

Abstract
Background:
The hedgehog signaling system (HHS) plays an important role in the regulation of cell proliferation and differentiation during the embryonic phases. However, little is known about the involvement of HHS in the malignant transformation of cells. This study aimed to detect the role of HHS in the malignant transformation of human bronchial epithelial (16HBE) cells.
Methods:
In this study, two microfluidic chips were designed to investigate cigarette smoke extract (CSE)-induced malignant transformation of cells. Chip A contained a concentration gradient generator, while chip B had four cell chambers with a central channel. The 16HBE cells cultured in chip A were used to determine the optimal concentration of CSE for inducing malignant transformation. The 16HBE cells in chip B were cultured with 12.25% CSE (Group A), 12.25% CSE + 5 μmol/L cyclopamine (Group B), or normal complete medium as control for 8 months (Group C), to establish the in vitro lung inflammatory-cancer transformation model. The transformed cells were inoculated into 20 nude mice as cells alone (Group 1) or cells with cyclopamine (Group 2) for tumorigenesis testing. Expression of HHS proteins was detected by Western blot. Data were expressed as mean ± standard deviation. The t-test was used for paired samples, and the difference among groups was analyzed using a one-way analysis of variance.
Results:
The optimal concentration of CSE was 12.25%. Expression of HHS proteins increased during the process of malignant transformation (Group B vs. Group A, F = 7.65, P < 0.05). After CSE exposure for 8 months, there were significant changes in cellular morphology, which allowed the transformed cells to grow into tumors in 40 days after being inoculated into nude mice. Cyclopamine could effectively depress the expression of HHS proteins (Group C vs. Group B, F = 6.47, P < 0.05) and prevent tumor growth in nude mice (Group 2 vs. Group 1, t = 31.59, P < 0.01).
Conclusions:
The activity of HHS is upregulated during the CSE-induced malignant transformation of 16HBE cells. Cyclopamine can effectively depress expression of HHS proteins in vitro and prevent tumor growth of the transformed cells in vivo.
Keywords: Hedgehog Signaling System, Lung Cancer, Malignant Transformation, Microfluidic Chip
摘要
背景:
Hedgehog信号系统(HHS) 在胚胎期对于调节细胞增殖和分化起到重要作用。然而,人们对于HHS在细胞恶性转化中的 作用还知之甚少。本研究的目的在于检测HHS在人支气管上皮细胞(16HBE)恶性转化中的作用。
方法:
在本研究中,设计了两个微流控芯片来完成香烟提取物(cigarette smoke extract,CSE)诱导的细胞恶性转化研究。芯片A含 有一个浓度梯度发生器 (concentration gradient generator) ,芯片B含有4个细胞培养池和一个中央通道。芯片A中的16HBE细胞 用于确定最佳CSE浓度,芯片B中培养的16HBE细胞用于(1) A组: 正常全培养基,(2) B组:12.25% CSE, (3) C组:12.25% CSE + 5μmol/L 环巴胺(cyclopamine) 培养8个月,建立肺部炎-癌转化体外模型。转化后的细胞接种到20只裸鼠中(1组)或 细胞+环巴胺 (2 组)检测成瘤性。成对样本比较使用t检验,多组间差异采用方差分析。
结果:
结果表明最佳CSE浓度为12.25%。HHS蛋白的表达在恶性转化过程中增加(B组 vs. A组, F=7.65, P<0.05)。CSE处 理8个月后,细胞形态发生明显变化,并且在植入裸鼠40天内能够长成肿块。环巴胺能够有效抑制HHS蛋白表达(B组 vs. A 组,F=6.47, P<0.05) 并抑制肿瘤生长(2组 vs. 1组, t=31.59, P<0.01)。
结论:
HHS活性在CSE诱导的细胞恶性转化过程中表达上调。环巴胺能够有效抑制HHS蛋白表达并阻止转化细胞长成肿瘤。
INTRODUCTION
As reported, lung cancer has the highest morbidity and mortality among all human malignancies.[1] Cigarette smoke is an important contributor to the development of lung cancer.[2] However, the cellular and molecular mechanisms underlying the malignant transformation processes in lung cancer are not entirely known at this time.[3,4,5]
The hedgehog signaling system (HHS) is an embryonic cellular signaling pathway which transfers a signal to the proper target cells to trigger the transformation of cells.[6] Hedgehog (HH) signaling consists of extracellular ligand proteins, including sonic HH (SHH), desert HH, Indian HH, transmembrane receptors (patched [Ptch1 and Ptch2] and smoothened [SMO]), nuclear transcription factors (glioma-associated oncogene homolog [GLI-1, GLI-2, and GLI-3]), and downstream target genes (vascular endothelial growth factor and matrix metalloproteinases).[7,8] The binding of Ptch1 to SHH relives the inhibitory effect on SMO, allowing it to activate the putative serine-threonine kinase Fused (Fu) and leading to the entry and integration of GLI-1 into the nucleus for target gene activation.[9] This signaling pathway also participates in many pathological processes in adults, such as oncogenesis.[10] The advent of molecular targeted therapies and molecular biology has renewed interest in the roles of the HHS in the development and metastasis of cancer.[6,7,8,11] However, little is known about the involvement of the HHS in the malignant transformation and tumorigenesis of human lung epithelial cells. For this reason, there is a need for an acceptable and validated in vitro model of smoke-induced lung cancer, as it would provide valuable insight into the mechanisms, diagnosis, prevention, and therapy of lung cancer.[12,13,14] The aim of this study was to detect the function of HH signaling during the process of cigarette smoke extract (CSE)-induced malignant transformation of 16HBE cells using a microfluidic chip detection platform. Chip A contained a concentration gradient generator (CGG) used to determine the optimal concentration of CSE, and microfluidic Chip B was used to assess the effects of long-term CSE stimulation on the transformation of 16HBE cells.
The microfluidic chip comes from one of the biomicrochip technologies. Microfluidic chips display consistent reaction conditions and synchronous multipoint detection, providing rapid and cost-effective results. The use of microfluidic chips for screening target chemical gradients is more efficient, economical, and convenient than traditional techniques. Microfluidic chips can be used in microbiological sample preparation, purification, and molecular biological detection.[15,16] They can also be used to provide the continuous flow of fresh nutrients for cultured cells.[17,18] At the same time, microfluidic chips may be used for high-throughput screening, which plays an important role in the field of drug discovery, single-cell analysis, and cell signaling pathway transduction. Microfluidic chips have significantly impacted the field of biomedicine.
This study aimed to detect the role of the HHS in the process of malignant transformation in human bronchial epithelial (16HBE) cells. After screening for the optimal CSE concentration, we established and examined the in vitro model of lung inflammatory-cancer transformation using microfluidic chips and performed tumorigenesis testing in nude mice. The investigation would demonstrate the activity of the HHS in CSE-induced malignant transformation of human lung epithelial cells, which might provide measures of intervention.
METHODS
Ethical approval
Animal experimental procedures were reviewed and approved by the Animal Experimental Ethics Committee of Dalian Medical University (Liaoning, China).
Animals
A total of 20 specific-pathogen-free (SPF) grade 28-day-old BALB/c-nu/nu mice were provided by the SPF Experimental Animal Center of Dalian Medical University, and all procedures were performed in a special laboratory for nude mice. At the end of the experiment, the mice were sacrificed by 10% chloral hydrate at 3.5 ml/kg weight, and the tumor was excised and stored in 4% formaldehyde.
Cigarette smoke extract-containing medium preparation
The smoke from two research grade cigarettes (University of Kentucky, KY, USA) was passed through serum-free RPMI-1640 medium in to a 50 ml conical tube and sterilized by filtrating through a 0.22 μm pore filter (Merck Millipore, Tullagreen, Carrigtwohill, Republic of Ireland) to obtain the CSE stock solutions [Figure 1]. The pH of the solutions was adjusted to 7.4, and an optical density of 0.43 ± 0.02 at 320 nm was defined as 100% CSE, using the 721 spectrometer (Changsha Qlong Instruments Co., Hunan, China). The freshly prepared CSE was used immediately.
Figure 1.

Device for preparation of CSE-containing medium. CSE-containing medium was prepared by passing the smoke from two research grade cigarettes through 50 ml of serum-free RPMI-1640 medium, followed by filtration through a 0.22-μm pore filter. CSE: Cigarette smoke extract.
Establishment of the microfluidic chip detection platforms
Briefly, the corresponding polydimethylsiloxane (PDMS) microfluidic chips were fabricated using the Sylgard 184 kit (DowCorning, Midland, TX, USA) with the standard soft lithography strategy. They were glued to glass slides that were pretreated with oxygen plasma (19.95 Pa, 50 W, 25 s). The prepared chips were treated with dehydrated alcohol triplicately and washed with distilled water before being autoclaved at 120°C and 103.4 kPa for 30 min.
Two types of microfluidic chips were manufactured for this study. Chip A was used to determine the optimal concentration of CSE for cell stimulation, while chip B was used for the long-term stimulation of cultured cells using the optimal concentration of CSE.[19] Chip A contained a CGG module as described previously,[20,21] which consisted of six main channels and 18 cell chambers (diameter 2 mm and height 100 μm) [Figure 2]. The CGG module included five cascaded-mixing stages. After the incorporation of the CGG module, microfluidic chip A could generate a concentration gradient of CSE (theoretical proportion 0:1:3:5:7:9) by controlling channel length and adjusting the flow rate of two merging solutions, as previously reported.[14]
Figure 2.

Structure of microfluidic chip A for screening CSE concentrations. Chip A was used to determine the optimal CSE concentration with (left) the general structure shown and (right) the actual chip (size 6 cm × 6 cm). The cells were cultured in the 18 cell chambers with CSE-containing culture medium added through the medium inlet. The medium was continuously exchanged with the fresh medium through a pump. CSE: Cigarette smoke extract; CGG: Concentration gradient generator.
Microfluidic chip B was designed to assess the effects of long-term CSE stimulation on cultured cells. It consisted of four cell chambers (length 2.0 mm, width 1 mm, and height 100 μm) and one central channel (length 15 mm, width 0.8 mm, and height 100 μm) [Figure 3]. There were 20 traffic channels (width 30 μm and height 100 μm) between each cell chamber and the one central channel.
Figure 3.

Structure of microfluidic chip B for assessing the malignant transformation of cells. Chip B was used to assess the long-term stimulation of cells using the optimal concentration of CSE. The general structure of chip B shows the central channel and four cell chambers (left), while depicts an actual picture of chip B (right; size 4.5 cm × 1.5 cm). CSE: Cigarette smoke extract.
Cell culture and intervention
A total of 1 × 106 16HBE cells/ml in complete medium (RPMI-1640 + 10% fetal bovine serum) were injected into the cell culture chambers through No. 1–6 inlets of chip A by a syringe pump with a constant flow rate of 8 μl/min for 10 s. The waste medium was aspirated from the outlet. After 2 h, the attached cells were supplied with the normal medium at a constant rate of 6 μl/min by a syringe pump, until 80% confluency was reached. The normal medium and CSE stock solutions were supplied into the channels by a syringe pump through the inlets. After treatment with or without CSE for 48 h, the cells were stained with 1 μg/ml Hoechst 33342 (Sigma Aldrich, MO, USA) for 30 min. Next, the cells were washed twice with phosphate-buffered saline (3 min each) and examined under a fluorescent microscope (Olympus IX71, Japan, magnification, ×200) using the 350 nm excitation wavelength and 461 nm emission wavelength. The optimal CSE concentration was defined as the highest possible concentration that did not inhibit cell growth (>80% cell viability).
For chip B, a total of 1 × 106 cells/ml 16HBE cells were injected into the cell culture chambers through No. 1–4 inlets of chip B by a syringe pump with a constant flow rate of 8 μl/min for 10 s. The waste liquid was aspirated from the outlet. After 2 h, the attached cells were supplied with the normal medium through the central channel at a constant rate of 6 μl/min by a syringe pump until 80% confluency was reached. Next, the chips with the cells were stimulated respectively with 12.25% CSE (Group A), 12.25% CSE + 5 μmol/L cyclopamine (LC Laboratories, MA, USA; Group B), or normal complete medium as control for 8 months of treatment (Group C). The cells were detached with 0.05% trypsin for 3 min. Cells from the 15th, 25th, 35th, and 45th generation were collected and stored in liquid nitrogen. Samples from each collected generation were examined for changes in cell morphology by microscopy [Figure 4] and protein expression by Western blot.
Figure 4.

Changes in cellular morphology in CSE-induced 16HBE cells. The morphological changes were observed under an inverted microscope (original magnification ×200) in the 16HBE cells stimulated with (a–d) 12.25% CSE or (e–h) 12.25% CSE + 5 μmol/L cyclopamine at the indicated generation. In the 45th generation of the 12.25% CSE-treated group, tumor-like cells were observed. The tumor-like cells were not found in the 12.25% CSE + 5 μmol/L cyclopamine group or the control group (i). CSE: Cigarette smoke extract.
Tumorigenicity assay of the differentiated cells cultured in the chips in nude mice
A total of 5 × 106 cells/mouse (0.1 ml, 5 × 107 cells/ml) of the 45th generation cells, with significant morphology changes after CSE stimulation, were subcutaneously implanted in the postmedian axillary fossa of 20 athymic nude mice (Group 1). The inoculation process was performed in an ultra-clean cabinet. Half of the mice were injected with 5 μl/week cyclopamine (5 μmol/L) in the original injection site 1 week after inoculation (Group 2). Tumor size was measured weekly until the diameter reached 1 cm. After about 40 days, the tumors were excised for pathological examination.
Statistical analysis
Data were expressed as the mean ± standard deviation (SD). The t-test was used for paired-samples, and the difference among groups was tested by the one-way analysis of variance (ANOVA) using SPSS 19.0 (SPSS Inc., Chicago, IL, USA).A value of P < 0.05 was considered statistically significant.
RESULTS
Optimal cigarette smoke extract concentration for 16HBE cellular transformation in chip A
In this study, two microfluidic chips were designed for investigating CSE-induced malignant transformation in the 16HBE cells. Chip A was used to determine the optimal concentration of CSE for cell stimulation, while chip B was used for the long-term stimulation of cultured cells using the optimal concentration of CSE. After treatment with or without CSE for 48 h in chip A, the 16HBE cells were stained with Hoechst 33342 to examine the cellular apoptotic rate under a fluorescent microscope. The results showed that CSE-induced 16HBE cell apoptosis was dose dependent, with lower doses (<12.28%) corresponding to higher cell survival (>80% cell viability) [Figure 5]. These data suggested that the optimal concentration of CSE was 12.25%, which was then used in chip B to investigate malignant transformation in 16HBE cells with long-term CSE stimulation.
Figure 5.
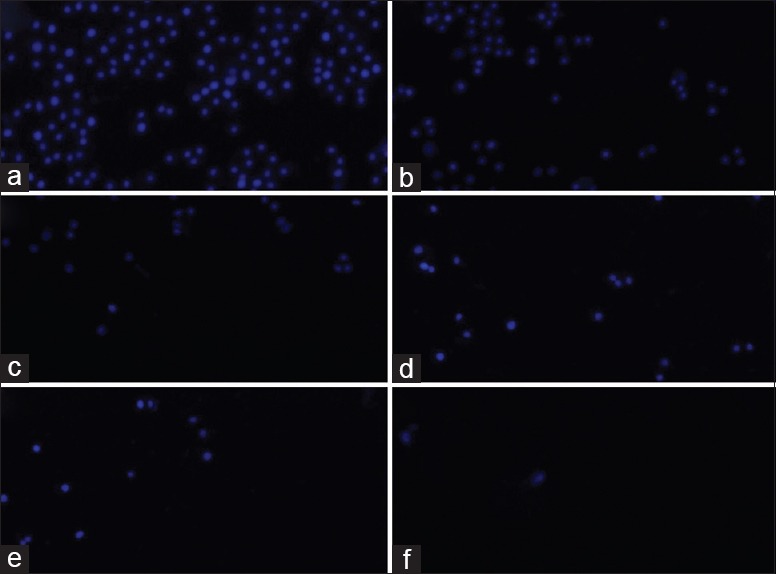
Different concentrations of CSE-induced 16HBE cell apoptosis (original magnification ×200). 16HBE cells were pretreated with (a) 91.88%, (b) 46.79%, (c) 19.86%, (d) 12.28%, (e) 2.37%, or (f) 0% CSE. After exposure to CSE, cell viability was assessed by an apoptosis assay. The blue stain indicated apoptotic cells, and the apoptosis rate was calculated by dividing the number of dead cells by the total number of cells. All experiments were performed in triplicate. CSE: Cigarette smoke extract.
16HBE cell malignant transformation induced by cigarette smoke extract on a microfluidic chip
During the stimulation with 12.25% CSE in chip B, an inverted microscope was used to monitor changes in cellular morphology intermittently to identify the transformed tumor cells. From the 15th generation, the cells were harvested once and preserved in liquid nitrogen every 10 generations until the 45th generation, at which time the cellular morphology had changed significantly [Figure 4a–d]. After stimulation with 12.25% CSE for 15 weeks, some cells displayed condensed nuclei and abnormal nuclear–cytoplasmic ratios, accompanied by atypical mitoses. As CSE exposure was prolonged, the abnormal cells increase accompanied by a loss of contact inhibition. Interestingly, these alterations were not apparent in the cells treated with cyclopamine. There were no morphological changes shown in the control cells after CSE induction. These results indicated that after the 45th generation, significant morphology changes were observed in the 16HBE cells; however, cyclopamine prohibited these changes [Figure 4e-h].
Alterations of the hedgehog signaling proteins during the malignant transformation of cells
To investigate the effect of HH signaling in the CSE-induced transformation of 16HBE cells, the expression of GLI-1 and SMO was analyzed in the CSE-induced 16HBE cells by Western blot. Nontreated cells and 12.25% CSE + 5 μmol/L cyclopamine-treated cells served as controls. The results revealed that both GLI-1 and SMO expression were significantly increased in the CSE-stimulated group from the 15th to 45th generation when compared to the nontreated groups. In addition, the tendency increased with long durations of CSE (Group B vs. Group A, F = 7.65, P < 0.05). In the meantime, the expression of GLI-1 and SMO, induced by CSE, was significantly prevented by the HHS inhibitor, cyclopamine (Group C vs. Group B, F = 6.47, P < 0.05), even though they were still higher than those of the nontreated cells [Figure 6]. Thus, the expression analysis of the HHS proteins verified that GLI-1 and SMO were upregulated by CSE stimulation, and cyclopamine treatment effectively decreased the CSE-induced upregulation of GLI-1 and SMO [Figure 7], indicating that HH signaling is likely involved in the CSE-induced malignant transformation of 16HBE cells.
Figure 6.

GLI-1 and SMO expression in the transformed cells. GLI-1 and SMO expression in the CSE-induced transformed cells at the (a) 15th, (b) 25th, (c) 35th, and (d) 45th generation was detected by Western blot. Nontreated (normal) and CSE + cycl-treated cells were used as controls. CSE: Cigarette smoke extract; Cycl: Cyclopamine; GLI: Glioma-associated oncogene homolog; SMO: Smoothened.
Figure 7.
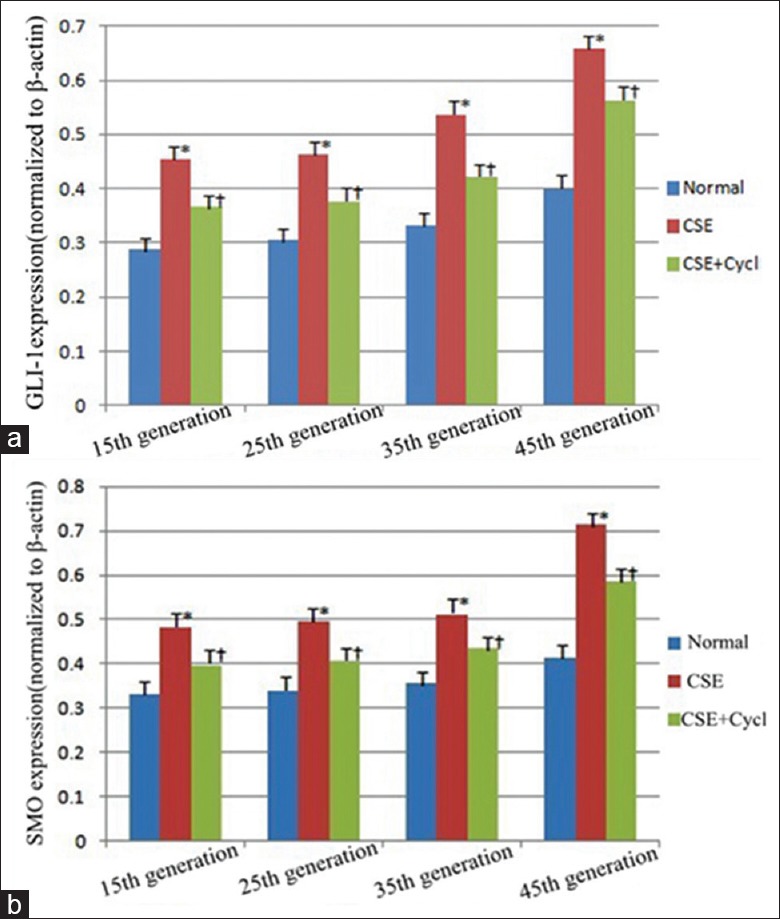
The trends of GLI-1 and SMO expression in the CSE-induced transformed cells at the 15th, 25th, 35th, and 45th generation. (a) Densitometric analysis of GLI-1 expression from the 15th to 45th generation in three independent assays. (b) Densitometric analysis of SMO expression from the 15th to 45th generation in three independent assays. The densitometric analyses were normalized to the corresponding β-actin. *P < 0.01 vs. the controls. †P < 0.05 vs. the controls. CSE: Cigarette smoke extract; Cycl: Cyclopamine; SMO: Smoothened; GLI: Glioma-associated oncogene homolog.
Tumorigenicity of the cigarette smoke extract-induced differentiated cells by subcutaneous inoculation in nude mice
To verify the properties of the CSE-induced transformed 16HBE cells, the 45th generation CSE-treated cells were subcutaneously inoculated into athymic nude mice. Forty days later, the inoculated cells grew into a lump about 1.0 cm in diameter [Figure 8a and 8b]. The histochemical stain of the lump indicated that the cells were transformed with condensed nuclei, abnormal nuclear–cytoplasmic ratios, atypical mitoses, and other cancer-like characteristics. Notably, local injection with 5 μmol/L cyclopamine inhibited tumor growth (Group 2 vs. Group 1, t = 31.59, P < 0.01). These data further verified that cyclopamine could significantly decrease the tumor growth in mice injected with transformed 16HBE cells [Figure 8c and 8d].
Figure 8.
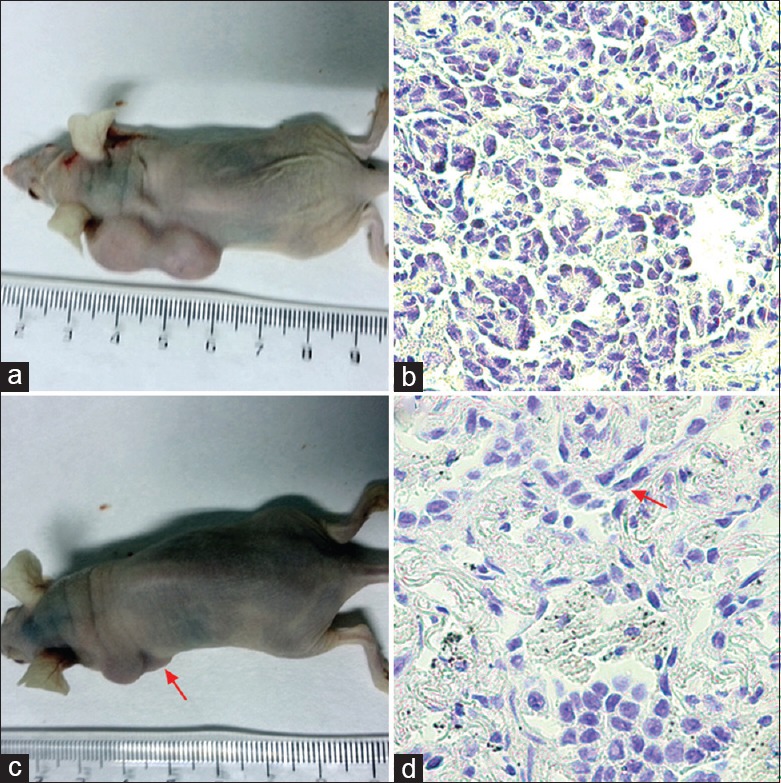
Subcutaneous inoculation of CSE-induced malignant cells in nude mice and intervention. The 45th generation of the CSE-treated (Group 1) and CSE + cyclopamine-treated cells (Group 2) was subcutaneously inoculated into athymic nude mice. (a and c) The cells formed a lump by 40 days postinoculation. (b and d) The structure of the inoculated cells from the CSE-treated and CSE + cyclopamine-treated cells (arrows) was shown by immunohistochemistry (original magnification ×200). CSE: Cigarette smoke extract.
Here, we reconfirmed the involvement of the HHS in cancer transformation using newly fabricated microfluidic chips in vitro and in vivo. Furthermore, we showed that cyclopamine significantly prevented the CSE-induced malignant transformation of 16HBE cells [Figure 8].
DISCUSSION
Cigarette smoke can result in human airway injury through acute inflammation and oxidative stress. The chronic inflammation caused by long-term smoke exposure may induce the transformation of normal bronchial epithelial (BE) cells to cancer cells. Schamberger et al.[22] studied CSE-induced primary human bronchial epithelial cell (PHBEC) transformation and discovered that smoke exposure led to a decrease in the number of ciliated cells, while the number of Clara and goblet cells increased. Stinn et al.[23] directly induced lung cancer in A/J mice using mainstream cigarette smoke. Therefore, it is believed that lung cancer and chronic obstructive pulmonary disease are smoking-related diseases, and that lung cancer is also a chronic inflammatory process.[24,25,26,27,28,29]
Since the discovery of the HH gene by Nüsslein-Volhard and Wieschaus in Drosophila in 1980,[6] researchers have investigated the role of HH signaling in many types of cancer.[30] Given the fact that HH signaling can regulate the differentiation of embryonic stem cells, it is also reasonable to suggest that HH signaling may play an important role in the mechanism of pulmonary inflammatory transformation.[8] Therefore, this study uses CSE as a long-term stimulation factor, to promote the malignant transformation of PHBECs from inflammation. This allowed us to establish an inflammatory-cancer transformation cell model and achieve a dynamic study of the role of HH signaling in the inflammatory-cancer transformation in vitro. To do this, we first constructed a device for extracting and absorbing cigarette smoke, which could complete the extraction and aseptic filtration process of smoke in a short period of time. This allowed us to use the smoke extract daily to stimulate the BE cells.
Cell culture is normally performed using cell culture flasks with static growing conditions, which is different from the multifaceted environment found in vivo. The traditional cell culture model was abandoned in this study for the advanced microfluidic chip technology, which integrated high throughput and dynamic feeding. As a powerful technology, microfluidic chips can be used to create program-controlled microenvironments, making them widely applicable to the biomedical community. The microfluidic technology has been utilized in many research disciplines to study various biological processes, including controllable cytokine gradients for neural progenitor cell differentiation and three-dimensional cell cultures for assessing the differentiation of mouse embryonic carcinoma cells.[31,32] Nevertheless, few studies have employed the microfluidic system for investigating CSE-induced malignant transformation in vitro. Therefore, there is an urgent need for an in vitro model of lung disease using BE cells.[33] Currently, there have been two commercially available models developed for normal human tracheal/BE (NHT/BE) cells, including EpiAirway® by MatTek and MucilAir™ by Epithelix. NHT/BE cells in both systems are maintained at the air–liquid interface. Other in vitro models have been applied for tissue culture.[33] The CSE-induced malignant transformation in vitro model, especially for HBEs, has seldom been reported. Here, we created a microfluidic chip system that has several advantages, including its simple operation, constant maintenance of conditions, and its low cost. Under the condition of simulating the cell microenvironment in vivo, the growth state of the cells was observed dynamically and the detection was completed in real time, suggesting that it has valuable commercial developmental potential. In this study, the liquid hydrodynamics principle was used in combination with the existing microfluidic chip technology from our laboratory.[14] Using safe, nontoxic, and cost-effective PDMS, chip A was created with a CGG capable of generating six different concentrations of CSE simultaneously. This allowed for the rapid detection of the optimal CSE concentration (12.25%) for cell stimulation, greatly reducing the workload and time commitment required of traditional techniques. Chip B was created to observe cellular morphology and biological changes in the cells exposed to long-term CSE stimulation. The use of the central channel helped improve the CSE stimulation efficiency and prevented CSE from blocking the tube, leading to a successful experiment.
The experimental results in this study demonstrate several things. (1) The self-made smoke extraction and filtration equipment were successfully employed for stimulating cultured cells. There was a large difference between the different CSE concentrations in the human BE cells, as the high dose of CSE (≥19.86%) promoted cell apoptosis, while the low dose of CSE (≤12.28%) showed less apoptosis and continued cell growth (P < 0.05). Therefore, it is reasonable to select 12.25% as the best concentration for CSE stimulation, which guarantees maximum stimulation, facilitates the accurate preparation of the solution, and reduces the difficulty of solution configuration. (2) Long-term exposure to CSE can cause 16HBE cell changes in morphology, cell polarity, contact inhibition of growth changes, and chromosome aberrations, suggesting that transformed cells are characteristic of malignant cells. (3) During continuous CSE exposure, the expression of SMO and GLI proteins in 16HBE cells increased significantly compared with those in the nonstimulation group, and their expression increased gradually with the prolonged CSE stimulation. However, the increase can be inhibited by the SMO inhibitor, cyclopamine. This suggested that HH signaling is upregulated in the lung inflammatory-cancer transformed cells. (4) Cyclopamine significantly decreased CSE-induced malignant transformation of 16HBE cells and tumorigenesis of the differentiated cells after subcutaneous inoculation into nude mice. The HHS may be an excellent biomarker for the development of new targeted therapies for the treatment of cancer and other related diseases.[34]
Compared to the traditional cell culture systems, the microfluidic chip technology combines a continuous biological culture and dynamic biomolecule detection platform, which can achieve efficient, accurate, and reliable monitoring of multiple variables using a single chip. In addition, the microfluidic chip technology is highly convenient and cost-effective.[31,32] In this study, the microfluidic chip provided an ideal platform for rapidly screening different concentrations of CSE, and made it possible to monitor the malignant transformation of human bronchial epithelial cells. In summary, our results suggest that the HH signaling activity is increased during the CSE-induced malignant transformation of human bronchial epithelial cells. Cyclopamine can effectively depress the expression of the HHS proteins and inhibit tumor formation when the transformed cells are inoculated into nude mice.
Financial support and sponsorship
This study was supported by grants from the Natural Science Foundation of China (No. 91129733, No. 81071228, and No. 81330060) and the Special Fund for Health-Scientific Research in the Public Interest Program from National Health and Family Planning Commission (No. 201202011).
Conflicts of interest
There are no conflicts of interest.
Footnotes
Edited by: Qiang Shi
REFERENCES
- 1.Beckett P, Tata LJ, Hubbard RB. Risk factors and survival outcome for non-elective referral in non-small cell lung cancer patients – Analysis based on the National Lung Cancer Audit. Lung Cancer. 2014;83:396–400. doi: 10.1016/j.lungcan.2013.10.010. doi: 10.1016/j.lungcan.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 2.Tsugane S. Tobacco smoking and cancer risk: Epidemiological evidence. Nihon Rinsho. 2013;71:390–6. [PubMed] [Google Scholar]
- 3.Zhou J, Zhu G, Huang J, Li L, Du Y, Gao Y, et al. Non-canonical GLI1/2 activation by PI3K/AKT signaling in renal cell carcinoma: A novel potential therapeutic target. Cancer Lett. 2016;370:313–23. doi: 10.1016/j.canlet.2015.11.006. doi: 10.1016/j.canlet.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 4.Lee Y, Kim HY, Lee SH, Lim KY, Lee GK, Yun T, et al. Clinical significance of heterogeneity in response to retreatment with epidermal growth factor receptor tyrosine kinase inhibitors in patients with lung cancer acquiring secondary resistance to the drug. Clin Lung Cancer. 2014;15:145–51. doi: 10.1016/j.cllc.2013.11.008. doi: 10.1016/j.cllc.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 5.Song Z, Zhang Y. Gefitinib and erlotinib for non-small cell lung cancer patients who fail to respond to radiotherapy for brain metastases. J Clin Neurosci. 2014;21:591–5. doi: 10.1016/j.jocn.2013.05.022. doi: 10.1016/j.cllc.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 6.Nüsslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature. 1980;287:795–801. doi: 10.1038/287795a0. [DOI] [PubMed] [Google Scholar]
- 7.Bai XY, Zhang XC, Yang SQ, An SJ, Chen ZH, Su J, et al. Blockade of hedgehog signaling synergistically increases sensitivity to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer cell lines. PLoS One. 2016;11:e0149370. doi: 10.1371/journal.pone.0149370. doi: 10.1371/journal.pone.0149370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bora-Singhal N, Perumal D, Nguyen J, Chellappan S. Gli1-mediated regulation of sox 2 facilitates self-renewal of stem-like cells and confers resistance to EGFR inhibitors in non-small cell lung cancer. Neoplasia. 2015;17:538–51. doi: 10.1016/j.neo.2015.07.001. doi: 10.1016/j.neo.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Szczepny A, Rogers S, Jayasekara WSN, Park K, McCloy RA, Cochrane CR, et al. The role of canonical and non-canonical hedgehog signaling in tumor progression in a mouse model of small cell lung cancer. Oncogene. 2017;36:5544–50. doi: 10.1038/onc.2017.173. doi: 10.1038/onc.2017.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanna A, Shevde LA. Hedgehog signaling: Modulation of cancer properies and tumor mircroenvironment. Mol Cancer. 2016;15:24. doi: 10.1186/s12943-016-0509-3. doi: 10.1186/s12943-016-0509-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Della Corte CM, Bellevicine C, Vicidomini G, Vitagliano D, Malapelle U, Accardo M, et al. SMO gene amplification and activation of the hedgehog pathway as novel mechanisms of resistance to anti-epidermal growth factor receptor drugs in human lung cancer. Clin Cancer Res. 2015;21:4686–97. doi: 10.1158/1078-0432.CCR-14-3319. doi: 10.1158/1078-0432.CCR-14-3319. [DOI] [PubMed] [Google Scholar]
- 12.Kim C, Chapman RS, Hu W, He X, Hosgood HD, Liu LZ, et al. Smoky coal, tobacco smoking, and lung cancer risk in Xuanwei, China. Lung Cancer. 2014;84:31–5. doi: 10.1016/j.lungcan.2014.01.004. doi: 10.1016/j.lungcan.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lam HC, Choi AM, Ryter SW. Isolation of mouse respiratory epithelial cells and exposure to experimental cigarette smoke at air liquid interface. J Vis Exp. 2011 doi: 10.3791/2513. pii: 2513 doi: 103791/2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li E, Xu Z, Liu F, Wang H, Wen J, Shao S, et al. Continual exposure to cigarette smoke extracts induces tumor-like transformation of human nontumor bronchial epithelial cells in a microfluidic chip. J Thorac Oncol. 2014;9:1091–100. doi: 10.1097/JTO.0000000000000219. doi: 10.1097/JTO.0000000000000219. [DOI] [PubMed] [Google Scholar]
- 15.Kim SM, Lee SH, Suh KY. Cell research with physically modified microfluidic channels: A review. Lab Chip. 2008;8:1015–23. doi: 10.1039/b800835c. doi: 10.1039/b800835c. [DOI] [PubMed] [Google Scholar]
- 16.Tanaka Y, Sato K, Shimizu T, Yamato M, Okano T, Kitamori T, et al. Biological cells on microchips: New technologies and applications. Biosens Bioelectron. 2007;23:449–58. doi: 10.1016/j.bios.2007.08.006. doi: 10.1016/j.bios.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 17.Wang S, Li E, Gao Y, Wang Y, Guo Z, He J, et al. Study on invadopodia formation for lung carcinoma invasion with a microfluidic 3D culture device. PLoS One. 2013;8:e56448. doi: 10.1371/journal.pone.0056448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu Z, Gao Y, Hao Y, Li E, Wang Y, Zhang J, et al. Application of a microfluidic chip-based 3D co-culture to test drug sensitivity for individualized treatment of lung cancer. Biomaterials. 2013;34:4109–17. doi: 10.1016/j.biomaterials.2013.02.045. doi: 10.1016/j.biomaterials.2013.02.045. [DOI] [PubMed] [Google Scholar]
- 19.Gruber W, Hutzinger M, Elmer DP, Parigger T, Sternberg C, Cegielkowski L, et al. DYRK1B as therapeutic target in hedgehog/GLI-dependent cancer cells with smoothened inhibitor resistance. Oncotarget. 2016;7:7134–48. doi: 10.18632/oncotarget.6910. doi: 10.18632/oncotarget.6910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gorojankina T. Hedgehog signaling pathway: A novel model and molecular mechanisms of signal transduction. Cell Mol Life Sci. 2016;73:1317–32. doi: 10.1007/s00018-015-2127-4. doi: 10.1007/s00018-015-2127-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manetti F, Petricci E. Evaluation of WO2014207069 A1: Multitarget hedgehog pathway inhibitors and uses thereof. Expert Opin Ther Pat. 2016;26:529–35. doi: 10.1517/13543776.2016.1132309. doi: 10.1517/13543776.2016.1132309. [DOI] [PubMed] [Google Scholar]
- 22.Schamberger AC, Staab-Weijnitz CA, Mise-Racek N, Eickelberg O. Cigarette smoke alters primary human bronchial epithelial cell differentiation at the air-liquid interface. Sci Rep. 2015;5:8163. doi: 10.1038/srep08163. doi: 10.1038/srep08163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stinn W, Berges A, Meurrens K, Buettner A, Gebel S, Lichtner RB, et al. Towards the validation of a lung tumorigenesis model with mainstream cigarette smoke inhalation using the A/J mouse. Toxicology. 2013;305:49–64. doi: 10.1016/j.tox.2013.01.005. doi: 10.1016/j.tox.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 24.Balkwill F, Mantovani A. Inflammation and cancer: Back to Virchow? Lancet. 2001;357:539–45. doi: 10.1016/S0140-6736(00)04046-0. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 25.Deng B, Li Y, Zhang Y, Bai L, Yang P. Helicobacter pylori infection and lung cancer: A review of an emerging hypothesis. Carcinogenesis. 2013;34:1189–95. doi: 10.1093/carcin/bgt114. doi: 10.1093/carcin/bgt114. [DOI] [PubMed] [Google Scholar]
- 26.Takahashi H, Ogata H, Nishigaki R, Broide DH, Karin M. Tobacco smoke promotes lung tumorigenesis by triggering IKKbeta- and JNK1-dependent inflammation. Cancer Cell. 2010;17:89–97. doi: 10.1016/j.ccr.2009.12.008. doi: 10.1016/j.ccr.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cho WC, Kwan CK, Yau S, So PP, Poon PC, Au JS, et al. The role of inflammation in the pathogenesis of lung cancer. Expert Opin Ther Targets. 2011;15:1127–37. doi: 10.1517/14728222.2011.599801. doi: 10.1517/14728222.2011.599801. [DOI] [PubMed] [Google Scholar]
- 28.Young RP, Hopkins RJ, Christmas T, Black PN, Metcalf P, Gamble GD, et al. COPD prevalence is increased in lung cancer, independent of age, sex and smoking history. Eur Respir J. 2009;34:380–6. doi: 10.1183/09031936.00144208. doi: 10.1183/09031936.00144208. [DOI] [PubMed] [Google Scholar]
- 29.Christopoulos A, Saif MW, Sarris EG, Syrigos KN. Epidemiology of active tuberculosis in lung cancer patients: A systematic review. Clin Respir J. 2014;8:375–81. doi: 10.1111/crj.12094. doi: 10.1111/crj.12094. [DOI] [PubMed] [Google Scholar]
- 30.Armas-López L, Zúñiga J, Arrieta O, Ávila-Moreno F. The hedgehog-GLI pathway in embryonic development and cancer: Implications for pulmonary oncology therapy. Oncotarget. 2017;8:60684–703. doi: 10.18632/oncotarget.19527. doi: 10.18632/oncotarget.19527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park JY, Kim SK, Woo DH, Lee EJ, Kim JH, Lee SH, et al. Differentiation of neural progenitor cells in a microfluidic chip-generated cytokine gradient. Stem Cells. 2009;27:2646–54. doi: 10.1002/stem.202. doi: 10.1002/stem.202. [DOI] [PubMed] [Google Scholar]
- 32.Kim C, Lee KS, Bang JH, Kim YE, Kim MC, Oh KW, et al. 3-dimensional cell culture for on-chip differentiation of stem cells in embryoid body. Lab Chip. 2011;11:874–82. doi: 10.1039/c0lc00516a. doi: 10.1039/c0lc00516a. [DOI] [PubMed] [Google Scholar]
- 33.BéruBé K, Aufderheide M, Breheny D, Clothier R, Combes R, Duffin R, et al. In vitro models of inhalation toxicity and disease. The report of a FRAME workshop. Altern Lab Anim. 2009;37:89–141. [PubMed] [Google Scholar]
- 34.Benvenuto M, Masuelli L, De Smaele E, Fantini M, Mattera R, Cucchi D, et al. In vitro and in vivo inhibition of breast cancer cell growth by targeting the hedgehog/GLI pathway with SMO (GDC-0449) or GLI (GANT-61) inhibitors. Oncotarget. 2016;7:9250–70. doi: 10.18632/oncotarget.7062. doi: 10.18632/oncotarget.7062. [DOI] [PMC free article] [PubMed] [Google Scholar]