

Abstract
PURPOSE
Wnt/β-catenin signaling is required for leukemic stem cell function. FLT3 mutations are frequently observed in acute myeloid leukemia (AML). Anomalous FLT3 signaling increases β-catenin nuclear localization and transcriptional activity. FLT3 tyrosine kinase inhibitors (TKIs) are used clinically to treat FLT3-mutated AML patients, but with limited efficacy. We investigated the anti-leukemia activity of combined Wnt/β-catenin and FLT3 inhibition in FLT3-mutant AML.
EXPERIMENTAL DESIGN
Wnt/β-catenin signaling was inhibited by the β-catenin/CBP antagonist C-82/PRI-724 or siRNAs, and FLT3 signaling by sorafenib or quizartinib. Treatments on apoptosis, cell growth, and cell signaling were assessed in cell lines, patient samples, and in vivo in immunodeficient mice by flow cytometry, western blot, RT-PCR, and CyTOF.
RESULTS
We found significantly higher β-catenin expression in cytogenetically unfavorable and relapsed AML patient samples and in the bone-marrow-resident leukemic cells compared to circulating blasts. Disrupting Wnt/β-catenin signaling suppressed AML cell growth, induced apoptosis, abrogated stromal protection, and synergized with TKIs in FLT3-mutated AML cells and stem/progenitor cells in vitro. The aforementioned combinatorial treatment improved survival of AML-xenografted mice in two in vivo models and impaired leukemia cell engraftment. Mechanistically, the combined inhibition of Wnt/β-catenin and FLT3 cooperatively decreased nuclear β-catenin and the levels of c-Myc and other Wnt/β-catenin and FLT3 signaling proteins. Importantly, β-catenin inhibition abrogated the microenvironmental protection afforded the leukemic stem/progenitor cells.
CONCLUSION
Disrupting Wnt/β-catenin signaling exerts potent activities against AML stem/progenitor cells and synergizes with FLT3 inhibition in FLT3-mutant AML. These findings provide a rationale for clinical development of this strategy for treating FLT3-mutated AML patients.
Keywords: stem/progenitor cells, Wnt/β-catenin, FLT3 mutant, AML, microenvironment
Introduction
Wnt signaling plays essential roles in regulating cell proliferation, survival, and differentiation. Activation of canonical Wnt signaling stabilizes β-catenin, resulting in its nuclear translocation and increased interactions with co-transcriptional regulator T-cell factor (TCF) 4/lymphoid enhancer factor 1 (1). Deregulation of Wnt/β-catenin signaling is linked to acute myeloid leukemia (AML) initiation and progression (2) and is required for leukemia stem cells (LSC) self-renewal and survival (3). Overexpression of β-catenin was reported to be an independent adverse prognostic factor for AML (4). In fact, β-catenin was found to be overexpressed in most AML samples and localized more frequently in the nucleus of bone marrow (BM) LSC compared with normal CD34+ cells (5). Thus, inhibition of β-catenin signaling represents a promising therapeutic strategy in AML (6, 7).
FMS-like tyrosine kinse-3 (FLT3) mutations, including internal tandem duplication (FLT3-ITD) and tyrosine kinase domain mutations (FLT3-TKD), are frequently observed in AML (8). These mutations cause ligand-independent activation of the tyrosine kinase and downstream signaling pathways including MAPK/ERK, JAK/STAT5, and PI3K/AKT, which in turn activate canonical Wnt signaling by stabilizing and increasing nuclear localization and transcriptional activity of β-catenin (9). Furthermore, aberrant β-catenin signaling contributes to FLT3-ITD-related myeloid transformation by TCF-dependent transcriptional activity, and FLT3 mutations also directly cooperate with Wnt signaling in AML (10).
FLT3 mutations are associated with poor prognosis in AML (11, 12). Consequently, FLT3 tyrosine kinase inhibitors (TKIs) have been developed to treat AML patients with FLT3 mutations. Unfortunately, their effects are often limited because of acquired FLT3 mutations, TKI-induced alternative signaling activation, microenvironment-mediated resistance, and their inability to eradicate LSC (13, 14). Thus, strategies to improve the efficacy of TKIs are needed for the therapy of FLT3-mutated AML.
Disrupting Wnt/β-catenin signaling may target AML and LSC/progenitor cells and has the potential of enhancing the activity of TKIs in FLT3-mutated AML. C-82 is a β-catenin specific inhibitor that binds to CBP, inhibits β-catenin/CBP interaction, and impedes Wnt/β-catenin-mediated cell proliferation, self-renewal, and survival in several hematological malignancies including ALL and CML (15–17). However, the anti-leukemia activity of C-82 in AML including the effects on AML stem/progenitor cells and the combinations of C-82 with TKIs in FLT3-mutated AML have not been investigated. In this study, we investigated the effects of C-82 on AML and LSC/progenitor cells and its combinatorial activity when paired with the FLT3-ITD directed TKI sorafenib or quizartinib. We demonstrate that C-82 exerted potent activity against AML cells and LSC and synergized with TKIs in FLT3-mutated AML in vitro and in vivo.
Materials and Methods
Protein determination by reverse-phase protein array
Expression of β-catenin protein in samples from newly diagnosed AML patients and paired newly diagnosed and relapsed or paired peripheral blood (PB) and BM samples in AML patients were determined by reverse-phase protein array (RPPA) as previously described (18). The patient populations, clinical characteristics, and sample preparation for RPPA were described previously (19). Antibody against β-catenin (#8480) was from Cell Signaling Technology (Danvers, MA).
Cells and cell culture
Molm13 cells were purchased from the German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany), and MV4-11 and KG-1 cells from the American Type Culture Collection (Manassas, VA). OCI-AML3 cells were provided by Dr. M. Minden (Ontario Cancer Institute, Ontario, Canada). Ba/F3 (Ba/F3-WT) or Ba/F3 cells stably transfected with human FLT3-ITD (Ba/F3-ITD), FLT3-D835G (Ba/F3-D835G), or D835Y (Ba/F3-D835Y) point mutations as previously described (20) were provided by Dr. D. Small (Johns Hopkins University School of Medicine, Baltimore, MD). Ba/F3 cells with FLT3-ITD plus N676D (Ba/F3-ITD+676) or Y842C mutation (Ba/F3-ITD+842) were generated as previously described (21). Cell lines were validated by STR DNA fingerprinting using the AmpF_STR Identifier Kit according to manufacturer’s instructions (Applied Biosystems, Cat#4322288). The STR profiles were compared to known ATCC fingerprints, and to the Cell Line Integrated Molecular Authentication database (CLIMA) version 0.1.200808 (http://bioinformatics.istge.it/clima/) (22). The STR profiles matched known DNA fingerprints or were identified as unique (OCI-AML3). Mycoplasma testing was performed using the PCR Mycoplasma Detection Kit from Applied Biological Materials (Richmond, BC, Canada) per manufacturer’s instructions. Authenticated and mycoplasma-free cells are stored under liquid nitrogen and are never kept in culture for more than 4 months. Cells were cultured in RPMI-1640 supplemented with 10% heat-inactivated fetal calf serum, 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. Ba/F3-WT cells were supplemented with 2 ng/ml murine interleukin-3 (PeproTech, Inc, Rocky Hill, NJ).
Primary samples were obtained after informed consent following the MD Anderson Cancer Center Institution Review Board approved protocols in accordance with Declaration of Helsinki. Mononuclear cells were purified by Ficoll-Hypaque (Sigma-Aldrich, St. Louis, MO) density-gradient centrifugation and cultured in α-MEM supplemented with 10% fetal calf serum. Table 1 summarizes patient clinical characteristics.
Table 1.
Characteristics of AML patients and experiments
| Pt No | Source | % Blasts | Disease status | FLT3 status | Cytogenetic | experiments |
|---|---|---|---|---|---|---|
| 1 | PB | 90 | New diagnosis | WT | 46,XY | C-82 |
| 2 | PB | 70 | New diagnosis | ITD | 46,XY | C-82 |
| 3 | PB | 93 | Relapse/refractory | WT | Complex karyotype | C-82 |
| 4 | PB | 74 | Relapse/refractory | WT | Complex karyotype | C-82 |
| 5 | PB | 84 | Relapse/refractory | WT | Complex karyotype | C-82 |
| 6 | BM | 74 | Relapse/refractory | WT | Complex karyotype | C-82 |
| 7 | PB | 78 | Relapse/refractory | WT | Complex karyotype | C-82 |
| 14 | BM | 52 | Primary refractory | ITD + D835 | 47,XY, +9 | C-82 + TKI |
| 15 | BM | 74 | New diagnosis | D835 | 47,XY,inv(16),+22,del(7) | C-82 + TKI |
| 16 | PB | 56 | Relapse/refractory | ITD | 46,XY | C-82 + TKI |
| 17 | BM | 83 | Relapse | D835 | 46,XY | C-82 + TKI |
| 18 | PB | 52 | New diagnosis | D835 | 46,XY | C-82 + TKI |
| 19 | PB | 72 | Relapse/refractory | D835 | 46,XY | C-82 + TKI |
| 20 | BM | 61 | Relapse | WT | 46,XX | C-82 + TKI |
| 21 | BM | 60 | New diagnosis | WT | 46,XX | C-82 + TKI |
| 22 | BM | 97 | Relapse | WT | 46,XY, t(1;3), del(7) | C-82 + TKI |
| 23 | BM | 78 | Relapse/refractory | ITD | 46,XY | Mouse experiments |
Pt No, patient number.
Gene silencing by siRNA
Molm13 and MV4-11 cells were electroporated with 750 nM of 4 individual or SMARTpool ON-TARGET plus CTNNB1 siRNAs of the four (Dharmacon, Lafayette, CO) using an Amaxa apparatus (Solution L, program Q-001) (Lonza, Walkersville, MD) following the manufacturer’s instructions. β-catenin silencing was confirmed by western blot analysis.
Cell viability and apoptosis assay
Ba/F3 cells (1×105 cells/ml), AML cell lines (2×105 cells/ml), and primary samples (5×105 cells/ml) were plated in 96-well plates with or without BM-derived mesenchymal stem cell (MSC) co-culture and treated with C-82, sorafenib (American Custom Chemicals Corporation; San Diego, CA) (or quizartinib; Selleck Chemicals; Houston, TX), or their combination. Cell viability was determined by the Trypan blue exclusion using a Vi-Cell XR Cell Counter (Beckman Coulter, Brea, CA). Apoptosis was estimated by flow cytometry after annexin V–Cy5.5 staining in the presence of 7-amino-actinomycin D (7-AAD). Apoptosis in primary samples was assessed in CD45+ and CD34+CD38− cells after the cells were incubated with CD34-PC5.5, CD38-PE Cy7, and CD45-APC H7 antibodies (BD Biosciences). Specific apoptosis was defined as:
Cell cycle distribution
Cells (1×106) were exposed to 10 μM EdU (Invitrogen, San Diego, CA) in a culture medium for 2 h, fixed in 4% formaldehyde, and processed with the Click-iT EdU Alexa Fluor 647 flow cytometry assay kit (Thermo Fisher Scientific, Waltham, MA) following the manufacturer’s instructions. After cells were treated with RNase and counterstained with 10 mg/ml propidium iodide (Sigma-Aldrich), DNA content was determined by flow cytometry (Beckman Coulter). Cell cycle distribution was analyzed using Kaluza software (Beckman Coulter).
Western blot analysis
Protein levels were determined by western blot as described previously (23) using the Odyssey Infrared Imaging System and the signals quantified by Odyssey software version 3.0 (LI-COR Biosciences, Lincoln, NE). Protein expression was normalized to the loading control. Cytoplasmic and nuclear fractions were isolated as previously described (24). Antibodies against β-catenin (#8480), phospho (p)-FLT3 (Tyr589/591) (#3464), p-ERK (Thr202/Tyr204) (#4376), p-AKT (Ser473) (#4058), AKT (#4685), c-Myc (#5605), STAT5 (#94205), and CD44 (#5640) were from Cell Signaling Technology; ERK2 (#sc-154) and FLT3 (#sc-479) from Santa Cruz Biotechnology (Dallas, TX); survivin (#AF886) from R&D systems (Minneapolis, MN); p-STAT5 (pY694) (#611965) from BD Biosciences (San Jose, CA). β-actin, α-tubulin (both from Sigma-Aldrich), or Lamin B1 (Cell Signaling Technology) was used as a loading control.
Protein determination by flow cytometry
Cells were incubated with LIVE/DEAD® Fixable Yellow Dead Cell Stains (Thermo Fisher Scientific) for 30 min in the dark, fixed with 4% paraformaldehyde, and permeabilized with 0.115% Triton X-100 in PBS. Cells were then stained with Fc-blocker (Miltenyi Biotec, San Diego, CA) at 4°C for 10 min, followed with CD34-PC5.5, CD38-APC (BD Biosciences), CD44-BV421 (BioLegend, San Diego, CA) and survivin-FITC (R&D Systems) antibodies and resuspended in 0.1% paraformaldehyde in PBS and analyzed using a Gallios flow cytometer (Beckman Coulter). Survivin and CD44 levels were expressed as the mean fluorescence intensity (MFI) difference of cells stained with the target antibodies and stained with IgG.
Confocal immunofluorescence microscopy
Cells were mounted onto slides, fixed with 4% paraformaldehyde, and permeabilized with 100% methanol. After cells were incubated with blocking solution (1% BSA/PBS), mouse anti-human CD44 and rabbit anti-human β-catenin monoclonal antibodies (Cell Signaling Technology) were added (1:200). Cells were washed, and secondary antibodies (Alexa 594-tagged donkey anti-mouse and Alexa 488-tagged donkey anti-rabbit; Life Technologies, Grand Island, NY) were added. Cells were stained with 4′6-diamidino-2-phenylindole (DAPI, Thermo Fisher Scientific). Single, central cellular plane, confocal images were collected using an Olympus FV1000 laser confocal microscope. Images were captured using the Olympus TIRF 100× 1.45 NA objective at the central plane of the cell as determined by the DAPI label. Images were loaded into 3I’s Slidebook Software (v5.5, Intelligent Imaging Innovations, Denver, CO). In Slidebook, cellular masking was performed to identify the nuclear and cytoplasmic space. All collected channels were used to create the whole cell mask and the DAPI marker was used to create the nuclear mask with the focal z resolution at ~0.3 microns. While multiple z-planes were not captured, any protein co-expressing (co-localizing) with DAPI would be determined to be located in the nucleus. By subtracting the nuclear mask from the whole cell mask, the cytoplasmic mask was created. The mean intensity for each marker was quantified in each mask (Nuclear, Cytoplasmic, and Whole cell) by SlideBook imaging software and compared to determine the amount of nuclear accumulation.
Real-time RT-PCR
RNA isolation and RT-PCR were performed as previously described (25) with minor modifications. The PCR amplification mixture (20 μl) contained cDNA, a primer pair of human Wnt signaling primer library for pathway PCR array (Real Time Primer, LLC, Elkins Park, PA), and SYBR Green PCR fast master mix (Thermo Fisher Scientific). The abundance of each transcript relative to that of B2M was calculated using the 2−ΔCt method, where ΔCt is the mean Ct of the transcript of interest minus the mean Ct of the B2M transcript.
Adhesion
Adhesion of leukemia cells to MSCs were determined as previous described (25). Briefly, leukemia cells were added to MSCs that were plated the night before and cultured for 24 h. Floating and attached leukemia cells were counted by flow cytometry after staining the cells with human CD45 antibody in the presence of counting beads (Life Technologies, Grand Island, NY). Adhesion of leukemia cells was defined as attached viable CD45+ cells/total viable leukemia cells.
In vivo studies
Animal experiments were performed in accordance with the MD Anderson Cancer Center Institutional Animal Care and Use Committee approved protocols. Molm13 cells (5×105) stably expressing a dual luciferase-GFP reporter (Molm13-GFP/Luc) were injected into NOD/SCIDIL2RγNull (NSG) mice, and cells from a FLT3-ITD mutated AML patient (no.23, Table 1) (2×106) collected from spleen of second generation patient-derived xenograft (PDX) in NOD/SCIDIL2RγNull-3/GM/SF (NSGS) mice were injected into NSGS mice via tail vein (both 6-8-wk-old, females; Jackson Laboratory, Bar Harbor, ME). After confirming engraftment either by imaging using the IVIS-200 noninvasive bioluminescence in vivo imaging system (Xenogen, Hopkinton, MA) or by flow cytometry measuring human CD45+ cells in mouse PB, mice were randomized to the following treatment groups (n=10/group): vehicle control, PRI-724 (C-82 pro-drug) (40 mg/kg) by subcutaneous mini-pump, sorafenib (5 mg/kg for NSG and 10 mg/kg for NSGS mice) by daily oral gavage, or PRI-724 plus sorafenib for 4 wk. Three mice/group were killed 2 h after dosing on 15th for NSG and 25th for NSGS mice of treatment days. Leukemia burden was assessed by flow cytometry, CyTOF mass cytometry, or H&E staining. Mice were monitored daily and survival time was recorded.
NSGS mice (7 to 8-wk old, females; Jackson Laboratory) were also injected via tail vein with the PDX cells (no.23, Table 1) (2×106) untreated or after ex vivo treatment with C-82 (1.0 μM), sorafenib (2.5 μM) or both for 48 h (n=6/group). Leukemia cell engraftment and progression were assessed by flow cytometry and survival was monitored.
CyTOF
BM cells from mice were labeled with metal-tagged antibodies for cell surface and intracellular proteins (supplementary Table 1) and analyzed using a CyTOF2 mass cytometer (Fluidigm, San Francisco, CA) (25, 26). The viable cells were gated with FlowJo software (Tree Star Inc., Ashland, OR) and exported. The exported FCS files were transferred into the spanning-tree progression analysis of density-normalized events (SPADE) software and analyzed as reported previously (27, 28).
Statistical analyses
Cell line experiments were conducted in triplicates. Results were expressed as means ± SEM unless otherwise stated. The combination index (CI) was determined by the Chou-Talalay method and expressed as the mean of CI values obtained at the 50%, 75%, and 90% effective doses (29). CI<1.0 was considered synergistic; =1.0 additive; and >1.0 antagonistic. Statistical analyses were performed using a two-tailed Student t-test or one-way analysis of variance (ANOVA) for multiple group comparisons. Mouse survival was estimated by the Kaplan-Meier method, and analyzed using log-rank statistics. P<0.05 was defined as statistically significant.
Results
High β-catenin expression is associated with unfavorable patient characteristics in AML and induced by the BM microenvironment
Using RPPA, we determined the expression of β-catenin in 511 newly-diagnosed AML patient samples. We found that high levels were associated with unfavorable cytogenetics (P<0.01), and β-catenin level was significantly higher in relapsed vs. paired newly diagnosed samples (n=47, P=0.043) (Supplementary Fig. 1A). Higher β-catenin expression was observed in BM than paired PB samples (n=140, P=0.0006) (Supplementary Fig. 1B), suggesting that BM microenvironment modulates β-catenin expression in AML cells. Consistent with this observation, co-culturing OCI-AML3 cells with MSCs (48 h) induced the expression of β-catenin and its target, the adhesion protein CD44 in leukemia cells, while inhibition of β-catenin with Wnt/β-catenin signaling antagonist C-82 suppressed the MSC-induced CD44 expression and the adhesion of OCI-AML3 cells to MSCs (Supplementary Fig. 1C). The BM stroma induction of β-catenin levels in AML cells was further demonstrated by co-culturing Molm13 and PB cells from primary patients (n=3) with MSCs (48 h) (Supplementary Fig. 1D).
Disrupting Wnt/β-catenin signaling induces apoptosis, suppresses cell growth, and inhibits β-catenin downstream targets in AML bulk and stem/progenitor cells
We treated several AML cell lines with C-82 (48 h) and observed marked apoptosis induction (EC50=0.82±0.04 μM and 0.79±0.02 μM) and cell growth suppression (IC50=0.42±0.06 μM and 0.39±0.02 μM) in OCI-AML3 and Molm13 cells (Fig. 1A), respectively. Although partially suppressing cell growth, C-82 only slightly induced apoptosis in KG1 cells at the doses used (Fig. 1A). Cell cycle analysis showed that the treatment decreased S-phase cells, while increasing cells in sub-G1 phase in OCI-AML3 and Molm13 consistent with growth suppression and apoptosis induction in these cells (Fig. 1A). C-82 also induced apoptosis (72 h) in AML blasts (n=7; EC50=1.90±0.28 μM and IC50=1.17±0.26 μM) and CD34+CD38− stem/progenitor cells (n=6; EC50=4.25±0.69 μM and IC50=2.11±0.04 μM) from AML patients (Table 1), and had minimal toxicity to normal BM CD34+ cells (Fig. 1B). C-82 treatment decreased the β-catenin/CBP downstream targets including survivin, c-Myc, and CD44 in Molm13, OCI-AML3, AML bulk, and CD34+CD38− stem/progenitor cells from patients, as determined by either western blot or flow cytometry in available samples (Fig. 1C). Representative FACS plots of survivin and CD44 are shown in Supplementary Fig. 2.
Fig. 1.
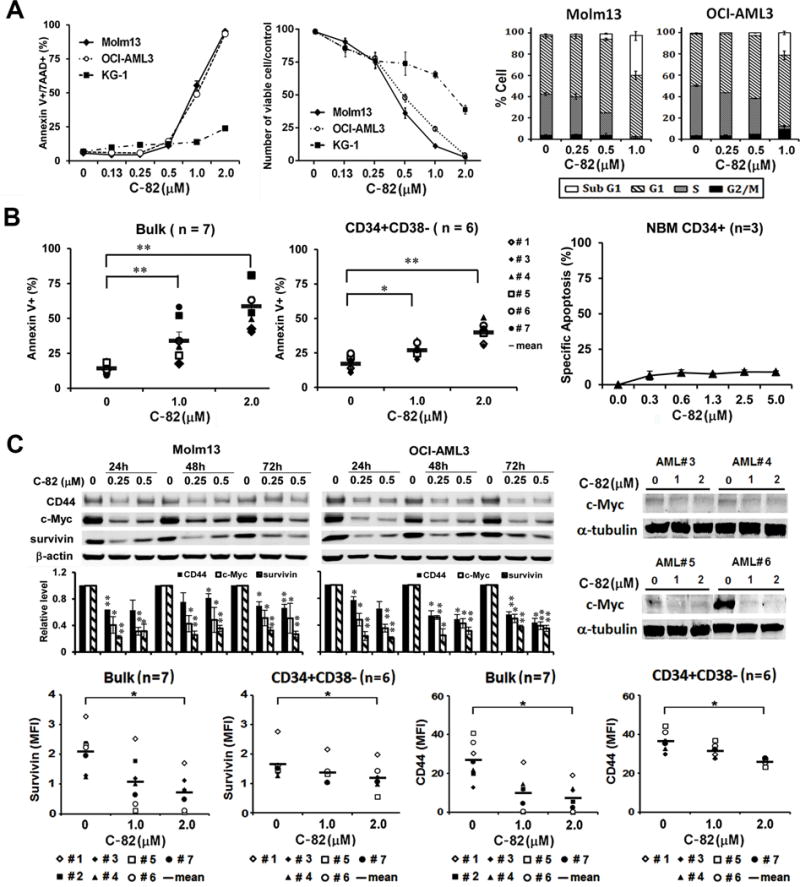
C-82 induces apoptosis, suppresses growth, and inhibits β-catenin targets in AML cells and stem/progenitor cells. (A) AML cell lines were treated with C-82 for 48 h. Apoptosis was measured by flow cytometry. Viable cell count was assessed by Trypan blue exclusion method. Cell cycle status was determined by flow cytometry. (B) AML patient and normal BM (NBM) samples were treated with C-82. Apoptosis in bulk and CD34+CD38− AML (72 h) and normal CD34+ (48 h) cells was measured by flow cytometry. (C) Molm13 and OCI-AML3 cells were treated for 24, 48, or 72 h and AML patient samples for 72 h with C-82. Survivin, CD44, and c-Myc expressions were determined by western blot or flow cytometry. For Molm13 and OCI-AML3 cells, the top panel shows a representative western blot and the lower panel the quantitative western blots of three independent experiments. *, P<0.05; **, P<0.01.
Combined inhibition of β-catenin and FLT3 signaling synergistically induces apoptosis in FLT3-mutated AML cells and CD34+CD38− AML stem/progenitor cells
In agreement with the finding that constitutive activation of FLT3 stimulates β-catenin pathway (9), we observed that Molm13 cells were very sensitive to C-82 treatment (Fig. 1A). To demonstrate that FLT3 mutations were indeed associated with β-catenin, we determined β-catenin expression and C-82 sensitivity in Ba/F3 cells without or with FLT3 mutations. We found that cells with FLT3 mutations expressed higher β-catenin and were generally more sensitive to C-82 than cell lines without FLT3-mutation (Fig. 2A). We then treated Ba/F3 cells and AML cell lines with C-82, sorafenib, and their combination. The combination synergistically induced apoptosis (CI<1) in Ba/F3-ITD, Ba/F3-D835G, Ba/F3-ITD+676, and even in the sorafenib-resistant Ba/F3-ITD+842 cells (Fig. 2B). The synergy was also observed in Molm13 and MV4-11 cells with FLT3-ITD mutation, even when they were co-cultured with human BM-derived MSCs (Fig. 2C). Similar results were obtained in Molm13 and MV4-11 cells treated with C-82 and the novel FLT3 inhibitor quizartinib (Supplementary Fig. 3). To further demonstrate that C-82-induced β-catenin inhibition contributes to the synergy in the combination, we inhibited β-catenin expression using 4 specific and pooled siRNAs in Molm13 and MV4-11 cells. Each siRNA was able to reduce β-catenin expression and the pool (SMARTpool) was slightly more effective (Supplementary Fig. 4). We next treated the cells with the SMARTpool siRNA (24 h) and then with sorafenib for additional 48 h. Fig. 2D shows that siRNA silencing of β-catenin enhanced sorafenib-induced cell death in these cells. No synergy was detected when C-82 was combined with sorafenib in FLT3-WT Ba/F3 or OCI-AML3 cells, while the synergy was observed when C-82 was combined with chemotherapeutic agent Ara-C in both Molm13 and OCI-AML3 cells (Supplementary Fig. 5). Importantly, synergy was also observed in bulk and CD34+CD38− AML cells from FLT3-mutated patients treated with C-82 and sorafenib with or without MSC co-culture (48 h) (even in a patient with ITD+D835 mutation, #14, Table 1), while cells from FLT3-WT AML patients were in general less sensitive to C-82, and resistant to sorafenib, and no synergy was observed in these cells with the combination treatment (Fig. 2E). Furthermore, either agent alone, or their combination, had limited cytotoxicity on normal BM cells (Supplementary Fig. 6).
Fig. 2.
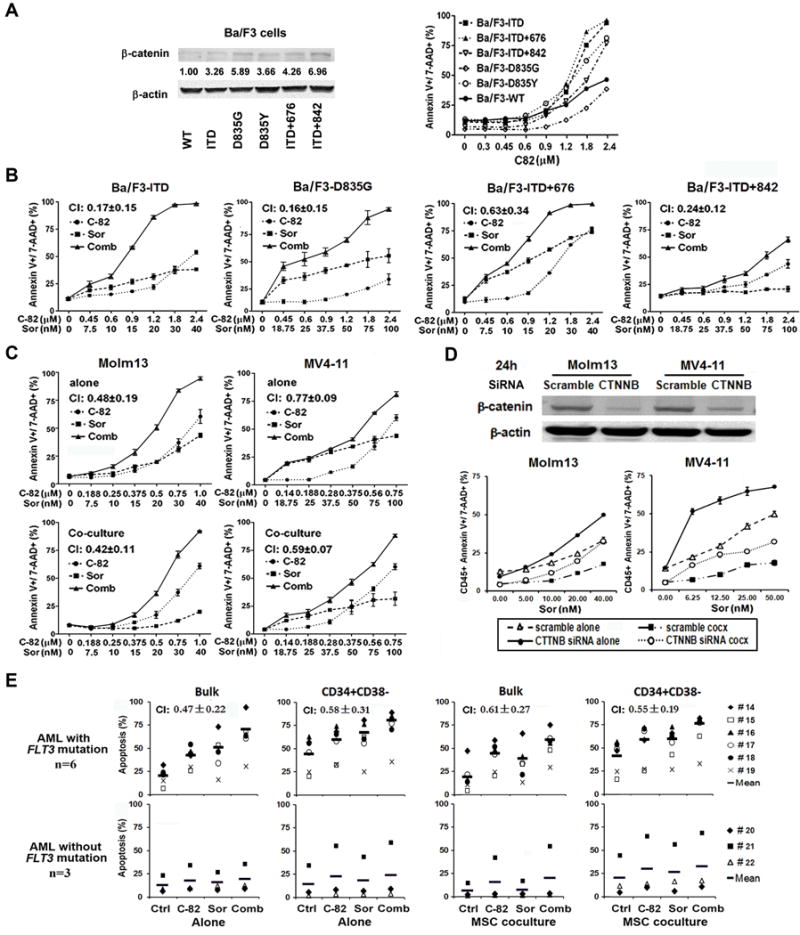
Combined inhibition of β-catenin and FLT3 synergistically induces apoptosis in cell lines, AML blasts, and CD34+CD38− AML stem/progenitor cells with FLT3 mutations. (A) Expression of β-catenin in Ba/F3 cells without or with FLT3 mutations determined by western blot and apoptosis in these cells treated with C-82 detected (24 h) by flow cytometry. (B) Apoptosis in FLT3-mutated Ba/F3 cells treated with C-82, sorafenib, or both (24 h). (C) Apoptosis in Molm13 and MV4-11 cells without (top) or with (bottom) MSC co-culture treated with C-82, sorafenib, or both (48 h). (D) Molm13 and MV4-11 cells were treated with CTNNB1 SMARTpool ON-TARGET plus siRNAs for 24 h, then with sorafenib. β-catenin silencing was confirmed by western blot. Apoptosis was measured after cells were treated with sorafenib for 48 h. (E) AML patient samples with or without FLT3 mutations were treated with C-82 (1.0 μM), sorafenib (2.5 μM), or both (48 h). Apoptosis was determined in bulk and CD34+CD38− cells. Apoptotic cells were assessed by flow cytometry. CI values were calculated. CI<1.0 indicated synergistic effect. cocx, co-culture; Ctrl, control; Sor, sorafenib; Comb, combination.
Combinations of C-82 and TKIs inhibit both β-catenin/CBP and FLT3 signaling and decreases β-catenin nuclear localization in AML cells
FLT3-ITD AML cells were treated with C-82, sorafenib, or both. Wnt signal pathway PCR array analysis revealed that both C-82 and sorafenib markedly suppressed the expression of multiple genes involved in the Wnt/β-catenin signaling pathway, including β-catenin (CTNNB1), CD44, survivin (BIRC5), cyclin D1 (CCND1), and c-Myc and several frizzled family receptors/co-receptors and Wnt ligands, such as FZD1, FZD3, FZD5, FZD6, low-density lipoprotein receptor (LDLR), WNT10B, and WNT3A (≥2-fold decreases) (indicated by arrows), but C-82 or the combination was more effective than sorafenib alone for most target inhibitions (Fig. 3A). Western blot confirmed that C-82, sorafenib, or the combination suppressed the expression of Wnt/β-catenin/CBP downstream targets such as c-Myc, survivin, and CD44 in Molm13 and MV4-11 cells (Fig. 3B). Sorafenib or quizartinib and the combination but not C-82 alone, effectively inhibited FLT3 signaling molecules including p-FLT3, p-STAT5, p-ERK, and p-AKT (Fig. 3B and 3C). Optimal inhibition of both β-catenin and FLT3 signaling was observed only when C-82 and TKIs were combined (Fig. 3A, 3B, and 3C). As expected, sorafenib did not affect signaling pathways in OCI-AML3 cells (Fig. 3D).
Fig. 3.
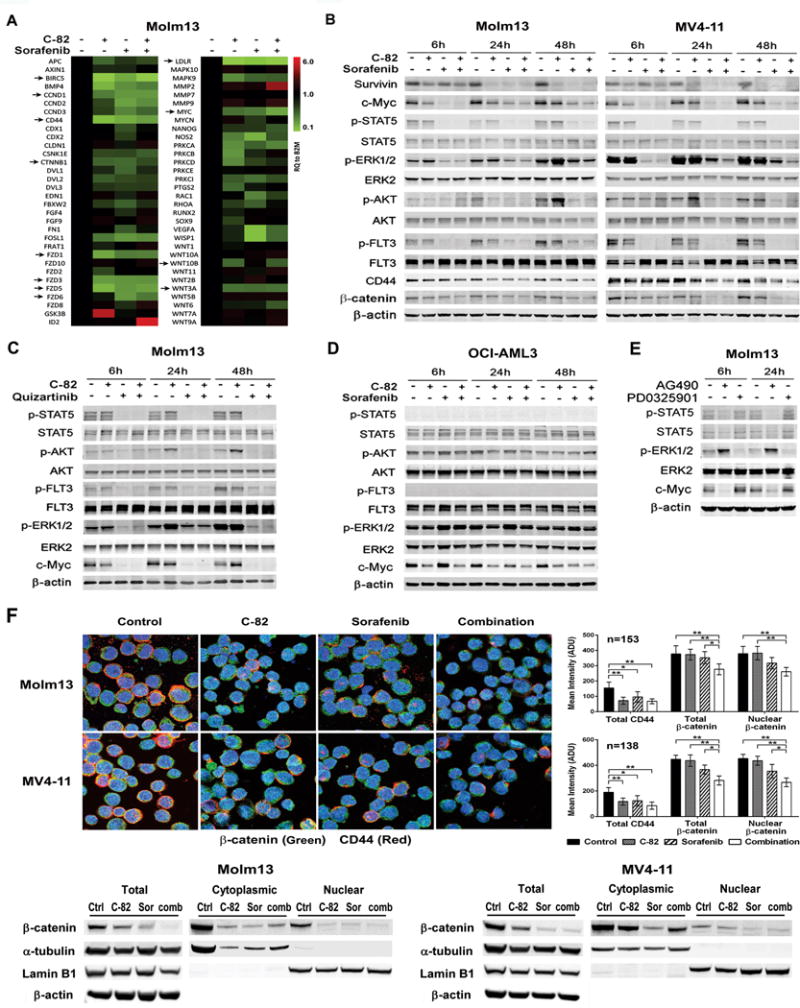
C-82 and TKI combination inhibits β-catenin/CBP and FLT3 signaling and reduces β-catenin nuclear localization in AML cells. (A) Molm13 cells were treated with C-82 (0.5 μM), sorafenib (20 nM), or both for 48 h, and RNA levels were determined by the Wnt signaling pathway PCR array (arrows indicate genes discussed in the text). (B) Expression of β-catenin/CBP and FLT3 signaling and downstream targets were analyzed by western blotting in Molm13 and MV4-11 cells treated with C-82 and sorafenib for 6, 24, 48 h. C-82 and sorafenib were 0.5 μM and 20 nM for Molm13 and 0.56 μM and 75 nM for MV4-11, respectively. (C) Molm13 cells were treated with C-82 (0.5 μM) and quizartinib (1.6 nM) and (D) OCI-AML3 cells were treated with C-82 (0.5 μM) and sorafenib (20 nM) for 6, 24, and 48 h. (E) Molm13 cells were treated with JAK/STAT inhibitor AG490 (100 μM) or MEK/ERK inhibitor PD0325901 (200 nM) for 6, 24 h. Protein expressions were determined by western blot analysis. (F) Confocal image (1000×) and quantifications illustrating expressions of β-catenin and CD44, and the nuclear localization of β-catenin (top) and β-catenin levels in total, cytoplasmic, and nuclear fractions by western blot analysis (bottom) in Molm13 or MV4-11 cells treated with C-82 (0.5 or 0.56 μM), sorafenib (20 or 75 nM), or both for 48 h. Ctrl, control; Sor, sorafenib; Comb, combination. *, P<0.05; **, P<0.01.
Because c-Myc is a β-catenin target, it was not surprising that C-82 reduced c-Myc levels in Molm13, MV4-11, and OCI-AML3 cells independent of their FLT3 mutational status (Fig. 3A–3D). Interestingly, TKIs potently decreased c-Myc levels in FLT3-ITD mutated AML cells, coinciding with potent inhibitions of p-ERK and p-STAT5 (Fig. 3B and 3C). This would suggest that, in addition to modulating c-Myc through regulation of Wnt/β-catenin, FLT3 may also regulate c-Myc level through p-ERK or p-STAT5 signaling.
To test this, we treated Molm13 cells with the STAT5 inhibitor AG490 or an ERK inhibitor PD0325901 (both from Selleck Chemicals), and found that AG490, but not PD0325901, suppressed c-Myc expression (Fig. 3E), suggesting that the TKI-induced c-Myc inhibition is mediated in part, through FLT3/STAT5 signaling. Furthermore, C-82 and sorafenib in combination inhibited the expression of total β-catenin and CD44, and also reduced the nuclear localization of β-catenin in Molm13 and MV4-11 cells by confocal analysis, and the decreased β-catenin nuclear localization was further confirmed by western blot (Fig. 3F) (images of each single staining are shown in Supplementary Fig. 7). These data indicated that the combination could disrupt both β-catenin/CBP and FLT3 signaling, and induce cell death more effectively in FLT3-mutated AML cells.
Inhibition of β-catenin enhances the anti-leukemia activity of TKIs against FLT3-mutated AML xenografts and impairs engraftment in immunodeficient mice
To test the effect of the combinatorial strategy in FLT3-mutated AML in vivo, we used two xenograft murine models. For the Molm13-GFP/Luc cell xenograft in NSG murine model (Fig. 4A), treatment with PRI-724 (C-82 pro-drug) or sorafenib inhibited leukemia growth, and the combination was the most effective in this regard, as determined by in vivo imaging analysis (Fig. 4B and 4C) and flow cytometric measurement of human CD45+ cells in mouse PB (Fig. 4D). Mice treated with PRI-724 (19 d, P=0.025) or sorafenib (28 d, P=0.0002) had a statistically significantly longer median survival compared with controls (17 d), and the combined treatment further prolonged the survival (30.5 d; P=0.0005 vs. PRI-724; P=0.0056 vs. sorafenib) in this aggressive xenograft AML model (Fig. 4E). Tissues were collected (n=3/group) 2 h after the 15-d treatments. The combination markedly decreased leukemia cells in the BM and spleen, as determined by flow cytometry, H&E staining, and reduced spleen sizes (Fig. 4F and 4G). Using CyTOF mass cytometry that can simultaneously measure cell surface and intracellular proteins, we determined the number of human CD45+ cells and protein expression in these cells in 15-d-treated BM samples. SPADE tree analysis showed that the combination greatly decreased human CD45+ populations (Fig. 4H) and the expression of β-catenin, CD44, c-Myc, p-FLT3, p-ERK, p-AKT, and p-STAT5 in these cells (Fig. 4I). Although reduction of survivin in Molm13 cells upon the combinatorial treatment was observed in vitro (Fig. 3B), we observed only slightly lower survivin levels, which did not reach statistical significance in vivo at the time samples were collected (2 h after treatments on day 15). There are several reasons why we did not see the same level of target reduction of the combination treatment in vitro and in vivo, such as i) the doses for in vivo and in vitro do not exactly translate, ii) it is much easier to perform a time course in vitro than in vivo, and iii) leukemia cells collected from mice lived in a very different microenvironment compared to cells in culture, as we have shown that the BM microenvironment induces β-catenin and its downstream targets. Of note, the survivin decrease was observed in BM samples collected from AML patients who had underwent phase I dose-escalation with PRI-724 (30).
Fig. 4.
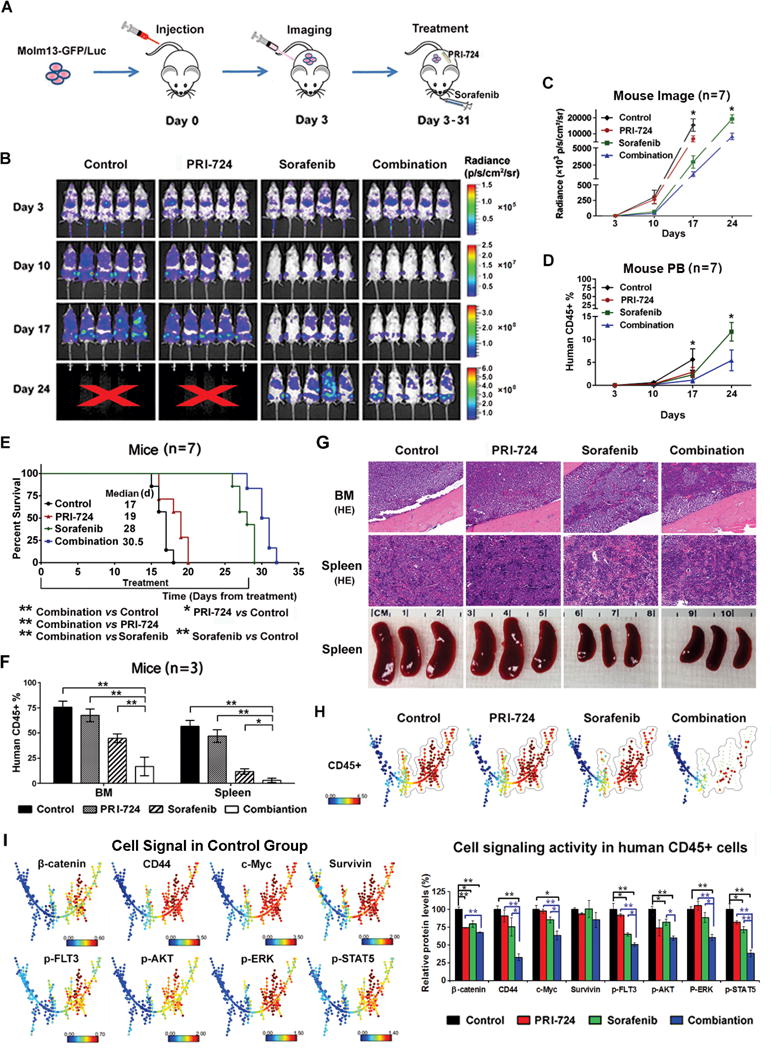
PRI-724 enhances sorafenib activity in Molm13-GFP/Luc xenograft NSG mice. (A) Experimental scheme. After confirmed engraftment of Molm13-GFP/Luc cells by imaging, mice were treated with RPI-724, sorafenib, or the combination for 4 wk. (B) Weekly images of mice using the IVIS-200 imaging system. X, no alive mice. (C) Imaging quantification by the IVIS-200 software. (D) Leukemia cells (human CD45+) in mouse PB determined by flow cytometry. (E) Survival curves. (F) Leukemia cells (human CD45+) in mouse BM and spleen detected by flow cytometry. (G) Mouse BM and spleen H&E (HE) staining (200×) and spleen sizes. (H) CyTOF/SPADE analysis of leukemia cells (human CD45+) in mouse BM samples after 15 d-treatments. (I) The expression of β-catenin targets and FLT3 downstream signal proteins in human CD45+ cells in BM of control mice presented in SPADE tree (left panel) and the quantitation of these proteins in various treatment groups (right panel). *, P<0.05; **, P<0.01.
For the PDX xenograft in the NSGS murine model (Fig. 5A), the combination treatment also showed the strongest anti-leukemia activity as demonstrated by flow cytometric measurement of human CD45+ cells in mouse PB (n=10/group) after a 3-wk treatment (Fig. 5B) and in BM and spleen cells collected from mice (n=3/group) after a 25-d treatment (Fig. 5C). Mice treated with PRI-724 or sorafenib had a significantly longer median survival (31 d, P=0.008 or 48 d, P=0.0003; respectively) compared with controls (29 d), and the combination further prolonged the survival (54 d; P=0.0005 vs. PRI-724; P=0.0067 vs. sorafenib) (Fig. 5D). An attempt to determine leukemia cell populations and the target protein expression by CyTOF in collected BM cells failed due to insufficient cells in sorafenib and the combination treatment groups. Results indicate that PRI-724 enhanced sorafenib activity in vivo against FLT3-mutated AML. Except for an initial slight body weight loss in sorafenib and the combination treatment groups, no significant drug-related toxicity was observed.
Fig. 5.
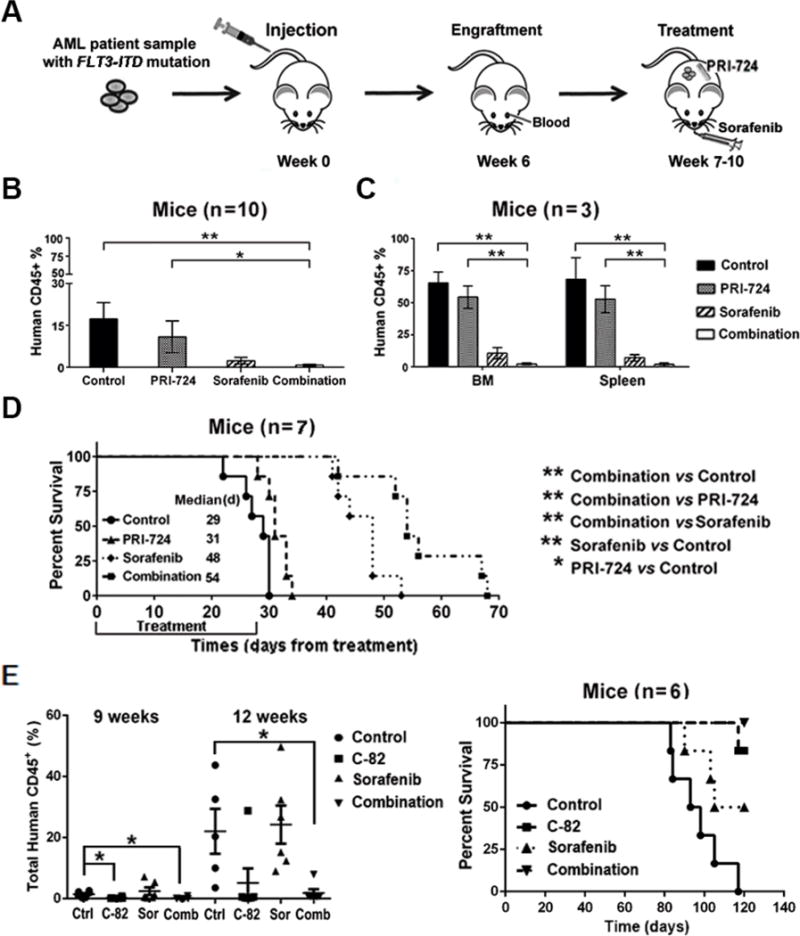
Inhibition of β-catenin enhances sorafenib activity in FLT3-ITD PDX cell xenograft NSGS mice and suppresses engraftment potential of the PDX cells. (A) Experimental scheme. After confirmed engraftment by flow cytometry of human CD45 positivity, NSGS mice were treated with RPI-724, sorafenib, and the combination for 4 wk. (B) Leukemia burden assessed by flow cytometry measuring human CD45+ cells in mouse PB after 3-wk treatments. (C) Leukemia burdens in mouse BM and spleen determined by flow cytometry after 25-d treatments. (D) Survival curves. (E) Engraftment of the ex vivo treated PDX cells and survival curve. The PDX cells (2×106) were untreated or treated with C-82 (1 μM), sorafenib, (2.5 μM), or both for 48 h and then injected into NSGS mice. Engraftment was assessed by flow cytometric measurement of human CD45 positivity at 9 and 12 wk post cell injection. Ctrl, control; Sor, sorafenib; Comb, combination. * P<0.05, ** P<0.01.
To further assess the potential of inhibition of β-catenin and its combination with TKIs on targeting FLT3-mutated AML stem/progenitor cells, we treated the above PDX cells with C-82, sorafenib, or both ex vivo, then injected the cells to NSGS mice. As shown in Figure 5E, mice injected with cells prior exposed to C-82 or the combination of C-82 and sorafenib, but not sorafenib alone, engrafted significantly slower, exhibited lower leukemia burden than the untreated controls. These animals also lived longer. At 120 d after cell injections, 5/6 or 3/6 mice injected with untreated or sorafenib-treated cells died, while 5/6 or 6/6 mice injected with C-82 or C-82 plus sorafenib-treated cells were alive (Figure 5E). The results support the notion that inhibition of β-catenin, but not FLT3-ITD tyrosine kinase, targets AML stem/progenitor cells.
Discussion
Besides its roles in AML initiation and progression, β-catenin signaling plays an essential role in the BM microenvironment supporting minimal residual disease and LSC maintenance, which are responsible for leukemia relapse (31). Inhibition of β-catenin is regarded as a promising strategy to eliminate LSC (32). Our finding of higher β-catenin expression in BM than in paired PB AML samples supports an important role for Wnt/β-catenin signaling in the leukemia BM microenvironment. Disruption of Wnt/β-catenin signaling by C-82 suppressed growth, induced apoptosis, and overcame stromal protection of AML cells and stem/progenitor cells. Moreover, the β-catenin downstream target CD44 is overexpressed in AML cells and reportedly plays an important role in chemoresistance (33). CD44 also regulates AML LSC homing to microenvironmental niches and maintaining a primitive phenotype (34). Hence, targeting CD44 not only increases chemosensitivity but also potentially eradicates AML LSC in BM niche (33, 35). In this regard, we showed that inhibition of Wnt/β-catenin signaling decreased CD44 expression in AML blasts and LSCs, inhibited adhesion of AML cells to MSCs, and induced death of AML cells co-cultured with MSCs.
Although highly heterogeneous, approximately 30% of AML patients have FLT3 mutations that contribute to disease progression and poor prognosis (36, 37). To overcome limitations and improve efficacy of FLT3 TKIs, novel combinatorial strategies are required including targeting multiple signaling pathways, disrupting BM microenvironment protection, and eliminating LSC (38, 39). FLT3 signaling is known to increase β-catenin nuclear localization and transcription activity (9, 40) and Wnt/β-catenin pathway to modulate the TKI sensitivity through GSK-3β-dependent mechanisms (41). Moreover, it was reported that FLT3 mutations cooperate with Wnt/β-catenin signaling transduction in AML cells and primary AML blasts (10). Interestingly, a recent study demonstrated that the microenvironment can confer sorafenib resistance in FLT3-mutated cells by activating integrinαvβ3/PI3K/AKT/GSKβ/β-catenin signaling pathway (42) further supporting the induction of β-catenin by BM microenvironment and suggesting the potential for combinatorial inhibition of FLT3 and β-catenin in FLT3-mutated AML. We demonstrated that inhibition of β-catenin by C-82 decreases the expression of CD44, an important LSC homing protein, which may contribute to the synergistic effect of C-82 and sorafenib in FLT3-mutated cells under in vitro leukemia-MSC co-culture conditions, and in vivo in mouse models, by disrupting leukemia-microenvironmental interactions.
Our initial study focused on investigating the effect of targeting β-catenin on AML cells and stem/progenitor cells and we observed higher sensitivity of FLT3-mutated cells to the β-catenin inhibitor C-82. After confirming higher levels of β-catenin in FLT3-mutated cells, which corresponded to C-82 sensitivity, we demonstrated that combined inhibition of β-catenin by C-82 and FLT3 by TKIs exerted synergistic anti-leukemic effects on FLT3-mutated AML cells and LSC/progenitor cells, in agreement with a recent report showing that a novel dual FLT3 and Wnt/β-catenin inhibitor SKLB-677, suppressed leukemia stem-like cells and had potent anti-leukemia activity in FLT3-mutated AML cells (43). Our study further demonstrated that inhibition of β-catenin impaired the engraftment of FLT3-ITD cells derived from an AML patient in NSGS mice and that the combined inhibition of β-catenin and FLT3 signaling significantly prolonged survival compared to inhibiting each target alone in mice xenografted with either a FLT3-mutated cell line or PDX cells. Importantly, PRI-724 was well tolerated showing an acceptable toxicity profile in a phase 1, dose-escalation study in patients with advanced AML (30). This study was recently completed suggesting that the combination has the potential for clinical development.
Mechanistic studies showed that C-82 and TKIs effectively inhibited Wnt/β-catenin targets and FLT3 downstream signaling, respectively; while the combination more potently inhibited both. FLT3-ITD in AML was reported to induce c-Myc, which is one of Wnt/β-catenin targets and an essential regulator of cell cycle and apoptosis (10). Depletion of β-catenin and c-Myc contributed to the synergistic effect of combined inhibition of β-catenin and histone deacetylase in FLT3-ITD mutations AML (1). We demonstrated that C-82 or TKI individually inhibited c-Myc expression in FLT3-mutated AML, and more so by their combination. Furthermore, we showed that, other than modulating β-catenin signaling, FLT3 inhibition directly suppressed c-Myc expression primarily through the FLT3-ITD/STAT5 signaling cascade.
Collectively, these findings provide a mechanistic basis for clinical development of this novel combinatorial strategy to overcome TKI resistance and improve outcomes in FLT3-mutated AML patients. Importantly, constitutive activation of multiple survival signaling pathways that can further activate Wnt/β-catenin signaling is common in many types of malignant cells. The combination strategy that targets both Wnt/β-catenin and other survival signaling pathways likely also applies to the treatment of other cancers.
Supplementary Material
Translational Relevance.
Aberrant Wnt/β-catenin signaling is associated with AML pathogenesis and required for leukemic stem cell survival and function. FLT3 is frequently mutated in AML and associated with poor prognosis. Anomalous FLT3 signaling further increases β-catenin signaling activity. FLT3 TKIs have been developed to treat AML patients with FLT3 mutations, but acquired FLT3 mutations, TKI-induced alternative signaling activation, microenvironment-mediated resistance, and inactivity against leukemia stem cells limit their efficacy. This study demonstrates that inhibition of Wnt/β-catenin signaling targets AML cells including stem/progenitor cells. Combined inhibition of Wnt/β-catenin and FLT3 cooperatively decreased nuclear β-catenin, the levels of Wnt/β-catenin, and FLT3 signaling proteins, and synergistically induced cell death in vitro and in vivo in two murine models and markedly impaired leukemia cell engraftment. These findings provide a mechanistic rationale for the clinical development of this combinatorial strategy for treating FLT3-mutated AML patients, a process that also targets successfully bone marrow microenvironment protected-leukemia stem/progenitor cells.
Acknowledgments
We thank Dr. Numsen Hail for assisting with the manuscript preparation and PRISM Pharma for providing C-82 and PRI-724.
Support: This work was supported by the Ryan Gibson Foundation and PRISM Pharma/Eisai research funding to BZC and by grants from the National Institutes of Health (CA055164) and the MD Anderson Cancer Center Support Grant (CA016672), Cancer Prevention Research Institute of Texas (CPRIT, RP121010), and the Paul and Mary Haas Chair in Genetics to MA.
Footnotes
Conflicts of interests: BZC received research funding from PRISM Pharma/Eisai.
References
- 1.Fiskus W, Sharma S, Saha S, Shah B, Devaraj SG, Sun B, et al. Pre-clinical efficacy of combined therapy with novel β-catenin antagonist BC2059 and histone deacetylase inhibitor against AML cells. Leukemia. 2015;29:1267–78. doi: 10.1038/leu.2014.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lane SW, Wang YJ, Lo Celso C, Ragu C, Bullinger L, Sykes SM, et al. Differential niche and Wnt requirements during acute myeloid leukemia progression. Blood. 2011;118:2849–56. doi: 10.1182/blood-2011-03-345165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dietrich PA, Yang C, Leung HH, Lynch JR, Gonzales E, Liu B, et al. GPR84 sustains aberrant β-catenin signaling in leukemic stem cells for maintenance of MLL leukemogenesis. Blood. 2014;124:3284–94. doi: 10.1182/blood-2013-10-532523. [DOI] [PubMed] [Google Scholar]
- 4.Ysebaert L, Chicanne G, Demur C, De Toni F, Prade-Houdellier N, Ruidavets JB, et al. Expression of β-catenin by acute myeloid leukemia cells predicts enhanced clonogenic capacities and poor prognosis. Leukemia. 2006;20:1211–6. doi: 10.1038/sj.leu.2404239. [DOI] [PubMed] [Google Scholar]
- 5.Gandillet A, Park S, Lassailly F, Griessinger E, Vargaftig J, Filby A, et al. Heterogeneous sensitivity of human acute myeloid leukemia to β-catenin down-modulation. Leukemia. 2011;25:770–80. doi: 10.1038/leu.2011.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim Y, Thanendrarajan S, Schmidt-Wolf IG. Wnt/β-catenin: a new therapeutic approach to acute myeloid leukemia. Leuk Res Treatment. 2011;2011:428960. doi: 10.4061/2011/428960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eaves CJ, Humphries RK. Acute myeloid leukemia and the Wnt pathway. N Engl J Med. 2010;362:2326–7. doi: 10.1056/NEJMcibr1003522. [DOI] [PubMed] [Google Scholar]
- 8.Schnittger S, Schoch C, Dugas M, Kern W, Staib P, Wuchter C, et al. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood. 2002;100:59–66. doi: 10.1182/blood.v100.1.59. [DOI] [PubMed] [Google Scholar]
- 9.Kajiguchi T, Chung EJ, Lee S, Stine A, Kiyoi H, Naoe T, et al. FLT3 regulates β-catenin tyrosine phosphorylation, nuclear localization, and transcriptional activity in acute myeloid leukemia cells. Leukemia. 2007;21:2476–84. doi: 10.1038/sj.leu.2404923. [DOI] [PubMed] [Google Scholar]
- 10.Tickenbrock L, Schwäble J, Wiedehage M, Steffen B, Sargin B, Choudhary C, et al. Flt3 tandem duplication mutations cooperate with Wnt signaling in leukemic signal transduction. Blood. 2005;105:3699–706. doi: 10.1182/blood-2004-07-2924. [DOI] [PubMed] [Google Scholar]
- 11.Kiyoi H, Ohno R, Ueda R, Saito H, Naoe T. Mechanism of constitutive activation of FLT3 with internal tandem duplication in the juxtamembrane domain. Oncogene. 2002;21:2555–63. doi: 10.1038/sj.onc.1205332. [DOI] [PubMed] [Google Scholar]
- 12.Al-Mawali A, Gillis D, Lewis I. Characteristics and prognosis of adult acute myeloid leukemia with internal tandem duplication in the FLT3 gene. Oman Med J. 2013;28:432–40. doi: 10.5001/omj.2013.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Daver N, Cortes J, Ravandi F, Patel KP, Burger JA, Konopleva M, et al. Secondary mutations as mediators of resistance to targeted therapy in leukemia. Blood. 2015;125:3236–45. doi: 10.1182/blood-2014-10-605808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li L, Osdal T, Ho Y, Chun S, McDonald T, Agarwal P, et al. SIRT1 activation by a c-MYC oncogenic network promotes the maintenance and drug resistance of human FLT3-ITD acute myeloid leukemia stem cells. Cell Stem Cell. 2014;15:431–46. doi: 10.1016/j.stem.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kida A, Kahn M. Hypoxia selects for a quiescent, CML stem/leukemia initiating-like population dependent on CBP/catenin transcription. Curr Mol Pharmacol. 2013;6:204–10. doi: 10.2174/1874467207666140219121219. [DOI] [PubMed] [Google Scholar]
- 16.Gang EJ, Hsieh YT, Pham J, Zhao Y, Nguyen C, Huantes S, et al. Small-molecule inhibition of CBP/catenin interactions eliminates drug-resistant clones in acute lymphoblastic leukemia. Oncogene. 2014;33:2169–78. doi: 10.1038/onc.2013.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou H, Mak PY, Mu H, Mak DH, Zeng H, Cortes J, et al. Combined inhibition of β-catenin and Bcr-Abl synergistically targets tyrosine kinase inhibitor-resistant blast crisis chronic myeloid leukemia blasts and progenitors in vitro and in vivo. Leukemia. 2017;31(10):2065–74. doi: 10.1038/leu.2017.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kornblau SM, Tibes R, Qiu YH, Chen W, Kantarjian HM, Andreeff M, et al. Functional proteomic profiling of AML predicts response and survival. Blood. 2009;113:154–64. doi: 10.1182/blood-2007-10-119438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carter BZ, Qiu YH, Zhang N, Coombes KR, Mak DH, Thomas DA, et al. Expression of ARC (apoptosis repressor with caspase recruitment domain), an antiapoptotic protein, is strongly prognostic in AML. Blood. 2011;117:780–7. doi: 10.1182/blood-2010-04-280503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tse KF, Allebach J, Levis M, Smith BD, Bohmer F, Small D. Inhibition of the transforming activity of FLT3 internal tandem duplication mutants from AML patients by a tyrosine kinase inhibitor. Leukemia. 2002;6:2027–36. doi: 10.1038/sj.leu.2402674. [DOI] [PubMed] [Google Scholar]
- 21.Zhang W, Gao C, Konopleva M, Chen Y, Jacamo RO, Borthakur G, et al. Reversal of acquired drug resistance in FLT3-mutated acute myeloid leukemia cells via distinct drug combination strategies. Clin Cancer Res. 2014;20:2363–74. doi: 10.1158/1078-0432.CCR-13-2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Romano P, Manniello A, Aresu O, Armento M, Cesaro M, Parodi B. Cell Line Data Base: structure and recent improvements towards molecular authentication of human cell lines. Nucleic Acids Res. 2009;37:D925–32. doi: 10.1093/nar/gkn730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carter BZ, Mak DH, Wang Z, Ma W, Mak PY, Andreeff M, et al. XIAP downregulation promotes caspase-dependent inhibition of proteasome activity in AML cells. Leuk Res. 2013;37:974–9. doi: 10.1016/j.leukres.2013.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacamo R, Chen Y, Wang Z, Ma W, Zhang M, Spaeth EL, et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-κB mediates chemoresistance. Blood. 2014;123:2691–702. doi: 10.1182/blood-2013-06-511527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carter BZ, Mak PY, Chen Y, Mak DH, Mu H, Jacamo R, et al. Anti-apoptotic ARC protein confers chemoresistance by controlling leukemia-microenvironment interactions through a NFκB/IL1β signaling network. Oncotarget. 2016;7:20054–67. doi: 10.18632/oncotarget.7911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bendall SC, Simonds EF, Qiu P, Amir el AD, Krutzik PO, Finck R, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332:687–96. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han L, Qiu P, Zeng Z, Jorgensen JL, Mak DH, Burks JK, et al. Single-cell mass cytometry reveals intracellular survival/proliferative signaling in FLT3-ITD-mutated AML stem/progenitor cells. Cytometry A. 2015;87:346–56. doi: 10.1002/cyto.a.22628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carter BZ, Mak PY, Mu H, Zhou HS, Mak DH, Schober W, et al. Combined Targeting of BCL-2 and BCR-ABL Tyrosine Kinase Eradicates Chronic Myeloid Leukemia Stem Cells. Sci Transl Med. 2016;8:355ra117. doi: 10.1126/scitranslmed.aag1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 30.Cortes JE, Carter BZ, Quintas-Cardama A, Inada T, Morita K, Fujimori M, et al. A phase I dose-escalation study of PRI-724, a CBP/β-catenin modulator in patients with advanced acute myeloid leukemia (AML) Haematologica. 2014;99(s1):222–3. abstract n. s648. [Google Scholar]
- 31.Hu K, Gu Y, Lou L, Liu L, Hu Y, Wang B, et al. Galectin-3 mediates bone marrow microenvironment-induced drug resistance in acute leukemia cells via Wnt/β-catenin signaling pathway. J Hematol Oncol. 2015;8:1. doi: 10.1186/s13045-014-0099-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Konopleva M, Tabe Y, Zeng Z, Andreeff M. Therapeutic targeting of microenvironmental interactions in leukemia: mechanisms and approaches. Drug Resist Updat. 2009;12:103–13. doi: 10.1016/j.drup.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang NS, Wei M, Ma WL, Meng W, Zheng WL. Knockdown of CD44 enhances chemosensitivity of acute myeloid leukemia cells to ADM and Ara-C. Tumour Biol. 2014;35:3933–40. doi: 10.1007/s13277-013-1523-3. [DOI] [PubMed] [Google Scholar]
- 34.Siapati EK, Papadaki M, Kozaou Z, Rouka E, Michali E, Savvidou I, et al. Proliferation and bone marrow engraftment of AML blasts is dependent on β-catenin signaling. Br J Haematol. 2011;152:164–74. doi: 10.1111/j.1365-2141.2010.08471.x. [DOI] [PubMed] [Google Scholar]
- 35.Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12:1167–74. doi: 10.1038/nm1483. [DOI] [PubMed] [Google Scholar]
- 36.Gilliland DG. Molecular genetics of human leukemias: new insights into therapy. Semin Hematol. 2002;39:6–11. doi: 10.1053/shem.2002.36921. [DOI] [PubMed] [Google Scholar]
- 37.Fröhling S, Schlenk RF, Breitruck J, Benner A, Kreitmeier S, Tobis K, et al. Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: a study of the AML Study Group Ulm. Blood. 2002;100:4372–80. doi: 10.1182/blood-2002-05-1440. [DOI] [PubMed] [Google Scholar]
- 38.Kindler T, Lipka DB, Fischer T. FLT3 as a therapeutic target in AML: still challenging after all these years. Blood. 2010;116:5089–102. doi: 10.1182/blood-2010-04-261867. [DOI] [PubMed] [Google Scholar]
- 39.Chang E, Ganguly S, Rajkhowa T, Gocke CD, Levis M, Konig H. The combination of FLT3 and DNA methyltransferase inhibition is synergistically cytotoxic to FLT3/ITD acute myeloid leukemia cells. Leukemia. 2016;30:1025–32. doi: 10.1038/leu.2015.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kajiguchi T, Katsumi A, Tanizaki R, Kiyoi H, Naoe T. Y654 of β-catenin is essential for FLT3/ITD-related tyrosine phosphorylation and nuclear localization of β-catenin. Eur J Haematol. 2012;88:314–20. doi: 10.1111/j.1600-0609.2011.01738.x. [DOI] [PubMed] [Google Scholar]
- 41.Jiang J, Griffin JD. Wnt/β-catenin pathway modulates the sensitivity of the mutant FLT3 receptor kinase inhibitors in a GSK-3β dependent manner. Genes Cancer. 2010;1:164–76. doi: 10.1177/1947601910362446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yi H, Zeng D, Shen Z, Liao J, Wang X, Liu Y, et al. Integrin alphavbeta3 enhances β-catenin signaling in acute myeloid leukemia harboring Fms-like tyrosine kinase-3 internal tandem duplication mutations: implications for microenvironment influence on sorafenib sensitivity. Oncotarget. 2016;7:40387–97. doi: 10.18632/oncotarget.9617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ma S, Yang LL, Niu T, Cheng C, Zhong L, Zheng MW, et al. SKLB-677, an FLT3 and Wnt/β-catenin signaling inhibitor, displays potent activity in models of FLT3-driven AML. Sci Rep. 2015;5:15646. doi: 10.1038/srep15646. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.