

Abstract
Amyloid aggregation of amyloid-beta (Aβ) and islet amyloid polypeptide (IAPP) is associated with Alzheimer’s disease (AD) and type-2 diabetes (T2D), respectively. With T2D being the risk factor for AD and the ability of IAPP to cross the blood-brain barrier, the co-aggregation of Aβ and IAPP has been explored to understand the cross-talk between the two diseases. Recent studies demonstrated that soluble IAPP could significantly accelerate the aggregation of Aβ while pre-formed amyloids of IAPP were poor “seeds” for Aβ aggregation. Here, we apply all-atom discrete molecular dynamics simulations to investigate possible molecular mechanisms for the accelerated co-aggregation of IAPP and Aβ42 comparing to Aβ42 aggregation alone, which was confirmed by the complementary thioflavin-T fluorescence assay. Our simulation results suggest that peptides in the mixture tend to form heterodimers as the first step towards their co-aggregation. Strong inter-peptide interactions with IAPP in the heterodimer shift the helical conformation of Aβ42 in its amyloidogenic central hydrophobic core, residues 16-22 (Aβ16-22), to the extended conformation ready to form β-sheets. Our study suggests that the unfolding of Aβ16-22 helix contributes to the aggregation free energy barrier and corresponds to the rate-limiting conformational change for Aβ42 aggregation. Therefore, we propose that soluble IAPP promotes the aggregation of Aβ42 by binding-induced conformational change of Aβ42 in its amyloidogenic core and thus reduced aggregation free energy barrier.
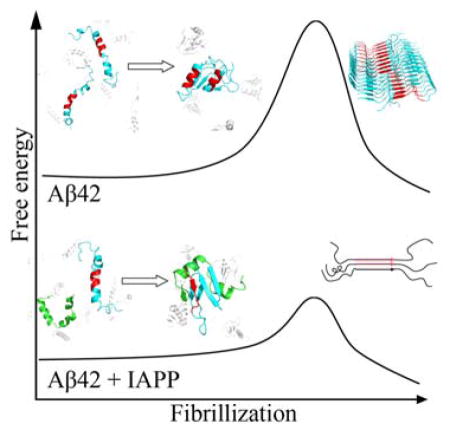
Graphical Abstract
Introduction
Accumulating clinical and epidemiological research suggest that type-2 diabetes (T2D) and Alzheimer’s disease (AD), two of the most common aging-associated diseases, are linked to each other.1–5 Both diseases have shared physiological processes, overlapping pathological abnormalities, and a relatively frequent appearance of mixed pathologies with T2D being found as a major risk factor for AD.6–11 A pathological hallmark common to both diseases is amyloid aggregation, where aberrant aggregation of amyloid-β (Aβ) and islet amyloid polypeptide (IAPP) is implicated in AD and T2D, respectively.12–15 Both peptides can self-assemble into amyloid fibrils16–18 and accumulate in human tissues as amyloid deposits. Given experimental evidences of IAPP being able to cross the blood-brain barrier19 and possibly expressed in sensory neurons,20 the cross- or co-aggregation of Aβ and IAPP has been explored to uncover the cross-talk between these two devastating diseases.21
Amyloid aggregation is a nucleation-dependent process with an “all-or-none” sigmoidal kinetics, where an initial lag phase of nucleation is followed by rapid elongation and saturation.22–24 While both Aβ and IAPP can form amyloid fibrils with similar characteristic of cross-β structures, the two peptides have distinctively different aggregation rates. Compared to Aβ of the same concentration, IAPP aggregates faster with significantly shorter lag phases in vitro. The lag phase can be shortened or eliminated by adding pre-formed amyloids, namely the “seeding” effect. Experiments in vitro have investigated the “cross-seeding” interactions between Aβ and IAPP,25,26 and revealed that amyloid of either one of the peptides can seed the aggregation of another, usually with a lower efficiency compared to the “self-seeding”.25 With experimentally-derived structural models of both Aβ and IAPP fibrils,27–31 molecular dynamic (MD) simulations have provided useful molecular insights into their cross-seeding interactions.11 In the absence of amyloid “seeds”, the aggregation of Aβ can also be accelerated with non-aggregated IAPP. The mixture of soluble Aβ and IAPP was found to have much shorter aggregation lag phase than Aβ alone but slightly longer than IAPP,32,33 although IAPP was also reported to delay the aggregation of Aβ.5 Based on the observation that the mixture aggregated in a similar manner as the self-aggregation of typical amyloidogenic peptides (e.g., in terms of aggregation kinetics, fibril morphology, and the ability to disrupt membranes), it was postulated that Aβ and IAPP form hetero-complexes first and the complexes subsequently aggregate.32 Experiments using electrospray ionization-ion mobility spectrometry-mass spectrometry (ESI-IMS-MS)-based method confirmed the formation of Aβ/IAPP hetero-complex and hetero-fibrils.33 Conformational changes upon the formation of hetero-complexes were also detected, but the molecular details and mechanisms of Aβ and IAPP co-aggregation remain unknown.
Here, we study the interactions between Aβ42 and IAPP, their formation of hetero-complexes, and the corresponding structures and conformational dynamics upon binding in order to understand their co-aggregation behaviors by combining atomistic discrete molecular dynamics (DMD) simulations with complementary thioflavin-T fluorescence assay (ThT). DMD is a special flavor of MD algorithm with high computational efficiency and predictive power, which has been extensively used by our group and others to study protein folding, protein dynamics, and amyloid aggregation.34–36 Our ThT assay first confirmed the previously reported acceleration of amyloid aggregation of Aβ/IAPP mixture compared to Aβ alone.32,33 Using DMD simulations, we identified the Aβ-IAPP heterodimer as the most populated hetero-complexes during the early stage of co-aggregation when mixed at equimolar ratio in silico. The residue-wise inter-peptide interactions revealed by DMD simulations are consistent with experimentally identified hot-spot regions for Aβ-IAPP binding.37 Our simulation results also suggested the helix unfolding of Aβ amyloidogenic region (Aβ16-22, a.k.a. the central hydrophobic core) as the major conformational change upon the formation of Aβ-IAPP heterodimer. The helical conformation of Aβ16-22 was more stable in Aβ42 dimer simulations. Using an approach proposed by the Wetzel group,23 analysis of Aβ42 aggregation kinetics by the ThT assay suggested that the rate-limiting step of Aβ42 aggregation was conformational changes taking place in approximately two peptides. Together with our computational results and previous reports of Aβ42 oligomerization,38,39 we propose an aggregation mechanism that the helix unfolding of Aβ16-22 corresponds to the major free energy barrier of Aβ42 aggregation, which can take place in various Aβ42 oligomers populated during the early stage of aggregation. Upon mixing with IAPP, the formation of Aβ-IAPP heterodimer induces the helix unfolding of Aβ16-22, which subsequently reduces the aggregation free energy barrier and accelerates the co-aggregation compared to Aβ42 alone. Our results offer a potential molecular mechanism for the increased AD risk in T2D patients as observed in clinical and epidemiological studies.
Results and Discussion
In vitro studies of Aβ42 and IAPP self- and cross-aggregation
Given different experimental results in the literature on the rates of Aβ/IAPP co-aggregation with respect to the self-aggregation of Aβ,5,32,33 we first applied the ThT fluorescence assay to study the self- and cross-aggregation of Aβ42 and IAPP (Methods). Mixtures of soluble Aβ42 and IAPP at equimolar ratio with different concentrations were tested (1.0, 2.0, 2.5, 3.0, 3.5, 4.0, and 5.0μM accordingly) and compared with control groups of individual peptides aggregating at the same concentrations. At least three repeats were done for each molecular system at a given concentration. All samples were subjected to the same shaking condition at 37°C to allow fast measurements of protein aggregation. Sigmoidal aggregation kinetics was observed in all peptide systems (e.g., Fig. S1), and the corresponding lag time and fibril elongation rate were obtained by fitting the data with an empirical sigmoidal function (Fig. 1A,B). Our results were consistent with previous experimental reports that IAPP is more aggregation prone than Aβ42 with shorter lag phases for all the tested concentrations.40,25 The lag time of the mixture was comparable to the IAPP alone and significantly reduced compared to Aβ42 alone, consistent with previous experimental reports that mixing of Aβ and IAPP retarded the rate of IAPP assembly while accelerated the fibril formation by Aβ.32,33 Although experimental studies in Ref.32 focused on the Aβ/IAPP co-aggregation in the presence of membrane, aggregations in the solution were also observed. Similarly, IAPP had higher aggregation elongation rates than Aβ at the same concentrations and the mixture had the corresponding values in between (Fig. 1B).
Figure 1.

In vitro studies of Aβ-IAPP cross- and self-association. Seven different concentrations from 1 to 5 μM were studied with each kinetic measurement performed at least in triplicate. The concentration dependence of (A) the aggregation lag time and (B) fibril elongation rate for Aβ-IAPP co-aggregation (blue), and the self-aggregation of Aβ (black) and IAPP (red). (C) For the aggregation of Aβ, the amount of aggregates during the initial phase of aggregation linearly depends on the square of time, t2. (D) The log-log plot of the slops in panel C vs. the peptide concentrations shows the power-law dependence. The linear fit in the log-log plot results into an exponent index of ~3.54s.
Although the ThT assay could not distinguish whether the signals were from IAPP fibrils, Abeta fibrils or the heterofibrils, the observation of peptide mixtures displaying single-sigmoidal aggregation kinetics in our experiments and others5,32,33 implied interactions between IAPP and Aβ42 during their co-aggregation. If each type of peptides aggregated independently, the aggregation kinetics of a peptide mixture should feature a simple superposition of two sigmoidal aggregation curves. Indeed, recent mass spectrometry experiments33 suggested the co-aggregation of the IAPP/Abeta hetero-complex and demonstrated the formation of heterofibrils. However, the structure and dynamics of these IAPP/Abeta hetero-complex is unknown.
Heterodimer formation is the first step towards the co-aggregation of Aβ42 and IAPP
We next performed all-atom DMD simulations of multiple Aβ42 and IAPP at 1:1 ratio to investigate the assembly dynamics of the peptide mixture. We studied two sets of molecular systems with one containing two Aβs and two IAPPs and another having four peptides of each species, where the same peptide concentration was maintained (Methods). For the smaller system, we performed 50 independent simulations with different initial inter-molecular distances and orientations at 300K. Each simulation lasted 200ns and thus an accumulative 10μs simulation was obtained. For the larger system, 20 independent simulations were performed with each lasted 100ns and a total of 2μs simulation time.
To capture the assembly dynamics, we calculated the number of atomic contacts between various peptides and also monitored the formation of peptide clusters. A peptide belonged to a cluster if it made contacts with at least one of the member peptides. When two Aβs and two IAPPs were mixed, the total number of contacts was dominated by the cross-species interactions between Aβ42 and IAPP, while the binding between Aβs and also between IAPPs was significantly smaller (Fig. S2). The number of Aβ-IAPP contacts increased rapidly in the early stage of simulations with obvious lag-time observed for both Aβ-Aβ and IAPP-IAPP interactions, suggesting an initial hetero-complexes formation between Aβ and IAPP in their mixture. For the peptide clustering analysis, we computed the time evolution of various cluster species weighted by the number of peptides in a cluster (Fig. 2A). We observed the rapid formation of Aβ-IAPP heterodimer following a decrease of initial Aβ42 and IAPP monomers. The heterodimer was the most populated species before they finally associated into the hetero-hexamer. A similar association dynamics was also observed for the larger system containing four Aβs and four IAPPs (Fig. 2B). The heterodimer was also the most populated species during the early assembly process, although other higher order oligomers (such as two IAPPs bound to one or three Aβs) were later observed before the appearance of hetero-octamer.
Figure 2.

The time evolution of peptide cluster species for Aβ-IAPP mixtures with molecular ratios of (A) 2:2 and (B) 4:4. The number of each species weighted by the number of peptides forming the cluster was averaged over the number of independent simulations. For simulations with 2:2 ratio, all possible cluster species were shown. In the case of 4:4 ratio, only the six most-populated cluster species were shown.
Hence, our simulations suggested that when Aβ42 and IAPP peptides were mixed at an equimolar ratio, the cross-species interaction dominates over their self-associations during their early assembly. The rapid formation of hetero-complexes could be explained by the opposite net charges of the two peptides (i.e., the net charges of Aβ and IAPP are −3e and +2e, respectively) and the relatively long-range electrostatic interactions under the physiological condition (the Debye length is ~10Å). Our results also indicated that the heterodimer of Aβ42 and IAPP is the major intermediate species populated towards their co-aggregation.
Computationally observed inter-peptide interactions for both cross- and self-association of Aβ42 and IAPP are consistent with previous experiments
The hot-spot regions for both cross- and self-association of Aβ42 and IAPP have been investigated experimentally.37 For experimental validation of our simulations, we performed DMD simulations of one Aβ42 with one IAPP mixture (i.e., cross-association), along with control simulations of two Aβs and two IAPPs (i.e., self-association). For each molecular system, we carried out 20 independent simulations at 300K with each run lasted for 600ns. We used the last 300ns trajectories to compute the residue-wise binding frequency maps (Fig. 3). From simulations of Aβ-IAPP cross-association, we identified several “hot regions” with high binding frequencies, including residues 17-24 and 27-42 in Aβ42, as well as residues 8-18 and 22-28 in IAPP (Fig. 3A). The same regions were also important for the self-association of Aβ42 and IAPP (Fig. 3B,C). Shared features were observed between cross- and self-associations. For instance, an “anti-parallel” binding pattern (i.e., perpendicular to the diagonal) due to interactions between C-terminal residues of Aβ was present in both Aβ-IAPP and Aβ-Aβ contact frequency maps (Fig. 3A,B). Similar patterns formed by the hot-spot residues of IAPP (e.g., 8-18 and 22-28) could also be found in both Aβ-IAPP and IAPP-IAPP maps (Fig. 3A,C). Our observations agree very well with previous experimental reports, in which the hot regions for cross- and self-associations were determined as residues 19-22, 27-32, 35-40 in Aβ, and residues 8-18, 22-28 in IAPP.37 These similarities suggest a common binding mechanism in cross- and self-associations of Aβ42 and IAPP, which may arise from their similarity in sequences.41–43 Both Aβ42 and IAPP are amyloidogenic with about 50% similarity in sequences, and the sequences with the highest degree of similarity are 15-21, 26-32 in Aβ42 and 10-16, 21-27 in IAPP (Fig. 3D), all of which are within or close to the hot regions determined both experimentally and computationally. Therefore, the regions important for both Aβ42 and IAPP self-associations also mediate their cross-association, and their cross- and self-associations could occur in a competitive manner.
Figure 3.

Identification of hot regions for inter-peptide interactions in both cross- and self-associations of Aβ and IAPP. Residue-wise contact frequency maps were computed for (A) Aβ and IAPP binding in heterodimer simulations, and self-association of (B) Aβ and (C) IAPP in dimer simulations. Histograms were also obtained to show the total contact frequency of each residue. (D) Sequence fragments with the highest degree of similarity between Aβ and IAPP were highlighted in orange and blue, while the domains experimentally-identified to be important for both their cross- and self-associations were highlighted in gray.
Conformational changes of Aβ42 and IAPP heterodimer upon cross-association
We further studied the structural properties of the Aβ-IAPP heterodimer. For comparison, we also performed simulations of Aβ42 and IAPP monomers and dimers, following the same protocol - i.e., 20 independent simulations of 600ns at 300K, with each of which the last 300ns trajectory was used to compute the secondary structure propensities (Fig. 4, Fig. S3). For the Aβ monomer, the amyloidogenic region (Aβ16-22) was partially helical, while the N- and C-termini mostly adopted β-sheet conformation (Fig. 4A, B). To evaluate whether the observed helical propensity in Aβ16-22 depended on the starting conformation (PDB ID: 1Z0Q44) where the solution NMR structure solved in the water/hexafluorosiopropanol mixture (70:30 volume ratio) contains partial helixes around the amyloidogenic Aβ16-22, we also performed similar simulations starting from the fully extended conformation (Fig. S4). We found that Aβ16-22 rapidly adopted the partially helical conformation and the helical propensities per residue were similar to simulations starting from the helical structure. Our observation of the partially helical structure in Aβ16-22 was also consistent with previous all-atom MD studies with different force fields.45,46 As for the IAPP monomer, we observed a stable helix in the N-terminus and the amyloidogenic IAPP22-29 mostly unstructured with a weak helical propensity (Fig. 4C,D and Fig. S3), consistent with a recent NMR study of isolated IAPP in solution.47 Compared to monomers, both Aβ42 and IAPP homodimers showed an overall increase of α-helix (Fig. 4A,C), consistent with many experimental observations of the increased α-helical contents during the early stage of amyloid aggregation of Aβ and IAPP before the final conversion to β-rich fibrils.48,49 Interestingly, the β-sheet content of Aβ16-22 in the homodimer was smaller than that in the monomer (Fig. 4B), but the β-sheet content of IAPP22-29 in the homodimer increased compared to the monomer (Fig. 4D). The differential propensities of Aβ42 and IAPP amyloidogenic cores to form β-sheet structures upon self-association might contribute to their different aggregation rates (Fig. 1A). In contrast, the heterodimer had the β-sheet propensity increased while helix propensity decreased for both Aβ and IAPP compared to the monomers, especially in the corresponding amyloidogenic regions (e.g., Aβ16-22 and IAPP22-29 in Fig. 4). Especially, the β-sheet propensity of IAPP22-29 was significantly increased in the heterodimer compared to IAPP monomer and homodimer (Fig. 4D). Given the importance of these amyloidogenic regions in forming the cross-β architecture in the mature amyloid fibrils,50 the observed conformational changes from α-helix to β-sheet in the heterodimer with respect to corresponding monomers and homodimers supports the promoted co-aggregation observed in experiments. Because of the relatively high helical propensities of Aβ16-22 and IAPP22-29 in monomers, homodimers and possibly high-order oligomers and the fact that unfolding of these helixes likely corresponds to the first step toward the formation of β-rich fibrils, we next focused our analysis on the helix unfolding of corresponding amyloidogenic regions of Aβ42 and IAPP in DMD simulations to capture the early events co-aggregation.
Figure 4.

The secondary structure propensity for each Aβ and IAPP residue in monomer, heterodimer, and homodimer. The ordered secondary structures of (A, C) helix propensity and (B, D) β-sheet were shown while the less ordered coil and turn were shown in Fig. S2. All data were averaged over the last half trajectories of 20 independent simulations, each of which lasted 600ns. Error bars denote the standard error of means (SEM).
Helix unfolding of Aβ16-22 is the major conformational change upon binding with IAPP in the heterodimer
To better understand the conformational dynamics of Aβ42 and IAPP upon association as the heterodimer, we further analyzed the simulation trajectories of the monomers and the heterodimer by focusing on their corresponding amyloidogenic regions. Following a similar approach in protein folding studies,51,52 we monitored the folding and unfolding of helixes in the amyloidogenic regions - i.e., Aβ16-22 and IAPP22-29 (Methods). During simulations of Aβ42 monomer, we could observe the dynamics of helix unfolding and refolding of the amyloidogenic Aβ16-22 (e.g., 22 unfolding and 9 refolding events), where the population of the helical state was approximately 61% (Fig. S5). In the presence of IAPP, the number of helix refolding events was drastically reduced (e.g., 18 unfolding and only 1 refolding events) and the population of the helical state was reduced to ~57%. In terms of IAPP22-29, the helix population was slightly changed from ~21% in monomer to ~20% in heterodimer, and the helix unfolding/refolding dynamics in heterodimer were also slower but still frequent (i.e., from 72/56 to 34/18). These observations indicated that the binding between Aβ42 and IAPP in the heterodimer increased the free energy barriers of helix unfolding/refolding of their corresponding amyloidogenic regions. For IAPP22-29, the barrier increase did not change the equilibrium between helical and non-helical conformations, and the frequent unfolding/refolding dynamics suggested that the computational observation was significant. The conformation dynamics of Aβ16-22, on the other hand, was shifted towards the non-helical state upon binding with IAPP. With the rare observation of helix refolding events of Aβ16-22 suggesting a slow conformational dynamics, the conformational equilibrium might not have been reached in our constant temperature simulations (e.g., up to 600ns in each independent simulation).
In order to better sample the conformational equilibrium of the heterodimer, we applied replica exchange DMD (REXDMD) simulations, where parallel simulations at different temperatures were performed with Monte-Carlo based periodic exchanges between replicas of neighboring temperatures.53 Although the interpretation of kinetics might be challenging compared to serial ones at constant temperatures, replica exchange simulations have been widely applied for enhanced sampling of thermodynamic equilibrium by overcoming energy barriers at high temperatures. We performed REXDMD simulations for Aβ42 monomer, IAPP monomer, and the heterodimer (Methods). Each replica of simulations lasted 200ns, and trajectories of the last 150ns were used in data analysis. Using the weighted histogram analysis method (WHAM),54 we computed the specific heat (Cv) as the function of temperature (Fig. S6A). A peak in the Cv plot usually corresponds to conformational transitions around the temperature with the heights indicating the transition cooperativity. To uncover the details of conformational transitions, we also computed the temperature dependence of the number of inter-peptide contacts in the heterodimer (Fig. S6B) and secondary structure contents of both Aβ and IAPP in monomers and heterodimer simulations (Fig. S6C,D). The weak Cv peak of IAPP around ~305K corresponded to the gradual and non-cooperative unfolding of helixes (Fig. S6D), while the peak of Aβ at a higher temperature (~325K) resulted from unfolding of both helices and strands (Fig. S6C). Remarkably, the heterodimer displayed a high Cv peak around ~322K mainly due to the dissociation of a large number of inter-peptide contacts (Fig. S6B). Compared to the monomers, the strong inter-peptide binding in the heterodimer significantly reduced the helical content of Aβ (mainly in the amyloidogenic region of Aβ16-22) and increased its β-sheet content (Fig. S6C). For IAPP, Aβ binding did not significantly change the conformational dynamics with only a slight increase of the β-sheet content (Fig. S6D). Therefore, our REXDMD simulations confirmed the IAPP-binding induced helix unfolding of Aβ16-22 as observed in constant temperature simulations (Fig. 4, Fig. S5).
To better characterize the conformational change of Aβ16-22 upon IAPP binding, we further computed the potential mean force (PMF, i.e., the effective free energy landscapes) of the fragment with respect to the number of residues in helical content and the end-to-end distance at 300K (Fig. 5, details see Methods). For both monomer and heterodimer, four major basins could be observed, corresponding to helical, partially helical and non-helical states with either compact or extended conformations (e.g., typical snapshot structures in Fig. 5). The partially unfolded state corresponded to the intermediate of helix unfolding and the saddles connecting basins denote the folding pathways. Compared to Aβ16-22 in the monomer, the binding with IAPP rendered the non-helical basins deeper, the helical and partially helical states shallower, and the saddle between the non-helical and the intermediate states broadened. Therefore, our results suggested the binding with IAPP in the heterodimer at 300K shifted the conformational equilibrium of Aβ16-22 from helix towards extended non-helical conformational states, ready to form β-sheets (e.g., the snapshots in Fig. 5).
Figure 5.

The conformational free energy landscape of the amyloidogenic Aβ16-22 at 300K in simulations of (A) the Aβ42 monomer and (B) the Aβ42-IAPP heterodimer. Using the WHAM analysis of the replica exchange simulation trajectories, the 2D PMF was computed as the function of the number of helical residue and end-to-end distance. The basins correspond to helical (a, α), partially helical intermediate (b, β), and non-helical (c, d and γ, δ) states. Aβ peptides were shown as cartoon in cyan and IAPP in green. The amyloidogenic region of Aβ16-22 was highlighted in red.
Potential roles of helix-unfolding of Aβ16-22 in Aβ42 self-aggregation and co-aggregation with IAPP
Based on a nucleated polymerization mechanism, the Wetzel group proposed an aggregation kinetics analysis approach to determine the critical number of monomers, n*, the conformational changes of which are rate-limiting for aggregation.22,23 Briefly, during the initial growth phase before the rapid elongation, the increase of aggregation linearly depends on the square of time, t2, and the slope is the power function of peptide concentration with the exponent index corresponding to n*+2. Given the fast aggregation of IAPP and IAPP-Aβ mixture and the resulting limited number of data points with high noise-to-signal ratio for detailed analysis of the initial phases, we only applied the above analysis to the aggregation of Aβ. We selected data corresponding to the initial phase of Aβ aggregation, and estimated the amount of aggregates based on the ThT fluorescence intensity signal (Methods). For each concentration, we determined the slope with respect to t2 (e.g., Fig. 1C). Base on the log-log plot of the slopes vs. concentrations (Fig. 1D), we obtained the exponent of ~3.5 and thus the value of n* ~1.5.
Many previous experiments have reported the observation of Aβ oligomers such as pentamers and hexamers during the initial phase of amyloid aggregation.55 Here, our aggregation kinetics analysis suggests that the rate-limiting step of Aβ42 aggregation requires the conformational changes of approximately 2 peptides, which can take place within the initially formed Aβ oligomers. Based on our simulation results, these rate-limiting conformational changes correspond to the unfolding of α-helix in Aβ16-22. The relatively slow aggregation of Aβ42 observed in vitro might result from the stabilization of the helical conformation in Aβ16-22 in the self-associated oligomers such as homodimers (Fig. 4). Upon binding with IAPP in the mixture, the strong inter-peptide interactions shift the conformational equilibrium of Aβ16-22 from helical to the extended conformation (Fig. 5), which in turn reduce the aggregation free energy barrier as in the isolated Aβ peptides and thus shorten the aggregation lag time (Fig. 1).
Conclusion
Combining all-atom DMD simulations with the complementary ThT assay, we investigated the molecular mechanism for the accelerated co-aggregation of Aβ42 and IAPP mixture compared to the self-aggregation of Aβ alone. Our simulation results suggest that the formation of Aβ-IAPP heterodimer is the first step towards their co-aggregation due to electrostatic attraction between the two peptides with opposite net charges. The computationally determined residue-wise inter-peptide interactions between Aβ and IAPP in the heterodimer agree very well with previous experimental results of the hotspot regions for their cross-association. Detailed analysis of our serial simulations at constant temperature and replica exchange simulations at multiple temperatures revealed that the strong inter-peptide interactions in the heterodimer of Aβ and IAPP introduce the unfolding of helix in the amyloidogenic region of Aβ16-22, which makes Aβ ready for aggregation. The helix of Aβ16-22 was otherwise stabilized in the homodimer of Aβ42, consistent with the experimentally observed accumulation of helical contents before the rapid conversion into β-rich aggregates.48,49
Our ThT experiments confirmed previous experimental reports that the lag phase of Aβ and IAPP co-aggregation is shorter than that of Aβ alone but only slightly longer than IAPP. The kinetics analysis based on the nucleated growth polymerization theory indicates that the critical number of peptides, whose conformational change is the rate-limit step for aggregation, is approximately 2. With experimental observations of various Aβ42 oligomers and the in-registered β-sheet structures of Aβ16-22 in the core of Aβ42 amyloid fibrils, our combined computational and experimental studies suggest that the nucleation of fibrillization corresponds to the helix unfolding of central hydrophobic cores (i.e., 16-22) of approximately two peptides in these oligomers and the subsequent formation of inter-peptide β-sheets in this region.50 Therefore, the binding of IAPP with Aβ in the mixture reduces the free energy barrier for Aβ aggregation - i.e., the helix unfolding of the central hydrophobic core of Aβ16-22, and thus promotes the co-aggregation with reduced lag time for aggregation nucleation. With experimental evidences of IAPP being able to cross the blood-brain barrier,19 the overproduction of IAPP in T2D, known as the hyperamylinemia, may result into increased IAPP concentration in the brain, which in turn promotes the formation of toxic aggregates with Aβ42 and increases the risk of AD. While further experimental studies are necessary to validate our predicted co-aggregation mechanism of IAPP and Aβ42 and potential contribution to the cross-talk between AD and T2D, we hope our study will stimulate further computational and experimental studies to unravel this intriguing phenomenon with high biological and medical relevance.
Materials and Methods
Discrete molecular dynamics (DMD) simulations
DMD is a special approach of molecular dynamics, in which discrete step functions instead of continuous potentials are used to model inter-atomic interactions.56 The step-function potentials are adapted from the continuous Medusa force field57 to model inter-atomic interactions. The non-bonded inter-atomic interactions include van der Waals (VDW), solvation, hydrogen bond and electrostatic terms. The VDW parameters were adopted from the CHARMM force field58 and the EEF1 implicit solvent model59 was used to model the solvation term. The hydrogen bond interactions were implicitly modeled with a reaction-like approach.60 The Debye-Hückel approximation is applied to model the screened electrostatic interactions with the Debye length assigned ~10Å. We used the Anderson’s thermostat to maintain the temperature, which is fixed at 300K except for the replica exchange simulations.61 To ensure sufficient sampling and avoid potential bias arise from starting configurations, we perform multiple independent simulations for each molecular system with different initial conditions, including randomized velocities, intermolecular distances and orientations.
Simulation setup
In all simulations, we used the Aβ42 (PDB ID: 1Z0Q) and IAPP (PDB ID: 2L86) structures obtained from PDB to construct our systems. Counter ions Cl− and Na+ were also introduced into systems to achieve a neutral charge condition if necessary. For both Aβ and IAPP peptides, we maintained the peptide concentration same as that of a single peptide in a cubic box with the dimension of 83Å in all simulations. The periodic boundary condition was used. In replica exchange simulations for Aβ and IAPP monomers, five replicas were running at 275, 295, 315, 335, and 355K respectively, while eight replicas running at 275, 290, 300, 310, 320, 330, 340, and 355K were performed to model the Aβ-IAPP heterodimer. The temperatures were chosen to ensure that the simulation exchange rates were between 30% to 70%. During the analysis of simulation trajectories, we used the DSSP program to compute protein secondary structures, and the weighted histogram analysis method (WHAM) to evaluate the temperature dependence of thermodynamic quantities.54,62
Folding/Unfolding analysis
For the characterization of helix unfolding and refolding, we based on two criterions: a helical to non-helical state conversion was considered if a structure contains at least five helical residues inside amyloidogenic region converted into one contains no more than two helical residues, and the conversion would be determined as valid and recorded only if the new state could last for no less than 0.5ns simulation time. Non-helical to helical state conversion was identified in a similar way. Structural percentage and the number of conformational change were recorded according to all simulation trajectories.
Calculation of the potential mean force (PMF)
The two-dimensional PMF (or effective free energy) was computed according to
| (1) |
where KB is the Boltzmann constant, T corresponds to the simulation temperature 300K, and P(Nhelical, Dend-to-end) is the probability of finding a Aβ16-22 segment containing Nhelical helical residues, with an end-to-end distance of Dend-to-end at the time.
Thioflavin-T Fluorescence Assay
To study the kinetics of Aβ42 and IAPP amyloid fibrils formation in vitro, human β-Amyloid (1-42) and IAPP were purchased from GeneScript (RP10017) and Sigma-Aldrich Co. LLC (D2162), respectively. The lyophilizaed powder of Aβ42 or IAPP were first solubilized in 100% hexafluorosiopropanol (HFIP) (Sigma) at ~1mM and sonicated at 30% power efficiency for 30s (Qsonica Q125) to break preformed aggregation. The concentration was then determined spectrophotometrically using a calculated extinction coefficient at 280nm. All the water buffer used in ThT assay was applied to HisTrap Column to remove trace amount bivalent ions and filtrated by 0.22 um filter. Considering the aggregation rate for both IAPP and Abeta42, while mixing samples we always kept adding Abeta42 firstly into buffer and immediately add IAPP prior to measurements. All dilution was made in HFIP before the measurements, and the final concentration of HFIP were kept consistent by adding equal volume of diluted Abeta42 and IAPP at various concentrations. The Aβ42 and IAPP pre-treated in this way were then used to prepare serial dilution of single compound samples or 1:1 mixtures of concentrations from 1 to 5μM or 1uM:1uM to 5uM:5uM in 100mM Tris-HCl (pH 7.5), 100mM NaCl, 200μM EDTA and 15μM thioflavin T (ThT). All buffers were pass through HiTrap Chelating HP Column (GE Healthcare) to remove trace amount of endogenous divalent cations. The assays were performed in sealed Corning 96 well clear bottom half area, nonbinding surface plates and measured by exciting the ThT in samples at 440nm and reading the emission at 485nm at a constant temperature of 37°C using BioTek Synergy H1 Hybrid Reader with continuous shaking at 425cpm between measurements. All kinetic measurements were performed at least in triplicate. The fluorescent ThT data with time data were fitted to an empirical equation to reproduce the change of fluorescent intensity ThT upon binding to the amyloid fibrils (Imax-I0), the apparent rate constant for fibrillization (k), and the lag time (tlag=t0-2/k) to represent the time of nucleation before detectable amyloid formation:
| (2) |
where I, I0 and Imax are the reading, initial and maximum fluorescence values, t0 is the time required to reach half change of intensity.
Nucleation Kinetics
The details describing the nucleated growth polymerization mechanism could be found elsewhere.22,23 The important kinetics feature of the initial phase of aggregation we examined here is the linear dependence of the aggregation content on the time square (t2), and the power-law dependence of the corresponding slope on the peptide concentration. The exponent index corresponds to n*+2, where n* is the critical number of peptides whose conformational changes are the rate-limiting step for aggregation nucleation. By fitting the ThT assay data with the sigmoidal equation (Eq. 2), we chose the initial phase with t<tlag and estimated the amount of aggregation by normalizing them based on concentration conditions, ρ(I-I0)/Imax, with ρ corresponding to the total peptide concentration.
Supplementary Material
Acknowledgments
The work is supported in part by NSF CAREER CBET-1553945 and NIH MIRA R35GM119691. The content is solely the responsibility of the authors and does not necessarily represent the official views of NIH and NSF.
Footnotes
The Supporting Information is available free of charge on the ACS Publications website: Supplementary Figures S1–S5.
References
- 1.Hofman A, Ott A, Breteler MM, Bots ML, Slooter AJ, van Harskamp F, van Duijn CN, Van Broeckhoven C, Grobbee DE. Atherosclerosis, Apolipoprotein E, and Prevalence of Dementia and Alzheimer’s Disease in the Rotterdam Study. Lancet Lond Engl. 1997;349(9046):151–154. doi: 10.1016/S0140-6736(96)09328-2. [DOI] [PubMed] [Google Scholar]
- 2.Crane PK, Walker R, Hubbard RA, Li G, Nathan DM, Zheng H, Haneuse S, Craft S, Montine TJ, Kahn SE, et al. Glucose Levels and Risk of Dementia. N Engl J Med. 2013;369(6):540–548. doi: 10.1056/NEJMoa1215740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de la Monte SM, Wands JR. Alzheimer’s Disease Is Type 3 Diabetes-Evidence Reviewed. J Diabetes Sci Technol. 2008;2(6):1101–1113. doi: 10.1177/193229680800200619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de la Monte SM, Tong M, Lester-Coll N, Plater M, Wands JR. Therapeutic Rescue of Neurodegeneration in Experimental Type 3 Diabetes: Relevance to Alzheimer’s Disease. J Alzheimers Dis JAD. 2006;10(1):89–109. doi: 10.3233/jad-2006-10113. [DOI] [PubMed] [Google Scholar]
- 5.Yan L-M, Velkova A, Tatarek-Nossol M, Andreetto E, Kapurniotu A. IAPP Mimic Blocks Aβ Cytotoxic Self-Assembly: Cross-Suppression of Amyloid Toxicity of Aβ and IAPP Suggests a Molecular Link between Alzheimer’s Disease and Type II Diabetes. Angew Chem Int Ed. 2007;46(8):1246–1252. doi: 10.1002/anie.200604056. [DOI] [PubMed] [Google Scholar]
- 6.Li L, Hölscher C. Common Pathological Processes in Alzheimer Disease and Type 2 Diabetes: A Review. Brain Res Rev. 2007;56(2):384–402. doi: 10.1016/j.brainresrev.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 7.Jackson K, Barisone GA, Diaz E, Jin L, DeCarli C, Despa F. Amylin Deposition in the Brain: A Second Amyloid in Alzheimer Disease? Ann Neurol. 2013;74(4):517–526. doi: 10.1002/ana.23956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oskarsson ME, Paulsson JF, Schultz SW, Ingelsson M, Westermark P, Westermark GT. In Vivo Seeding and Cross-Seeding of Localized Amyloidosis: A Molecular Link between Type 2 Diabetes and Alzheimer Disease. Am J Pathol. 2015;185(3):834–846. doi: 10.1016/j.ajpath.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 9.Yan L-M, Velkova A, Kapurniotu A. Molecular Characterization of the Hetero-Assembly of β-Amyloid Peptide with Islet Amyloid Polypeptide. Curr Pharm Des. 2014;20(8):1182–1191. doi: 10.2174/13816128113199990064. [DOI] [PubMed] [Google Scholar]
- 10.Qiu WQ, Wallack M, Dean M, Liebson E, Mwamburi M, Zhu H. Association between Amylin and Amyloid-β Peptides in Plasma in the Context of Apolipoprotein E4 Allele. PLOS ONE. 2014;9(2):e88063. doi: 10.1371/journal.pone.0088063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baram M, Atsmon-Raz Y, Ma B, Nussinov R, Miller Y. Amylin–Aβ Oligomers at Atomic Resolution Using Molecular Dynamics Simulations: A Link between Type 2 Diabetes and Alzheimer’s Disease. Phys Chem Chem Phys. 2016;18(4):2330–2338. doi: 10.1039/c5cp03338a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiti F, Dobson CM. Protein Misfolding, Functional Amyloid, and Human Disease. Annu Rev Biochem. 2006;75(1):333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 13.Soto C. Unfolding the Role of Protein Misfolding in Neurodegenerative Diseases. Nat Rev Neurosci. 2003;4(1):49–60. doi: 10.1038/nrn1007. [DOI] [PubMed] [Google Scholar]
- 14.Sipe JD, Benson MD, Buxbaum JN, Ikeda S, Merlini G, Saraiva MJM, Westermark P. Nomenclature 2014: Amyloid Fibril Proteins and Clinical Classification of the Amyloidosis. Amyloid. 2014;21(4):221–224. doi: 10.3109/13506129.2014.964858. [DOI] [PubMed] [Google Scholar]
- 15.Westermark P. Aspects on Human Amyloid Forms and Their Fibril Polypeptides. FEBS J. 2005;272(23):5942–5949. doi: 10.1111/j.1742-4658.2005.05024.x. [DOI] [PubMed] [Google Scholar]
- 16.Knowles TPJ, Vendruscolo M, Dobson CM. The Amyloid State and Its Association with Protein Misfolding Diseases. Nat Rev Mol Cell Biol. 2014;15(6):384–396. doi: 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- 17.Eisenberg D, Jucker M. The Amyloid State of Proteins in Human Diseases. Cell. 2012;148(6):1188–1203. doi: 10.1016/j.cell.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, Thompson MJ, Balbirnie M, Wiltzius JJW, McFarlane HT, et al. Atomic Structures of Amyloid Cross-[Beta] Spines Reveal Varied Steric Zippers. Nat Lond. 2007;447(7143):453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 19.Banks WA, Kastin AJ, Maness LM, Huang W, Jaspan JB. Permeability of the Blood-Brain Barrier to Amylin. Life Sci. 1995;57(22):1993–2001. doi: 10.1016/0024-3205(95)02197-q. [DOI] [PubMed] [Google Scholar]
- 20.Mulder H, Leckström A, Uddman R, Ekblad E, Westermark P, Sundler F. Islet Amyloid Polypeptide (Amylin) Is Expressed in Sensory Neurons. J Neurosci Off J Soc Neurosci. 1995;15(11):7625–7632. doi: 10.1523/JNEUROSCI.15-11-07625.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fawver JN, Ghiwot Y, Koola C, Carrera W, Rodriguez-Rivera J, Hernandez C, Dineley KT, Kong Y, Li J, Jhamandas J, et al. Islet Amyloid Polypeptide (IAPP): A Second Amyloid in Alzheimer’s Disease. Curr Alzheimer Res. 2014;11(10):928–940. doi: 10.2174/1567205011666141107124538. [DOI] [PubMed] [Google Scholar]
- 22.Ferrone F. [17] Analysis of Protein Aggregation Kinetics. Methods Enzymol. 1999;309:256–274. doi: 10.1016/s0076-6879(99)09019-9. [DOI] [PubMed] [Google Scholar]
- 23.Chen S, Ferrone FA, Wetzel R. Huntington’s Disease Age-of-Onset Linked to Polyglutamine Aggregation Nucleation. Proc Natl Acad Sci. 2002;99(18):11884–11889. doi: 10.1073/pnas.182276099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wetzel R. Nucleation of Huntingtin Aggregation in Cells. Nat Chem Biol. 2006;2(6):297–298. doi: 10.1038/nchembio0606-297. [DOI] [PubMed] [Google Scholar]
- 25.O’Nuallain B, Williams AD, Westermark P, Wetzel R. Seeding Specificity in Amyloid Growth Induced by Heterologous Fibrils. J Biol Chem. 2004;279(17):17490–17499. doi: 10.1074/jbc.M311300200. [DOI] [PubMed] [Google Scholar]
- 26.Moreno-Gonzalez I, Edwards G, III, Salvadores N, Shahnawaz M, Diaz-Espinoza R, Soto C. Molecular Interaction between Type 2 Diabetes and Alzheimer’s Disease through Cross-Seeding of Protein Misfolding. Mol Psychiatry. 2017 doi: 10.1038/mp.2016.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tycko R. Molecular Structure of Amyloid Fibrils: Insights from Solid-State NMR. Q Rev Biophys. 2006;39(1):1–55. doi: 10.1017/S0033583506004173. [DOI] [PubMed] [Google Scholar]
- 28.Luca S, Yau W-M, Leapman R, Tycko R. Peptide Conformation and Supramolecular Organization in Amylin Fibrils: Constraints from Solid-State NMR. Biochemistry (Mosc) 2007;46(47):13505–13522. doi: 10.1021/bi701427q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiao Y, Ma B, McElheny D, Parthasarathy S, Long F, Hoshi M, Nussinov R, Ishii Y. Aβ(1-42) Fibril Structure Illuminates Self-Recognition and Replication of Amyloid in Alzheimer’s Disease. Nat Struct Mol Biol. 2015;22(6):499–505. doi: 10.1038/nsmb.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lührs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Döbeli H, Schubert D, Riek R. 3D Structure of Alzheimer’s Amyloid-β(1–42) Fibrils. Proc Natl Acad Sci U S A. 2005;102(48):17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu J-X, Qiang W, Yau W-M, Schwieters CD, Meredith SC, Tycko R. Molecular Structure of β-Amyloid Fibrils in Alzheimer’s Disease Brain Tissue. Cell. 2013;154(6):1257–1268. doi: 10.1016/j.cell.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seeliger J, Weise K, Opitz N, Winter R. The Effect of Aβ on IAPP Aggregation in the Presence of an Isolated β-Cell Membrane. J Mol Biol. 2012;421(2):348–363. doi: 10.1016/j.jmb.2012.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Young ML, Mahood AR, Saunders CJ, Tu L-H, Raleigh PD, Radford ES, Ashcroft EA. Insights into the Consequences of Co-Polymerisation in the Early Stages of IAPP and Aβ Peptide Assembly from Mass Spectrometry. Analyst. 2015;140(20):6990–6999. doi: 10.1039/c5an00865d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meral D, Urbanc B. Discrete Molecular Dynamics Study of Oligomer Formation by N-Terminally Truncated Amyloid β-Protein. J Mol Biol. 2013;425(12):2260–2275. doi: 10.1016/j.jmb.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dagliyan O, Tarnawski M, Chu P-H, Shirvanyants D, Schlichting I, Dokholyan NV, Hahn KM. Engineering Extrinsic Disorder to Control Protein Activity in Living Cells. Science. 2016;354(6318):1441–1444. doi: 10.1126/science.aah3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emperador A, Orozco M. Discrete Molecular Dynamics Approach to the Study of Disordered and Aggregating Proteins. J Chem Theory Comput. 2017;13(3):1454–1461. doi: 10.1021/acs.jctc.6b01153. [DOI] [PubMed] [Google Scholar]
- 37.Andreetto E, Yan L-M, Tatarek-Nossol M, Velkova A, Frank R, Kapurniotu A. Identification of Hot Regions of the Aβ–IAPP Interaction Interface as High-Affinity Binding Sites in Both Cross- and Self-Association. Angew Chem Int Ed. 2010;49(17):3081–3085. doi: 10.1002/anie.200904902. [DOI] [PubMed] [Google Scholar]
- 38.Walsh DM, Selkoe DJ. Aβ Oligomers – a Decade of Discovery. J Neurochem. 2007;101(5):1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 39.Hung LW, Ciccotosto GD, Giannakis E, Tew DJ, Perez K, Masters CL, Cappai R, Wade JD, Barnham KJ. Amyloid-β Peptide (Aβ) Neurotoxicity Is Modulated by the Rate of Peptide Aggregation: Aβ Dimers and Trimers Correlate with Neurotoxicity. J Neurosci. 2008;28(46):11950–11958. doi: 10.1523/JNEUROSCI.3916-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oskarsson ME, Paulsson JF, Schultz SW, Ingelsson M, Westermark P, Westermark GT. In Vivo Seeding and Cross-Seeding of Localized Amyloidosis. Am J Pathol. 2015;185(3):834–846. doi: 10.1016/j.ajpath.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 41.Uversky VN, Fink AL. Conformational Constraints for Amyloid Fibrillation: The Importance of Being Unfolded. Biochim Biophys Acta BBA - Proteins Proteomics. 2004;1698(2):131–153. doi: 10.1016/j.bbapap.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 42.Kapurniotu A. Amyloidogenicity and Cytotoxicity of Islet Amyloid Polypeptide. Pept Sci. 2001;60(6):438–459. doi: 10.1002/1097-0282(2001)60:6<438::AID-BIP10182>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 43.Wiltzius JJW, Sievers SA, Sawaya MR, Eisenberg D. Atomic Structures of IAPP (Amylin) Fusions Suggest a Mechanism for Fibrillation and the Role of Insulin in the Process. Protein Sci. 2009;18(7):1521–1530. doi: 10.1002/pro.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tomaselli S, Esposito V, Vangone P, van Nuland NAJ, Bonvin AMJJ, Guerrini R, Tancredi T, Temussi PA, Picone D. The Alpha-to-Beta Conformational Transition of Alzheimer’s Abeta-(1-42) Peptide in Aqueous Media Is Reversible: A Step by Step Conformational Analysis Suggests the Location of Beta Conformation Seeding. Chembiochem Eur J Chem Biol. 2006;7(2):257–267. doi: 10.1002/cbic.200500223. [DOI] [PubMed] [Google Scholar]
- 45.Man VH, Nguyen PH, Derreumaux P. High-Resolution Structures of the Amyloid-β 1-42 Dimers from the Comparison of Four Atomistic Force Fields. J Phys Chem B. 2017;121(24):5977–5987. doi: 10.1021/acs.jpcb.7b04689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carballo-Pacheco M, Strodel B. Comparison of Force Fields for Alzheimer’s A B42: A Case Study for Intrinsically Disordered Proteins. Protein Sci Publ Protein Soc. 2017;26(2):174–185. doi: 10.1002/pro.3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Camargo DCR, Tripsianes K, Buday K, Franko A, Göbl C, Hartlmüller C, Sarkar R, Aichler M, Mettenleiter G, Schulz M, et al. The Redox Environment Triggers Conformational Changes and Aggregation of HIAPP in Type II Diabetes. Sci Rep. 2017;7:44041. doi: 10.1038/srep44041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kayed R, Bernhagen J, Greenfield N, Sweimeh K, Brunner H, Voelter W, Kapurniotu A. Conformational Transitions of Islet Amyloid Polypeptide (IAPP) in Amyloid Formation in Vitro11Edited by R. Huber. J Mol Biol. 1999;287(4):781–796. doi: 10.1006/jmbi.1999.2646. [DOI] [PubMed] [Google Scholar]
- 49.Kirkitadze MD, Condron MM, Teplow DB. Identification and Characterization of Key Kinetic Intermediates in Amyloid β-Protein Fibrillogenesis11Edited by F. Cohen. J Mol Biol. 2001;312(5):1103–1119. doi: 10.1006/jmbi.2001.4970. [DOI] [PubMed] [Google Scholar]
- 50.Colvin MT, Silvers R, Ni QZ, Can TV, Sergeyev I, Rosay M, Donovan KJ, Michael B, Wall J, Linse S, et al. Atomic Resolution Structure of Monomorphic Aβ42 Amyloid Fibrils. J Am Chem Soc. 2016;138(30):9663–9674. doi: 10.1021/jacs.6b05129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khorasanizadeh S, Peters ID, Roder H. Evidence for a Three-State Model of Protein Folding from Kinetic Analysis of Ubiquitin Variants with Altered Core Residues. Nat Struct Mol Biol. 1996;3(2):193–205. doi: 10.1038/nsb0296-193. [DOI] [PubMed] [Google Scholar]
- 52.Ding F, Dokholyan NV, Buldyrev SV, Stanley HE, Shakhnovich EI. Direct Molecular Dynamics Observation of Protein Folding Transition State Ensemble. Biophys J. 2002;83(6):3525–3532. doi: 10.1016/S0006-3495(02)75352-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sugita Y, Okamoto Y. Replica-Exchange Molecular Dynamics Method for Protein Folding. Chem Phys Lett. 1999;314(1):141–151. [Google Scholar]
- 54.Kumar S, Rosenberg JM, Bouzida D, Swendsen RH, Kollman PA. THE Weighted Histogram Analysis Method for Free-Energy Calculations on Biomolecules. I. The Method. J Comput Chem. 1992;13(8):1011–1021. [Google Scholar]
- 55.Bitan G, Kirkitadze MD, Lomakin A, Vollers SS, Benedek GB, Teplow DB. Amyloid Beta -Protein (Abeta) Assembly: Abeta 40 and Abeta 42 Oligomerize through Distinct Pathways. Proc Natl Acad Sci U S A. 2003;100(1):330–335. doi: 10.1073/pnas.222681699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Westermark P, Li Z-C, Westermark GT, Leckström A, Steiner DF. Effects of Beta Cell Granule Components on Human Islet Amyloid Polypeptide Fibril Formation. FEBS Lett. 1996;379(3):203–206. doi: 10.1016/0014-5793(95)01512-4. [DOI] [PubMed] [Google Scholar]
- 57.Ding F, Dokholyan NV. Emergence of Protein Fold Families through Rational Design. PLoS Comput Biol. 2006;2(7):e85. doi: 10.1371/journal.pcbi.0020085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: A Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J Comput Chem. 1983;4(2):187–217. [Google Scholar]
- 59.Lazaridis T, Karplus M. Effective Energy Functions for Protein Structure Prediction. Curr Opin Struct Biol. 2000;10(2):139–145. doi: 10.1016/s0959-440x(00)00063-4. [DOI] [PubMed] [Google Scholar]
- 60.Ding F, Borreguero JM, Buldyrey SV, Stanley HE, Dokholyan NV. Mechanism for the Alpha-Helix to Beta-Hairpin Transition. Proteins. 2003;53(2):220–228. doi: 10.1002/prot.10468. [DOI] [PubMed] [Google Scholar]
- 61.Andersen HC. Molecular Dynamics Simulations at Constant Pressure and/or Temperature. J Chem Phys. 1980;72(4):2384–2393. [Google Scholar]
- 62.Kabsch W, Sander C. Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers. 1983;22(12):2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.