

Addendum to: Nature Communications 10.1038/s41467-017-00777-0; published online 26 September 2017
Supplementary Note 4 | Structural information of the DFT calculation
The DFT calculations in the main text use crystal structure information determined by x-ray diffraction (XRD) measurements of La4Ni3O10. The room temperature XRD measurements on all samples were consistent with mixed phases of orthorhombic and monoclinic symmetry. However, the in-plane lattice constants of these two phases differ by <1%, leading to minimal influence on band structure calculations. Furthermore, any impact on the experimental Fermi surface attributable to such deviations would be obscured by broadening of the bands measured by ARPES. Therefore, the DFT calculations adopt the higher symmetry orthorhombic crystal structure with space group Cmca and lattice constants a = 5.417 Å, c = 5.468 Å, b = 27.962 Å, with some additional relaxation of the internal coordinates. The detailed atomic coordinates are presented in Supplementary Table 1.
Supplementary Table 1 | The detailed atomic coordinates from the DFT calculation
| Atom | Wyckoff position | Coordinates |
|---|---|---|
| La1 | 8f | (0, 0.4326, 0.0014) |
| La2 | 8f | (0, 0.3016, 0.0099) |
| Ni1 | 4a | (0, 0, 0) |
| Ni2 | 8f | (0, 0.1394, 0.0024) |
| O1 | 8e | (0.25, 0.4921, 0.25) |
| O2 | 8f | (0, 0.0704, 0.0501) |
| O3 | 8e | (0.25, 0.3664, 0.25) |
| O4 | 8f | (0, 0.2161, 0.9641) |
| O5 | 8e | (0.25, 0.1461, 0.25) |
A comprehensive plot of the DFT results for this orthorhombic structure is shown in Supplementary Figure 4.
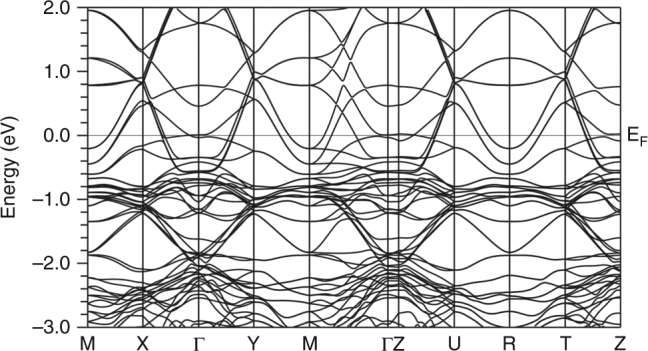
Supplementary Figure 4 | DFT band structure of orthorhombic La4Ni3O10 along more directions and over a wider energy range than is shown in the main text