

Abstract
Objective:
Exposure to halogens, such as chlorine or bromine, results in environmental and occupational hazard to the lung and other organs. Chlorine is highly toxic by inhalation, leading to dyspnea, hypoxemia, airway obstruction, pneumonitis, pulmonary edema, and acute respiratory distress syndrome (ARDS). Although bromine is less reactive and oxidative than chlorine, inhalation also results in bronchospasm, airway hyperresponsiveness, ARDS, and even death. Both halogens have been shown to damage the systemic circulation and result in cardiac injury as well. There is no specific antidote for these injuries since the mechanisms are largely unknown.
Data Sources:
This review was based on articles published in PubMed databases up to January, 2018, with the following keywords: “chlorine,” “bromine,” “lung injury,” and “ARDS.”
Study Selection:
The original articles and reviews including the topics were the primary references.
Results:
Based on animal studies, it is found that inhaled chlorine will form chlorine-derived oxidative products that mediate postexposure toxicity; thus, potential treatments will target the oxidative stress and inflammation induced by chlorine. Antioxidants, cAMP-elevating agents, anti-inflammatory agents, nitric oxide-modulating agents, and high-molecular-weight hyaluronan have shown promising effects in treating acute chlorine injury. Elevated free heme level is involved in acute lung injury caused by bromine inhalation. Hemopexin, a heme-scavenging protein, when administered postexposure, decreases lung injury and improves survival.
Conclusions:
At present, there is an urgent need for additional research to develop specific therapies that target the basic mechanisms by which halogens damage the lungs and systemic organs.
Keywords: Acute Lung Injury, Acute Respiratory Distress Syndrome, Bromine, Chlorine
摘要
目的:
卤素气体(如氯或溴),可导致环境污染和职业伤害(肺和其他器官损伤)。吸入氯气具有强烈毒性,可致呼吸困难、低氧血症、气道梗阻、肺部炎症、肺水肿和急性呼吸窘迫综合征(ARDS)。尽管溴气的反应性和氧化性弱于氯气,但吸入溴 气同样导致支气管痉挛、气道高反应性、ARDS甚至死亡。卤素气体还可损伤全身循环系统并造成心脏损伤。目前其诱发损伤 的机制尚不明了,临床上尚未有特异性解毒剂。
数据来源:
本文综述2018年1月前收录于Pubmed数据库的相关文章,关键词如下:氯气;溴气;肺损伤;急性呼吸窘迫综合征。
研究选择:
主要参考文献为相关的论著和综述。
结果:
动物实验表明:氯气吸入后形成氯衍生氧化产物,导致毒性反应。因此,潜在的治疗靶点为氯气引起氧化应激和炎症 反应。抗氧化剂、c-AMP调节剂、抗炎药物、一氧化氮调节剂和高分子透明质酸对急性氯损伤有治疗作用。而体内游离血红 素水平升高则和吸入溴气诱发的肺损伤相关。吸入溴气后给予可清除体内血红素的血红素结合蛋白,可明显降低肺损伤和提 高生存率。
结论:
目前尚需要大量基础和临床研究寻找可用于治疗吸入卤素气体后诱发的肺和气体脏器损伤的特异性药物。
INTRODUCTION
Acute respiratory distress syndrome (ARDS) was first described by Ashbaugh et al. in 1967.[1] In 2012, the Berlin definition was developed to diagnose patients with ARDS and classify them into three independent categories, which is essential for research and clinical practice.[2] Despite almost 50 years of basic and clinical research, the fundamental mechanisms that initiate and propagate this lung injury have not been defined completely. Although many hypotheses had been developed, they are often difficult to be tested in humans due to too many clinical variables. Thus, halogen inhalation-induced lung injury animal model is used to mimic acute lung injury and ARDS in human posthalogens exposure.
Halogens are widely used in commercial applications around the world; however, they are extremely toxic and present a significant threat to human health when released accidently or intentionally as chemical weapons during acts of terrorism. In 2005, approximately 54,900 kg of chlorine (Cl2) was accidentally released in a local mill in Graniteville, South Carolina, USA. Eight victims died of asphyxiation or acute respiratory failure on site immediately and 71 people were severely injured and presented with pulmonary complaints (predominant restrictive lung function pattern);[3] some of them also presented with dermal, ocular, otorhinolaryngeal, cardiac, or gastrointestinal injuries. Altogether 529 people were treated and released from emergency departments.[4] Eight to ten months after the event, cough and shortness of breath were still persistent in those patients.[5] Preliminary investigation demonstrates that Cl2-exposed mill workers experienced accelerated FEV1 decline in the 18 months after the incident.[6] Bromine (Br2), like Cl2, may also be accidentally released during transportation or industrial accidents. For instance, in 2011, a major accident occurred during Br2 transportation by train, and the accidentally released Br2 resulted in 42 people being hospitalized and more than 200 individuals sought medical care in Chelyabinsk, Russia.
Currently, the systemic pathophysiological changes resulting from halogens exposure and the mechanisms by which they damage the lung and the other organs were not fully understood; furthermore, there is no specific antidote for exposed individuals. Treatments following halogen exposure are mainly supportive, focusing on symptom alleviating, such as bronchospasm, pulmonary edema, and airway obstruction, using humidified oxygen, β2 agonists, and antibiotics for potential infections.[7] In severe cases with ARDS, respiratory support with intubation and positive-pressure ventilation may be necessary.[8] Thus, animal model of halogen inhalation provides us an opportunity to elucidate its basic mechanisms and to test novel therapeutic agents to reduce morbidity and mortality.
Cl2 INHALATION-INDUCED LUNG INJURY
Physicochemical properties
Cl2 is a water-soluble, yellow-green gas. It is an extremely reactive element and a strong oxidizing agent. It is an essential chemical widely used in industrial facilities, such as plastic manufacturing, water treatment, waste sanitation, paper industry, and pharmaceutical development. Human exposure could occur through releasing from industrial accidents, spilling during transportation, encountering in swimming pools, or mixing of domestic cleaning products. Although most exposures are accidental, Cl2 has been used as a chemical warfare agent since World War I.
Characteristics of injury
Cl2 irritates eyes, skins, and respiratory systems, and is highly toxic by inhalation, resulting in pulmonary damage from the upper airways to the alveolar compartment, causing dyspnea, hypoxemia, airway obstruction, pneumonitis, pulmonary edema, and acute lung injury in human.[9] The severity of injury depends primarily on duration and dose of exposure.
Animal models in multiple species including mice, rats, rabbits, pigs, sheep, and dogs[10,11,12] have been developed to elucidate mechanisms of Cl2 toxicity and to test novel therapeutic agents. Acute effects of Cl2 inhalation documented in animals include dyspnea or altered breathing patterns,[13] hypoxemia,[14] inflammation and mitochondrial damage,[10,11,13,15] fluid leak and pulmonary edema,[14] pulmonary hypertension, airway hyperresponsiveness (AHR),[10,16,17] lipid peroxidation,[13,18] impaired surfactants function,[14,19] changes in gene expression associated with increased susceptibility to acute lung injury,[20] induction of unfolded protein response signaling,[21] and pulmonary cell apoptosis or necrosis.[10,11,14,22] Subacute effects of Cl2 inhalation include abnormal epithelial repair,[16] mucus overproduction,[16] airway fibrosis,[22,23,24] and decreased lung function, such as persistent AHR or airway obstruction.[16,23,25,26] Systemic injuries, such as systemic vasculature dysfunction[27] and extensive cardiac injury,[28,29] have also been reported.
Mechanisms of toxicity
The toxicity of Cl2 is mainly due to oxidative stress following exposure. When inhaled, Cl2 first reacts with antioxidants in lung epithelial lining fluid.[30] Soluble Cl2 reacts with water to generate hypochlorous acid and hydrochloric acid following depletion of antioxidants. These products are highly reactive and will oxidize plasmalogens, which are enriched in lung and surfactant, to form chlorinated lipids (Cl-lipids), 2-chloropalmitaldehyde and 2-chlorostearalddehyde. These 2-chloro-lipids are targets for neutrophilic attack,[31] or could be further oxidized to the corresponding 2-chloropalmitic acid and 2-chlorostraric acid or be reduced to the respective 2-chlorofatty alcohols, which are considered proinflammatory [Figure 1]. Furthermore, studies showed that Cl-lipids could initiate acute or long-lasting injury due to their reaction with protein side chains, DNA, and lipids of the cells that lining the airway epithelium.[30] For instance, chloramines, a by-product of Cl2, could activate inflammatory cascades through stimulation of mitogen-activated protein kinase[32] and activation of nuclear factor-kappaB via IκBα oxidation, causing inflammatory cells infiltration in alveolar space.[33] In addition, chloramines could inhibit Na+-dependent alveolar fluid clearance and result in pulmonary edema.[34]
Figure 1.
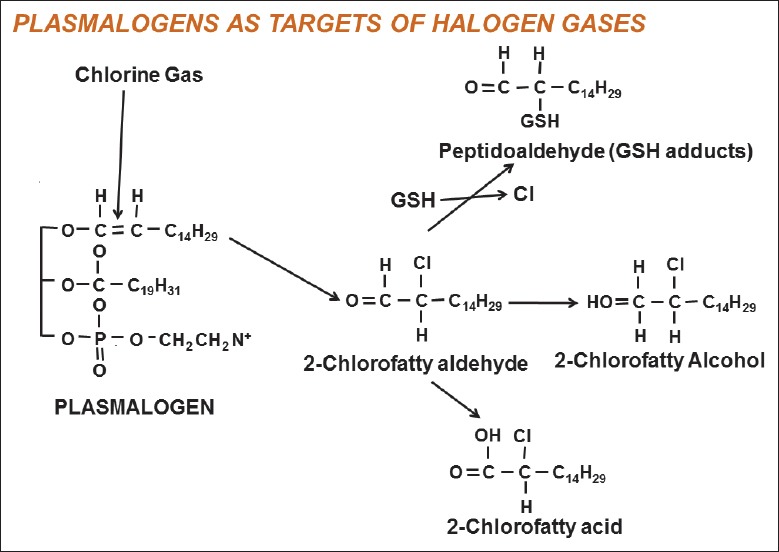
Plasmalogen-derived Cl2 oxidation products. The vinyl ether bond of plasmalogens is targeted by Cl2 resulting in 2-chlorofatty aldehyde production including 2-Cl-Pald and 2-Cl-Sald. The 2-chlorofatty aldehydes are either oxidized to the 2-chlorofatty acids, 2-Cl-PA and 2-Cl-SA, or reduced to the 2-chlorofatty alcohols, 2-chloropalmitoyl alcohol and 2-chlorostearoyl alcohol. Alternatively, nucleophilic attack of 2-chlorofatty aldehydes by GSH results in either palmitaldehyde or stearaldehyde GSH adduct formation. GSH: Glutathione. Source: Ford DA, Honavar J, Albert CJ, Duerr MA, Oh JY, Doran S, et al. Formation of chlorinated lipids post-chlorine gas exposure. J Lipid Res 2016;57:1529-40.
Possible therapeutic agents
Multiple therapeutic agents have shown promising effects in treating acute lung injury induced by Cl2 inhalation in animal models. As mentioned above, Cl2 toxicity is mainly caused by oxidative stress to tissues; thus, antioxidants including dimethylthiourea,[18] compound AEOL 10150 (a metalloporphyrin catalytic antioxidant),[35] N-acetyl cysteine,[36] aerosolized ascorbate, and deferoxamine[16] have been shown to alleviate Cl2-induced injury in animal models of different species.
Cyclic AMP-elevating agents have been shown to protect against Cl2-induced lung injury too. Arformoterol mitigates the Cl2 toxicity on airway reactivity and alveolar fluid clearance by increasing lung cyclic AMP level.[17] Rolipram inhibits degradation of the intracellular signaling molecule cyclic AMP, which alleviates pulmonary edema, inflammation, and AHR.[37]
Anti-inflammatory agents such as high-dose dexamethasone could reduce the acute inflammation and AHR;[26] mometasone and budesonide have a dose-dependent inhibition of neutrophil influx into lung tissues and of the number of neutrophils in lung lavage fluid;[38] formulations of rolipram, triptolide (a natural plant product with anti-inflammatory properties), and budesonide were developed for treatment of Cl2-induced acute lung injury by intramuscular injection;[39] aerosolized terbutaline and budesonide improved lung function post-Cl2 exposure in pigs;[40] low-molecular-weight hyaluronan (HA), formed by the degradation of high-molecular-weight HA (HMW-HA) by reactive intermediates, was detected in lung lavage fluid and in the peribronchial tissue of Cl2-exposed mice [Figure 2]; it activates RhoA and Ca2+ channels of airway smooth muscle cells, increasing their contractility and thus increases AHR. Intranasal administration of HMW-HA reversed the airway and distal lung injury in Cl2-exposed mice.[41] Neutrophil depletion abolished Cl2-induced AHR in large conducting airways and prevented increases in antioxidant gene expression and nuclear factor (erythroid-derived 2)-like 2 (NRF2) nuclear translocation;[42] heparin, which has both anti-inflammatory and anti-coagulant properties, reduced the disturbance in pulmonary coagulopathy and inflammation, thereby ameliorating acute lung injury post Cl2-exposure.[43] Recently, it is found that transient receptor potential (TRP) ion channels are also critical in the initiation and progression of acute lung injury due to Cl2 and other toxic inhalational agents. Postexposure treatment with inhibitors of TRP vanilloid, an ion channel expressed in pulmonary endothelial cells, had anti-inflammatory effects in Cl2-exposed mice and inhibited vascular leakage, AHR, and increase in elastance, therefore improving blood oxygen saturation.[44]
Figure 2.

Role of hyaluronan in Cl2-induced lung injury. (a) Detection of HA and IαI, a binding partner of HA, in the BALF of Cl2-exposed mice. Mice were exposed to Cl2 (400 ppm for 30 min) and returned to air. HA was measured in the BALF by ELISA at the indicated times. *,†P < 0.01 compared with air and the value to its left at the same time point, respectively. (b) Agar gel electrophoresis of HA. Lane 1, HA Mega-HA Ladder (Hyalose); lane 2, Select-HA Hi-Ladder; lane 3, HA; lane 4, HA exposed to Cl2 (400 ppm for 30 min) and stored at −4°C for 24 h; lane 5, sonicated HA; lane 6, Select-HA LoLadder, (c) agar gel electrophoresis of concentrated BALF from air and Cl2 exposed mice. Lane 1, Select-HA HiLadder; lane 2, Select-HA LoLadder; lane 3, 95% air-5% CO2 (Air); lanes 4 and 5, immediately post-Cl2; lane 6, 6 h post-Cl2, lane 7, 24 h post-Cl2; lane 8, as in lane 7 but the BALF was treated with hyaluronidase, which degrades HA. In all cases, proteins were visualized with Stains-All (Sigma). (d-f) representative image of mouse airways in naive state (d) or 6 h (e) and 24 h (f) after Cl2 exposure. Increased HA staining (green, arrows) at 24 h in the peribronchial area surrounding airway smooth muscle cells (×200). HA: Hyaluronan; IαI: Inter-α-trypsin-inhibitor; BALF: Bronchoalveolar lavage fluid. Source: Lazrak A, Creighton J, Yu Z, Komarova S, Doran SF, Aggarwal S, et al. Hyaluronan mediates airway hyperresponsiveness in oxidative lung injury. Am J Physiol Lung Cell Mol Physiol 2015;308:L891-903.
Nitric oxide-generating agents, such as sodium nitrite, resulted in lower protein levels in lung lavage fluid, significant reduction in the intensity of the apoptosis cells, restoration of normal lung wet-to-dry weight ratios, and improved postexposure survival;[13,45] 1400W, a potent, selective inducible nitric oxide synthase inhibitor abrogated the Cl2-induced AHR.[10]
Br2-INDUCED LUNG INJURY
Physicochemical properties
Br2, another toxic industrial chemical, is a brown liquid, which can readily evaporate to form a highly noxious gas that irritates the eyes, skin, respiratory system, as well as central nervous system.[46] Compared with Cl2, Br2 is a weaker oxidizing agent and less reactive.
Characteristics of injury
Br2 and hypobromous acid (HOBr), its hydrolysis product, are strong oxidants which will react with antioxidants present in the lung epithelial lining fluid initially after inhalation. If antioxidant stores are depleted, Br2 and HOBr will continue to react with plasma membranes of lung epithelial cells to form reactive intermediates, such as brominated lipids, which will cause injury to distal sites. Inflammatory response due to Br2 exposure worsens the initial pulmonary and systemic injury, which will in turn amplify lung damage due to the released inflammatory mediators.
Exposure to Br2 may lead to local and systemic damage. Contact with the skin results in lesions with brownish discoloration, tissue necrosis, and skin vesicles.[47] Depending on dose and duration, the inhalation of Br2 could lead to a variety of pulmonary symptoms, such as cough, dyspnea, hypoxia, or even death due to respiratory failure in adults.[46] Patients are at risk of developing reactive airway dysfunction and pulmonary fibrosis.[46,48] If exposed during pregnancy, risks of fetal growth restriction, fetal death, preterm delivery,[49] and cardiovascular birth defects are increased in mice model.[50] Br2-exposed neonatal mice also had decreased weight and impaired alveolar development.[51] Animal studies indicate that Br2 inhalation in early life, in pregnancy, or in adult period resulted in persistent inflammation and lung dysfunction.
Role of heme in Br2-induced lung injury
Recent animal study has shown that significant lung injury occurred within 24 h post-Br2 exposure (600 ppm for 30 min), which is characterized by increased protein exudates, inflammatory cell infiltration in alveolar space, and disruption of the airway parenchyma. Br2 gas inhalation also increases AHR following methacholine challenge and pulmonary edema. Moreover, 80% of the exposed C57BL/6 mice dead within 10 days postexposure.[52]
Heme oxygenase (HO)-1, the inducible isoform of HO, plays a vital role in defense against oxidant-induced lung injury during ARDS. HO-1 catalyzes the first and rate-limiting step in heme degradation into equimolar amounts of iron, carbon monoxide, and biliverdin. Heme is necessary for biologic processes and serves as a functional group in proteins. However, excessive heme catalyzes the formation of free radicals, resulting in oxidative stress and cellular injury. Significant increasing levels of free heme were detected in bronchoalveolar lavage fluid, lung tissues, and plasma of Br2-exposed mice, most likely originating from ruptured red blood cells. Postexposure administration of hemopexin, a heme-scavenging protein, decreased lung injury and improved survival.[52] Studies also demonstrated that humanized transgenic mice overexpressing HO-1 were significantly protected from exposure to Br2.[52] These are the first studies delineating the pathogenesis of Br2 toxicity in respiratory diseases and demonstrated the critical role of heme in ARDS caused by Br2 [Figure 3]. Thus, therapeutic approaches that reduce heme levels such as hemopexin or pharmacological induction of HO-1 may prove to be useful in treating ARDS secondary to Br2 inhalation.
Figure 3.
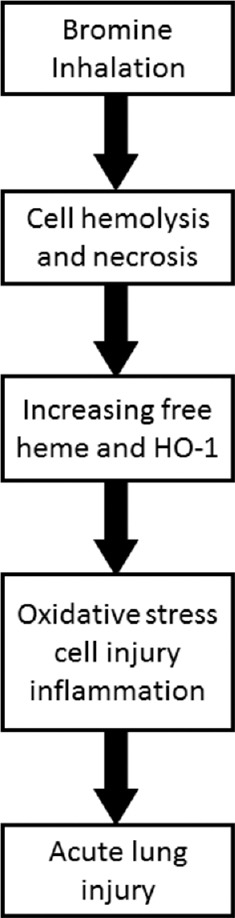
Mechanism of bromine (Br2) inhalation-induced lung injury. Br2 inhalation increases cell hemolysis and necrosis, resulting in elevated free heme in plasma, bronchoalveolar lavage fluid, lung tissue, and elevated HO-1 levels in lung tissue as well. Excessive heme catalyzes the formation of free radicals, resulting in oxidative stress and cellular injury as well as inflammation. This acute lung injury (ALI) increases lung permeability and impairs respiratory function. HO-1: Heme oxygenase-1; ALI: Acute lung injury.
CONCLUSIONS
With the worldwide usage of halogens, exposure becomes a significant health threat to public; however, there has been less effective treatment. Animal studies have demonstrated that oxidative stress as well as inflammation plays a critical role in halogen induced-lung injury and ARDS for which many countermeasures are developed to mitigate those injuries. The current challenges of developing effective therapies for halogen inhalation induced-lung injury include translating promising findings in preclinical models into treatment of clinical patients and developing interventions for chronic symptoms. However, these pharmacologic agents may need long time to be approved for clinical use. Thus, more small- and large-animal models mimicking human exposure are required to fully elucidate the complex pathology of halogen toxicity and to develop effective novel treatments which could be administrated feasibly after exposure.
Financial support and sponsorship
This work was supported by grants from the National Institutes of Environmental Health (NIEHS) 5U01ES02645802 and 1U01ES02769701 (S.M.), and Foundation for Anesthesia Education and Research (FAER, 2015) research fellowship (W.S.).
Conflicts of interest
There are no conflicts of interest.
Footnotes
Edited by: Li-Min Chen and Yuan-Yuan Ji
REFERENCES
- 1.Ashbaugh DG, Bigelow DB, Petty TL, Levine BE. Acute respiratory distress in adults. Lancet. 1967;2:319–23. doi: 10.1016/S0140-6736(67)90168-7. [Google Scholar]
- 2.ARDS Definition Task Force, Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, et al. Acute respiratory distress syndrome: The Berlin definition. JAMA. 2012;307:2526–33. doi: 10.1001/jama.2012.5669. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- 3.Balte PP, Clark KA, Mohr LC, Karmaus WJ, Van Sickle D, Svendsen ER, et al. The immediate pulmonary disease pattern following exposure to high concentrations of chlorine gas. Pulm Med. 2013;2013:325869. doi: 10.1155/2013/325869. doi: 10.1155/2013/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Sickle D, Wenck MA, Belflower A, Drociuk D, Ferdinands J, Holguin F, et al. Acute health effects after exposure to chlorine gas released after a train derailment. Am J Emerg Med. 2009;27:1–7. doi: 10.1016/j.ajem.2007.12.006. doi: 10.1016/j.ajem.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clark KA, Chanda D, Balte P, Karmaus WJ, Cai B, Vena J, et al. Respiratory symptoms and lung function 8-10 months after community exposure to chlorine gas: A public health intervention and cross-sectional analysis. BMC Public Health. 2013;13:945. doi: 10.1186/1471-2458-13-945. doi: 10.1186/1471-2458-13- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clark KA, Karmaus WJ, Mohr LC, Cai B, Balte P, Gibson JJ, et al. Lung function before and after a large chlorine gas release in graniteville, South Carolina. Ann Am Thorac Soc. 2016;13:356–63. doi: 10.1513/AnnalsATS.201508-525OC. doi: 10.1513/AnnalsATS.201508-525OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uemura K, Isono M, Kagohashi K, Hasegawa R, Satoh H. Bronchial damage and diffuse alveolar hemorrhage following chlorine gas inhalation: A case report. Exp Ther Med. 2017;14:5126–8. doi: 10.3892/etm.2017.5161. doi: 10.3892/etm.2017.5161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matos AM, Oliveira RR, Lippi MM, Takatani RR, Oliveira WF. Use of noninvasive ventilation in severe acute respiratory distress syndrome due to accidental chlorine inhalation: A case report. Rev Bras Ter Intensiva. 2017;29:105–10. doi: 10.5935/0103-507X.20170015. doi: 10.5935/0103-507X.20170015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malo JL, L'archevêque J, Castellanos L, Lavoie K, Ghezzo H, Maghni K, et al. Long-term outcomes of acute irritant-induced asthma. Am J Respir Crit Care Med. 2009;179:923–8. doi: 10.1164/rccm.200810-1550OC. doi: 10.1164/rccm.200810-1550OC. [DOI] [PubMed] [Google Scholar]
- 10.Martin JG, Campbell HR, Iijima H, Gautrin D, Malo JL, Eidelman DH, et al. Chlorine-induced injury to the airways in mice. Am J Respir Crit Care Med. 2003;168:568–74. doi: 10.1164/rccm.200201-021OC. doi: 10.1164/rccm.200201-021OC. [DOI] [PubMed] [Google Scholar]
- 11.Tian X, Tao H, Brisolara J, Chen J, Rando RJ, Hoyle GW, et al. Acute lung injury induced by chlorine inhalation in C57BL/6 and FVB/N mice. Inhal Toxicol. 2008;20:783–93. doi: 10.1080/08958370802007841. doi: 10.1080/08958370802007841. [DOI] [PubMed] [Google Scholar]
- 12.Musah S, Schlueter CF, Humphrey DM, Jr, Powell KS, Roberts AM, Hoyle GW. Acute lung injury and persistent small airway disease in a rabbit model of chlorine inhalation. Toxicol Appl Pharmacol. 2017;315:1–11. doi: 10.1016/j.taap.2016.11.017. doi: 10.1016/j.taap.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yadav AK, Doran SF, Samal AA, Sharma R, Vedagiri K, Postlethwait EM, et al. Mitigation of chlorine gas lung injury in rats by postexposure administration of sodium nitrite. Am J Physiol Lung Cell Mol Physiol. 2011;300:L362–9. doi: 10.1152/ajplung.00278.2010. doi: 10.1152/ajplung.00278.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leustik M, Doran S, Bracher A, Williams S, Squadrito GL, Schoeb TR, et al. Mitigation of chlorine-induced lung injury by low-molecular-weight antioxidants. Am J Physiol Lung Cell Mol Physiol. 2008;295:L733–43. doi: 10.1152/ajplung.90240.2008. doi: 10.1152/ajplung.90240.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jurkuvenaite A, Benavides GA, Komarova S, Doran SF, Johnson M, Aggarwal S, et al. Upregulation of autophagy decreases chlorine-induced mitochondrial injury and lung inflammation. Free Radic Biol Med. 2015;85:83–94. doi: 10.1016/j.freeradbiomed.2015.03.039. doi: 10.1016/j.freeradbiomed.2015.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fanucchi MV, Bracher A, Doran SF, Squadrito GL, Fernandez S, Postlethwait EM, et al. Post-exposure antioxidant treatment in rats decreases airway hyperplasia and hyperreactivity due to chlorine inhalation. Am J Respir Cell Mol Biol. 2012;46:599–606. doi: 10.1165/rcmb.2011-0196OC. doi: 10.1165/rcmb.2011-0196OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song W, Wei S, Liu G, Yu Z, Estell K, Yadav AK, et al. Postexposure administration of a {beta}2-agonist decreases chlorine-induced airway hyperreactivity in mice. Am J Respir Cell Mol Biol. 2011;45:88–94. doi: 10.1165/rcmb.2010-0226OC. doi: 10.1165/rcmb.2010-0226OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McGovern TK, Powell WS, Day BJ, White CW, Govindaraju K, Karmouty-Quintana H, et al. Dimethylthiourea protects against chlorine induced changes in airway function in a murine model of irritant induced asthma. Respir Res. 2010;11:138. doi: 10.1186/1465-9921-11-138. doi: 10.1186/1465-9921-11- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Massa CB, Scott P, Abramova E, Gardner C, Laskin DL, Gow AJ, et al. Acute chlorine gas exposure produces transient inflammation and a progressive alteration in surfactant composition with accompanying mechanical dysfunction. Toxicol Appl Pharmacol. 2014;278:53–64. doi: 10.1016/j.taap.2014.02.006. doi: 10.1016/j.taap.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leikauf GD, Pope-Varsalona H, Concel VJ, Liu P, Bein K, Berndt A, et al. Integrative assessment of chlorine-induced acute lung injury in mice. Am J Respir Cell Mol Biol. 2012;47:234–44. doi: 10.1165/rcmb.2012-0026OC. doi: 10.1165/rcmb.2012-0026OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li C, Weng Z, Doran SF, Srivastava RK, Afaq F, Matalon S, et al. Chlorine induces the unfolded protein response in murine lungs and skin. Am J Respir Cell Mol Biol. 2013;49:197–203. doi: 10.1165/rcmb.2012-0488RC. doi: 10.1165/rcmb.2012-0488RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O'Koren EG, Hogan BL, Gunn MD. Loss of basal cells precedes bronchiolitis obliterans-like pathological changes in a murine model of chlorine gas inhalation. Am J Respir Cell Mol Biol. 2013;49:788–97. doi: 10.1165/rcmb.2012-0369OC. doi: 10.1165/rcmb.2012-0369OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mo Y, Chen J, Schlueter CF, Hoyle GW. Differential susceptibility of inbred mouse strains to chlorine-induced airway fibrosis. Am J Physiol Lung Cell Mol Physiol. 2013;304:L92–102. doi: 10.1152/ajplung.00272.2012. doi: 10.1152/ajplung.00272.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Musah S, Chen J, Hoyle GW. Repair of tracheal epithelium by basal cells after chlorine-induced injury. Respir Res. 2012;13:107. doi: 10.1186/1465-9921-13-107. doi: 10.1186/1465-9921-13- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jonasson S, Koch B, Bucht A. Inhalation of chlorine causes long-standing lung inflammation and airway hyperresponsiveness in a murine model of chemical-induced lung injury. Toxicology. 2013;303:34–42. doi: 10.1016/j.tox.2012.10.022. doi: 10.1016/j.tox.2012.10.022. [DOI] [PubMed] [Google Scholar]
- 26.Jonasson S, Wigenstam E, Koch B, Bucht A. Early treatment of chlorine-induced airway hyperresponsiveness and inflammation with corticosteroids. Toxicol Appl Pharmacol. 2013;271:168–74. doi: 10.1016/j.taap.2013.04.037. doi: 10.1016/j.taap.2013.04.037. [DOI] [PubMed] [Google Scholar]
- 27.Honavar J, Samal AA, Bradley KM, Brandon A, Balanay J, Squadrito GL, et al. Chlorine gas exposure causes systemic endothelial dysfunction by inhibiting endothelial nitric oxide synthase-dependent signaling. Am J Respir Cell Mol Biol. 2011;45:419–25. doi: 10.1165/rcmb.2010-0151OC. doi: 10.1165/rcmb.2010-0151OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zaky A, Bradley WE, Lazrak A, Zafar I, Doran S, Ahmad A, et al. Chlorine inhalation-induced myocardial depression and failure. Physiol Rep. 2015;3 doi: 10.14814/phy2.12439. pii: e12439 doi: 1014814/phy212439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahmad S, Ahmad A, Hendry-Hofer TB, Loader JE, Claycomb WC, Mozziconacci O, et al. Sarcoendoplasmic reticulum Ca(2+) ATPase. A critical target in chlorine inhalation-induced cardiotoxicity. Am J Respir Cell Mol Biol. 2015;52:492–502. doi: 10.1165/rcmb.2014-0005OC. doi: 10.1165/rcmb.2014-0005OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Squadrito GL, Postlethwait EM, Matalon S. Elucidating mechanisms of chlorine toxicity: Reaction kinetics, thermodynamics, and physiological implications. Am J Physiol Lung Cell Mol Physiol. 2010;299:L289–300. doi: 10.1152/ajplung.00077.2010. doi: 10.1152/ajplung.00077.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duerr MA, Aurora R, Ford DA. Identification of glutathione adducts of α-chlorofatty aldehydes produced in activated neutrophils. J Lipid Res. 2015;56:1014–24. doi: 10.1194/jlr.M058636. doi: 10.1194/jlr.M058636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Midwinter RG, Vissers MC, Winterbourn CC. Hypochlorous acid stimulation of the mitogen-activated protein kinase pathway enhances cell survival. Arch Biochem Biophys. 2001;394:13–20. doi: 10.1006/abbi.2001.2530. doi: 10.1006/abbi.2001.2530. [DOI] [PubMed] [Google Scholar]
- 33.Midwinter RG, Cheah FC, Moskovitz J, Vissers MC, Winterbourn CC. IkappaB is a sensitive target for oxidation by cell-permeable chloramines: Inhibition of NF-kappaB activity by glycine chloramine through methionine oxidation. Biochem J. 2006;396:71–8. doi: 10.1042/BJ20052026. doi: 10.1042/BJ20052026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song W, Wei S, Zhou Y, Lazrak A, Liu G, Londino JD, et al. Inhibition of lung fluid clearance and epithelial Na+ channels by chlorine, hypochlorous acid, and chloramines. J Biol Chem. 2010;285:9716–28. doi: 10.1074/jbc.M109.073981. doi: 10.1074/jbc.M109.073981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McGovern T, Day BJ, White CW, Powell WS, Martin JG. AEOL10150: A novel therapeutic for rescue treatment after toxic gas lung injury. Free Radic Biol Med. 2011;50:602–8. doi: 10.1016/j.freeradbiomed.2010.12.001. doi: 10.1016/j.freeradbiomed.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wigenstam E, Koch B, Bucht A, Jonasson S. N-acetyl cysteine improves the effects of corticosteroids in a mouse model of chlorine-induced acute lung injury. Toxicology. 2015;328:40–7. doi: 10.1016/j.tox.2014.12.008. doi: 10.1016/j.tox.2014.12.008. [DOI] [PubMed] [Google Scholar]
- 37.Chang W, Chen J, Schlueter CF, Rando RJ, Pathak YV, Hoyle GW, et al. Inhibition of chlorine-induced lung injury by the type 4 phosphodiesterase inhibitor rolipram. Toxicol Appl Pharmacol. 2012;263:251–8. doi: 10.1016/j.taap.2012.06.017. doi: 10.1016/j.taap.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen J, Mo Y, Schlueter CF, Hoyle GW. Inhibition of chlorine-induced pulmonary inflammation and edema by mometasone and budesonide. Toxicol Appl Pharmacol. 2013;272:408–13. doi: 10.1016/j.taap.2013.06.009. doi: 10.1016/j.taap.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoyle GW, Chen J, Schlueter CF, Mo Y, Humphrey DM, Jr, Rawson G. Development and assessment of countermeasure formulations for treatment of lung injury induced by chlorine inhalation. Toxicol Appl Pharmacol. 2016;298:9–18. doi: 10.1016/j.taap.2016.03.001. doi: 10.1016/j.taap.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang J, Zhang L, Walther SM. Administration of aerosolized terbutaline and budesonide reduces chlorine gas-induced acute lung injury. J Trauma. 2004;56:850–62. doi: 10.1097/01.ta.0000078689.45384.8b. [DOI] [PubMed] [Google Scholar]
- 41.Lazrak A, Creighton J, Yu Z, Komarova S, Doran SF, Aggarwal S, et al. Hyaluronan mediates airway hyperresponsiveness in oxidative lung injury. Am J Physiol Lung Cell Mol Physiol. 2015;308:L891–903. doi: 10.1152/ajplung.00377.2014. doi: 10.1152/ajplung.00377.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McGovern TK, Goldberger M, Allard B, Farahnak S, Hamamoto Y, O'Sullivan M, et al. Neutrophils mediate airway hyperresponsiveness after chlorine-induced airway injury in the mouse. Am J Respir Cell Mol Biol. 2015;52:513–22. doi: 10.1165/rcmb.2013-0430OC. doi: 10.1165/rcmb.2013-0430OC. [DOI] [PubMed] [Google Scholar]
- 43.Zarogiannis SG, Wagener BM, Basappa S, Doran S, Rodriguez CA, Jurkuvenaite A, et al. Postexposure aerosolized heparin reduces lung injury in chlorine-exposed mice. Am J Physiol Lung Cell Mol Physiol. 2014;307:L347–54. doi: 10.1152/ajplung.00152.2014. doi: 10.1152/ajplung.00152.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Balakrishna S, Song W, Achanta S, Doran SF, Liu B, Kaelberer MM, et al. TRPV4 inhibition counteracts edema and inflammation and improves pulmonary function and oxygen saturation in chemically induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2014;307:L158–72. doi: 10.1152/ajplung.00065.2014. doi: 10.1152/ajplung.00065.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Honavar J, Doran S, Oh JY, Steele C, Matalon S, Patel RP, et al. Nitrite therapy improves survival postexposure to chlorine gas. Am J Physiol Lung Cell Mol Physiol. 2014;307:L888–94. doi: 10.1152/ajplung.00079.2014. doi: 10.1152/ajplung.00079.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Woolf A, Shannon M. Reactive airways dysfunction and systemic complaints after mass exposure to bromine. Environ Health Perspect. 1999;107:507–9. doi: 10.1289/ehp.99107507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sagi A, Baruchin AM, Ben-Yakar Y, Kon M, Eyal A, Mahler D, et al. Burns caused by bromine and some of its compounds. Burns Incl Therm Inj. 1985;11:343–50. doi: 10.1016/0305-4179(85)90097-x. doi: 10.1016/0305-4179(85)90097-X. [DOI] [PubMed] [Google Scholar]
- 48.Fredenburgh LE, Perrella MA, Mitsialis SA. The role of heme oxygenase-1 in pulmonary disease. Am J Respir Cell Mol Biol. 2007;36:158–65. doi: 10.1165/rcmb.2006-0331TR. doi: 10.1165/rcmb.2006-0331TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Horton BJ, Luben TJ, Herring AH, Savitz DA, Singer PC, Weinberg HS, et al. The effect of water disinfection by-products on pregnancy outcomes in two southeastern US communities. J Occup Environ Med. 2011;53:1172–8. doi: 10.1097/JOM.0b013e31822b8334. doi: 10.1097/JOM.0b013e31822b8334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lambert JA, Carlisle MA, Lam A, Aggarwal S, Doran S, Ren C, et al. Mechanisms and treatment of halogen inhalation-induced pulmonary and systemic injuries in pregnant mice. Hypertension. 2017;70:390–400. doi: 10.1161/HYPERTENSIONAHA.117.09466. doi: 10.1161/HYPERTENSIONAHA.117.09466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jilling T, Ren C, Yee A, Aggarwal S, Halloran B, Ambalavanan N, et al. Exposure of neonatal mice to bromine impairs their alveolar development and lung function. Am J Physiol Lung Cell Mol Physiol. 2018;314:L137–43. doi: 10.1152/ajplung.00315.2017. doi: 10.1152/ajplung.00315.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aggarwal S, Lam A, Bolisetty S, Carlisle MA, Traylor A, Agarwal A, et al. Heme attenuation ameliorates irritant gas inhalation-induced acute lung injury. Antioxid Redox Signal. 2016;24:99–112. doi: 10.1089/ars.2015.6347. doi: 10.1089/ars.2015.6347. [DOI] [PMC free article] [PubMed] [Google Scholar]