK9/MAP2K4-dependent mechanisms. In summary, our study supports a role for NEK9 and MAP2K4 in mediating buparlisib resistance and demonstrates the value of unbiased omic analyses in uncovering resistance mechanisms to targeted therapy.
INTRODUCTION
Triple negative breast cancer (TNBC) is an aggressive tumor subtype representing approximately 15% of breast cancer diagnoses. Due to the lack of hormone receptor expression and HER2/neu gene amplification, patients with TNBC do not benefit from ER or HER2 targeted therapies. In addition, resistance to chemotherapy is common and is associated with a poor prognosis. The median survival of patients with relapsed TNBC is in the range of 1-2 years [1]. Aberrant activation of PI3K signaling is frequently observed in TNBC as a result of genetic or epigenetic alterations, including PIK3CA mutation, PTEN mutation/loss and INPP4B loss, together observed in up to 35% of TNBC [2, 3], which theoretically should generate sensitivity to PI3K inhibitors [4]. However, single agent PI3K pathway inhibition has shown limited efficacy in TNBC [2, 3, 5]. To further therapeutic progress, response predictors and resistance mechanisms need to be identified. Unfortunately, biomarker studies on clinical trial samples are typically limited due to the difficulty in obtaining sufficient tumor material and the inability to perform serial biopsies [6, 7]. Patient-derived xenograft (PDX) models provide a useful preclinical platform for this purpose [8, 9], with demonstrated fidelity in capturing the mutational profiles, drug responsiveness, and molecular heterogeneity typical of human breast cancer [10–13].
The pan-PI3K inhibitor buparlisib (NVP-BKM120) targets all of the class I PI3-kinase isoforms (p110α/β/δ/γ) [14] and is in clinical trials for a variety of cancer types [15, 16], including advanced TNBC (NCT01629615). To date, there are no clinically validated biomarkers predictive of response to PI3K inhibitors. Previous studies have largely focused on genomic alterations, such as PIK3CA mutations, with inconsistent results [16]. Since genetic changes do not necessarily reflect protein or pathway activity [6], we hypothesized that investigations at the levels of the proteome and the phosphorylation status of proteins, a key post-translational modification central to cellular signal transduction [7], could potentially identify aberrant signaling pathways or proteins associated with treatment response or resistance [17–19].
In this study, we exposed TNBC PDX tumors to buparlisib or vehicle treatment and performed multi-omic characterization of tumor samples with the goal of identifying cellular signaling events associated with sensitivity and resistance to treatment. Each sample was analyzed using liquid chromatography-high resolution tandem mass spectrometry (LC-MS/MS) to generate deep, global proteome and phosphoproteome data that was integrated with information from kinase and transcriptome profiling. This rich data set was then correlated with phenotypic responses to provide new insights into intrinsic and adaptive resistance mechanisms to PI3K inhibition.
MATERIALS AND METHODS
Treatments of patient-derived triple negative breast cancer xenograft models
A schematic workflow of the study is presented in Fig. 1A. Six TNBC PDX models with moderate to high PI3K pathway activity (Supplementary Fig. 1) were selected from the Washington University Human in Mouse (WHIM) PDX collection [10] (WHIMs 2, 4, 6, 12, 21 and 30). Supplementary Table 1 summarizes the characteristics of the patients’ tumors from which the models were derived. These models have been shown to be genetically stable over multiple xenograft passages and retain the genetic characteristics of the original tumor [10, 11]. The responses of the 6 PDX models to buparlisib were assessed by treating tumor-bearing mice (n = 5-9 mice per treatment group, 2 tumors per mouse) with either vehicle or buparlisib, 30mg/kg by oral gavage on days 1-5 each week for 24-32 days. Tumor growth rates, calculated using a method adapted from Hather et al. [20] (Fig. 1B), showed a spectrum of responsiveness, with the most sensitive being WHIM4 and WHIM30, and the most resistant being WHIM2 and WHIM12 (Fig. 1C). Such variable response made this an ideal sample set for the investigation of potential resistance mechanisms to PI3K inhibition.
Figure 1. Long-term treatment of PDX models shows varying responses to buparlisib.
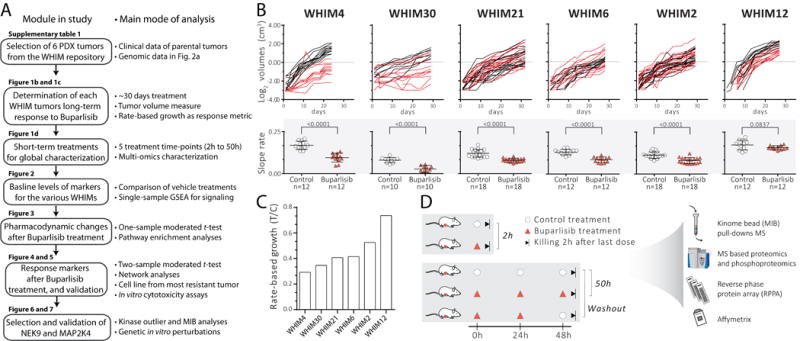
A, workflow of the present study. GSEA = gene set enrichment analysis. B, growth curves of buparlisib-treated and control-treated tumors. Red lines indicate buparlisib treatment and black lines represents control- (vehicle) treated animals. Lower panel (shaded grey) shows individual slopes (growth rate) and the comparison between buparlisib treatment and controls (bars are means and whiskers are standard deviations). C, resulting rate-based growth (treatment over control; T/C) indicating response to buparlisib for each PDX tumor/WHIM model, plotted from the most sensitive (WHIM4) to most resistant (WHIM12) PDX model. D, global characterization study design: 5 different treatment groups per tumor model. Tumors were collected at hour 2 or 50 (buparlisib/vehicle administered at hours 0, 24 and 48). A total of 30 samples for the 6 PDX models were analyzed with the -omic techniques listed to the right side.
To assess global changes at early time points, early passage PDX models were propagated in the 4th mammary fat pad of female NU/J homozygous mice (Jackson lab, Cat No. 2019) after clearance of mouse mammary tissues, as previously described [21]. Treatment began when tumors reached approximately 0.5 cm in diameter. Analyses examined changes induced 2 hours after either a single dose (“2 hours”) or 3 consecutive daily oral gavage doses (“50 hours”) of buparlisib (30mg/kg) or vehicle. The earlier, 2-hour time point was selected to capture acute signaling changes, while the later 50-hour time point was designed to allow sufficient duration of therapy to observe effects on cell proliferation and apoptosis markers (~2 days). A 5th treatment arm (“washout”), consisting of two consecutive daily doses of buparlisib on days 1 and 2, followed by vehicle on day 3, was also included to assess the stability of signaling changes after buparlisib withdrawal (Fig. 1D). Details of treatment response measurements and calculations can be found in extended materials and methods.
mRNA Gene Expression Data
RNA was extracted from cryopulverized PDX tumor tissue via homogenization in Trizol (Thermo Scientific, 15596018) followed by chloroform extraction, isopropanol precipitation, 70% ethanol wash, and elution in Milli-Q water. The eluted RNA was then treated with DNase (Qiagen, 79254) and purified using the RNeasy MinElute Cleanup Kit (Qiagen, 74204). Purified total RNA samples were then assayed for quality using a BioRad Experion instrument. Transcriptional profiling of buparlisib-treated PDX models was completed on Agilent Whole Human Gene Expression 4×44 K microarrays (Agilent Technologies, G4112F). Seventy-five nanograms of high-quality total RNA from both a PDX tumor biopsy and a universal reference sample (Stratagene) enriched with equal amounts of RNA from the MCF7 and ME16C cell lines were used to synthesize cDNA. Next, cDNA was amplified via T7 polymerase in vitro transcription with either cy5-CTP (Perkin Elmer, NEL581001) incorporation for the PDX sample or cy3-CTP (Perkin Elmer, NEL580001) incorporation for the reference sample using Agilent Technologies’ Low Input Quick Amp Labeling Kit (1590-2308). Amplified, labeled tumor and reference cRNA (825 ng) were then co-hybridized to the microarray, washed, and dried according to Agilent’s Two-Color Microarray-Based Gene Expression Analysis protocol (version 5.0.1). Processed arrays were scanned with an Agilent Microarray Scanner (G2505B), and probe data were extracted from the scanned image using Agilent’s Feature Extraction software version 9.5.3.1. Transcriptional profiling yielded a total of 30,484 quantified probes across all 6 PDX models and 5 conditions, corresponding to an average of 14,048 genes per tumor. Microarray data is available through the Gene Expression Omnibus (GEO) database, reference number GSE98824.
Reversed phase protein arrays
Reversed phase protein array (RPPA) assays were carried out as described previously [22], with minor modifications. Protein lysates were prepared from PDX tumors with modified tissue protein extraction reagent (TPER; Pierce) and a cocktail of protease and phosphatase inhibitors (Roche Life Science). The lysates were diluted into 0.5 mg/mL of total protein in SDS sample buffer and denatured on the same day. The Aushon 2470 Arrayer (Aushon BioSystems) with a 40-pin (185 μm) configuration was used to spot samples and control lysates onto nitrocellulose-coated slides (Grace Bio-labs) using an array format of 960 lysates/slide (2,880 spots/slide). The slides were processed and probed with a set of 204 antibodies, including several antibodies against total and phosphorylated proteins of the PI3K pathway using an automated slide stainer (Autolink 48, Dako). Each slide was incubated with one specific primary antibody and a negative control slide was incubated with antibody diluent instead of primary antibody. Primary antibody binding was detected using a biotinylated secondary antibody followed by streptavidin-conjugated IRDye680 fluorophore (LI-COR Biosciences). Total protein content of each spotted lysate was assessed by fluorescent staining with Sypro Ruby Protein Blot Stain according to the manufacturer’s instructions (Molecular Probes). All RPPA data is available through the GEO, reference number GSE100567. Details can be found in extended materials and methods.
Global proteome and phosphoproteome
For relative quantification of the global proteome and phosphoproteome by LC-MS/MS, cryo-pulverized PDX tumor samples were lysed at 4°C using an 8M urea-based lysis buffer. Protein concentrations were determined using the Pierce BCA assay. Extracted proteins were reduced with 5 mM dithiothreitol and alkylated with 10 mM iodoacetamide before being digested with LysC and trypsin overnight at room temperature. Both digestion steps were performed at a 1:50 enzyme-to-protein ratio. Resulting peptides were desalted on C18 SepPak columns (Waters, 100mg/1cc), and 666 μg peptide aliquots were dried down using a SpeedVac Concentrator (Savant SC210A). Desalted peptides were then labeled with 6-plex tandem mass tag (TMT6, Thermo Fisher Scientific) reagents according to the manufacturer’s instructions. TMT labeling was quenched and each TMT6-plex was combined so that each plex comprised one PDX model’s 5 treatments and a common internal reference (composed of equal amounts of digested material from all 30 samples). The internal reference was used to link all 6 TMT6-plexes after data acquisition. Each combined TMT6-plex was desalted on a C18 SepPak column (Waters, 500mg/6cc) and fractionated by high-pH reversed phase off-line chromatography into 24 fractions. Of each fraction, 5 % of material was removed and analyzed for its proteome, while the remaining 95 % was concatenated into 12 fractions and enriched for phosphopeptides using immobilized metal affinity chromatography (Supplementary Fig. 2A). In union, 10,401 proteins and 66,300 phosphosites were quantified by LC-MS/MS (Q Exactive, Thermo Fisher; searched using Spectrum Mill, Agilent) across all 30 samples, with an average of 10,027 quantified proteins and 35,412 fully quantified and sequence-localized phosphosites per sample. The abundance of each phosphosite was normalized to its corresponding protein abundance to better represent the actual change in phosphorylation status at a given site. All mass spectra contributing to this study can be downloaded in the original instrument vendor format from: https://cptac-data-portal.georgetown.edu/cptacPublic/ for the study name: Buparlisib treated patient-derived xenograft tumors of triple negative breast cancer. Details can be found in extended materials and methods.
Single-shot LC-MS/MS analysis of vehicle treated WHIM tumors and the WHIM12-derived cell line
WHIM12-derived cells were cultivated as described in Materials and Methods: In vitro validations; WHIM3 and WHIM12 patient-derived xenograft cell lines. A snap-frozen cell line pellet was lysed using the same lysis protocol as used for the WHIM tumor samples. One microgram of desalted peptides for all 2-hour vehicle-treated WHIM tumors (as well as the 50 hours vehicle treatment for WHIM12), and 1 μg from the WHIM12-derived cell line were analyzed using LC-MS/MS over a 110-minute method (Q Exactive, Thermo Fisher). Data-dependent acquisition was performed using Xcalibur QExactive v2.1. In total, 5,990 proteins were quantified by LC-MS/MS (searched using Max Quant [23]) across all 8 samples. Details can be found in extended materials and methods.
Detection of mRNA variants and splice isoforms by mass spectrometry
We mapped peptides that spanned splice junctions, had altered coding sequence due to a frameshift, or had a new protein C-terminus resulting from introduction of a novel stop codon. Searchable WHIM-specific sequence databases were created with the QUILTS software (quilts.fenyolab.org) [17, 24] using RNA-seq data for all 6 WHIM models from a previous publication [12]. Briefly, QUILTS takes junctions from RNA-seq data and enumerates all the resulting changes to protein sequences to create tumor specific protein sequence databases. The human RefSeq release 60 protein database (version 20130727) was used as a reference for the hg19 proteome and genome. FDR thresholding was kept stringent during these searches to decrease the risk of identifying false positive peptide spectrum matches.
Mass spectrometry-based kinome profiling
For a focused view on the kinome, we lysed an aliquot of the cryo-pulverized tumor tissue from all 30 samples using multiplexed kinase inhibitor bead (MIB [25]) lysis buffer, and extracted proteins through sonication (Covaris S220X). The protein content was determined by the Advanced Protein Assay (Cytoskeleton). MIB slurry was prepared to contain the following: 11.7 % of Shokat, PP58, Purvalanol B, UNC-21474 and 17.7 % VI-16832, CTx-0294885 and buparlisib. The aliquots of slurry and tissue lysate were tumbled overnight on a Mini Lab roller. Beads were washed and peptides were eluted using MIB elution buffer containing 2 % SDS, 100 mM DTT and 100 mM Tris, pH 7.6. The samples were digested using the enhanced filter-aided sample preparation method with the enzyme trypsin. Resulting peptides were extracted on solid phase and concentrations were measured using the Pierce Quantitative Fluorometric Peptide Assay Kit. Peptides were labeled with TMT6 reagents according to the manufacturer’s instructions. Each TMT6-plex comprised one PDX model’s 5 treatments and a common internal reference (composed of equal amounts of digested material from all 30 samples) to link all 6 TMT6-plexes (Supplementary Fig. 2B). Using the MIB approach coupled to mass spectrometry (Q-Exactive™ Plus Hybrid Quadrupole-Orbitrap™ Plus) we quantified 348 total kinases across all samples and 257 kinases on average per sample. All mass spectra contributing to this study can be downloaded in the original instrument vendor format from: https://cptac-data-portal.georgetown.edu/cptacPublic/ for the study name: Buparlisib treated patient-derived xenograft tumors of triple negative breast cancer. Details can be found in extended materials and methods.
Data availability
To support data availability and research transparency, we provide an Omics Data Browser. This tool provides researchers with the means to interrogate the data pertaining to this study with their genes of interest (provided in capitals, e.g. PIK3CA) and to retrieve graphical representations of the quantitative data, similar to the heatmap shown in Fig. 2. The underlying data in this Omics Data Browser comprises the mRNA Gene Expression, RPPA, global proteome and phosphoproteome and the mass spectrometry based kinome profiling data. The accessible data includes all WHIM tumors and all treatments, with global LC-MS/MS-based phosphoproteomic data normalized to respective protein levels. The Omics Data Browser can be retrieved at http://prot-shiny-vm.broadinstitute.org:3838/BKM120/ (with the password [BKM120viewer!]).
Figure 2. Genomic alterations and expression levels of key PI3K markers and signaling activities by different omic analysis of vehicle-treated PDX tumors.

A, copy number variation (CNV) and mutational status of PI3K pathway components from the whole exome sequencing data [12]. B, standardized expression levels of selected markers compared among the transcriptome (mRNA), global proteome (MS prot), global phosphoproteome (MS amino acid and site #), RPPA protein (RPPA prot), RPPA phosphosites (RPPA amino acid and site #) and kinome pulldown data (MIB). Selected PI3K markers show varying biology between different PDX tumors. Expression levels are z-scored. Both vehicle treatments (2 hours and 50 hours) are shown for each PDX tumor. C, a single sample gene set enrichment analysis for individual vehicle-treated tumors across all platforms shows enrichment for a selected set of gene-sets/pathways. Both vehicle treatments (2 hours and 50 hours) are shown for each PDX model. Good correlation for each PI3K marker and gene-set/pathway was observed between -omic types, with striking differences between the various PDX animals.
Statistical tests
Only markers identified as human (Homo sapiens) were considered for analyses to better represent changes in the tumor as opposed to the stroma (Mus musculus). Markers were filtered to include those present in ≥ 50% of the data (i.e. in 15 or more of the 30 samples) in order to reduce noise and increase power of analyses. Details on one- and two-sample moderated t-tests, PEST motifs identifier, GeNets pathway enrichment analyses, Kinase outlier analysis, Pearson correlation of mRNA and protein levels, gene-set enrichment analysis, and principal component analyses can be found in extended materials and methods.
In vitro validations
Chemicals
Buparlisib (Catalog no. CT-BKM120) was purchased from Chemietek. Afuresertib (Catalog no. S7521), AZD2014 (Catalog no.S2783), omipalisib (Catalog no. S2658), trametinib (Catalog no. S2673), MK2206 (Catalog no. S1078), and MK8669 (Catalog no. S1022) were purchased from Selleckchem. All drugs were prepared in stock solution of 10 mM in dimethyl sulfoxide (DMSO; Sigma) for in vitro experiments.
In vitro clonogenic assay
For clonogenic assay, cells were seeded at a density of 1,000 cells per well in 6-well plates in RPMI-1640 medium with 10 % FBS for 24 hours. Cells were then treated with buparlisib at 1 uM concentration or DMSO for 6 days. The colonies were fixed with 4 % paraformaldehyde and stained with 0.05 % crystal violet in methanol.
In vitro cytotoxic assay
For cytotoxic assay, WHIM12 cells were seeded at a density of 3,000 cells per well in 96-well plates in HuMEC medium with 5 % FBS for 24 hours. Cells were treated with different drugs either alone or in combination with buparlisib with concentrations ranging from 0-1 uM. Each experiment was repeated twice in triplicate. Synergistic or additive activity between different inhibitors and buparlisib was determined by calculating combination index values using Compu Syn software.
WHIM3 and WHIM12 patient-derived xenograft cell lines
The WHIM3 and WHIM12 models were previously characterized and generated by engrafting the primary breast tumor of a patient with metastatic TNBC into the humanized mammary fat pad of NOD/SCID mice [10, 26]. Cell lines were derived from the corresponding PDX models. WHIM3 cells were grown in RPMI-1640 medium supplemented with 10 % FBS, 1 % glutamine, and 1 % penicillin-streptomycin, while WHIM12 cells were grown in HuMEC medium (Gibco) supplemented with 5 % FBS, 1 % glutamine, and 1 % penicillin-streptomycin.
Short-hairpin RNA (shRNA) knockdown
Infection of WHIM12 cells: when growing cells reached approximately 50 % confluency, they were infected with control lentivirus (pLKO. Puro) or pLKO. Puro-expressing PIK3CA, NEK9 and MAP2K4-specific shRNA (Genome Institute, Washington University) constructs. Cells were infected in the presence of 4 μg/mL polybrene. After infection, cells were cultured for 48 hours followed by drug selection with 5 μg/ml puromycin (Invitrogen). Drug-resistant colonies were selected and expanded. NEK9 and MAP2K4 protein expression were validated in stably selected knockdown clones by western blotting.
Construction of PIK3CA Mutant expression construct
The mutant PIK3CA mammalian expression vector containing an in-frame deletion of amino acids V.105-R.108, and a change of amino acid E109 to T105, was constructed by PCR amplification from cDNA of the WHIM12 PDX cell line and sub-cloned into the pCDH1. Puro expression vector using NheI and BamHI sites. All clones were sequence-verified in Washington University sequencing facility. Infection of WHIM3 and MCF10A cells: MCF10A cells were grown in HuMEC medium (Gibco) supplemented with 10 % FBS, 1 % glutamine, and 1 % penicillin-streptomycin. When growing cells reached approximately 50 % confluency, they were infected with control lentivirus (pCDH1. Puro) or pCDH1. Puro-expressing PIK3CA in-frame deletion mutant construct. Cells were infected in the presence of 4 μg/mL polybrene. After infection, cells were cultured for 48 hours followed by drug selection with 5 μg/mL puromycin (Invitrogen). Drug-resistant colonies were selected and expanded.
Antibodies
Primary antibodies were rabbit anti-phospho-AKTS473, rabbit anti-AKT, rabbit anti-phospho-S6S240/244, rabbit anti-S6, rabbit anti-phospho-ERK1/2T202/204, rabbit anti-ERK1/2, rabbit anti-MAP2K4, anti-phospho-MAP2K4S257/T261, rabbit anti-p110α and rabbit anti-p85 (Cell Signaling), mouse anti-survivin, mouse anti-NEK9 and rabbit anti-β-actin (Santa Cruz). Secondary horseradish peroxidase (HRP)-conjugated anti-rabbit and anti-mouse antibodies were from Jackson ImmunoResearch Laboratories.
Western blotting
Cells were seeded in a 6-well plate at a density of 0.5 × 106, adhered overnight, followed by treatment with DMSO (0.1 %) and buparlisib at 1 uM concentration for 24 hours. Cells were harvested after 24 hours and lysed with 50 μL buffer containing 50 mmol/L Tris-HCl, pH 7.5, 150 mmol/L NaCl, 2 mmol/L EDTA, 1 % Triton, 1 mmol/L phenylmethylsulfonylfluoride, and Protease Inhibitor Cocktail (Sigma) for 20 min on ice. Lysates were cleared at 10,000 rpm for 15 min, boiled, separated on 12 % SDS gels, and transferred to a nitrocellulose membrane followed by overnight incubation with primary antibodies against pAKT473, AKT, pS6S240/244, S6, pERK1/2T202/204, ERK1/2, NEK9, MAP2K4, survivin (BRIC5) and β-actin. Protein bands were visualized after 1 hour incubation with HRP-conjugated secondary antibodies and development with ECL (GE Healthcare).
Immunohistochemistry
Immunohistochemistry for pAKT S473 was as described in our previous publication [21].
Co-immunoprecipitation
MCF10A (WT-PIK3CA), MCF10A (H1047R-PIK3CA mutant) and WHIM12 (PIK3CA mutant) were cultured in RPMI-FBS (10 %) and serum-free medium for 24 hours. After 24 hours, cells were lysed in RIPA buffer. Proteins (500 μg) were incubated with 5 μg of anti-p85 (Cell Signaling) antibodies at 4°C overnight, followed by incubation with 50 μl of slurry-Dynaprotein G magnetic beads at 4°C for 4 h with gentle shaking. Beads were collected by magnetic separator and washed three times with PBST. Proteins were eluted with 2× SDS buffer and separated on 8-12 % SDS-PAGE, followed by western blot. Rabbit IgG (5 μg) was used as a negative control.
RESULTS
TNBC PDX models differ in their baseline PI3K signaling
Somatic mutation profiles of the 6 TNBC PDX models by whole exome sequencing done previously [10, 12] demonstrated significant inter-tumor heterogeneity (Fig. 2A). Except for TP53, which is mutated in 5 of the 6 PDX models, the majority of the mutated genes were unique for specific PDX models. Interestingly, WHIM30 carries a BRCA1 p.E1410* deleterious mutation. All remaining WHIM PDX models have wild-type BRCA1. Notable PI3K pathway genomic alterations include a PIK3CA amplification in WHIM4, an INPP4B amplification in WHIM2, an INPP4B p.Q814K mutation in WHIM30, and an AKT3 p.C307Y mutation in WHIM6 (Fig. 2A). In addition, analysis of the sequencing data using the Pindel-C tool [27] uncovered a previously undetected complex in/del frameshift that results in a 4 amino acid deletion (105-108; VGNR) and a change of E109 to T105 in the p85 binding domain of PIK3CA in WHIM12. This mutation was not reported in the previous studies of WHIM12 [10, 12] where PIK3CA was annotated as WT. Those studies focused on PIK3CA hotspot mutations with known recurrence (E545 and H1047 hotspots), missing the more complex mutation presented in this study, which also needed more advanced mutation calling tools [27] to be annotated.
Correlation analyses comparing results from the various molecular characterization techniques showed a moderate to high correlation of markers (Supplementary Fig. 2C), indicating both coherence and complementarity with the multi-omic approach. Using principal component analyses of the vehicle-treated PDX tumors, the 6 WHIM models showed substantial inter-tumor heterogeneity at baseline with respect to their global transcriptional, proteomic and phosphoproteomic profiles (Supplementary Fig. 2D). Specific PI3K pathway components were highly variable, indicating differential PI3K signaling among WHIM models at baseline. PTEN loss at the protein level was noted in WHIM6, 12, 21 and 30, whereas PIK3CA amplification in WHIM4 was associated with overexpression at both RNA and protein levels (Fig. 2B). High inter-tumor heterogeneity was also supported at pathway levels using single sample gene-set enrichment analysis of data from all platforms. A strong enrichment for MAPK signaling in addition to AKT signaling was observed in WHIM12 (Fig. 2C).
EIF4B and PALLD are both known to be involved in PI3K signaling and are among 13 mRNA splice forms identified at the peptide level that are significantly differentially expressed at baseline between the most resistant and sensitive WHIMs (two-sample moderated t-test using vehicle treatments; Supplementary File 1). EIF4B and PALLD both show lower expression in the resistant models compared to the sensitive models at baseline. These 13 differentially regulated splice forms are involved in SIG Regulation of the Actin Cytoskeleton by Rho GTPases, PID E-cadherin Stabilization Pathway, PID Syndecan 1 Pathway, KEGG Leukocyte Transendothelial Migration and KEGG Tight Junction (GeNets, MSigDB C2:CP, Bonferroni adjusted p-value ≤ 0.05).
Buparlisib-induced pharmacodynamic changes were enriched for PI3K pathway modulation across PDX models
We set out to identify pharmacodynamic markers that changed in a common fashion across all 6 PDX models after buparlisib treatment in the global transcriptome, proteome and phosphoproteome. On a global level, regulated markers differed between treatment time points and across platforms (Fig. 3A). In contrast to the transcriptome and proteome, in which few or no significant changes (adjusted p-value ≤ 0.1) were observed until the 50 hours’ time point, the phosphoproteome responded rapidly, with 40 phosphosites significantly changed at 2 hours. Interestingly, the changes in the global phosphoproteome at 2 hours and 50 hours did not completely overlap, and no changes were detected in the washout samples, indicating a highly dynamic and reversible regulation of the phosphoproteome by buparlisib. As expected, significant changes in transcriptome and proteome continued to be observed in the washout samples, indicating their delayed and/or relatively durable responses to buparlisib.
Figure 3. Pharmacodynamic changes after buparlisib treatment show essential differences between the transcriptome, proteome and phosphoproteome.
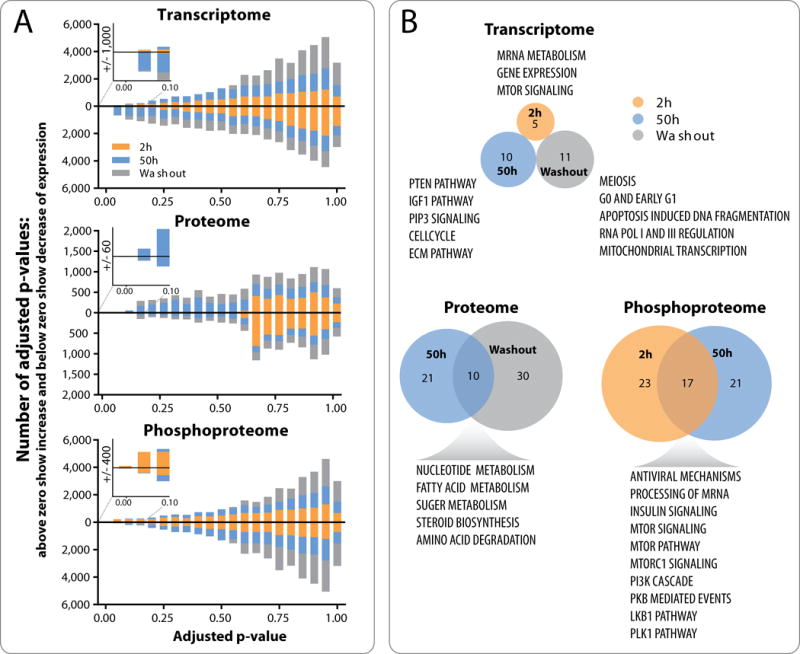
A, one-sample moderated t-tests identify markers that have an average difference from zero across all tumors. Lake plot graphs show stacked histograms of Benjamini-Hochberg adjusted p-values when analyzing samples collected at 2 hours, 50 hours and washout separately. Bars above zero represent markers with increased expression, while bars below zero represent markers with decreased expression. Inserted zooms show number of markers with an adjusted p-value ≤ 0.1 for each platform. B, Pathway enrichment analyses were performed using the GeNets networking tool against the Molecular Signatures Database (C2:CP). Venn diagrams show overlap of significantly enriched pathways (Bonferroni adjusted p-value ≤ 0.05) that are summarized in the text. No significant markers were identified in proteome space for 2 hours buparlisib treatment or in phosphoproteome space for washout treatment and these are therefore not included in the pathway enrichment measurements.
On a pathway level, buparlisib-induced changes in the transcriptome related to PIP3, PTEN, and mTOR signaling and several phases of the cell cycle, as well as to gene expression/transcription. Significant proteins in pathways related to nucleic acid, sugar, fatty acid, and amino acid metabolism as well as to steroid biosynthesis were upregulated by buparlisib treatment across all of the PDX models. In the phosphoproteome, buparlisib induced changes related to PI3K, AKT and mTOR signaling as well as the crosstalking PLK1 (Polo-Like Kinase 1) pathway (Fig. 3B and Supplementary File 2).
We next investigated specific phosphosites that are potential pharmacodynamic markers of buparlisib. T246 in AKT1S1 (AKT1 Substrate 1; Supplementary Fig. 3A) is a known AKT1 substrate [28, 29]. Ten phosphorylation sites including T246 were quantified on AKT1S1 in our global phosphoproteome data. Four of these sites indicated pharmacodynamic regulation after buparlisib treatment, with T246 showing the greatest decrease in phosphorylation, consistent with prior observations [28, 29].
Additional phosphosites that were strongly regulated across all tumors include EIF4EBP1 (Eukaryotic Translation Initiation Factor 4E Binding Protein 1; Supplementary Fig. 3B) and RPS6KB1 (Ribosomal Protein S6 Kinase B1; Supplementary Fig. 3C), both involved in PI3K signaling, as well as BABAM1 (BRISC And BRCA1 A Complex Member 1; Supplementary Fig. 3D) and PKMYT1 (Protein Kinase, Membrane Associated Tyrosine/Threonine 1; Supplementary Fig. 3E), involved in PLK1 signaling and cell cycle regulation. The largest number of pharmacodynamically-regulated phosphosites mapped to the well-known cell cycle-associated protein Ki-67 (Marker of Proliferation Ki-67; Supplementary Fig. 3F). The inhibitory effect of buparlisib on PI3K signaling was validated by RPPA analysis of PI3K pathway markers including pAKT (S473 and T308), pmTOR (S2448) and pRPS6KB1 (T389; Supplementary Fig. 4A) and by immunohistochemistry of pAKT (S473 (Supplementary Fig. 4B). Notably, significant residual pAKT activity and increased MAPK/MEK signaling were observed in the RPPA data set in the most resistant WHIM12 model (Supplementary Fig. 4A).
Though tangential to the main analysis, a novel and interesting finding of the study was that the majority of pharmacodynamically-regulated phosphosites that mapped to Ki-67 fell within, or close to, predicted PEST motifs (rich in proline [P], glutamic acid [E], serine [S], and threonine [T]) of Ki-67 (Supplementary Fig. 5B). These pharmacodynamic changes correlated with Ki-67 protein levels (Supplementary Fig. 5C), linking PEST phosphorylation of Ki-67 with regulation of its protein abundance in response to buparlisib treatment. The entire list of potential pharmacodynamic markers can be found in Supplementary File 3.
PI3K, PLK1 and MAPK/MEK family members distinguish the most buparlisib-sensitive tumors from the most resistant tumors
To identify differential response markers associated with tumor growth, we compared buparlisib-induced changes in the proteome and phosphoproteome of the two most sensitive PDX models (WHIM4 and WHIM30) with those of the two most resistant models (WHIM2 and WHIM12), normalizing all drug-treated samples to their vehicle controls. WHIM6 and WHIM21 were excluded for this analysis since they showed intermediate responses. The significance of differential markers was determined by a two-sample moderated t-test. Combining the 50 hours buparlisib treatment with the washout treatment samples increased the statistical power and prioritized the identification of relatively stable markers. With an adjusted p-value ≤ 0.05, 108 protein markers and 340 phosphosites were differentially expressed between the resistant and the sensitive tumors (Fig. 4A and 4B). These proteins are involved in pathways related to the Complement System, the Endosomal Vacuolar Pathway and Coagulation, as well as the MTA3 Pathway (Supplementary File 5) that has previously been shown to be down-regulated in ER-negative breast tumors [30]. Furthermore, a Connectivity Map (CMAP; https://clue.io/cmap) analysis showed that proteins that are significantly and relatively higher in the sensitive tumors after buparlisib treatment are very similar to the transcriptional changes that are observed in multiple cell lines (from various tissue origins, including breast) after treatment with many PI3K and mTOR inhibitors. In contrast, the same CMAP analysis showed that proteins that are relatively high in the resistant tumors show similar traits to a diverse set of treatments (Supplementary Fig. 6). Pathway enrichment analyses using significantly regulated phosphoproteins (that is, with phosphosites collapsed to their median expression for each corresponding phosphoprotein) showed a strong connection to PI3K/AKT/mTOR signaling (Supplementary File 5). A heatmap based on a curated list of PI3K-associated markers derived from the KEGG PI3K pathway (hsa04151) and the crosstalking MAPK/MEK and PLK1 pathways is shown across all WHIM models for phosphosites differentially regulated between the sensitive and resistant PDX models (Fig. 4C). Signaling between a subset of these markers indicates possible strong crosstalk between these pathways (PLK1/PI3K/MAPK/MEK) after buparlisib treatment (Fig. 4D).
Figure 4. Many differentially expressed markers are involved in PI3K signaling.
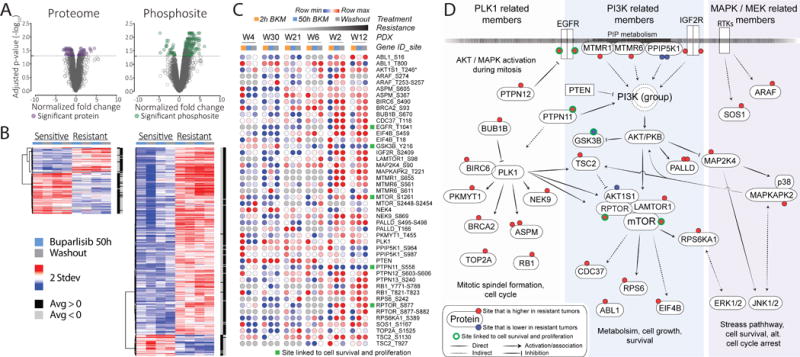
A, volcano plots of proteins and phosphosites, in which fold changes are plotted versus the -log10 of their adjusted p-values. Fold changes are based on a two sample moderated t-test identifying response markers between the two most sensitive and the two most resistant tumors and p-values are adjusted using the Benjamini-Hochberg approach. Fifty hours buparlisib and washout treatments were used as replicates. B, heatmaps respectively represent significantly differentiated proteins and phosphosites (adjusted p-value ≤ 0.05). C, significant response markers that are differentially expressed relative to vehicle treatments between the two most resistant and the two most sensitive PDX tumors are plotted for all PDX tumors. Green squares indicate phosphosites linked to cell survival and proliferation. D, a schematic network that puts the significant markers in panel C into the context of their signaling. These markers highlight the PLK1, PI3K and MAPK/MEK pathways (shaded colors). Rectangular gene boxes indicate significant changes on protein level, as opposed to regulated phosphosites (red and blue circles).
Important differentially regulated sites included T1041 on EGFR, which is known to be phosphorylated with EGF stimulation [31], consistent with feedback upregulation by buparlisib in resistant tumors. Persistent PI3K signaling was reflected by the phosphorylation of site S1261 on mTOR, which promotes cell growth [32], site S877 on RPTOR, known to be phosphorylated during mitosis [33], and a number of other downstream PI3K signaling components including RPS6A1 and EIF4B. PTPN11 (SHP2) is an additional interesting finding since this protein has been shown to interact with the regulatory subunit of PI3K, promoting the p85-p110 phosphorylation that is seen during proliferative cell states [34]. After buparlisib treatment, a loss of phosphorylation was observed on PTPN11 S558. While the function of this exact site has not been previously described, our data indicate a link to buparlisib resistance. A family member of PTPN11 is PTPN12 (PTP-PEST), which is considered a tumor suppressor and inhibits several receptor tyrosine kinases in some breast cancers [35]. An increase of phosphorylation was observed on PTPN12 after buparlisib treatment, most notably a dual phosphorylation on sites S603-S606, which is located C-terminally on PTPN12 and overlaps with a well-known PEST motif [36]. Additionally, phosphorylation at Y216 of GSK3B was decreased. This site is involved in altered apoptosis [37] and cell growth [38], among other cellular mechanisms. In our study GSK3B Y216 showed less phosphorylation in the resistant tumors compared to the sensitive tumors after buparlisib treatment, indicating little or no induction of programmed cell death. Evidence of MAPK/MEK activation, such as increased phosphorylation of the ERK downstream target RSK1 after buparlisib, was also prominent in resistant tumors. Taken together, the regulation of these sites indicates that cell growth and proliferation are active in the most resistant tumors despite buparlisib treatment, and that the resistance is, in part, driven by failure to inhibit PI3K downstream signaling as well as related crosstalking pathways such as those involved in MAPK/MEK and PLK1 signaling.
Validation of crosstalk between PI3K, EGFR, and MAPK signaling pathways mediating buparlisib resistance
For in vitro validation of candidate biomarkers associated with buparlisib resistance, a cell line was derived from an early passage of the WHIM12 PDX model. In single-shot LC-MS/MS analyses the WHIM12 tumor (vehicle-treated sample) and cell line clustered well in the space of ca. 6,000 proteins (Supplementary Fig. 7A), separating from all other vehicle-treated PDX models in the first principal component. In the second component, additional distinct cell line-dependent features were evident. More importantly, the WHIM12 cell line maintained the tumor’s functional resistance to buparlisib, as well as being resistant to AKT inhibitors (Afuresertib and MK2206) and an mTOR inhibitor (MK8669; Supplementary Fig. 7B).
To test the hypothesis that persistent PI3K/AKT/mTOR signaling and crosstalk with EGFR and MAPK/MEK signaling pathways contribute to resistance to buparlisib, we selected a set of clinical grade inhibitors that target highly regulated (phospho-) proteins or markers associated with PI3K/AKT/mTOR signaling hubs and evaluated their effects alone or in combination with buparlisib in the WHIM12 cell line (Fig. 5A). Consistent with the importance of EGFR and MAPK/MEK in mediating buparlisib resistance, the combination of buparlisib and either the MEK inhibitor trametinib or the EGFR inhibitor erlortinib showed synergistic anti-tumor effects on the WHIM12 cell line (Fig. 5B; Supplementary Fig. 7C). Furthermore, testing a hypothesis based on persistent PI3K/AKT/mTOR activation in buparlisib-resistant PDX models, we demonstrated that combining buparlisib with either an AKT inhibitor (afuresertib) or an mTOR inhibitor (AZD2014) led to significantly decreased viability of the WHIM12 cell line (Fig. 5C and 5D). Additional combinations of buparlisib with MK2206 (an AKT inhibitor) and/or MK8669 (an mTOR inhibitor) likewise showed increased anti-tumor effects in the WHIM12 cell line (Fig. 5E). These combinatorial effects were further exemplified by the highly selective and potent dual p110α/β/δ/γ and mTORC1/2 inhibitor omipalisib, which showed a strong cytotoxic effect on its own (Fig. 5F).
Figure 5. A WHIM12-derived cell line is sensitive to regimens that combine PI3K inhibition with agents that target PI3K downstream signaling components or the MEK/ERK pathway.
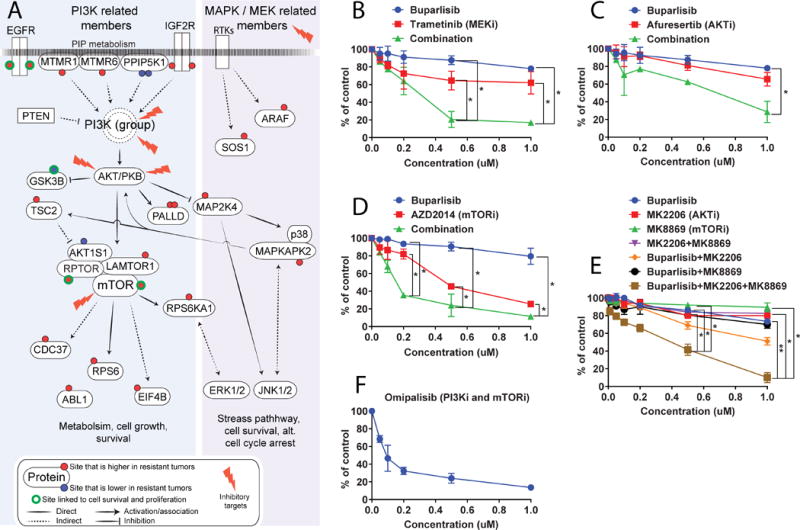
A, a schematic network indicates targets for in vitro validation. B, buparlisib in combination with the MEK inhibitor trametinib shows synergistic decrease of cell viability of the WHIM12 cell line. C and D, cells were treated with buparlisib alone or in combination with a PI3K or mTOR inhibitor, respectively, showing synergistic effects on the WHIM12-derived cell line. E, a combination of PI3K and/or mTOR inhibition leads to synergistic decrease of cell viability. F, the dual PI3K and mTOR inhibitor omipalisib shows a marked decrease in cell viability on its own. B-F, mean and error bars (standard deviations) of cell viability treated with indicated drug(s) relative to control from three independent experiments are shown; * ≤ 0.05 and ** ≤ 0.01 as significance.
A NEK9-MAP2K4 signaling axis mediates resistance to buparlisib in concert with a p85 binding domain mutation of PIK3CA
As protein and lipid kinases are common drug targets, we performed a kinase outlier analysis in xenograft tumors to identify kinase phosphorylation events significantly enriched relative to the background distribution of the breast tumor phosphoproteome [29], then determined the subset enriched in buparlisib-resistant tumors compared with sensitive tumors based on a Benjamini-Hochberg corrected Fisher’s exact test probability as previously described [17]. The analysis again highlighted members of the MAPK and PLK1 pathways. For example, outliers enriched in the most resistant tumors (WHIM2 and WHIM12; Fig. 6A) included MAPK6, MAPK7, MAPK9, MAP2K4 and NEK9 (NIMA Related Kinase 9). NEK9 and MAP2K4 are of particular interest as they represent hubs of the PLK1 and MAPK signaling pathways, respectively, and were also identified as significantly more phosphorylated after buparlisib treatment in the global phosphoproteome (Fig. 4C). Additionally, a focused analysis of the kinome MIB data showed baseline upregulation of NEK9 in WHIM12, and also demonstrated MAP2K4 upregulation in the resistant tumors after buparlisib treatment (Fig. 6B).
Figure 6. Identification of NEK9 and MAP2K4 in association with buparlisib resistance.
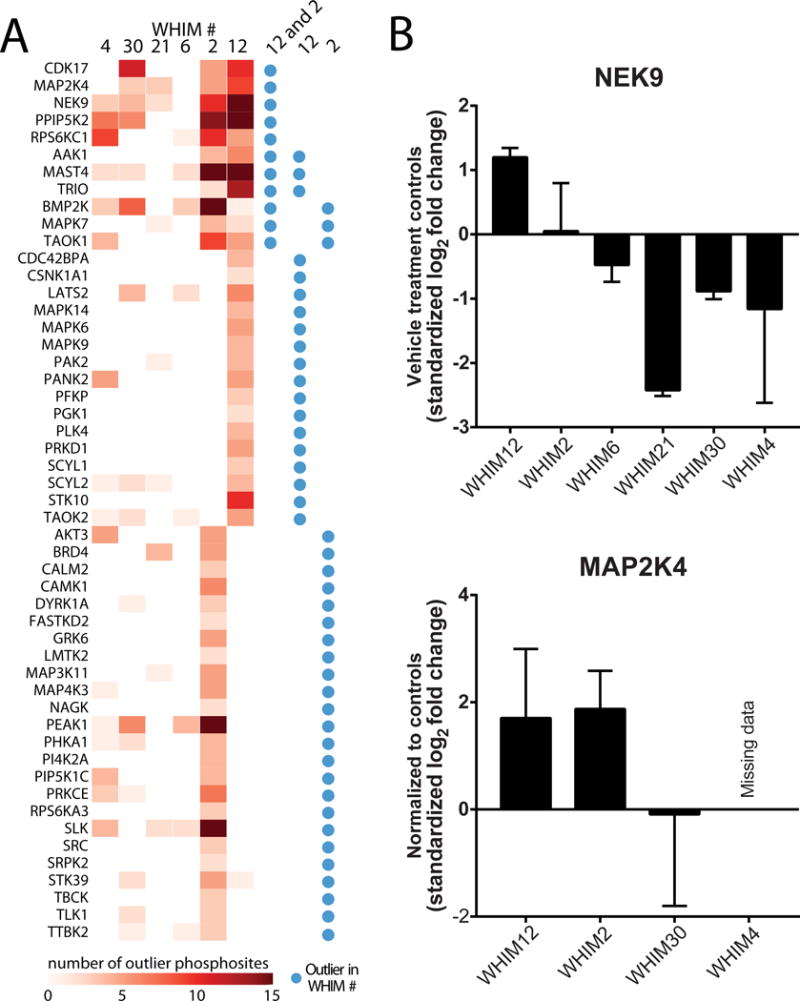
A, an outlier expression analysis of the global phosphoproteome found 51 kinases to be enriched in the resistant group (WHIM2 and WHIM12), WHIM12 alone, or WHIM2 alone. Shading indicates the number of phosphosite outliers identified in the specified gene. Blue circles annotate outlier kinase events by sensitivity group (Resistant: WHIM12, WHIM2 versus non-resistant: W6, W21, W30, W4) or resistant sample (WHIM12 versus all others or WHIM2 versus all others), identified based on enrichment (≥ 50% samples containing phosphosite outlier, ≤ 5% FDR using Benjamini-Hochberg adjusted p-values). B, upper panel: MIB data of baseline NEK9 levels. Lower panel: MAP2K4 after buparlisib treatment normalized to respective controls (bars are mean and whiskers are standard deviations). No MAP2K4 peptides were detected in WHIM4’s kinase-enriched MIB samples (missing data/undetected).
To evaluate their contribution to buparlisib resistance, we knocked down NEK9 and MAP2K4 using specific shRNAs in the WHIM12-derived cell line (Fig. 7A). Knockdown of either gene significantly inhibited WHIM12 cell growth and increased sensitivity to buparlisib (Fig. 7B-E). Mechanistic analysis revealed that while buparlisib treatment led to the expected feedback upregulation of pERK1/2 levels in the vector control cells, NEK9 or MAP2K4 knockdown abolished that upregulation, indicating their potential roles in regulating the crosstalk between PI3K and ERK signaling. Furthermore, treatment with buparlisib led to much lower levels of pAKT, pS6 and survivin (BRIC5) in NEK9 and MAP2K4 knockdown cells compared to control cells (Fig. 7F and 7G, respectively). Interestingly, the baseline levels of pERK and pAKT/pS6 were also consistently lower in the NEK9 knockdown cells, consistent with the reduced cell proliferation at baseline observed by the colony formation assay (Fig. 7B).
Figure 7. In vitro target validation of NEK9 and MAP2K4 in association with buparlisib resistance.
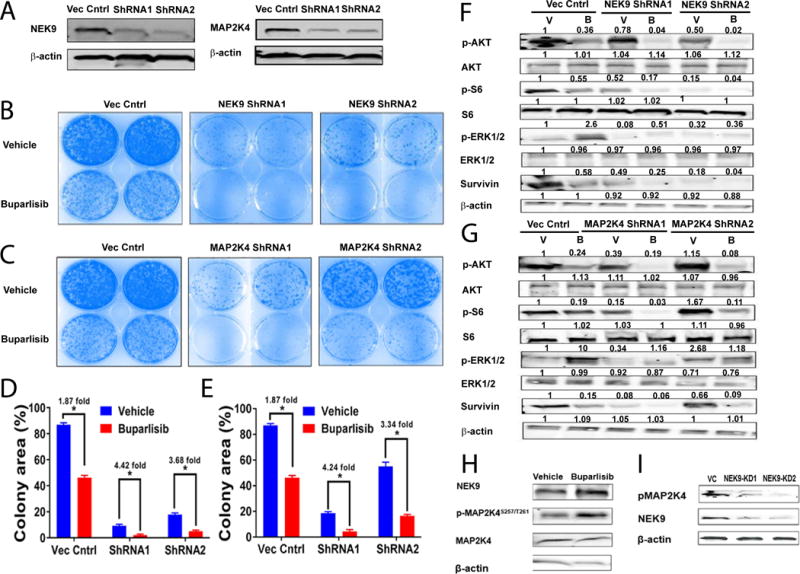
A, Western blot showing knockdown of either NEK9 or MAP2K4 in the WHIM12 cell line. B and C, colony formation assays of WHIM12 cells with NEK9 or MAP2K4 knocked down, respectively. D and E, quantified growth of colony formation assays in panel B (NEK9) and C (MAP2K4), respectively. F, Western blot of a set of relevant PI3K markers after NEK9 knockdown in the WHIM12 cell line – with or without buparlisib treatment. G, Western blot of a set of relevant markers after MAP2K4 knockdown in the WHIM12 cell line – with or without buparlisib treatment. H, NEK9 protein levels and MAP2K4 S257/T261 phosphorylation (pMAP2K4) after buparlisib treatment. I, NEK9 knockdown in the WHIM12 cell line leads to loss of MAP2K4 S257/T261 phosphorylation. V: Vehicle; B: buparlisib; VC: Vector Control; KD1: knockdown clone 1; KD2: knockdown clone 2. * p ≤ 0.05. All analyses performed on a cell line derived from the WHIM12 xenograft.
To determine whether the NEK9/MAPK axis-mediated resistance mechanism was specific to buparlisib in the WHIM12-derived cell line, we evaluated the effect of this axis on sensitivity to other PI3K inhibitors. Knockdown of either NEK9 or MAP2K4 increased sensitivity to BYL719 (a p110α specific inhibitor), AZD6482 (a p110β specific inhibitor) and GDC-0941 (a pan-PI3K inhibitor) as compared to vector control cells in WHIM12 (Supplementary Fig. 7D-F). We also observed an increased sensitivity to buparlisib after NEK9 and MAP2K4 knockdown in MDA-MB-231 and BT549 TNBC cells (Supplementary Fig. 7G and 7H).
A direct kinase-substrate relationship between NEK9 and MAP2K4 is suggested by the NEK9 substrate specificity sequence SXXXT [39], which matches that of MAP2K4 at S257 and T261, and supported by the association between NEK9 upregulation and increased phosphorylation of MAP2K4 S257/T261upon buparlisib treatment in the WHIM12 cell line (Fig. 7H). Importantly, NEK9 knockdown clearly reduced phosphorylation of MAP2K4 S257/T261 (Fig. 7I). The global phosphorylation data also showed MAP2K4 site S257 to be differentially regulated between the resistant and sensitive PDX models, albeit with a modest adjusted p-value of 0.10 (Supplementary File 4). These data provide further mechanistic insight into NEK9 and MAP2K4 interactions in mediating cancer cell proliferation and resistance to buparlisib.
A prominent genetic feature of WHIM12 is the complex indel frameshift mutation located in a mutation hotspot in the p85-binding domain of PIK3CA (Supplementary Fig. 8A and 8B). An antibody against p85 was able to pull down p110α using tumor lysates from the PIK3CA wild type WHIM4 and WHIM30, but not WHIM12, indicating an effect on interaction with p85. Furthermore, hot-spot mutations in the helical and catalytic domain of PIK3CA (MFC10A cells) did not affect the interaction of p85-p110α under either serum free or serum supplemented conditions. While in the WHIM12 derived cell line the p85-p110α binding was present under serum free conditions, but lost when adding serum. This suggests that the interaction might be central to p110α activation by growth factors in WHIM12 (Supplementary Fig. 8C-E). To further investigate its impact on buparlisib responsiveness, we transfected the WHIM12 mutant form of PIK3CA into two buparlisib-sensitive, PIK3CA wild type cell lines, MCF10A and a PDX/WHIM3 derived cell line [26]. Expression of the WHIM12 PIK3CA mutation led to a partial resistance to buparlisib in both cell lines (Supplementary Fig. 9A-D). This is in contrast to an MCF10A line expressing the PIK3CA E545K mutation, which remained sensitive to buparlisib (Supplementary Fig. 9A). Basal levels of pAKT and pERK1/2 protein were also increased in PIK3CA mutant-overexpressing WHIM3 cells compared to vector control cells (Supplementary Fig. 9E). Up to 2-fold higher levels of pERK1/2 were observed post-buparlisib treatment in cells expressing the WHIM12 mutant form of PIK3CA compared to the vehicle control. Conversely, knocking down PIK3CA in WHIM12 increased the sensitivity to buparlisib (Supplementary Fig. 10A and 10B). This effect correlated to a reduction, rather than up-regulation, in the levels of pERK1/2 upon buparlisib treatment (Supplementary Fig.10C). Taken together these results show an important role of this novel PIK3CA mutation in mediating buparlisib resistance in the WHIM12 tumor.
We next examined the effect of the p85 binding domain mutation in PIK3CA on NEK9/MAP2K4 protein expression. A mild but consistent NEK9 increase was observed in the WHIM3 line when overexpressing the PIK3CA mutation (Supplementary Fig. 9E). More strikingly, PIK3CA knockdown in the WHIM12 cell line led to significant reduction of baseline and post-treatment NEK9 and MAP2K4 levels (Supplementary Fig. 10C), indicating a relationship between this specific PIK3CA mutation, buparlisib resistance and the NEK9-MAP2K4 axis.
In summary, these results are consistent with our findings of a resistance mechanism to buparlisib in WHIM12, involving persistent activation of PI3K signaling and crosstalk to EGFR, MAPK/MEK and PLK1 pathways. We also showed central roles for the NEK9 kinase as an effector of PI3K signaling and upstream regulator of MAP2K4 phosphorylation and buparlisib resistance, dependent on a complex PIK3CA mutation. While most of these results pertain to WHIM12, the most resistant tumor, the relationship of NEK9 and MAP2K4 to buparlisib resistance may be more general given that buparlisib resistance also showed a NEK9/MAP2K4 dependency in several other cell lines.
DISCUSSION
In this study, we characterized the molecular response of 6 TNBC PDX models to buparlisib, a pan-PI3K inhibitor being tested for the treatment of several solid tumors [14–16], using unbiased quantitative transcriptomics, proteomics and phosphoproteomics (Fig. 1D), and uncovered novel markers of PI3K inhibition and potential resistance mechanisms. As is common for studies emphasizing depth of coverage over sample throughput, statistical power is challenging. We nevertheless benefited from a deep and systemic view of treatment-related events.
Analyses of baseline, control-treated models showed extensive heterogeneity independent of the analytical platform. This heterogeneity was evident on both marker and pathway levels (Fig. 2B and 2C; Supplementary Fig. 2D). Tumor heterogeneity, together with the low number of samples in this study, impeded our ability to identify a unified mechanism of buparlisib resistance with high confidence. However, markers shared between omic platforms showed a moderate positive correlation, with correlations between mRNA and protein levels very similar to two previous human cancer studies [29, 40] and a recent large PDX baseline study that included our 6 models [12] (Supplementary Fig. 2C).
Using the RNA-seq data from the PDX baseline study described above [12], we were able to identify mRNA splice isoforms that are translated and differentially expressed between sensitive and resistant tumors. These splice forms include known PI3K members, indicating that there is already an intrinsic priming of PI3K signaling that differs between the buparlisib sensitive and resistant tumors. Interestingly, the identified splice forms EIF4B and PALLD were additionally found to be significantly more phosphorylated in the resistant tumors after treatment (Fig. 4C), further highlighting their roles in PI3K signaling and buparlisib response.
Amidst the baseline heterogeneity, we sought to identify changes that were in agreement across all tumors after buparlisib treatment. The global pattern of pharmacodynamic changes showed a dynamic phosphoproteome, compared to changes that were slower but more durable in the transcriptome and proteome (Fig. 3A). Phosphoproteome changes were heavily associated with PI3K-related signaling (Fig. 3B). Remarkably, no phosphosite markers were stable in the washout treatment arm; all phosphosite changes common across all 6 tumors were evanescent, and likely dependent on continuous administration of buparlisib. In sum, buparlisib treatment yielded direct but transient effects on the phosphoproteome; an “instantaneous” impact commensurate with the treatment-specific qualities we would expect of PI3K, AKT and mTOR signaling. The temporal dynamics of buparlisib are of relevance to clinical use, as dosing and treatment plans should maximize buparlisib efficacy while minimizing adverse side effects [16]. We therefore examined pharmacodynamic response markers of buparlisib treatment in more detail. We identified the established pharmacodynamic marker T246 site on AKT1S1 [28, 29] (Supplementary Fig. 3A), providing an important biological quality control for our study, since AKT1S1 is a well-known substrate for AKT1 and T246 phosphorylation is known to be lost upon PI3K inhibition. We have advanced the current understanding by showing that this marker is transient in nature and that there are several other sites on AKT1S1 that show similar dynamics to T246 after buparlisib treatment/PI3K inhibition. Additional markers that showed pharmacodynamic regulation are also involved in PI3K signaling as well as PLK1 signaling and the modulation of several sites is accentuated at the 50 hours’ time point. An unanticipated, and to our knowledge previously undescribed finding was the dynamic phosphorylation of Ki-67 and its relationship to protein expression. The Ki-67 index, or MIB1 index, is a common clinical test that indicates cell proliferation, although Ki-67’s exact function is uncertain. Our LC-MS/MS data revealed an increase of phosphorylation in regions on Ki-67 that are rich in PEST-motifs, which often induce protein degradation [41]. Phosphorylation of these sites was observed in all WHIM tumors except for WHIM6, before any decrease was seen for the Ki-67 protein level. Interestingly, WHIM6 was also the only tumor that did not show any increase in Ki-67 phosphorylation upon buparlisib treatment. Therefore, monitoring Ki-67 phosphorylation status might serve as an earlier and more sensitive marker of decreased proliferation than Ki-67 protein level expression.
Response markers differentiating the most sensitive from the most resistant PDX tumors were evident in the proteome, phosphoproteome and kinome results. Changes in the phosphoproteome were strongly associated with PI3K signaling, with some specific markers indicating continuously active PI3K signaling in the most resistant tumors even after buparlisib treatment (Fig. 4). However, connections to MAPK/MEK and PLK1 signaling were also observed, and MAPK/MEK signaling is well known to confer resistance to PI3K inhibition (reviewed in Castellano and Downward [42]). A key role for MAPK member MAP2K4 in PI3K crosstalk signaling is further supported by loss of function mutations in MAP2K4 and MAP3K1 in some luminal breast cancers [26], which are often sensitive to PI3K/AKT/mTOR inhibition [43]. Further study is warranted in luminal breast cancer to investigate the interplay of MAP2K4/MAP3K1 mutation and sensitivity to PI3K inhibitor efficacy in clinical trials. Many markers are also higher in the sensitive tumors (WHIM4 and WHIM30) compared to the resistant tumors (WHIM12 and WHIM2) after buparlisib treatment. Proteomic markers that are induced after 50 hours’ treatment (and at washout) include PTEN, and are involved in the Complement System, Signaling via EGFR, and WNT Pathway and CTNNB1 Signaling (Supplementary Table 2). Additionally, PIK3CA is amplified at the DNA, RNA and proteome levels in WHIM4 (Fig. 2A and 2B), which could, taken together, partly explain this model’s sensitivity to buparlisib, in contrast to the mutation of PIK3CA in WHIM12 that appears to induce resistance.
Differential NEK9 and MAP2K4 phosphorylation were evident in the two most resistant WHIM models (Fig. 4C), a finding validated by our focused kinome analyses (Fig. 6A and 6B). NEK9 is an understudied kinase that is primarily thought to play a role in mitotic progression [44]. We established the functional significance of these kinases in buparlisib resistance by demonstrating decreased colony formation of WHIM12 in vitro, and restoration of sensitivity to buparlisib upon knockdown of either kinase (Fig. 7B-E). Our data indicated that these kinases affect the regulation of PI3K signaling and crosstalk resistance mechanisms, including the MAPK/ERK pathway, in a buparlisib-dependent manner. However, we cannot exclude that these observations of resistance may reflect a general overall resistance to drug treatment, and may not represent a specific mechanism of resistance to buparlisib. Knocking out NEK9 and MAP2K4 in the WHIM12-derived cell line and then treating those cells with BYL719, AZD6482, and GDC-0941, which are p110α specific-, p110β specific- and pan-PI3K inhibitors, respectively, showed increased sensitivity to inhibitor treatment (Supplementary Fig. 7D-F). We also observed an increase in sensitivity to buparlisib after knockdown of either NEK9 or MAP2K4 in MDA-MB213 and BT549 cell lines (Supplementary Fig. 7G and 7H). Additionally, our CMAP analysis showed that proteins that are high in the resistant tumors compared to the sensitive tumors show similar behavior to a wide range of inhibitors (IKK, PKC, CDK and HDAC inhibitors among others; Supplementary Fig. 6). Taken together, these results indicate a more general resistance mechanism controlled by NEK9 and MAP2K4 in TNBC. Furthermore, prior literature has reported that both NEK9 and MAP2K4 are associated with an increase in drug resistance for several other drugs and cancers [45–49], but not to buparlisib treatment in TNBC, which we show here for the first time.
Additionally supportive of an oncogenic role for NEK9 and MAP2K4 were changes in survivin. Survivin is highly expressed in most cancers and is associated with chemotherapy resistance [50], and a clear correlation between NEK9/MAP2K4 levels and survivin expression was observed in this study (Fig. 7F and 7G). While there is no established link between NEK9 and pMAP2K4, NEK9 has substrate specificity SXXXT [39], matching the S257/T261 motif of MAP2K4. When silencing NEK9 in the WHIM12-derived cell line we observed a loss of MAP2K4 phosphorylation at sites S257/261 (Fig. 7I), supporting a direct kinase-substrate relationship. Further suggesting the centrality of these kinases in PI3K resistance signaling is the fact that the p85-binding mutant form of PIK3CA induced NEK9 and a partial buparlisib resistance, while loss of PIK3CA led to a loss of NEK9 and MAP2K4 and an increased sensitivity to buparlisib (Supplementary Fig. 9 and Supplementary Fig. 10). Our data further suggests that the mutant form of PIK3CA described herein is less bound to the p85 regulatory subunit in the most resistant tumor (WHIM12) compared to the sensitive tumors (WHIM4 and WHIM30; Supplementary Fig. 8C-D), highlighting the possibility of buparlisib being, in part, a p85-dependent PI3K inhibitor. Taken together, these data point toward an oncogenic role for a novel NEK9-MAP2K4 signaling axis in PI3K-dysregulated breast cancer, and nominate these proteins as possible hubs in buparlisib resistance in TNBC. NEK9 and MAP2K4 should be further evaluated, possibly as therapeutic targets in PI3K inhibition regimens.
The discovery of poorly characterized kinases, such as NEK9, as potentially important regulators of breast cancers illustrates the value of integrated omics approaches that include mass spectrometry-based phosphoproteomics in addressing translational questions. Our systematic exploration of the impact of PI3K inhibition in high fidelity, in vivo models of TNBC supports the relevance of PI3K pathway and potential combination strategies to overcome resistance in a subset of TNBC. The in-depth multi-omic approach provides the clinical and research communities with a wealth of data that may help to advance strategies for patient selection and precision treatment options for this deadly disease.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
Integrative phospho-proteogenomic analyses is used to determine intrinsic resistance mechanisms of triple negative breast tumors to PI3K inhibition.
Acknowledgments
We would like to thank J. Qiao and L. C. Tang for their lab expertise and help related to the global proteome and phosphoproteome. The expert technical assistance with enriched kinome analysis by R. Connors and Dr. Y. Mi is gratefully acknowledged. We also want to thank Drs. K. Rajapakshe, C. Coarfa, and Q. Mo for data processing and normalization on RPPA data. We thank F. Jia, M. G. Costello and K. Holloway from the Antibody-based Proteomics Core/Shared Resource for their excellent technical assistant in performing RPPA experiments. We would like to thank our breast cancer patient advocates Ms. L. Facer and Ms. L. Blath for their input on the study concept.
This work was supported by the grants U24CA160034, U24CA210986 and U01CA214125 to S.A. Carr. from the US National Institutes of Health as part of the National Cancer Institute Clinical Proteomics Tumor Analysis Consortium Initiative. This work was also supported in part by the NCI Cancer Proteome Characterization Center at Washington University and University of North Carolina (U24CA160035; R.R. Townsend. and M.J. Ellis), the Cancer Prevention and Research Institute of Texas Proteomics and Metabolomics Core Facility Support Award (RP170005, D.P. Edwards and S. Huang) and NCI Cancer Center Support Grant to Antibody-based Proteomics Core/Shared Resource (P30CA125123; D.P. Edwards and S. Huang), the Proteomics Shared Resource at Washington University and the Siteman Cancer Center (NCI P30 CA091842; R.R. Townsend), Susan G. Komen for the Cure (IIR13263475, Susan G. Komen for the Cure; C. Ma and S. Li), and A Sister’s Hope (C. Ma).
Footnotes
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
References
- 1.Hudis CA, Gianni L. Triple-negative breast cancer: an unmet medical need. Oncologist. 2011;16(Suppl 1):1–11. doi: 10.1634/theoncologist.2011-S1-01. [DOI] [PubMed] [Google Scholar]
- 2.Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486(7403):395–99. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marty B, Maire V, Gravier E, Rigaill G, Vincent-Salomon A, Kappler M, et al. Frequent PTEN genomic alterations and activated phosphatidylinositol 3-kinase pathway in basal-like breast cancer cells. Breast Cancer Res. 2008;10(6):R101. doi: 10.1186/bcr2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De P, Sun Y, Carlson JH, Friedman LS, Leyland-Jones BR, Dey N. Doubling down on the PI3K-AKT-mTOR pathway enhances the antitumor efficacy of PARP inhibitor in triple negative breast cancer model beyond BRCA-ness. Neoplasia. 2014;16(1):43–72. doi: 10.1593/neo.131694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol. 2013;10(3):143–53. doi: 10.1038/nrclinonc.2013.10. [DOI] [PubMed] [Google Scholar]
- 6.Crystal AS, Shaw AT, Sequist LV, Friboulet L, Niederst MJ, Lockerman EL, et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science. 2014;346(6216):1480–86. doi: 10.1126/science.1254721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Riley NM, Coon JJ. Phosphoproteomics in the Age of Rapid and Deep Proteome Profiling. Anal Chem. 2016;88(1):74–94. doi: 10.1021/acs.analchem.5b04123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hidalgo M, Amant F, Biankin AV, Budinská E, Byrne AT, Caldas C, et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 2014;4(9):998–1013. doi: 10.1158/2159-8290.CD-14-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosfjord E, Lucas J, Li G, Gerber HP. Advances in patient-derived tumor xenografts: from target identification to predicting clinical response rates in oncology. Biochem Pharmacol. 2014;91(2):135–43. doi: 10.1016/j.bcp.2014.06.008. [DOI] [PubMed] [Google Scholar]
- 10.Li S, Shen D, Shao J, Crowder R, Liu W, Prat A, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 2013;4(6):1116–30. doi: 10.1016/j.celrep.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ding L, Ellis MJ, Li S, Larson DE, Chen K, Wallis JW, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464(7291):999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang KL, Li S, Mertins P, Cao S, Gunawardena HP, Ruggles KV, et al. Proteogenomic integration reveals therapeutic targets in breast cancer xenografts. Nat Commun. 2017;8:14864. doi: 10.1038/ncomms14864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med. 2015;21(11):1318–25. doi: 10.1038/nm.3954. [DOI] [PubMed] [Google Scholar]
- 14.Brachmann SM, Kleylein-Sohn J, Gaulis S, Kauffmann A, Blommers MJ, Kazic-Legueux M, et al. Characterization of the mechanism of action of the pan class I PI3K inhibitor NVP-BKM120 across a broad range of concentrations. Molecular cancer therapeutics. 2012;11(8):1747–57. doi: 10.1158/1535-7163.MCT-11-1021. [DOI] [PubMed] [Google Scholar]
- 15.Rodon J, Brana I, Siu LL, De Jonge MJ, Homji N, Mills D, et al. Phase I dose-escalation and -expansion study of buparlisib (BKM120), an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. Invest New Drugs. 2014;32(4):670–81. doi: 10.1007/s10637-014-0082-9. [DOI] [PubMed] [Google Scholar]
- 16.Ma CX, Luo J, Naughton M, Ademuyiwa F, Suresh R, Griffith M, et al. A Phase I Trial of BKM120 (Buparlisib) in Combination with Fulvestrant in Postmenopausal Women with Estrogen Receptor-Positive Metastatic Breast Cancer. Clin Cancer Res. 2016;22(7):1583–91. doi: 10.1158/1078-0432.CCR-15-1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534(7605):55–62. doi: 10.1038/nature18003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang B, Wang J, Wang X, Zhu J, Liu Q, Shi Z, et al. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014;513(7518):382–7. doi: 10.1038/nature13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang H, Liu T, Zhang Z, Payne SH, Zhang B, McDermott JE, et al. Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell. 2016;166(3):755–65. doi: 10.1016/j.cell.2016.05.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hather G, Liu R, Bandi S, Mettetal J, Manfredi M, Shyu WC, et al. Growth rate analysis and efficient experimental design for tumor xenograft studies. Cancer Inform. 2014;13(Suppl 4):65–72. doi: 10.4137/CIN.S13974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu S, Li S, Guo Z, Luo J, Ellis MJ, Ma CX. Combined targeting of mTOR and AKT is an effective strategy for basal-like breast cancer in patient-derived xenograft models. Mol Cancer Ther. 2013;12(8):1665–75. [Google Scholar]
- 22.Chang CH, Zhang M, Rajapakshe K, Coarfa C, Edwards D, Huang S, et al. Mammary Stem Cells and Tumor-Initiating Cells Are More Resistant to Apoptosis and Exhibit Increased DNA Repair Activity in Response to DNA Damage. Stem Cell Reports. 2015;5(3):378–91. doi: 10.1016/j.stemcr.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26(12):1367–72. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 24.Ruggles KV, Tang Z, Wang X, Grover H, Askenazi M, Teubl J, et al. An Analysis of the Sensitivity of Proteogenomic Mapping of Somatic Mutations and Novel Splicing Events in Cancer. Mol Cell Proteomics. 2016;15(3):1060–71. doi: 10.1074/mcp.M115.056226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell. 2012;149(2):307–21. doi: 10.1016/j.cell.2012.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma CX, Cai S, Li S, Ryan CE, Guo Z, Schaiff WT, et al. Targeting Chk1 in p53-deficient triple-negative breast cancer is therapeutically beneficial in human-in-mouse tumor models. J Clin Invest. 2012;122(4):1541–52. doi: 10.1172/JCI58765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ye K, Wang J, Jayasinghe R, Lameijer EW, McMichael JF, Ning J, et al. Systematic discovery of complex insertions and deletions in human cancers. Nat Med. 2016;22(1):97–104. doi: 10.1038/nm.4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andersen JN, Sathyanarayanan S, Di Bacco A, Chi A, Zhang T, Chen AH, et al. Pathway-based identification of biomarkers for targeted therapeutics: personalized oncology with PI3K pathway inhibitors. Sci Transl Med. 2010;2(43):43ra55. doi: 10.1126/scitranslmed.3001065. [DOI] [PubMed] [Google Scholar]
- 29.Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534(7605):55–62. doi: 10.1038/nature18003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fujita N, Jaye DL, Kajita M, Geigerman C, Moreno CS, Wade PA. MTA3, a Mi-2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell. 2003;113(2):207–19. doi: 10.1016/s0092-8674(03)00234-4. [DOI] [PubMed] [Google Scholar]
- 31.Tong J, Taylor P, Peterman SM, Prakash A, Moran MF. Epidermal growth factor receptor phosphorylation sites Ser991 and Tyr998 are implicated in the regulation of receptor endocytosis and phosphorylations at Ser1039 and Thr1041. Mol Cell Proteomics. 2009;8(9):2131–44. doi: 10.1074/mcp.M900148-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Acosta-Jaquez HA, Keller JA, Foster KG, Ekim B, Soliman GA, Feener EP, et al. Site-specific mTOR phosphorylation promotes mTORC1-mediated signaling and cell growth. Mol Cell Biol. 2009;29(15):4308–24. doi: 10.1128/MCB.01665-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vazquez-Martin A, Cufí S, Oliveras-Ferraros C, Menendez JA. Raptor, a positive regulatory subunit of mTOR complex 1, is a novel phosphoprotein of the rDNA transcription machinery in nucleoli and chromosomal nucleolus organizer regions (NORs) Cell Cycle. 2011;10(18):3140–52. doi: 10.4161/cc.10.18.17376. [DOI] [PubMed] [Google Scholar]
- 34.Breitkopf SB, Yang X, Begley MJ, Kulkarni M, Chiu YH, Turke AB, et al. A Cross-Species Study of PI3K Protein-Protein Interactions Reveals the Direct Interaction of P85 and SHP2. Sci Rep. 2016;6:20471. doi: 10.1038/srep20471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun T, Aceto N, Meerbrey KL, Kessler JD, Zhou C, Migliaccio I, et al. Activation of multiple proto-oncogenic tyrosine kinases in breast cancer via loss of the PTPN12 phosphatase. Cell. 2011;144(5):703–18. doi: 10.1016/j.cell.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Playford MP, Lyons PD, Sastry SK, Schaller MD. Identification of a filamin docking site on PTP-PEST. J Biol Chem. 2006;281(45):34104–12. doi: 10.1074/jbc.M606277200. [DOI] [PubMed] [Google Scholar]
- 37.Kotliarova S, Pastorino S, Kovell LC, Kotliarov Y, Song H, Zhang W, et al. Glycogen synthase kinase-3 inhibition induces glioma cell death through c-MYC, nuclear factor-kappaB, and glucose regulation. Cancer Res. 2008;68(16):6643–51. doi: 10.1158/0008-5472.CAN-08-0850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coluccia AM, Vacca A, Dunach M, Mologni L, Redaelli S, Bustos VH, et al. Bcr-Abl stabilizes beta-catenin in chronic myeloid leukemia through its tyrosine phosphorylation. EMBO J. 2007;26(5):1456–66. doi: 10.1038/sj.emboj.7601485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu J, Rho HS, Newman RH, Zhang J, Zhu H, Qian J. PhosphoNetworks: a database for human phosphorylation networks. Bioinformatics. 2014;30(1):141–2. doi: 10.1093/bioinformatics/btt627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang B, Wang J, Wang X, Zhu J, Liu Q, Shi Z, et al. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014;513(7518):382–87. doi: 10.1038/nature13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garcia-Alai MM, Gallo M, Salame M, Wetzler DE, McBride AA, Paci M, et al. Molecular basis for phosphorylation-dependent, PEST-mediated protein turnover. Structure. 2006;14(2):309–19. doi: 10.1016/j.str.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 42.Castellano E, Downward J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer. 2011;2(3):261–74. doi: 10.1177/1947601911408079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Almstedt K, Schmidt M. Targeted Therapies Overcoming Endocrine Resistance in Hormone Receptor-Positive Breast Cancer. Breast Care (Basel) 2015;10(3):168–72. doi: 10.1159/000405017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fry AM, O’Regan L, Sabir SR, Bayliss R. Cell cycle regulation by the NEK family of protein kinases. J Cell Sci. 2012;125(Pt 19):4423–33. doi: 10.1242/jcs.111195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith SC, Petrova AV, Madden MZ, Wang H, Pan Y, Warren MD, et al. A gemcitabine sensitivity screen identifies a role for NEK9 in the replication stress response. Nucleic Acids Res. 2014;42(18):11517–27. doi: 10.1093/nar/gku840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang L, Pan Y, Dai JL. Evidence of MKK4 pro-oncogenic activity in breast and pancreatic tumors. Oncogene. 2004;23(35):5978–85. doi: 10.1038/sj.onc.1207802. [DOI] [PubMed] [Google Scholar]
- 47.Phadke M, Remsing Rix LL, Smalley I, Bryant AT, Luo Y, Lawrence HR, et al. Dabrafenib inhibits the growth of BRAF-WT cancers through CDK16 and NEK9 inhibition. Mol Oncol. 2017 doi: 10.1002/1878-0261.12152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cooper MJ, Cox NJ, Zimmerman EI, Dewar BJ, Duncan JS, Whittle MC, et al. Application of multiplexed kinase inhibitor beads to study kinome adaptations in drug-resistant leukemia. PLoS One. 2013;8(6):e66755. doi: 10.1371/journal.pone.0066755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jones CL, Gearheart CM, Fosmire S, Delgado-Martin C, Evensen NA, Bride K, et al. MAPK signaling cascades mediate distinct glucocorticoid resistance mechanisms in pediatric leukemia. Blood. 2015;126(19):2202–12. doi: 10.1182/blood-2015-04-639138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fukuda S, Pelus LM. Survivin, a cancer target with an emerging role in normal adult tissues. Mol Cancer Ther. 2006;5(5):1087–98. doi: 10.1158/1535-7163.MCT-05-0375. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
To support data availability and research transparency, we provide an Omics Data Browser. This tool provides researchers with the means to interrogate the data pertaining to this study with their genes of interest (provided in capitals, e.g. PIK3CA) and to retrieve graphical representations of the quantitative data, similar to the heatmap shown in Fig. 2. The underlying data in this Omics Data Browser comprises the mRNA Gene Expression, RPPA, global proteome and phosphoproteome and the mass spectrometry based kinome profiling data. The accessible data includes all WHIM tumors and all treatments, with global LC-MS/MS-based phosphoproteomic data normalized to respective protein levels. The Omics Data Browser can be retrieved at http://prot-shiny-vm.broadinstitute.org:3838/BKM120/ (with the password [BKM120viewer!]).
Figure 2. Genomic alterations and expression levels of key PI3K markers and signaling activities by different omic analysis of vehicle-treated PDX tumors.
A, copy number variation (CNV) and mutational status of PI3K pathway components from the whole exome sequencing data [12]. B, standardized expression levels of selected markers compared among the transcriptome (mRNA), global proteome (MS prot), global phosphoproteome (MS amino acid and site #), RPPA protein (RPPA prot), RPPA phosphosites (RPPA amino acid and site #) and kinome pulldown data (MIB). Selected PI3K markers show varying biology between different PDX tumors. Expression levels are z-scored. Both vehicle treatments (2 hours and 50 hours) are shown for each PDX tumor. C, a single sample gene set enrichment analysis for individual vehicle-treated tumors across all platforms shows enrichment for a selected set of gene-sets/pathways. Both vehicle treatments (2 hours and 50 hours) are shown for each PDX model. Good correlation for each PI3K marker and gene-set/pathway was observed between -omic types, with striking differences between the various PDX animals.