

Abstract
The melanocortin system has five receptors and antagonists of the central melanocortin receptors (MC3R, MC4R) are postulated to be viable therapeutics for disorders of negative energy balance such as anorexia, cachexia, and failure to thrive. Agouti-related protein (AGRP) is an antagonist of the MC3R and an antagonist/inverse agonist of the MC4R. Biophysical NMR based structural studies have demonstrated that the active sequence of this hormone, Arg-Phe-Phe, is located on an exposed β-hairpin loop. It has previously been demonstrated that the macrocyclic octapeptide scaffold c[Pro1-Arg2-Phe3-Phe4-Asn5-Ala6-Phe7-DPro8] is 16-fold less potent than AGRP at the mMC4R. Herein, it was hypothesized that the Phe7 position may be substituted to produce more potent and/or selective melanocortin receptor antagonist ligands based on this template. A ten-member library was synthesized that substituted small (Gly), polar (Ser), acidic (Asp), basic (Lys), aliphatic (Leu, Nle, and Cha), and aromatic (Trp, Tyr, hPhe) amino acids to explore potential modifications at the Phe7 position. The most potent mMC4R antagonist contained a Nle7 substitution, was equipotent to the lead ligand, 200-fold selective for the mMC4R over the mMC3R, and caused a significant increase in food intake when injected intrathecally (i.t.) into male mice. Three compounds possessed sigmoidal dose-response inverse agonist curves at the mMC5R, while the remaining seven decreased cAMP production from basal levels at 100 μM concentrations. These findings will add to the knowledge base towards the development of potent and selective probes to study the role of the melanocortin system in diseases of negative energy balance, and can be useful in the design of molecular probes to examine the physiological functions of the mMC5R.
Keywords: AGRP, structure-activity relationships, melanocortin receptors, IT, food intake
TOC image
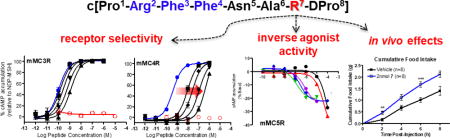
Introduction
The melanocortin system is comprised of five G protein-coupled receptors (GPCRs, MC1-5R)1–8 that have been associated with a wide range of physiological functions ranging from skin pigmentation9 to energy homeostasis.10–14 The melanocortin receptors couple to G proteins and signal primarily by stimulating the adenylate cyclase pathway, thereby increasing intracellular cyclic AMP (cAMP) upon stimulation by agonist compounds. Endogenous agonists for the melanocortin receptors include the adrenocorticotropic hormone (ACTH) and α-, β-, and γ-melanocyte stimulating hormones (MSHs). These peptides are produced via posttranslational processing of the proopiomelanocortin (POMC) gene transcript.15–17 Notably, this receptor system also contains naturally occurring antagonists: agouti signaling protein (ASP) and agouti-related protein (AGRP).18–22 The MC3R and MC4R have been investigated as promising targets for anti-obesity drugs due to their integral roles in regulating food intake and energy homeostasis.10–14 Energy homeostasis is modulated by the MC3R,10, 13–14, 23 MC4R,11–12, 24 POMC-derived agonists,25–26 ASP,11 and AGRP.19 To date there have been numerous synthetic peptide and small molecule ligands developed for the study of the melanocortin system, as reviewed by Ericson et al.27
Transgenic mice overexpressing AGRP weigh more than wild type control mice,28 and administration of AGRP via intracerebroventricular (icv) injection in mice increases food intake.14, 29 It has been proposed that self-starvation and physical hyperactivity in rats, induced via food restriction in the presence of running wheels, may be the result of insufficient suppression of central melanocortin receptor activity.30 The effects of self-starvation in rats can be alleviated by central administration of AGRP.30–31 A human single nucleotide polymorphism (SNP) in AGRP (A67T) has been identified in patients with anorexia nervosa.32 Therefore diseases which produce a negative energy balance such as anorexia nervosa, cachexia, and failure to thrive in children may be alleviated through treatment with central melanocortin receptor antagonists.30–31, 33–35
Using recombinant protein, human (h)AGRP was originally found to be a functional antagonist of endogenous agonist α-MSH at the human MC3R (hMC3R) and hMC4R, and did not have antagonist activity at the hMC1R.19, 21 It was later reported that a truncated version of human AGRP, hAGRP(83-132) possesses inverse agonist activity at the hMC4R as well as a mouse (m) MC4R mutated to possess constitutive activity.36–37
Studies utilizing recombinant human agouti first showed that hASP is a competitive antagonist of α-MSH activity at both the mouse and human MC1R, and the human MC4R.38 Later studies were performed on a synthesized ASP variant, hASP(80-132, Q115Y,S124Y) (ASP-YY), which changed two residues from ASP to the homologous residues contained in AGRP to aid in proper folding.39 In AGRP, these residues (Y109,Y118) are located within β-sheets that immediately flank the active loop sequence, and are hypothesized to confer stability to the protein through aromatic interactions, of which the ASP native residues (Q115,S124) are apparently incapable.39 Agouti-YY was found to inhibit α-MSH activity at the hMC1R, hMC3R, and hMC4R with functional antagonist potencies of 4.0, 2.6, and 0.5 nM respectively. These functional potencies are similar to the binding affinities of ASP or other ASP variants, in which these peptides are > 3-fold more potent binders at the MC4R than the MC3R.40–42 Agouti (ASP) has also been shown to possess inverse agonist activity at the hMC4R.43
The decapeptide H-Tyr-c[Cys-Arg-Phe-Phe-Asn-Ala-Phe-Cys]-Tyr-OH (AGRP[109-118]), containing the active loop sequence of AGRP, is a micromolar agonist at the MC1R (EC50 = 3 μM), possesses an antagonist pA2 value of 6.8 at the mMC4R, and does not possess an antagonist potency at the mMC3R that is quantifiable at the highest concentrations of antagonist assayed.44 Truncations of H-Tyr-c[Cys-Arg-Phe-Phe-Asn-Ala-Phe-Cys]-Tyr-OH to an octapeptide are therefore hypothesized to confer selectivity toward the MC4R as compared to the MC3R, with the caveat that some of these peptides may possess agonist activity at the MC1R.45
In the effort to create selective MC4R antagonist ligands that are equipotent or more potent than the native hormone AGRP, further structure-activity relationship (SAR) studies have been performed around the active loop sequence c[Cys-Arg-Phe-Phe-Asn-Ala-Phe-Cys].46 These studies provide important SAR information for the advancement of AGRP-based molecular probes and therapeutic design. The cyclic structure of this peptide is necessary for binding, as a linear derivative of this peptide containing serine isosteric replacements (H-Ser-Arg-Phe-Phe-Asn-Ala-Phe-Ser-OH) was unable to bind the MC3R and MC4R at up to 20000 nM concentrations.47 Previous studies have suggested that the Arg-Phe-Phe tripeptide sequence is important for potent antagonist ligands.47–48 Structural studies of AGRP and “miniAGRP” have indicated that the active loop adopts a β-hairpin conformation, stabilized by five disulfide bonds.49–51 The ability of heterochiral proline residues to induce β-hairpin turns in macrocyclic peptides has been previously established.52–55 Previous studies on truncated AGRP macrocycles replacing Cys110 and Cys117 with a head-to-tail cyclized DPro-Pro motif have hypothesized that the resulting macrocyclic peptide may better mimic the β-hairpin loop contained in the native hormone, as these peptides show increased potency relative to H-Tyr-c[Cys-Arg-Phe-Phe-Asn-Ala-Phe-Cys]-Tyr-NH2 at the mMC4R (sequence = c[Pro1-Arg2-Phe3-Phe4-Asn5-Ala6-Phe7-DPro8], 1, pA2 = 7.7).46
Previous SAR studies performed on this scaffold have focused on the Phe3, Phe4, Asn5, and Ala6 positions, and the results for these compounds assayed at the mMC4R are summarized in Figure 1.46, 56 Replacing Phe3 and Phe4 in this macrocyclic template with other aromatic amino acid derivatives (including Bip, Phg, and hPhe) decreases or ablates potency at the mMC3R and the mMC4R with the exception of Nal(1′), which is equipotent to the lead compound.46 Nearly all of these Phe3- and Phe4-substituted macrocycles are micromolar MC1R agonists, and are unable to stimulate the MC5R (with the exception of the Anc4-substituted peptide, which is a micromolar agonist at the mMC5R). The SAR around the Asn5 position suggests that this position is more amenable to change. In one study, replacing this residue with multiple different amino acids (Gly, Dap, Dab, Orn, Lys, Arg) resulted in ligands that were equipotent or more potent antagonists than the lead ligand at the mMC3R and mMC4R.46 Four of these substitutions possessed some agonist activity at the mMC1R at 100 μM concentrations, and all were unable to activate the MC5R.46 A subsequent study has replaced Asn5 with a larger variety of amino acids (Ala, Abu, Ser, Thr, Asp, Glu, DDap, His, Nle, Leu, Val, Phe, and Trp) in order to elaborate on the SAR around this position.56 All of these peptides showed quantifiable antagonist potency at the mMC4R: two were more potent than the Asn5 substitution (containing DDap and His substitutions), while the remainder possessed decreased potency. Notably, many of these ligands also possessed mMC5R inverse agonist activity, a novel pharmacology at this receptor.56 This study also examined the Ala6 position, replacing this residue with Asp, Glu, Lys, His, Phe, Ser, Leu, and Gly residues. All but one of these compounds, possessing a Ser substitution, resulted in decreased potency compared to the lead ligand at the mMC4R. Many of these peptides also possessed inverse agonist activity at the mMC5R, though notably the Lys6-substituted peptide possessed some agonist activity.56 During this study it was noted that the Arg5 and Ser6 substitutions corresponded to residues contained in the ASP active loop. Therefore, the Phe7 was substituted with an Ala to elaborate on the hypothesis that ASP residues may be substituted into this macrocyclic octapeptide template to generate potent mMC4R antagonist ligands. Notably, this Ala7-substituted peptide was also equipotent to the lead compound.56
Figure 1.
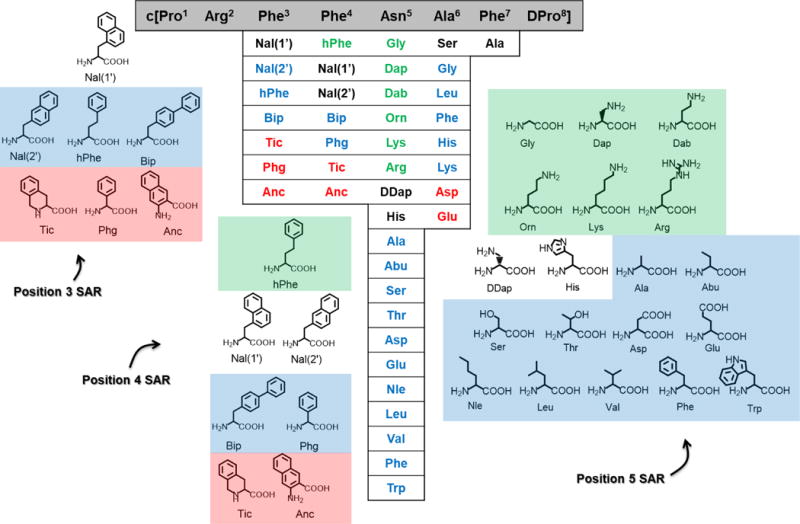
Summary of the structure-activity relationship observed for this octapeptide scaffold at the mMC4R, as published previously.46, 56 In this figure, the sequence of the lead scaffold is shown in the black bar. Below each position is a listing of the different substitutions that have been tested at that position with a color scheme. Compounds containing substitutions shown in green possess increased potency compared to the lead scaffold, compounds containing substitutions shown in black are equipotent to the lead scaffold, compounds containing substitutions shown in blue possess decreased potency compared to the lead scaffold, and compounds possessing substitutions shown in red did not possess antagonist potency that could be observed at the highest concentrations used in that assay. The studies examining the Phe3, Phe4, and Asn5 positions utilize unnatural amino acids, and the structures of the amino acids are provided and colored as described above.
Both Ala7 and Phe7 substitutions have been reported to possess equipotent mMC4R antagonist potency, despite the difference between the methyl side chain of Ala and the phenyl side chain of the Phe. Due to these differences, the present study focused on what properties of the Phe side chain were important for the observed antagonist potency. While Ala can be thought of as removal of a side chain, it is also the smallest side chain that may be considered aliphatic. It was hypothesized aliphatic or aromatic substitutions at the Phe7 position may possess antagonist potency at the mMC4R, and that changing this residue to various aliphatic or aromatic amino acids will afford ligands that are more potent than the lead ligand or have unique pharmacological profiles. The present study explores the SAR around the Phe7 position of the macrocyclic template and tests this hypothesis through the synthesis and pharmacological screening of Phe7-substituted macrocyclic ligands.
Results and Discussion
Peptide Synthesis and Characterization
The peptides reported herein were synthesized in a semi- and fully automated fashion using standard fluorenylmethoxycarbonyl (Fmoc) methodology.57–58 Following synthesis, peptides were purified using semi-preparative reverse phase high-pressure liquid chromatography (RP-HPLC). Purity was confirmed to be >95% via analytical RP-HPLC using two diverse solvent systems, and the peptide mass confirmed using matrix-assisted laser desorption ionization (MALDI) time of flight (TOF) mass spectrometry (University of Minnesota LeClaire-Dow Instrumentation Facility). Analytical characterization data for these peptides may be found in Table 1.
Table 1.
Analytical characterization data for peptides.
| Compound ID | KAF# | Sequence | HPLC RT (system 1) |
HPLC RT (system 2) |
Purity % | M+1 (calculated) |
M+1 (observed) |
|---|---|---|---|---|---|---|---|
| 2 | KAF2039-1 | c[Pro-Arg-Phe-Phe-Asn-Ala-Gly-DPro] | 10.5 | 18.9 | >95% | 888.0 | 887.9 |
| 3 | KAF2039-3 | c[Pro-Arg-Phe-Phe-Asn-Ala-Ser-DPro] | 14.8 | 24.3 | >98% | 918.0 | 918.7 |
| 4 | KAF2039-4 | c[Pro-Arg-Phe-Phe-Asn-Ala-Lys-DPro] | 14.2 | 23.3 | >98% | 959.1 | 959.0 |
| 5 | KAF2039-5 | c[Pro-Arg-Phe-Phe-Asn-Ala-Asp-DPro] | 15.1 | 25.3 | >95% | 946.1 | 946.9 |
| 6 | KAF2039-6 | c[Pro-Arg-Phe-Phe-Asn-Ala-Leu-DPro] | 17.4 | 28.3 | >98% | 944.1 | 944.1 |
| 7 | KAF2039-7 | c[Pro-Arg-Phe-Phe-Asn-Ala-Nle-DPro] | 17.4 | 28.7 | >95% | 944.1 | 944.5 |
| 8 | KAF2039-9 | c[Pro-Arg-Phe-Phe-Asn-Ala-Trp-DPro] | 18.2 | 28.4 | >98% | 1017.2 | 1017.5 |
| 9 | KAF2039-10 | c[Pro-Arg-Phe-Phe-Asn-Ala-Tyr-DPro] | 16.1 | 25.5 | >98% | 994.1 | 994.8 |
| 10 | KAF2039-11 | c[Pro-Arg-Phe-Phe-Asn-Ala-Cha-DPro] | 19.1 | 30.6 | >98% | 984.2 | 984.5 |
| 11 | KAF2039-13 | c[Pro-Arg-Phe-Phe-Asn-Ala-hPhe-DPro] | 18.3 | 29.4 | >98% | 992.2 | 992.9 |
HPLC RT = peptide retention time in solvent system 1 (10% acetonitrile in 0.1% trifluoroacetic acid in water and a gradient of 90% acetonitrile over 35 minutes) or solvent system 2 (10% methanol in 0.1% trifluoroacetic acid in water and a gradient of 90% methanol over 35 minutes). An analytical Vydac C18 column (Vydac 218TP104) was used with a flow rate of 1.5 mL/min. The peptide purity was determined by HPLC at a wavelength of 214 nm.
Compounds were first assayed as agonists in HEK293 cells stably expressing the mMC1R, mMC3R, mMC4R, or mMC5R using the AlphaScreen cAMP Assay.59–61 None of the library compounds possessed agonist activity at the mMC3R or mMC4R at up to 100 μM concentrations, so antagonist potencies were measured. Competitive antagonist activity was measured using a Schild analysis at the mMC3R and mMC4R with the synthetic, non-selective, potent melanocortin agonist NDP-MSH.62 Fold-change calculations were performed using the Ki values derived from the Schild analysis [pA2 = −log(Ki)].
Library Design and Structure-Activity Relationship
Herein, it was hypothesized that the possession of aliphatic or aromatic side chains in the 7 position of the macrocyclic template is important for MC4R antagonist activity of the peptide, and this was tested through removal of the side chain by replacing this residue with Gly7 (KAF2039-1). This was also tested through replacing this residue with an Ala, which has been previously reported (KAF3094).56 Additionally, to test this hypothesis, the Phe7 residue was substituted with a hydrophilic amino acid (Ser) and charged amino acids (Lys and Asp) (KAF2039-3, KAF2039-4, and KAF2039-5). Due to the hydrophobic nature of the phenyl side chain in the lead peptide, it was hypothesized that these substitutions would decrease the potency compared to the lead ligand. Aliphatic amino acids Leu and Nle were used to replace the Phe7 residue (KAF2039-6, KAF2039-7) to test the hypothesis that the Ala7-substitution previously reported contributes to the activity of 1 through aliphatic interactions. The hypothesis that either aliphatic or aromatic amino acid substitutions could result in potent mMC4R antagonist ligands was tested further by replacing the Phe7 residue with other aromatic amino acids such as Trp and Tyr (KAF2039-9, KAF2039-10), and by removing the aromaticity of this side chain through replacement of the Phe7 residue with cyclohexylalanine (Cha) (KAF2039-11). Finally, to test the hypothesis that the side chain length of the Phe7 residue is important for the activity of the lead ligand (1), the side chain of this residue was elongated by one methylene unit (hPhe) (KAF2039-13). The amino acids used in this study are illustrated in Figure 2.
Figure 2.
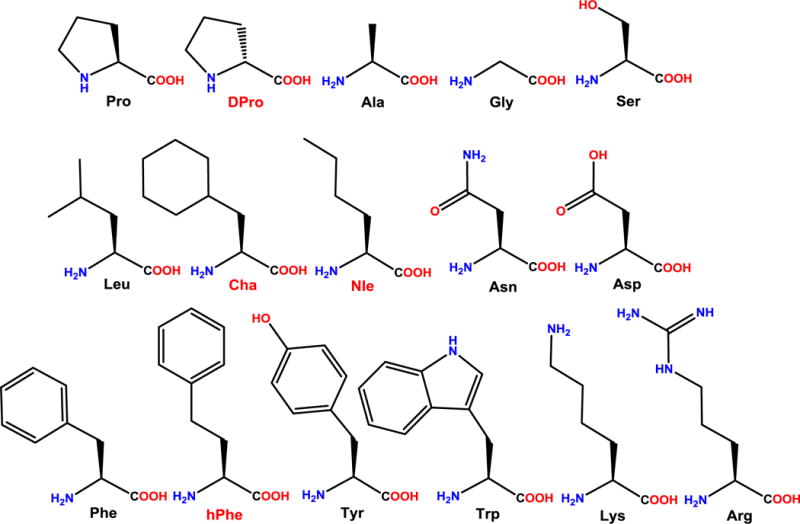
Structures of the amino acids used in this study with three-letter amino acid abbreviations. The three-letter abbreviations for unnatural or uncommon amino acids have been highlighted in red.
The pharmacological results for these experiments are summarized in Table 2. The pharmacology at the mMC3R and mMC4R of the three most potent compounds at the mMC4R are illustrated in Figure 3. The Gly7-substituted compound was 110-fold less potent at the mMC4R than 1, supporting the hypothesis that the possession of an aromatic or aliphatic sidechain is important for the activity of the lead ligand. Polar amino acids decreased antagonist potency at the mMC4R. The Ser7-substituted compound was 10-fold less potent than 1, 40-fold decreased potency was observed for Lys7, and the antagonist potency was not measurable for the Asp7 substitution at the concentrations used in this study. These results support the hypothesis that the hydrophobic nature of the native Phe7 sidechain in 1 is important for receptor activity. Lengthening the Phe residue by one methylene unit (KAF2039-13) produced a compound which is 8-fold less potent than 1 at the mMC4R. The aliphatic amino acids investigated in this study yielded different results. The Nle7-substituted compound (KAF2039-7) was equipotent to 1 at the mMC4R, while KAF2039-6 was 15-fold less potent than 1 at the MC4R. The aromaticity of the Phe7 ring in 1 is important, as substitution with a cyclohexyl ring (Cha7) decreases potency 16-fold compared to the lead compound. Substitution of the Phe7 side chain with Tyr7 and Trp7 (KAF2039-10, KAF2039-9) yielded compounds that were equipotent to the lead ligand at the mMC4R.
Table 2.
Pharmacology of the macrocyclic AGRP-based peptides modified in the Phe7 position [Pro1-Arg2-Phe3-Phe4-Asn5-Ala6-Phe7-DPro8].
| Compound ID | KAF# | R7 | Agonist | Antagonist | Antagonist | Inverse Agonist | ||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| mMC1R | mMC3R | mMC4R | mMC5R | |||||
| Agonist EC50 (nM) |
pA2 | Antagonist Ki (nM) |
pA2 | Antagonist Ki (nM) |
Inverse Agonist EC50 (nM) |
|||
| NDP-MSH | 0.010±0.003 | EC50 = 0.06±0.01 nM | EC50 = 0.35±0.06 nM | EC50 = 0.10±0.01 nM | ||||
| hAGRP(87-132)a | > 100,000 | 8.9±0.2 | 1.3 | 9.4±1.0 | 0.40 | > 100,000 | ||
| 1b | MDE5108-10c | Phe | 25% @ 100 μM | 6.3±0.1 | 500 | 8.2±0.1 | 6.3 | 130 (−10%) |
|
| ||||||||
| 2 | KAF2039-1 | Gly | 60% @ 100 μM | < 5.5 | 6.17±0.04 | 680 | −25% @ 100 μM | |
| 3 | KAF2039-3 | Ser | 35% @ 100 μM | 6.0±0.2 | 1,000 | 7.2±0.2 | 63 | −55% @ 100 μM |
| 4 | KAF2039-4 | Lys | > 100,000 | 5.7±0.1 | 2,000 | 6.60±0.06 | 250 | −40% @ 100 μM |
| 5 | KAF2039-5 | Asp | 40% @ 100 μM | < 5.5 | < 5.5 | −25% @ 100 μM | ||
| 6 | KAF2039-6 | Leu | > 100,000 | < 5.5 | 7.03±0.09 | 93 | −35% @ 100 μM | |
| 7 | KAF2039-7 | Nle | > 100,000 | 6.1±0.1 | 790 | 8.4±0.2 | 4.0 | −35% @ 100 μM |
| 8 | KAF2039-9 | Trp | partial agonist 1,100 ± 200 (50%) |
6.67±0.07 | 210 | 8.2±0.2 | 6.3 | 150 (−20%) |
| 9 | KAF2039-10 | Tyr | > 100,000 | 6.23±0.05 | 590 | 8.00±0.06 | 10 | −40% @ 100 μM |
| 10 | KAF2039-11 | Cha | > 100,000 | 5.8±0.2 | 1,600 | 7.0±0.03 | 100 | 780 (−20%) |
| 11 | KAF2039-13 | hPhe | > 100,000 | 6.5±0.3 | 320 | 7.30±0.06 | 50 | 290 (−20%) |
| 12b | KAF3094 | Ala | > 100,000 | 6.1±0.2 | 790 | 8.2±0.1 | 6.3 | −25% @ 100 μM |
Compounds were assayed in duplicate replicates and values are expressed as the mean ± the standard error of the mean (SEM) of at least 3 independent experiments. “Partial agonist”: partial agonist activity, with a maximal percent activated listed in parenthesis. “X% @ 100 μM”: Compounds that partially stimulated the receptor but were unable to generate a sigmoidal dose-response at 100 μM concentrations are listed by their percent activation at 100 μM. “> 100,000”: Compounds which showed no agonist activity at 100 μM are listed as > 100,000. “< 5.5”: The use of <5.5 indicates that no antagonist potency was observed in the highest concentration ranged assayed (10,000, 5,000, 1,000, and 500 nM). “X (-X%)”: For compounds that displayed inverse agonist activity and appeared to generate a sigmoidal dose-response, the average apparent potency at the inflection point and maximal decrease in basal activity at the plateau is listed. “-X% @ 100 μM”: For all other compounds with inverse agonist activity, the percent decrease observed in receptor activity compared to basal at 100 μM is provided. The pA2 values were determined using the Schild analysis with agonist NDP-MSH (pA2 = −log[Ki]).
This peptide has been previously published.48
These peptides have been previously reported.56
Figure 3.
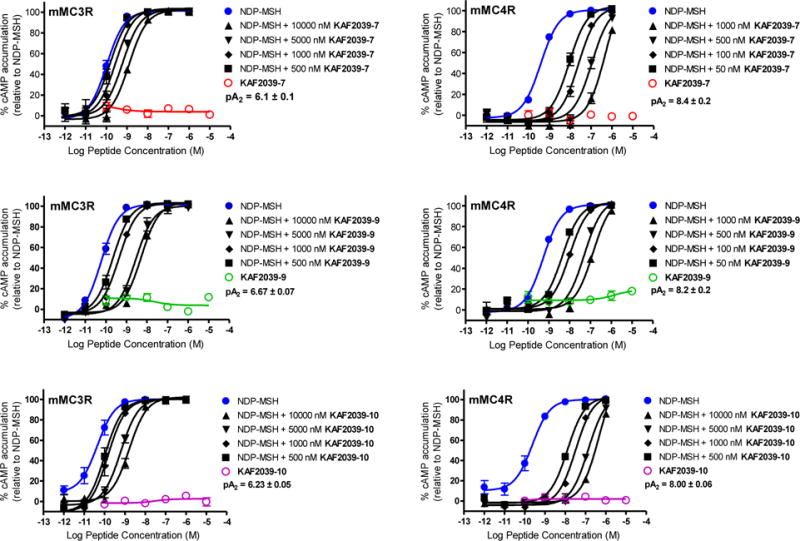
Illustrations of the in vitro antagonist (mMC3R, mMC4R) pharmacology of 7, 8, and 9. A Schild antagonist experimental strategy was implemented using the agonist NDP-MSH. Data were normalized to an NDP-MSH response as has been previously reported.59–61
The results obtained from this study support the hypothesis that the activity of the lead ligand c[Pro-Arg-Phe-Phe-Asn-Ala-Phe-DPro] (1) at the MC4R as mediated through Phe7 is due to aromatic interactions, but that aliphatic substitutions are also tolerated in this position. It is interesting that the Leu7 and Cha7 substitutions both resulted in approximately 15-fold decreased potency compared to the lead ligand, while the Nle7 substitution was equipotent to 1. These two amino acids (Leu7 and Cha7) possess aliphatic sidechains that are branched on the γ-carbon, while the Nle7 residue possesses a linear side chain. If the aliphatic interactions formed by the Nle7 side chain are sufficient for producing antagonist activity, it might be expected that the interactions by Leu7 and Cha7 would be as well. It would therefore be anticipated that these compounds (KAF2039-6, KAF2039-11) would be equipotent to the Nle7-substituted compound (KAF2039-7). However, this was not what was observed. Interestingly in ASP, the residue at the analogous 7 position is not a Phe, and is an Ala. The Ala7-substituted compound (KAF3094) was also equipotent to 1 at the mMC4R, as previously reported.56 Therefore, it is possible that two distinct types of amino acids in the 7 position (aromatic, aliphatic) are equipotent the MC4R, and that branching at the γ-carbon may be unfavorable in forming putative ligand-receptor interactions for aliphatic substitutions.
Most of the substitutions examined in this study showed a reduced potency compared to the lead ligand at the MC3R. Antagonist activity was not observed for the Gly7-, Asp7-, or Leu7-containing peptides (KAF2039-1, KAF2039-5, KAF2039-6) at the concentrations examined in this study at the mMC3R. The Lys7- and Cha7-containing peptides (KAF2039-4, KAF2039-11) resulted in decreased potency compared to 1 (4-fold and 3-fold, respectively), and Nle7-, Trp7-, Tyr7-, and hPhe7-containing peptides (KAF2039-7, KAF2039-9, KAF2039-10, KAF2039-13) were all equipotent to 1 at the mMC3R.
The lead ligand is 80-fold more selective at the mMC4R than the mMC3R. Of the compounds that showed quantifiable potency at both the mMC3R and mMC4R, two possessed fold selectivities between 30 and 80. The Tyr7-containing peptide was 60-fold more potent at the mMC4R than the mMC3R, and the Trp-containing peptide was 30-fold more potent at the mMC4R than the mMC3R. Notably, the Nle7-substituted peptide was 200-fold more potent at the mMC4R than the mMC3R, an increase in selectivity compared to 1. Additionally, the Ala7-substituted peptide (KAF3094) was previously reported to possess 130-fold selectivity for the mMC4R over the mMC3R.56 Above, it was postulated that two distinct types of amino acids at the 7 position of the macrocyclic template (linear aliphatic, aromatic) are capable of possessing equipotent antagonist activity at the mMC4R. These data suggest that the linear aliphatic substitutions may possess different pharmacological profiles compared to the aromatic substitutions. The active loop of ASP (sequence = −c[Cys1−Arg2−Phe3−Phe4−Arg5−Ser6−Ala7−Cys8] −) contains an Ala residue in the 7 position, and studies on ASP-YY suggest that ASP is more selective for the MC4R (5-fold) than AGRP (equipotent).39, 48 It is possible that the linear aliphatic substitutions in this position, which are more ASP-like, may be used in order to increase mMC4R selectivity against the mMC3R as opposed to aromatic substitutions, which are more AGRP-like. The remainder of the macrocyclic peptides resulted in decreased mMC3R/mMC4R selectivity compared to the lead ligand; the Ser7-containing peptide was 16-fold more potent at the mMC4R, the Lys7-containing peptide was 8-fold more potent at the mMC4R, the Cha7-containing peptide was 16-fold more potent at the mMC4R, and the hPhe7 -containing peptide was 6-fold more potent at the mMC4R.
Four of the ligand studied displayed agonist activity at the mMC1R. The Gly7-, Ser7-, and Asp7-containing peptides (KAF2039-1, KAF2039-3, KAF2039-5) all were able to partially stimulate the MC1R at 100 μM concentrations. The Trp7-containing peptide KAF2039-9 was a partial agonist at the mMC1R (EC50 = 1,100 ± 200 nM), and resulted in 50% receptor activation. The pharmacology of this peptide at the mMC1R is illustrated in Figure 4.
Figure 4.
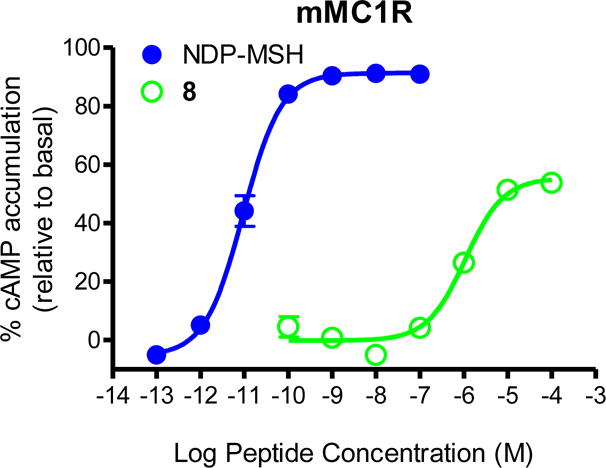
Illustration of the in vitro partial agonist pharmacology of 8 at the mMC1R. These data were normalized to an NDP-MSH response as has been previously reported.59–61
Similar to a previous report, all the Phe7 side chain modifications examined in this study resulted in MC5R inverse agonist activity (Table 2, Figure 5).56 Three compounds (KAF2039-9, KAF2039-11, and KAF2039-13) appeared to generate sigmoidal dose-response curves with inverse agonist activities of 20%, which is shown in Figure 5. The sigmoidal shape of these curves allowed the calculation of apparent inverse agonist potencies (the inflection point on the sigmoidal dose-response curve). The Trp7- and hPhe7-containing peptides were equipotent to each other, while the Cha7-containing peptide was 5-fold less potent than the most potent inverse agonist at the mMC5R (KAF2039-9). Both the Trp7 and hPhe7-containing peptides possess aromatic moieties, and based on these results it appears that this functionality may be important for generating mMC5R inverse agonist ligands. Many ligands appeared to display inverse agonist efficacy, but did not plateau to generate a sigmoidal dose-response. These compounds are listed by the percent cAMP accumulation observed relative to basal at 100 μM. The two types of pharmacological results described above (apparent sigmoidal dose-response, and inverse agonist activity at 100 μM concentrations) observed for select compounds are illustrated in Figure 5. The Gly7- and Asp7- containing peptides possessed similar percent decreases in cAMP accumulation (-25%) compared to KAF2039-9, KAF2039-11, and KAF2039-13. The Leu7- and Nle7-containing peptides possessed percent decreases in cAMP accumulation of –35% relative to basal. The Ser7-, Lys7-, and Tyr7-containing peptides (KAF2039-3, KAF2039-4, and KAF2039-10) all showed the most prevalent MC5R inverse agonist pharmacology, with −55%, −40%, and −40% decreases in cAMP accumulation respectively. Interestingly, the ligands possessing the greatest decrease from basal levels at 100 μM concentrations at the mMC5R (KAF2039-3, KAF2039-4, and KAF2039-10) contain hydrogen bond donors. It may be speculated that the Tyr7- and Lys7-containing peptides orient a hydrogen bond donor-proton for a productive interaction, yielding an inverse agonist pharmacology of ~40%. A shorter hydrogen bond donor yields a greater inverse agonist response, as KAF2039-3 possessed a 55% inverse agonist pharmacology. This SAR, combined with previously reported MC5R inverse agonism,56 is a promising start in the development of MC5R inverse agonist peptides which are selective, potent, and possess percent decreases in cAMP from basal of >55%.56 Such ligands could be used to study the activity of the MC5R by way of a pseudo conditional knockdown of MC5R activity.
Figure 5.
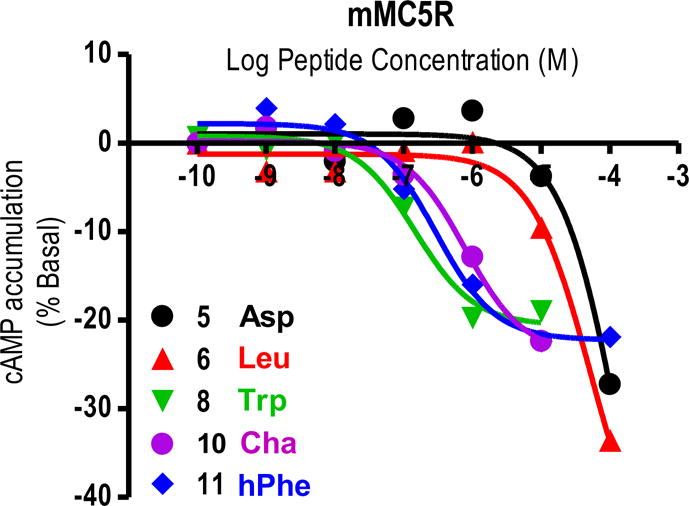
Illustration of the in vitro inverse agonist pharmacology of 5, 6, 8, 10, 11 at the mMC5R. The three-letter amino acid abbreviation for the amino acid in the Phe7 position is provided. Two different pharmacological results were obtained from these studies. For some compounds, a sigmoidal dose-response curve was observed from which an apparent potency and percent cAMP accumulation change from basal can be calculated. Other compounds did not plateau, and for these compounds a percent cAMP accumulation change from basal at 100 μM concentrations is listed.
Animal Studies
As discussed above, MC1R agonism is commonly observed in MC4R antagonist ligands. There have been a few ligands reported in literature that are >100-fold selective for the MC4R over the MC3R, but these ligands possess MC1R agonist activity as reviewed by Ericson et al.27 One compound reported herein, KAF2039-7, possesses selectivity for the MC4R over the MC3R (200-fold) and does not possess MC1R agonist activity, thereby possessing a more selective pharmacological profile at the melanocortin receptors. Due to its potency at the mMC4R (pA2 = 8.4±0.2) and selectivity for the mMC4R over the mMC3R (approximately 200-fold), KAF2039-7 was selected as a candidate for animal studies in mice to examine the potential in vivo effects of this scaffold series (shown in Figure 6 and Figure 7). Intrathecal (IT) administration of melanocortin compounds into the spinal cord for the study of metabolic disorder is new to the field, and may provide some benefits as opposed to intracerebroventricular (ICV) injection into mice.63 For example, it has been proposed that IT administration may result in a more sensitive in vivo response compared to ICV administration.63 Male mice injected IT with 2 nmol KAF2039-7 showed a statistically significant increase in food intake at 2 and 6 hours post-injection, and exhibited a prolonged trend of increased food intake up to 72 hours post-injection (Figure 6). There was an overall effect of KAF2039-7 treatment on mouse body weight compared to vehicle control, with treated animals weighing more than control animals (Figure 7).
Figure 6.
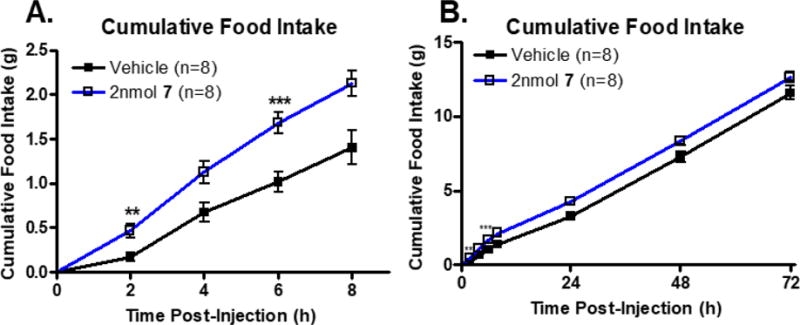
A stock of 7 was prepared using a 20% solutol solution (final concentration of 10nmol/μL). On days of experimentation, the 7 stock was diluted using sterile ddH2O to the desired concentration of 2nmol/5μL. A. Cumulative food intake of male WT mice receiving 2 nmol 7 in 5 μL ddH2O vs. 5 μL of vehicle via IT injection. Male mice injected IT with 7 ate significantly more food t=2h and 6h post-injection than male mice injected with vehicle; **p<0.01, ***p<0.001. Data shown as mean ± SEM. B. Cumulative food intake of male WT mice receiving 2 nmol 7 in 5 μL ddH2O vs. 5 μL of vehicle via cannula. Data shown as mean ± SEM.
Figure 7.
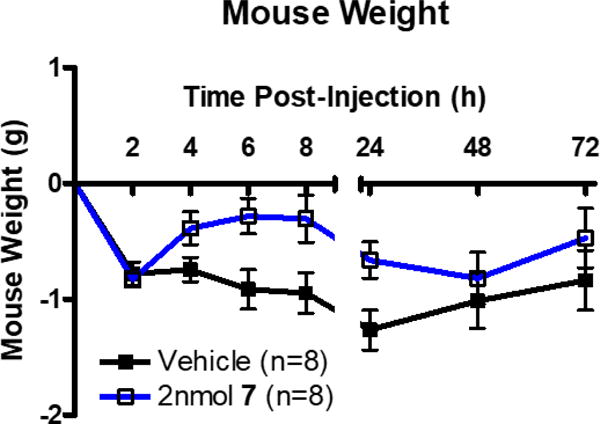
A stock of 7 was prepared in 20% solutol to a final concentration of 10nmol/μL. On days of experimentation, the 7 stock was diluted using sterile ddH2O to the desired concentration of 2nmol/5μL. Difference in mouse weight from t=0h of male WT mice receiving 2 nmol 7 in 5 μL ddH2O vs. 5 μL of vehicle via cannula. Data shown as mean ± SEM.
Previously, it was demonstrated that IT administration of 2 nmol AGRP(86-132) statistically increased the average daily food intake for 2 days post-injection.63 Mice treated with KAF2039-7 showed a strong trend in increased food intake for up to 72-hours. By fine-tuning the properties of these macrocyclic octapeptides, future AGRP-mimetics may be able to display the same potency and duration of action of AGRP(86-132) despite the truncation of 34 residues, an important step in the development of probes and therapeutic leads for the treatment of disease states with negative energy balance.
Conclusions
Overall, these studies advance the development of potent and selective antagonists at the melanocortin 4 receptors that lack MC1R agonism and identify a position in the macrocyclic template c[Pro-Arg-Phe-Phe-Asn-Ala-Phe-DPro] that can be modified to generate peptides possessing high nanomolar mMC5R inverse agonism. These ligands, and derivatives thereof, can make interesting probes for the investigation of the physiological role(s) of the MC5R. This study also produced one ligand (KAF2039-7) that was equipotent to the lead ligand (1), and was 200-fold more selective for the mMC4R over the mMC3R without possessing MC1R agonist activity. When injected IT into male mice, this peptide causes a significant increase in food intake at 2 and 6 hours post-injection, with a trend exhibited out until 72 hours post-injection. Potent and selective antagonists are useful pharmacological probes in understanding the differential roles of the mMC3R and mMC4R in body weight management and energy homeostasis, and afford a deeper understanding of the melanocortin system. The insights provided by these data will be useful in the future development of therapeutics to treat anorexia, cachexia, or other diseases of negative energy balance such as failure to thrive.
Experimental Section
Peptide Synthesis
All peptides were synthesized using flourenyl-9-methoxycarbonyl (Fmoc) methodology on a H-Pro-CTC Resin (0.67 meqiv/g substitution) purchased from Peptides International (Louisville, KY).57–58 Coupling reagents O-benzotriazol-1-yl-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU), (benzotriazolyloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP), and 1-hydroxybenzotriazole (HOBt) were purchased from Peptides International (Louisville, KY). Amino acids Fmoc-DPro, Fmoc-Gly, Fmoc-Ser(tBu), Fmoc-Lys(Boc), Fmoc-Asp(OtBu), Fmoc-Nle, Fmoc-DPhe, Fmoc-Trp(Boc), Fmoc-Tyr(tBu), Fmoc-cyclohexylalanine (Fmoc-Cha), Fmoc-Ala, Fmoc-Asn(Trt), Fmoc-Phe, and Fmoc-Arg(Pbf), which were purchased from Peptides International (Louisville, KY). The Fmoc-homophenylalanine (Fmoc-hPhe) was purchased from Synthetech (Albany, OR). Dichloromethane (DCM), N,N-Dimethylformamide (DMF), methanol, acetonitrile, and anhydrous ethyl ether were purchased from Fisher (Fair Lawn, NJ). Trifluoroacetic acid (TFA), piperidine, dimethyl sulfoxide (DMSO), N-N-Diisopropylethylamine (DIEA) and triisopropylsilane (TIS) were purchased from Sigma-Aldrich (St. Lous, MO). All reagents were ACS grade or better, and were used without purification.
Peptides were synthesized in parallel at a 0.075 mmol scale on a H-Pro-CTC-resin using a semi-automated synthesizer (LabTech 1, Advanced Chemtech, Lousiville, KY). Resin was swelled in dimethylformamide (DMF) for at least one hour prior to the first coupling. The first two amino acids (DPro, and variable) were coupled using the semi-automated synthesizer in a 16-well Teflon reaction block. Fmoc protected amino acid (3.1 eqiv), HBTU (3 equiv), and DIEA (5 equiv) were added, and the solution was agitated for 1 hour. The presence of a free secondary amine was monitored using a chloranil test.64 This procedure was repeated if necessary. Next, the amino acid was deprotected with 20% piperidine in DMF (1 × 2 min, 1 × 18 min). Deprotection was monitored again using a chloranil test. The second amino acid was coupled using the same procedure as described above. Subsequent amino acids were coupled on an automated synthesizer (Vantage, Advanced Chemtech, Lousville, KY). The following procedure was used: (i) wash with DMF (4 mL for 2 min × 3), (ii) deprotect with 20% piperidine in DMF (4 mL × 5 min, 4 mL × 20 min), (iii) wash with DMF (4 mL for 2 min × 3), (iv) couple with Fmoc amino acid (3.1 equiv, dissolved in DMF), HBTU (3 equiv, dissolved in DMF), and DIEA (5 equiv) for 1hr, (iv) empty well block and repeat coupling procedure, (v) proceed to coupling of next amino acid using steps i-iv. Following synthesis, the N-terminal Fmoc amines were deprotected with 20% piperidiene in DMF (2 mL/well, 1 × 2 min, 1 × 18 min) and dried in vacuo after washing with methanol.
Peptides were cleaved from solid support using 1% trifluoroacetic acid (TFA) in dichloromethane (DCM) by rinsing the resin 4 times with 2 mL of cleavage solution for 1.5 minutes. Peptides were precipitated with cold ether and pelleted via centrifugation (4,000 rpm at 4°C for 4 minutes, ThermoScientific Sorvall Legend XTR). The supernatant was decanted, and the peptides were then dried in vacuo in the presence of desiccant overnight. For the coupling of the C-terminus (Pro1 in the macrocyclic template) to the N-terminus (Arg2 in the macrocyclic template) to create the macrocycle, peptides were dissolved in DCM (1 mM), and cyclized using BOP (3 equiv), HOBt (3 equiv), and DIEA (6 equiv), stirring the solution overnight at room temperature. The peptides were dried under reduced pressure for a minimum of 1 hour, and 5 mL of a 95:2.5:2.5 TFA:H2O:TIS solution were added at room temperature for 2 hours. The peptides were then concentrated via evaporation under an increased pressure of N2(g) and precipitated with cold ether. Peptides were pelleted via centrifugation at 4,000 rpm at 4°C for 4 minutes, and the supernatant was decanted. The final products were dried in vacuo in the presence of desiccant.
Peptides were purified via reverse phase high pressure liquid chromatography (RP-HPLC, Shimadzu) on a semi-preparative C18 column (Vydac 218TP1010, 1 cm × 25 cm) using acetonitrile and 0.1% TFA in H2O. Analytical data was then collected using an analytical C18 column (Vydac 218TP, 4.6 mm × 250 mm) on a Shimadzu chromatography system equipped with a photodiode array detector in two different solvent systems: acetonitrile/0.1% TFA in H2O, and methanol/0.1% TFA in H2O to confirm purity > 95%. Molecular weights were then confirmed using matrix-assisted laser desorption ionization (MALDI) and time of flight (TOF) mass spectrometry analysis (AB-Sciex 5800 MALDI/TOF-MS, LeClaire-Dow Instrumentation Facility, University of Minnesota), using a cyano-4-hydroxycinnamic acid (CCA) matrix. Peptides were assayed as TFA salts.
AlphaScreen™ cAMP Functional Assay
The AlphaScreen™ cAMP assay (PerkinElmer Life Sciences, Cat #6760625M) was used to measure the functional cAMP potencies of peptides at HEK293 cells stably expressing the individual melanocortin receptors (mMC1R, mMC3R, mMC4R, and mMC5R). The assay was performed according to manufacturer instructions, and as previously described.59–61
Cells were 70-90% confluent at the start of the assay. Growth medium was aspirated and cells were rinsed with 1 mL Gibco® Versene solution, then 1 mL fresh Versene solution was added. Cells were incubated at 37°C until cells had detached from the plate, then pelleted via centrifugation (800 rpm, 5 minutes; Sorvall™ Legend™ XTR centrifuge, swinging bucket rotor). The medium was aspirated and the cell pellet was resuspended a second time in Dulbecco’s phosphate buffered saline solution (DPBS 1× [−] without calcium and magnesium chloride, Gibco ® Cat#14190-144). A 10 μL of each cell line was removed for counting, during which the remaining cells were centrifuged a second time as described above. Cells were counted by adding 10 μL Trypan blue dye (BioRad) and counted manually using a hemocytometer. After centrifugation, the DPBS was aspirated and the pellet was resuspended in a solution of freshly-made stimulation buffer [Hank’s Balanced Salt Solution (HBSS 10× (−) sodium bicarbonate) and (−) phenol red, Gibco®), 0.5 mM isobutylmethylxanthine (IBMX), 5 mM HEPES buffer solution (1M, Gibco®), 0.1% bovine serum albumin (BSA) in Milli-Q water, pH = 7.4] to a concentration of 10,000 cells per μL. An acceptor bead solution was created by diluting the acceptor bead stock solution (5 mg/mL anti-cAMP acceptor beads in stimulation buffer) to a concentration of 0.1 mg/mL in stimulation buffer. The acceptor bead solution was added to the cells such that there were 0.5 μg anti-cAMP acceptor beads in each cell line.
Next, 10 μL of the cell/acceptor bead solution per well was added to an Opti-384 plate using a 16 channel pipettor. This was repeated for each cell line. To each well 5 μL of compound was added so that, when diluted with the 10 μL cell/acceptor bead solution, the compound reached the desired concentration(s). Compounds were run in duplicate replicates, and each cell line had a positive control (10−4 M forskolin) and negative control (plain stimulation buffer, instead of compound). The 384-well plate was sealed with a cover slip, covered with aluminum foil, and incubated at room temperature in a dark desk drawer for 2 hours. A biotinylated cAMP/Streptavidin-coated donor bead working solution was prepared by diluting the stock solution of donor beads (5 mg/mL) and cAMP biotinylated tracer (1 μL with 1× PBS 14190-144) in a freshly prepared lysis buffer [10% Tween-20, 5 mM HEPES buffer solution (1M, Gibco®), 0.1% bovine serum albumin (BSA) in Milli-Q water, pH = 7.4] such that the final solution contained 0.5 μg of donor beads and 0.62 μmol biotinylated cAMP. This donor bead/biotinylated cAMP solution was allowed to incubate in a dark desk drawer at room temperature for a minimum of 30 minutes.
After the incubation period for the cells/acceptor beads/compound solution was completed, 10 μL of the donor bead/biotinylated cAMP mixture was added to each well in a room containing a green light using a multichannel pipettor, mixing well. The plate was then re-sealed with the cover slip, covered in aluminum foil, and allowed to incubate in a dark desk drawer at room temperature for another 2 hours. Following the second incubation period, the plate was inserted into an Enspire™ Alpha plate reader and read using a pre-normalized assay protocol set by the manufacturer.
Cell Data Analysis
The data collected were analyzed using PRISM software (v4.0, GraphPad Inc.). Agonist potency was evaluated by calculating the EC50 values using non-linear regression analysis with the PRISM software. Antagonist potency was determined using a Schild analysis [pA2 = −log(Ki)] and the agonist ligand NDP-MSH.62 Because this assay is loss-of-function, mMC1R, mMC3R, and mMC4R data were normalized to represent a percent response relative to control ligand (NDP-MSH), as has been reported previously, unless otherwise specified.59–61 For illustrative purposes, mMC1R partial agonist activity (seen in Figure 4) was normalized to a basal response to yield a percent difference from basal activity. For illustrative and data analysis purposes, inverse agonist activity (seen in Figure 5) for the mMC5R was also normalized to a basal response to yield a percent difference from basal activity.
Animals
This study was conducted in accordance with the guidelines set up by the Institutional Animal Care and Use Committee (IACUC) at the University of Minnesota. Male Wildtype (WT) mice (mixed 129/Sv×C57BL/6J background) derived from an in house breeding colony were used throughout this experiment as previously reported in literature.14, 23, 61, 63, 65 Each mouse was individually housed in standard polycarbonate conventional cages provided by the University of Minnesota’s Research Animal Resources (RAR). Mice were singly housed in order to adequately measure food intake of individual mice during experiments. At the beginning of this experiment, mice were age-matched at 24 weeks old. Research lab staff performed weekly cage changes. Mice had ad libitum access to normal chow (Harlan Teklad 2018 Diet: 18.6% crude protein, 6.2% crude fat, 3.5% crude fiber, with energy density of 3.1kcal/g) and water. Mice were maintained on a revered 12-hour light/dark cycle (lights off at 12:00pm) and housed in a temperature-controlled room at 23°-25°C. Mice were monitored daily to assess health.
Mouse Feeding Studies
All mouse feeding experiments were designed following a crossover, non-fasting (nocturnal) paradigm. Compound (KAF2039-7) or vehicle was administered IT in a single injection 2 hours before lights out (t=0h). Intrathecal injections were performed as previously described.63, 66–68 Mouse body weight and food weight was recorded in 2, 4, 6, 8, 24, 48 and 72 hours post-injection following a single IT injection. Mice were given 7 days between treatments to re-establish pre-treatment feeding behavior and body weight. Eight male WT mice were used for the whole feeding study.
Compounds
A stock of KAF2039-7 was prepared using a 20% solutol (Kolliphor HS 15; Sigma) solution (final concentration of 10nmol/μL) and stored at −20°C. On days of experimentation, the KAF2039-7 stock was diluted using sterile ddH2O to the desired concentration of 2nmol/5μL. The vehicle was created using identical volumes of 20% solutol to sterile ddH2O as the experimental compound.
Animal Data Analysis
Primary dependent variables were: (i) food intake; and (ii) body weight. Food intake and body weight was analyzed a two-factor within-subject Analysis of Variance (ANOVA) with between session variables of compound and the within-subject variable of time. To identify sources of significant interactions at specific time-points, follow-up independent sample t-tests with Bonferroni correction was performed. Graphpad Prism was used to graph data. Data was analyzed using Statistical Package for the Social Science Software (SPSS) and was represented as the mean ± error with p < 0.05 indicating significance.
Cumulative Food Intake Statistics
To examine the effect of drug (7/Vehicle) on food intake during the first 72 hours post-injection, a two-factor repeated measures Analysis of Variance (ANOVA) was performed with drug as the between-subject factor and time as the within-subject factor. Results showed that there was a main effect of drug (F1,14 = 7.017; p = 0.019). A follow-up independent sample t-test with Bonferroni correction (to control for multiple comparisons) revealed that mice that were administered with 2nmol of 7 ate significantly more 2 (t14 = −3.145; p = 0.007) and 6 (t14 = −4.030; p = 0.001) hours post-injection compared to vehicle controls.
Mouse Weight Statistics
To analyze the effect of 2 nmol 7 on changes in mouse weight, a two-factor repeated measure ANOVA was performed, with drug as the between-subject factor and time as the within-subject factor. Results indicated that there was a main effect of time (F6,84 = 2.578; p = 0.024) and a main effect of drug (F1,14 = 2.938; p = 0.045). A follow-up independent sample t-test with Bonferroni correction did not show significance between treatment at specific time points.
Acknowledgments
Funding Sources. This work has been supported by the NIH Grant R01DK091906 (C.H.-L.). M.D.E. is a recipient of an NIH F32 Postdoctoral Fellowship (F32DK108402). K.A.F. is a recipient of a 2018 Bighley Fellowship and a 2018 Roswell Fellowship. We would also like to acknowledge the receipt of a 2017 Wallin Neuroscience Discovery Fund Award.
Abbreviations
- ACTH
adrenocorticotropin hormone
- Fmoc
9-fluorenylmethoxycarbonyl
- AGRP
agouti-related protein
- GPCR
G-protein coupled receptor
- cAMP
cyclic 5′-adenosine monophosphate
- MC1R
melanocortin-1 receptor
- MC2R
melanocortin-2 receptor
- MC3R
melanocortin-3 receptor
- MC4R
melanocortin-4 receptor
- MC5R
melanocortin-5 receptor
- MSH
melanocyte stimulating hormone
- POMC
proopiomelanocortin
- α-MSH
alpha-melanocyte stimulating hormone
- β-MSH
beta-melanocyte stimulating hormone
- γ-MSH
gamma-melanocyte stimulating hormone
- μM
micromolar
- NDP-MSH (4-Norleucine-7-D-Phenylalanine)
Ac-Ser-Tyr-Ser-Nle-Glu-His-DPhe-Arg-Trp-Gly-Lys-Pro-Val-NH2
- Nle
norleucine
- Cha
cyclohexylalanine
- hPhe
homophenylalanine
- RP-HPLC
reverse-phase high-pressure liquid chromatography
- SAR
structure-activity relationships
- SNP
single nucleotide polymorphism
- IT
intrathecal
Footnotes
Author Contributions. K.A.F., M.D.E., and C.H.-L. designed the research. K.A.F., K.T.F., D.N.A., M.M.L., and S.L.W. performed experiments. K.A.F. and C.H.-L. analyzed the data. K.A.F. wrote the manuscript with the help of C.H.-L.
Conflict of Interest. The authors declare no competing financial interests.
References
- 1.Chhajlani V, Muceniece R, Wikberg JES. Molecular Cloning of a Novel Human Melanocortin Receptor. Biochem Biophys Res Commun. 1993;195:866–873. doi: 10.1006/bbrc.1993.2125. [DOI] [PubMed] [Google Scholar]
- 2.Chhajlani V, Wikberg JES. Molecular Cloning and Expression of the Human Melanocyte Stimulating Hormone Receptor cDNA. FEBS Lett. 1992;309:417–420. doi: 10.1016/0014-5793(92)80820-7. [DOI] [PubMed] [Google Scholar]
- 3.Gantz I, Konda Y, Tashiro T, Shimoto Y, Miwa H, Munzert G, Watson SJ, DelValle J, Yamada T. Molecular Cloning of a Novel Melanocortin Receptor. J Biol Chem. 1993;268:8246–8250. [PubMed] [Google Scholar]
- 4.Gantz I, Miwa H, Konda Y, Shimoto Y, Tashiro T, Watson SJ, DelValle J, Yamada T. Molecular Cloning, Expression, and Gene Localization of a Fourth Melanocortin Receptor. J Biol Chem. 1993;268:15174–15179. [PubMed] [Google Scholar]
- 5.Gantz I, Shimoto Y, Konda Y, Miwa H, Dickinson CJ, Yamada T. Molecular Cloning, Expression, and Characterization of a Fifth Melanocortin Receptor. Biochem Biophys Res Commun. 1994;200:1214–1220. doi: 10.1006/bbrc.1994.1580. [DOI] [PubMed] [Google Scholar]
- 6.Griffon N, Mignon V, Facchinetti P, Diaz J, Schwartz JC, Sokoloff P. Molecular Cloning and Characterisation of the Rat Fifth Melanocortin Receptor. Biochem Biophys Res Commun. 1994;200:1007–1014. doi: 10.1006/bbrc.1994.1550. [DOI] [PubMed] [Google Scholar]
- 7.Mountjoy K, Robbins L, Mortrud M, Cone R. The Cloning of a Family of Genes that Encode the Melanocortin Receptors. Science. 1992;257:1248–1251. doi: 10.1126/science.1325670. [DOI] [PubMed] [Google Scholar]
- 8.Roselli-Rehfuss L, Mountjoy KG, Robbins LS, Mortrud MT, Low MJ, Tatro JB, Entwistle ML, Simerly RB, Cone RD. Identification of a Receptor for γ Melanotropin and Other Proopiomelanocortin Peptides in the Hypothalamus and Limbic System. Proc Natl Acad Sci. 1993;90:8856–8860. doi: 10.1073/pnas.90.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lerner AB, McGuire JS. Effect of ɑ- and β-Melanocyte Stimulating Hormones on the Skin Colour of Man. Nature. 1961;189:176–179. doi: 10.1038/189176a0. [DOI] [PubMed] [Google Scholar]
- 10.Chen AS, Marsh DJ, Trumbauer ME, Frazier EG, Guan XM, Yu H, Rosenblum CI, Vongs A, Feng Y, Cao L, Metzger JM, Strack AM, Camacho RE, Mellin TN, Nunes CN, Min W, Fisher J, Gopal-Truter S, MacIntyre DE, Chen HY, Van der Ploeg LHT. Inactivation of the Mouse Melanocortin-3 Receptor Results in Increased Fat Mass and Reduced Lean Body Mass. Nat Genet. 2000;26:97–102. doi: 10.1038/79254. [DOI] [PubMed] [Google Scholar]
- 11.Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of Melanocortinergic Neurons in Feeding and the Agouti Obesity Syndrome. Nature. 1997;385:165–168. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 12.Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Targeted Disruption of the Melanocortin-4 Receptor Results in Obesity in Mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 13.Butler AA, Kesteson RA, Khong K, Cullen MJ, Pelleymounter MA, Dekoning J, Baetscher M, Cone RD. A Unique Metalolic Syndrome Causes Obesity in the Melanocortin-3 Receptor-Deficient Mouse. Endocrinology. 2000;141:3518–3521. doi: 10.1210/endo.141.9.7791. [DOI] [PubMed] [Google Scholar]
- 14.Irani BG, Xiang Z, Yarandi HN, Holder JR, Moore MC, Bauzo RM, Proneth B, Shaw AM, Millard WJ, Chambers JB, Benoit SC, Clegg DJ, Haskell-Luevano C. Implication of the Melanocortin-3 Receptor in the Regulation of Food Intake. Eur J Pharmacol. 2011;660:80–87. doi: 10.1016/j.ejphar.2010.10.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakanishi S, Inoue A, Kita T, Inoue A, Nakamura M, Chang ACY, Cohen SN, Numa S. Nucleotide Sequence of Cloned cDNA for Bovine Corticotropin-β-Lipotropin Precursor. Nature. 1979;278:423–427. doi: 10.1038/278423a0. [DOI] [PubMed] [Google Scholar]
- 16.Eipper BA, Mains RE. Structure and Biosynthesis of Pro-Adrenocorticotropin/Endorphin and Related Peptides. Endocr Rev. 1980;1:1–27. doi: 10.1210/edrv-1-1-1. [DOI] [PubMed] [Google Scholar]
- 17.Smith AI, Funder JW. Proopiomelanocortin Processing in the Pituitary, Central Nervous System, and Peripheral Tissues. Endocr Rev. 1988;9:159–179. doi: 10.1210/edrv-9-1-159. [DOI] [PubMed] [Google Scholar]
- 18.Bultman SJ, Michaud EJ, Woychik RP. Molecular Characterization of the Mouse Agouti Locus. Cell. 1992;71:1195–1204. doi: 10.1016/s0092-8674(05)80067-4. [DOI] [PubMed] [Google Scholar]
- 19.Ollmann MM, Wilson BD, Yang YK, Kerns JA, Chen Y, Gantz I, Barsh GS. Antagonism of Central Melanocortin Receptors in Vitro and in Vivo by Agouti-Related Protein. Science. 1997;278:135–138. doi: 10.1126/science.278.5335.135. [DOI] [PubMed] [Google Scholar]
- 20.Blanchard SG, Harris CO, Ittoop O, Nichols JS, Parks DJ, Truesdale AT, Wilkison WO. Agouti Antagonism of Melanocortin Binding and Action in the B16F10 Murine Melanoma Cell Line. Biochemistry. 1995;34:10406–10411. doi: 10.1021/bi00033a012. [DOI] [PubMed] [Google Scholar]
- 21.Fong TM, Mao C, MacNeil T, Kalyani R, Smith T, Weinberg D, Tota MR, Van der Ploeg LHT. ART (Protein Product of Agouti-Related Transcript) as an Antagonist of MC-3 and MC-4 Receptors. Biochem Biophys Res Commun. 1997;237:629–631. doi: 10.1006/bbrc.1997.7200. [DOI] [PubMed] [Google Scholar]
- 22.Miller MW, Duhl DM, Vrieling H, Cordes SP, Ollmann MM, Winkes BM, Barsh GS. Cloning of the Mouse Agouti Gene Predicts a Secreted Protein Ubiquitously Expressed in Mice Carrying the Lethal Yellow Mutation. Genes Dev. 1993;7:454–467. doi: 10.1101/gad.7.3.454. [DOI] [PubMed] [Google Scholar]
- 23.Rowland NE, Schaub JW, Robertson KL, Andreasen A, Haskell-Luevano C. Effect of MTII on Food Intake and Brain c-fos in Melanocortin-3, Melanocortin-4, and Double MC3 and MC4 Receptor Knockout Mice. Peptides. 2010;31:2314–2317. doi: 10.1016/j.peptides.2010.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Farooqi IS, Keogh JM, Yeo GSH, Lank EJ, Cheetham T, O’Rahilly S. Clinical Spectrum of Obesity and Mutations in the Melanocortin 4 Receptor Gene. N Engl J Med. 2003;348:1085–1095. doi: 10.1056/NEJMoa022050. [DOI] [PubMed] [Google Scholar]
- 25.Krude H, Biebermann H, Luck W, Horn R, Brabant G, Gruters A. Severe Early-Onset Obesity, Adrenal Insufficiency and Red Hair Pigmentation Caused by POMC Mutations in Humans. Nat Genet. 1998;19:155–157. doi: 10.1038/509. [DOI] [PubMed] [Google Scholar]
- 26.Yaswen L, Diehl N, Brennan MB, Hochgeschwender U. Obesity in the Mouse Model of Pro-Opiomelanocortin Deficiency Responds to Peripheral Melanocortin. Nat Med. 1999;5:1066–1070. doi: 10.1038/12506. [DOI] [PubMed] [Google Scholar]
- 27.Ericson MD, Lensing CJ, Fleming KA, Schlasner KN, Doering SR, Haskell-Luevano C. Bench-Top to Clinical Therapies: A Review of Melanocortin Ligands from 1954 to 2016. Biochim Biophys Acta, Mol Basis Dis. 2017;1863:2414–2435. doi: 10.1016/j.bbadis.2017.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Graham M, Shutter JR, Sarmiento U, Sarosi I, Stark KL. Overexpression of AGRT Leads to Obesity in Transgenic Mice. Nat Genet. 1997;17:273–274. doi: 10.1038/ng1197-273. [DOI] [PubMed] [Google Scholar]
- 29.Ebihara K, Ogawa Y, Katsuura G, Numata Y, Masuzaki H, Satoh N, Tamaki M, Yoshioka T, Hayase M, Matsuoka N, Aizawa-Abe M, Yoshimasa Y, Nakao K. Involvement of Agouti-Related Protein, an Endogenous Antagonist of Hypothalamic Melanocortin Receptor, in Leptin Action. Diabetes. 1999;48:2028–2033. doi: 10.2337/diabetes.48.10.2028. [DOI] [PubMed] [Google Scholar]
- 30.Kas MJH, van Dijk G, Scheurink AJW, Adan RAH. Agouti-Related Protein Prevents Self-Starvation. Mol Psychiatry. 2003;8:235–240. doi: 10.1038/sj.mp.4001206. [DOI] [PubMed] [Google Scholar]
- 31.Adan RAH, Hillebrand JJG, De Rijke C, Nijenhuis W, Vink TOM, Garner KM, Kas MJH. Melanocortin System and Eating Disorders. Ann NY Acad Sci. 2003;994:267–274. doi: 10.1111/j.1749-6632.2003.tb03189.x. [DOI] [PubMed] [Google Scholar]
- 32.Vink T, Hinney A, van Elburg AA, van Goozen SH, Sandkuijl LA, Sinke RJ, Herpertz-Dahlmann BM, Hebebrand J, Remschmidt H, van Engeland H, Adan RA. Association Between an Agouti-Related Protein Gene Polymorphism and Anorexia Nervosa. Mol Psychiatry. 2001;6:325–328. doi: 10.1038/sj.mp.4000854. [DOI] [PubMed] [Google Scholar]
- 33.Ge YL, Ohta T, Driscoll DJ, Nicholls RD, Kalra SP. Anorexigenic Melanocortin Signaling in the Hypothalamus is Augmented in Association with Failure-to-Thrive in a Transgenic Mouse Model for Prader–Willi Syndrome. Brain Res. 2002;957:42–45. doi: 10.1016/s0006-8993(02)03583-7. [DOI] [PubMed] [Google Scholar]
- 34.Marks DL, Butler AA, Turner R, Brookhart G, Cone RD. Differential Role of Melanocortin Receptor Subtypes in Cachexia. Endocrinology. 2003;144:1513–1523. doi: 10.1210/en.2002-221099. [DOI] [PubMed] [Google Scholar]
- 35.Marks DL, Ling N, Cone RD. Role of the Central Melanocortin System in Cachexia. Cancer Res. 2001;61:1432–1438. [PubMed] [Google Scholar]
- 36.Haskell-Luevano C, Monck EK. Agouti-Related Protein Functions as an Inverse Agonist at a Constitutively Active Brain Melanocortin-4 Receptor. Regul Pept. 2001;99:1–7. doi: 10.1016/s0167-0115(01)00234-8. [DOI] [PubMed] [Google Scholar]
- 37.Nijenhuis WAJ, Oosterom J, Adan RAH. AgRP(83–132) Acts as an Inverse Agonist on the Human-Melanocortin-4 Receptor. Mol Endocrinol. 2001;15:164–171. doi: 10.1210/mend.15.1.0578. [DOI] [PubMed] [Google Scholar]
- 38.Lu D, Willard D, Patel IR, Kadwell S, Overton L, Kost T, Luther M, Chen W, Woychik RP, Wilkison WO, Cone RD. Agouti Protein is an Antagonist of the Melanocyte-Stimulating-Hormone Receptor. Nature. 1994;371:799–802. doi: 10.1038/371799a0. [DOI] [PubMed] [Google Scholar]
- 39.McNulty JC, Jackson PJ, Thompson DA, Chai B, Gantz I, Barsh GS, Dawson PE, Millhauser GL. Structures of the Agouti Signaling Protein. J Mol Biol. 2005;346:1059–1070. doi: 10.1016/j.jmb.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 40.Kiefer LL, Veal JM, Mountjoy KG, Wilkison WO. Melanocortin Receptor Binding Determinants in the Agouti Protein. Biochemistry. 1998;37:991–997. doi: 10.1021/bi971913h. [DOI] [PubMed] [Google Scholar]
- 41.Yang Y-k, Dickinson CJ, Zeng Q, Li JY, Thompson DA, Gantz I. Contribution of Melanocortin Receptor Exoloops to Agouti-Related Protein Binding. J Biol Chem. 1999;274:14100–14106. doi: 10.1074/jbc.274.20.14100. [DOI] [PubMed] [Google Scholar]
- 42.Yang Y-k, Thompson DA, Dickinson CJ, Wilken J, Barsh GS, Kent SBH, Gantz I. Characterization of Agouti-Related Protein Binding to Melanocortin Receptors. Mol Endocrinol. 1999;13:148–155. doi: 10.1210/mend.13.1.0223. [DOI] [PubMed] [Google Scholar]
- 43.Chai BX, Neubig RR, Millhauser GL, Thompson DA, Jackson PJ, Barsh GS, Dickinson CJ, Li JY, Lai YM, Gantz I. Inverse Agonist Activity of Agouti and Agouti-Related Protein. Peptides. 2003;24:603–609. doi: 10.1016/s0196-9781(03)00104-9. [DOI] [PubMed] [Google Scholar]
- 44.Haskell-Luevano C, Monck EK, Wan YP, Schentrup AM. The Agouti-Related Protein Decapeptide (Yc[CRFFNAFC]Y) Possesses Agonist Activity at the Murine Melanocortin-1 Receptor. Peptides. 2000;21:683–689. doi: 10.1016/s0196-9781(00)00194-7. [DOI] [PubMed] [Google Scholar]
- 45.Patel MP, Fabersunne CSC, Yang Y-k, Kaelin CB, Barsh GS, Millhauser GL. Loop Swapped Chimeras of the Agouti-related Protein (AgRP) and the Agouti Signaling Protein (ASIP) Identify Contacts Required for Melanocortin 1 Receptor (MC1R) Selectivity and Antagonism. J Mol Biol. 2010;404:45–55. doi: 10.1016/j.jmb.2010.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ericson MD, Wilczynski A, Sorensen NB, Xiang Z, Haskell-Luevano C. Discovery of a β-Hairpin Octapeptide, c[Pro-Arg-Phe-Phe-Dap-Ala-Phe-DPro], Mimetic of Agouti-Related Protein(87–132) [AGRP(87–132)] with Equipotent Mouse Melanocortin-4 Receptor (mMC4R) Antagonist Pharmacology. J Med Chem. 2015;58:4638–4647. doi: 10.1021/acs.jmedchem.5b00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tota MR, Smith TS, Mao C, MacNeil T, Mosley RT, Van der Ploeg LHT, Fong TM. Molecular Interaction of Agouti Protein and Agouti-Related Protein with Human Melanocortin Receptors. Biochemistry. 1999;38:897–904. doi: 10.1021/bi9815602. [DOI] [PubMed] [Google Scholar]
- 48.Wilczynski A, Wang XS, Joseph CG, Xiang Z, Bauzo RM, Scott JW, Sorensen NB, Shaw AM, Millard WJ, Richards NG, Haskell-Luevano C. Identification of Putative Agouti-Related Protein(87−132)-Melanocortin-4 Receptor Interactions by Homology Molecular Modeling and Validation Using Chimeric Peptide Ligands. J Med Chem. 2004;47:2194–2207. doi: 10.1021/jm0303608. [DOI] [PubMed] [Google Scholar]
- 49.McNulty JC, Thompson DA, Bolin KA, Wilken J, Barsh GS, Millhauser GL. High-Resolution NMR Structure of the Chemically-Synthesized Melanocortin Receptor Binding Domain AGRP(87−132) of the Agouti-Related Protein. Biochemistry. 2001;40:15520–15527. doi: 10.1021/bi0117192. [DOI] [PubMed] [Google Scholar]
- 50.Bolin KA, Anderson DJ, Trulson JA, Thompson DA, Wilken J, Kent SBH, Gantz I, Millhauser GL. NMR Structure of a Minimized Human Agouti Related Protein Prepared by Total Chemical Synthesis. FEBS Lett. 1999;451:125–131. doi: 10.1016/s0014-5793(99)00553-0. [DOI] [PubMed] [Google Scholar]
- 51.Jackson PJ, McNulty JC, Yang YK, Thompson DA, Chai B, Gantz I, Barsh GS, Millhauser GL. Design, Pharmacology, and NMR Structure of a Minimized Cystine Knot with Agouti-Related Protein Activity. Biochemistry. 2002;41:7565–7572. doi: 10.1021/bi012000x. [DOI] [PubMed] [Google Scholar]
- 52.Späth J, Stuart F, Jiang L, Robinson JA. Stabilization of a β-Hairpin Conformation in a Cyclic Peptide Using the Templating Effect of a Heterochiral Diproline Unit. Helv Chim Acta. 1998;81:1726–1738. [Google Scholar]
- 53.Favre M, Moehle K, Jiang L, Pfeiffer B, Robinson JA. Structural Mimicry of Canonical Conformations in Antibody Hypervariable Loops Using Cyclic Peptides Containing a Heterochiral Diproline Template. J Am Chem Soc. 1999;121:2679–2685. [Google Scholar]
- 54.Jiang L, Moehle K, Dhanapal B, Obrecht D, Robinson JA. Combinatorial Biomimetic Chemistry: Parallel Synthesis of a Small Library of β-Hairpin Mimetics Based on Loop III from Human Platelet-Derived Growth Factor B. Helv Chim Acta. 2000;83:3097–3112. [Google Scholar]
- 55.Robinson JA. β-Hairpin Peptidomimetics: Design, Structures and Biological Activities. Acc Chem Res. 2008;41:1278–1288. doi: 10.1021/ar700259k. [DOI] [PubMed] [Google Scholar]
- 56.Ericson MD, Freeman KT, Schnell SM, Fleming KA, Haskell-Luevano C. Structure–Activity Relationship Studies on a Macrocyclic Agouti-Related Protein (AGRP) Scaffold Reveal Agouti Signaling Protein (ASP) Residue Substitutions Maintain Melanocortin-4 Receptor Antagonist Potency and Result in Inverse Agonist Pharmacology at the Melanocortin-5 Receptor. J Med Chem. 2017;60:8103–8114. doi: 10.1021/acs.jmedchem.7b00856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carpino LA. The 9-Fluorenylmethoxycarbonyl Function, a New Base-Sensitive Amino-Protecting Group. J Am Chem Soc. 1970;92:5748–5749. [Google Scholar]
- 58.Carpino LA, H GY. The 9-Fluorenylmethoxycarbonyl Amino-Protecting Group. J Org Chem. 1972;37:3404–3409. [Google Scholar]
- 59.Singh A, Tala SR, Flores V, Freeman K, Haskell-Luevano C. Synthesis and Pharmacology of α/β3-Peptides Based on the Melanocortin Agonist Ac-His-dPhe-Arg-Trp-NH2 Sequence. ACS Med Chem Lett. 2015;6:568–572. doi: 10.1021/acsmedchemlett.5b00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ericson MD, Schnell SM, Freeman KT, Haskell-Luevano C. A Fragment of the Escherichia coli ClpB Heat-Shock Protein is a Micromolar Melanocortin 1 Receptor Agonist. Bioorg Med Chem Lett. 2015;25:5306–5308. doi: 10.1016/j.bmcl.2015.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lensing CJ, Freeman KT, Schnell SM, Adank DN, Speth RC, Haskell-Luevano C. An in Vitro and in Vivo Investigation of Bivalent Ligands That Display Preferential Binding and Functional Activity for Different Melanocortin Receptor Homodimers. J Med Chem. 2016;59:3112–3128. doi: 10.1021/acs.jmedchem.5b01894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schild HO. pA, A New Scale for the Measurement of Drug Antagonism. Br J Pharmacol Chemother. 1997;120:29–46. doi: 10.1111/j.1476-5381.1947.tb00336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Adank DN, Lunzer MM, Lensing CJ, Wilber SL, Gancarz AM, Haskell-Luevano C. Comparative In Vivo Investigation of Intrathecal and Intracerebroventricular Administration with Melanocortin Ligands MTII and AGRP into Mice. ACS Chem Neurosci. 2017 doi: 10.1021/acschemneuro.7b00330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Christensen T. Qualitative Test for Monitoring Coupling Completeness in Solid-Phase Peptide-Synthesis Using Chloranil. Acta Chem Scand, Ser B. 1979;33:763–766. [Google Scholar]
- 65.Lensing CJ, Adank DN, Doering SR, Wilber SL, Andreasen A, Schaub JW, Xiang Z, Haskell-Luevano C. Ac-Trp-DPhe(p-I)-Arg-Trp-NH2, a 250-Fold Selective Melanocortin-4 Receptor (MC4R) Antagonist over the Melanocortin-3 Receptor (MC3R), Affects Energy Homeostasis in Male and Female Mice Differently. ACS Chem Neurosci. 2016;7:1283–1291. doi: 10.1021/acschemneuro.6b00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Le Naour M, Lunzer MM, Powers MD, Portoghese PS. Opioid Activity of Spinally Selective Analogues of N-naphthoyl-β-naltrexamine in HEK-293 Cells and Mice. J Med Chem. 2012;55:670–677. doi: 10.1021/jm200902v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lunzer MM, Portoghese PS. Selectivity of δ- and κ-opioid Ligands Depends on the Route of Central Administration in Mice. J Pharmacol Exp Ther. 2007;322:166–171. doi: 10.1124/jpet.107.120279. [DOI] [PubMed] [Google Scholar]
- 68.Hylden JL, Wilcox GL. Intrathecal Morphine in Mice: a New Technique. Eur J Pharmacol. 1980;67:313–316. doi: 10.1016/0014-2999(80)90515-4. [DOI] [PubMed] [Google Scholar]