


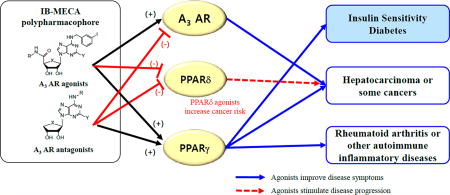
Abstract
A3 adenosine receptor (AR) ligands including A3 AR agonist, N6-(3-iodobenzyl)adenosine-5′-N-methyluronamide (1a, IB-MECA) were examined for adiponectin production in human bone marrow mesenchymal stem cells (hBM-MSCs). In this model, 1a significantly increased adiponectin production, which is associated with improved insulin sensitivity. However, A3 AR antagonists also promoted adiponectin production in hBM-MSCs, indicating that the A3 AR pathway may not be directly involved in the adiponectin promoting activity. In a target deconvolution study, their adiponectin-promoting activity was significantly correlated to their binding activity to both peroxisome proliferator activated receptor (PPAR) γ and PPARδ. They functioned as both PPARγ partial agonists and PPARδ antagonists. In the diabetic mouse model, 1a and its structural analogues A3 AR antagonists significantly decreased the serum levels of glucose and triglyceride, supporting their antidiabetic potential. These findings indicate that the polypharmacophore of these compounds may provide therapeutic insight into their multipotent efficacy against various human diseases.
Graphical abstract
INTRODUCTION
The concept of polypharmacology defines a single drug molecule that can simultaneously modulate multiple drug targets to treat complex diseases with polygenic etiology.1,2 Recently, a single drug molecule interacting with multiple kinases in a network of dysregulated cellular pathway in cancer cells has provided experimental evidence to demonstrate the effectiveness of the polypharmacology approach.3,4 However, most current polypharmacology cases in the drug discovery field have been limited to anticancer kinase inhibitors, affecting virtually the same protein families. The feasibility of applying polypharmacology clinically should be supported by a variety of single molecules simultaneously targeting multiple protein families with a direct causal relationship with a multietiological complex disease.
Phenotype-based approaches have been successfully proven as a viable alternative to a defined molecular target-based approach in drug discovery.4,5 For complex chronic diseases with multifactorial genetic and epigenetic etiologies, such as type II diabetes and obesity, a phenotype-based pharmacological assay has several advantages over target-based assays. To develop antidiabetic and antiobesity drugs, a phenotypic assay based on the adipogenesis model of human bone marrow mesenchymal stem cells (hBM-MSCs) has been studied by simultaneously measuring adiponectin production and lipid accumulation.6–9 Adiponectin, an adipocytokine mainly produced in the adipocytes, has been used as a diagnostic biomarker for metabolic diseases. For example, the ratio of serum adiponectin to leptin in patients with type II diabetes is lower than that in the healthy population.10,11 Notably, recombinant adiponectin showed therapeutic benefits in various animal models of human metabolic diseases.10–12 In fact, sulfonylurea-type antidiabetic drugs and peroxisome proliferator activated receptor (PPAR) γ agonists increase adiponectin biosynthesis and lipid accumulation in hBM-MSC-based phenotypic assay system.7,9,13,14 In addition, nonsteroidal anti-inflammatory drugs (NSAIDs) such as aspirin, ibuprofen, and indomethacin also increase adiponectin production and lipid droplet development during adipogenesis in hBM-MSCs.9,15 At higher concentrations, ibuprofen and indomethacin directly bind to PPARγ, which explains their pharmacological effect on adiponectin production during adipogenesis.16 In contrast, aspirin does not directly bind to PPARγ, and the molecular targets associated with the effect of aspirin on adipogenesis are not fully understood. Therefore, chemical compounds discovered from phenotypic assays require additional study to identify and validate their direct molecular targets; this process is defined as drug target deconvolution.3–5
Extracellular adenosine regulates various biological functions by acting on a G-protein-coupled receptor (GPCR) family of adenosine receptors (ARs), which consist of four subtypes: A1, A2A, A2B, and A3 ARs.17 The downstream effectors of ARs are coupled with the cyclic adenosine monophosphate (cAMP) mediated intracellular signaling pathway. A1 and A3 ARs inhibit intracellular cAMP signaling, whereas A2A and A2B receptors promote it. Adenosine plays a role in many physiological functions such as circulation, renal blood flow, cardiac rhythm, lipolysis, immune function, and angiogenesis. Importantly, the modulation of AR functions by their specific agonists or antagonists has pharmacological significance in inflammatory diseases, neurodegenerative diseases, metabolic diseases, and cancers. Adenosine and its chemical derivatives are also used clinically in the diagnosis of supraventricular tachycardia or administered as antiarrhythmic agents.18,19
Several reports indicate that adenosine plays a role in mammalian adipogenesis and osteogenesis. Caffeine, a major ingredient of coffee, tea, and many commercial soft drinks, is classified as an AR antagonist.20 Caffeine inhibits adipogenesis in adipose-tissue-derived MSCs,21 and adenosine affects the differentiation lineage commitment of hMSCs into adipocytes or osteoblasts.22,23 The results of studies in AR knockout mice indicate that AR modulators have therapeutic potential for diabetes and obesity.24 In a high-fat-diet mouse model, A2B AR upregulation was correlated with insulin receptor substrate 2 (IRS-2) expression, indicating that A2B AR is a potential drug target in human metabolic diseases.25 Currently, the role of ARs in adipogenesis in hMSCs is not fully understood.
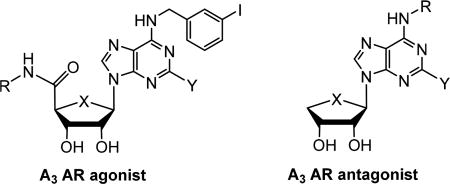
In the present study, we evaluated the effect of various AR agonists 1a–d and 2a–d and antagonists 3a–d on adipogenesis in hBM-MSCs to elucidate the subtype specific roles of ARs (Figure 1). From this study, we discovered that a specific A3 AR agonist, N6-(3-iodobenzyl)adenosine-5′-N-methyluronamide (1a, IB-MECA)17,26 and related A3 AR ligands promoted adiponectin production during adipogenesis in hBM-MSCs. In the structure–activity relationship (SAR), the adiponectin promoting activity of A3 AR ligands tested was not correlated to their A3 AR binding affinities. It was found that 1a and related A3 AR ligands have polypharmacological characteristics of an A3 AR modulator, a PPARγ partial agonist, and a PPARδ antagonist in the target deconvolution of their adiponectin promoting activity in hBM-MSCs. Herein, we report the polypharmacology of A3 AR ligands acting as PPARγ partial agonists and PPARδ antagonists.
Figure 1.

Structures of A3 AR ligands used in this study.
RESULTS AND DISCUSSION
Synthesis of A3 AR Agonists 2a–d and A3 AR Antagonists 3a–d
A3 AR agonists 2a–d were synthesized as shown in Scheme 1, according to our previously published procedure.27 2,3-Isoproplylidene-d-ribonolactone (4) was converted to 2,3-isoproplylidene-l-lyxonolactone (5) via the mesylation followed by intramolecular relactonization of the product of aqueous potassium hydroxide (KOH) cleavage of the d-ribonolactone ring.28 Benzoylation of 5 followed by reduction with sodium borohydride (NaBH4) afforded diol 6, which was converted to 4-thiosugar 7 by mesylation and cyclization of resulting dimesylate with sodium sulfide (Na2S).
Scheme 1.

Synthesis of N6-(3-Iodobenzyl)-4′-thioadenosine Derivatives 2a–d27,a
aReagents and conditions: (a) BzCl, pyridine, CH2Cl2, rt, 12 h, 92%; (b) NaBH4, MeOH, 0 °C to rt, 3 h, 90%; (c) MsCl, Et3N, DMAP, CH2Cl2, 0 °C, 30 min; (d) Na2S·9H2O, DMF, 100 °C, 15 h, 42% for 2 steps from 6; (e) mCPBA, CH2Cl2, −78 °C, 1 h, 95%; (f) 6-chloropurine, Et3N, TMSOTf, CH3CN, DCE, rt to 80 °C, 4 d, 53%; (g) R2NH2, Et3 N, EtOH, rt, 24 h; (h) 80% AcOH, 70 °C, 12 h; (i) TBSOTf, pyridine, 50 °C, 5 h; (j) NaOMe, MeOH, rt, 4 h, 82% for 3 steps from 8; (k) PDC, DMF, rt, 20 h; (l) RNH2, EDC, HOBt, DIPEA, CH2Cl2, rt, 15 h; (m) TBAF, THF, rt, 1 h.
Oxidation of 7 with meta-chloroperbenzoic acid (mCPBA) followed by Pummerer-type condensation of the resulting sulfoxide with 6-dichloropurine in the presence of trimethylsilyl trifluoromethanesulfonate (TMSOTf) afforded the N6-(3-iodobenzyl)amino derivative 8 after treating the condensed product with 3-iodobenzylamine. Hydrolysis of 2,3-acetonide of 8, protection of resulting diol with tert-butyldimethylsilyl (TBS) group, and removal of the benzoyl group yielded 9. Oxidation of 9 with pyridinium dichromate (PDC) in DMF yielded the acid, which was coupled with various amines such as ethylamine, cyclopropylamine, 1-cyclopropylmethamine, and cyclobutylamine in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and 1-hydroxybenzotriazole (HOBt) to afford various 5′-uronamides 2a–d27 after desilylation.
A3 AR antagonists 3a–d were synthesized from d-mannose according to our previously published procedure, as illustrated in Scheme 2.29 Treatment of d-mannose with 2,2-dimethoxypropane under acidic conditions gave the diacetonide, which was reduced with NaBH4 followed by mesylation of resulting diol that afforded the dimesylate 10. Cyclization of 10 with Na2S in DMF followed by selective hydrolysis of 5,6-acetonide yielded diol 11. Treatment of 11 with excess lead(IV) tetraacetate gave the glycosyl donor 12. Condensation of 12 with 6-chloropurine and 2,6-dichloropurine in the presence of TMSOTf as a Lewis acid afforded 6-chloropurine derivative 13a and 2,6-dichloropurine derivative 13b, respectively, after the removal of the isopropylidene group. Treatment of 13a with 3-iodo-, 3-chloro-, and 2-chlorobenzylamines yielded the final 3a–c, respectively, while 13b was converted to 3d by treating with 3-iodobenzylamine.
Scheme 2.

Synthesis of Truncated 4′-Thioadenosine Derivatives 3a–d29,a
aReagents and conditions: (a) 2,2-dimethoxypropane, camphosulfonic acid, acetone, rt, 15 h, 95%; (b) NaBH4, EtOH, rt, 2 h, 92%; (c) MsCl, Et3N, CH2Cl2, rt, 1 h, 94%; (d) Na2S, DMF, 80 °C, 15 h, 78%; (e) 60% AcOH, rt, 2 h, 81%; (f) Pb(OAc)4, EtOAc, rt, 15 h, 60%; (g) 6-chloropurine for 13a, 2,6-dichloropurine for 13b, ammonium sulfate, HMDS, 170 °C, 15 h, then TMSOTf, DCE, rt to 80 °C, 3 h; (h) 2 N HCl, THF, rt, 15 h; (i) RNH2, Et3 N, EtOH, rt, 1–3 d.
Effects of 1a on Adiponectin Production during Adipogenesis in hBM-MSCs
To determine whether AR signaling affects adipogenesis in hBM-MSCs, an endogenous ligand adenosine, A1 AR agonist, 2-chloro-N6-cyclopentyladenosine (14, CCPA),30 a nonspecific AR agonist, 5′-(N-ethylcarboxamido) adenosine (15, NECA),31 A2A AR agonist, 2-p-(2-carboxyethyl)phenethylamino-5′-N-ethylcarboxamidoadenosine hydrochloride hydrate (16, CGS21680),32 or A3 AR agonist 1a26 was added to the cells along with the insulin, dexamethasone and 3-isobutyl-1-methylxanthine (IDX) adipogenesis-inducing medium (Figure 2A). Caffeine, a nonspecific AR antagonist, was evaluated in parallel. A3 AR agonist 1a significantly promoted adiponectin production during adipogenesis in hBM-MSCs compared to that of the IDX control, whereas adenosine, 14, 15, or 16 had no significant effect. Consistent with a previous report in the literature,21 1 mM caffeine inhibited adipogenesis in hMSCs. Regarding lipid accumulation, 1a increased the number and size of lipid droplets in differentiated adipocytes compared with that in the IDX control, whereas caffeine decreased them (Figure 2B). Compound 1a upregulated adiponectin production during adipogenesis in hBM-MSCs in a concentration-dependent manner (Figure 2C).
Figure 2.

Effects of AR signaling modulators on adipogenesis in hBM-MSCs. (A) hBM-MSCs were grown under IDX conditions and/or co-treated with adenosine, caffeine, 14, 15, 16, or 1a. (B) ORO staining was performed to estimate lipid droplets on the 7th day in culture. (C) On the 7th day in culture, the supernatant was harvested and ELISA was performed to measure the levels of adiponectin accumulated in the supernatants over 48 h. Results are the mean ± SD of three measurements using hBM-MSCs from three independent donors (n = 3, three independent experiments): (*) p ≤ 0.05 and (**) p ≤ 0.01.
In preadipocyte studies in the human AR-transfected murine osteoblast precursor cell line 7F2, the AR agonists 14 and 15 increased adipocyte differentiation by 20–30%.22 However, in the adipogenesis model of hBM-MSCs, both 14 and 15 did not significantly promote adipogenesis in comparison with that in the control (Figure 1), suggesting that the AR signaling pathways differ between hMSCs and the murine 7F2 cell line. AR subtypes show transitional expression profile changes after the induction of adipocyte differentiation from preadipocytes.33 In the human AR-transfected murine 7F2 system, A1 AR overexpression promotes adipogenesis whereas A2 AR overexpression suppresses it.22 Mammalian adipogenesis involves the lineage commitment of MSCs to preadipocytes, establishment of the adipogenic lineage, and terminal differentiation into functional adipocytes.34 Each AR subtype may have different roles in adipogenesis regulation depending on the differentiated stage of MSCs. Therefore, the difference between the pharmacological effects of AR agonists on hBM-MSCs and those on the human AR-transfected murine osteoblast precursor cell line 7F2 may be partly explained by different lineage commitment stages for adipogenesis or osteogenesis.
Independency of A3 AR Signaling on 1a-Induced Upregulation of Adiponectin Production in hBM-MSCs
A3 AR agonists such as 1a and its 2-chloro derivative 1b35 have been studied as novel therapeutics to treat rheumatoid arthritis or myocardial ischemia–reperfusion injury.17,36 We have reported the results of structure–activity relationship on novel A3 AR agonists and antagonists, whose pharmacophore was structurally related to 1a (Table 1).27,29
Table 1.
Adiponectin-Secreting Activity of 1a and Related A3 AR Ligandsa
| ||||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| adiponectin (pg/mL)c | ||||||||
|
|
||||||||
| compd | X | Y | R |
Ki (hA1AR) (nM)b or % displacement at 1 µM |
Ki (hA2AAR) (nM)b or % displacement at 1 µM |
Ki (hA3AR) (nM)b or % displacement at 1 µM |
20 µM | 4 µM |
| A3 AR agonists | ||||||||
| 1a | O | H | CH3 | 51.2 ± 5.1 | 2910 ± 580 | 1.8 ± 0.7 | 216 ± 12** | 117 ± 20 |
| 1b | O | Cl | CH3 | 222 ± 22 | 5360 ± 2470 | 1.4 ± 0.3 | 603 ± 10** | 221 ± 10** |
| 1c | S | H | CH3 | 20.2 ± 2.9 | 475 ± 144 | 0.3 ± 0.1 | 343 ± 20** | 126 ± 14 |
| 1d | S | Cl | CH3 | 193 ± 46 | 223 ± 36 | 0.38 ± 0.07 | 738 ± 59** | 131 ± 10 |
| 2a | S | H | CH2CH3 | 5.4 ± 0.3 | 57.6 ± 6.9 | 0.42 ± 0.22 | 242 ± 4** | 118 ± 38 |
| 2b | S | H | cyclopropyl | 9.27 ± 0.83 | 15.2 ± 2.6 | 3.03 ± 0.23 | 249 ± 26** | 174 ± 5** |
| 2c | S | H | cyclopropyl-CH2 | 159 ± 40 | 1600 ± 80 | 2.16 ± 0.29 | 490 ± 16** | 242 ± 11** |
| 2d | S | H | cyclobutyl | 23.6 ± 4.2 | 122 ± 62 | 1.17 ± 0.16 | 633 ± 39** | 323 ± 30** |
| A3 AR antagonists | ||||||||
| 3a | S | Cl | 3-I-Bn | 2490 ± 940 | 341 ± 75 | 4.16 ± 0.5 | 857 ± 69** | 192 ± 19* |
| 3b | S | Cl | 3-Cl-Bn | 38% | 18% | 1.66 ± 0.9 | 442 ± 5** | 196 ± 1** |
| 3c | S | Cl | 2-Cl-Bn | 13% | 1600 ± 135 | 25.8 ± 6.3 | 268 ± 14** | 107 ± 6 |
| 3d | S | H | 3-Cl-Bn | 860 ± 210 | 440 ± 110 | 1.5 ± 0.4 | 247 ± 7** | 163 ± 12* |
| IDX control | 100 ± 6 | |||||||
| glibenclamide | 1000 ± 193** | 838 ± 67** | ||||||
Results are the mean ± SD of three measurements using hBM-MSCs from three independent donors (n = 3, three independent experiments):
p ≤ 0.05 and
p ≤ 0.01.
Binding affinities of 1a and related A3 AR ligands to human A1, A2A, and A3 AR were previously reported.27,29
In the IDX medium, 1a and related A3 AR ligands were included to induce adipogenesis in hBM-MSCs. On the 7th day in culture, cell culture supernatants were harvested and ELISA was performed to measure levels of adiponectin.
In order to confirm the specific association of the A3 AR signaling pathway with the regulation of adipogenesis in hBM-MSCs, we investigated the effects of both A3 AR agonists and antagonists on adiponectin production. A3 AR agonists 1a–c and 2a–d significantly promoted adiponectin production in hBM-MSCs (Table 1). Notably, A3 AR antagonists 3a–d also increased adiponectin production in the same phenotypic assay. At a concentration of 20 µM, compound 3a was the most potent promoter of adipogenesis among the tested A3 AR agonists and antagonists (Figure 3A and Table 1). These results suggest that A3 AR signaling is not associated with adiponectin-promoting activity during adipogenesis in hBM-MSCs. To address this question, we evaluated the effects of the A3 AR ligands, (1S,2R,3S,4R,5S)-4-[6-[[(3-chlorophenyl)methyl]-amino]-2-[2-(3,4-difluorophenyl)ethynyl]-9H-purin-9-yl]-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide (17, MRS5698)37 and N-[2-(2-furanyl)-8-propyl-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-yl]-N′-(4-methoxyphenyl)-urea (18, MRE3008F20),38 which are chemically different from 1a, on adipogenesis in hBM-MSCs (Figure 3B). Unlike 1a and related A3 AR ligands, the A3 AR agonist 17 and A3 AR antagonist 18 had no effect on adipogenesis in hBM-MSCs. Next, we determined whether a pharmacological correlation existed between the adiponectin-promoting activity and A3 AR binding affinity values of 1a and related A3 AR ligands. The correlation coefficient between the adiponectin-promoting activity at 20 µM and Ki values of 1a-related A3 AR ligands was 0.04 (p = 0.54), which indicates no statistically significant association between the two variables (Figure 3C). When the A1 or A2A AR binding affinity of these compounds were compared, no significant correlation was observed (Figure 3D and Figure 3E). In this regard, the lack of structure–activity relationship between AR binding affinity and the adiponectin-promoting activity of 1a and its related A3 AR ligands suggests that other molecular targets are associated with the effect of 1a on adipogenesis in hBM-MSCs.
Figure 3.

Evaluation of adiponectin-promoting activity of 1a and related A3 AR ligands. (A) 1a, 1b, 1c, or 3a was co-treated with the IDX medium in hBM-MSCs. On the 7th day in culture, cell culture supernatants were harvested and ELISA was performed to measure levels of adiponectin. (B) The effects of A3 AR agonists 1a, 1c, and 17 and A3 AR antagonist 18 on adiponectin production were evaluated. The pharmacological correlation between A1 AR (C), A2A AR (D), A3 AR (E) binding Ki value at 1 µM and adiponectin levels was analyzed. Values represent mean ± SD (n = 3, three independent experiments): (*) p ≤ 0.05 and (**) p ≤ 0.01.
Specific Binding of 1a and Related A3 AR Ligands to PPARγ and PPARδ
Nuclear receptors (NRs) like PPARα, PPARγ, PPARδ, liver X receptor (LXR)α, LXRβ, and glucocorticoid receptor (GR) play roles in mammalian adipogenesis.39 In order to identify molecular targets directly associated with the phenotypic activity of 1a and related A3 AR ligands in promoting adiponectin production during adipogenesis of hBM-MSCs, we investigated the effects on NR binding or coactivation (Figure 4A).
Figure 4.

TR-FRET NRs binding or coactivator assays with 1a and related A3 AR ligands. (A) TR-FRET competitive binding assays with 1a and 1b at 4 µM as ligands of PPARα, PPARγ, PPARδ, and GR were performed. In TR-FRET LXRα and LXRβ coactivator assays, 1a and 1b at 4 µM were evaluated to determine whether transactivation occurred. The positive controls were 19 for PPARα, 21 for PPARγ, 22 for PPARδ, dexamethasone for GR, and 20 for LXRα and LXRβ. DMSO in buffer was used as a blank control. (B) The kinase activity was evaluated by measuring the γ-32P-ATP incorporation to CDK complexes. The inhibitory effects of 1a and 1c on the phosphorylation of CDK5/p25 and CDK5/p35 were tested at each Km ATP concentration. DMSO was included in each negative control. Values were expressed in terms of percentage compared to each positive control. The TR-FRET-based competitive binding activities of troglitazone, 1a, 1b, 1c, and 3a to PPARα (C), PPARγ (D), and PPARδ (E) were evaluated. Pearson’s correlation coefficients (r2) between the binding affinities to PPARγ at 20 µM (F) or 4 µM (G) and relative adiponectin levels in the cell culture supernatants were calculated with RStudio software. Correlation coefficients between the binding affinities to PPARδ at 2 µM (H) or 0.4 µM (I) and relative adiponectin levels were calculated in the same way. Results are the mean ± SD of three independent experiments: (*) p ≤ 0.05 and (**) p ≤ 0.01.
Compounds 1a and 1b were evaluated in the preliminary target identification study because they have been clinically investigated for various diseases such as cancers and rheumatoid arthritis.17,36 In the time-resolved fluorescence resonance energy transfer (TR-FRET) based receptor-binding assay, 4 µM 1a and 1b competitively replaced the binding of the labeled PPARδ ligand by 26% and 75%, respectively (Figure 4A). Compound 1b also replaced the binding of the labeled PPARγ ligand by 59%. Both compounds had no significant effects on PPARα, LXRα/β, or GR, compared to their positive controls, 2-(4-(2-(1-cyclohexanebutyl)-3-cyclohexylureidoethyl)phenylthio)-2-methylpropionic acid (19, GW7647),40 N-(2,2,2-trifluoroethyl)-N-[4-[2,2,2-trifluoro-1-hydroxy-1-(trifluoromethyl)ethyl]phenyl]benzenesulfonamide (20, T0901317),41 and dexamethasone, respectively (Figure 4A). In addition, cyclin-dependent kinase 5 (CDK5) was recently reported to regulate adipogenesis by affecting PPARγ phosphorylation.42 We also evaluated the effect of 1a and 1b on CDK5 activity because purine nucleoside structure of these compounds may affect kinase activity. Neither 1a nor 1b affected CDK activity at concentrations up to 20 µM (Figure 4B). Therefore, both PPARγ and PPARδ may contribute to the ability of 1a and related A3 AR ligands to promote adiponectin production, which can be used as the measure of insulin sensitivity.12 Compound 1a and related A3 AR ligands can bind to PPARs at higher concentrations. Next, we analyzed the concentration–response relationship of 1a and related A3 AR ligands in terms of their binding activity to PPARα, PPARγ, and PPARδ (Figure 4C–E, Table 2). In the PPARα binding assay, 1a, 1b, 1c, and 3a did not replace over 50% of the binding activity of the labeled PPARα ligand up to 20 µM, which was much less potent than 19 (Figure 4C, Table 2).
Table 2.
TR-FRET PPAR Binding Activity of 1a and Related A3 ARa
| compd | PPARαb% displacement at 20 µM | Ki (PPARγc) or % displacement at 20 µM | Ki (PPARδd) or % displacement at 20 µM |
|---|---|---|---|
| A3 AR agonists | |||
| 1a | 16.0% | 22.2% | 48.3% |
| 1b | 46.5% | 2.18 ± 0.32 nM | 0.43 ± 0.03 nM |
| 1c | 13.5% | 46.9% | 2.54 ± 0.15 nM |
| 1d | 37.8% | 4.60 ± 0.26 nM | 0.16 ± 0.09 nM |
| 2a | 29.3% | 37.1% | 3.15 ± 0.47 nM |
| 2b | 37.6% | 49.1% | 2.98 ± 0.39 nM |
| 2c | 16.4% | 1.83 ± 0.25 nM | 2.37 ± 0.66 nM |
| 2d | 11.7% | 0.17 ± 0.06 nM | 2.61 ± 0.72 nM |
| A3 AR antagonists | |||
| 3a | 35.6% | 3.42 ± 0.47 nM | 0.00483 ± 0.00023 nM |
| 3b | 4.50% | 40.8% | 0.0102 ± 0.0091 nM |
| 3c | 19.2% | 19.2% | 0.62 ± 0.14 nM |
| 3d | 18.5% | 18.5% | 49.1% |
Values represent the mean expression ± SD (three independent experiments):
p ≤ 0.05 and
p ≤ 0.01.
The Ki of the positive control 19 was 0.0541 ± 0.0089 in the parallel study.
The Ki of the positive control 21 in the parallel experiment was 0.0497 ± 0.0115. Troglitazone and glibenclamide showed 96.2% and 77.5% binding to PPARγ at 10 µM, respectively.
The Ki of the positive control 22 was 0.0480 ± 0.0014 in the parallel experiment. Troglitazone and glibenclamide bound to PPARδ 23.4% and 21.5% at 10 µM, respectively, compared to 22.
In the PPARγ analysis, 1b and 3a displayed significant competitive binding activity in a concentration-dependent manner (Figure 4D, Table 2), while 1c showed very weak binding activity. The Ki values of 1b and 3a were 2.18 and 3.42, respectively, but not as potent as the PPARγ agonist, N-(2-benzoylphenyl)-O-[2-(methyl-2-pyridinylamino)ethyl]-l-tyrosine hydrochloride (21, GW1929)43 (Figure 4D). Consistent with the literature,13 glibenclamide, a sulfonylurea antidiabetic drug, showed PPARγ binding activity in the TR-FRET-based assay (Figure 4D). Compared to PPARα and PPARγ binding activities, 1a and most related A3 AR ligands competitively displaced the labeled PPARδ ligand in a concentration-dependent manner (Figure 4E, Table 2). Importantly, compounds 3a and 3b exhibited maximal PPARδ binding activity, which was compared to that of a PPARδ agonist, 2-[2-methyl-4-[[[4-methyl-2-[4-(trifluoromethyl)phenyl]-5-thiazolyl]methyl]thio]phenoxy]acetic acid (22, GW501516)44 (Figure 4E). The Ki values for PPARδ binding of compounds 3a and 3b, which were identified as A3 AR antagonists, were 4.83 and 10.2 nM, respectively (Figure 4E, Table 2). Notably, a significant correlation was observed between the level of PPARγ ligand replacement at 20 and 4 µM and the adiponectin-promoting activity in hBM-MSCs (Figure 4F and Figure 4G). Regarding the correlation coefficients between PPARδ binding affinity and adiponectin-promoting activity, significant associations were observed with 1a and related A3 AR ligands at both 2 and 0.4 µM concentrations (Figure 4H and Figure 4I). Therefore, in affecting adipogenesis of hBM-MSCs, the effects of 1a and related A3 AR ligands on PPARδ occurred at lower concentrations than those for PPARγ. These results suggest that 1a and related A3 AR ligands promote adiponectin production in hBM-MSCs by modulating the activity of PPARγ and PPARδ, not A3 AR. Structure–activity relationships for PPAR activity were also analyzed. Among A3 AR agonists tested, 2-Cl substitution (1b and 1d) showed better binding activities at PPARγ and PPARδ than the 2-H substitution (1a and 1c), while more bulky alkyl or cycloalkyl substituent at the 5′-uronamides increased the binding activity at PPARγ. For PPARδ binding activity, compound 1d was the most potent inhibitor among A3 AR agonists. Extension of the 5′-uronamide group with alkyl or cycloalkyl substituents did not reduce the inhibitor potency at PPARδ compared to 1c. For A3 AR antagonists 3a–d, they generally displayed poor binding activity at PPARα and PPARγ but showed very strong binding activity at the PPARδ, among which N6-3-iodobenzyl derivative 3a was the most potent. All tested A3 AR agonists and antagonists were devoid of binding activity at PPARα. Structurally, this polypharmacophore encompasses adenosine-5′-uronamides and its 4′-truncated derivatives that also have bulky hydrophobic substitutions at the N6 position, such as halobenzyl.
Polypharmacophore of 1a To Bind A3 AR, PPARγ, and PPARδ
Next, we investigated whether 1a and related A3 AR ligands were PPAR agonists or antagonists. To determine a functional outcome for the effect of 1a, 1c, and 3a on PPARγ, we performed a luciferase-reporter PPARγ transactivation assay.45 We observed that 10 µM 1a, 1c, and 3a increased PPARγ transactivation by 29.9%, 46.9%, and 54.3%, respectively (Figure 5A).
Figure 5.

PPARγ and PPARδ transactivation activity of 1a, 1c, and 3a. (A) For PPARγ transactivation assay, CV-1 cells were transiently co-transfected with the PPARγ expression vector and the PPARγ responsive elements (PPRE) luciferase reporter and then treated with troglitazone, 1a, 1c, or 3a. (B) TR-FRET PPARδ coactivator assay was performed using fluorescein-C33 coactivator peptide. 22 and 23 were used as PPARδ antagonist and PPARδ agonist, respectively. Results are the mean ± SD of three measurements (n = 3, three independent experiments): (*) p ≤ 0.05 and (**) p ≤ 0.01.
Compound 3a (30 µM) did not achieve maximal PPARγ transactivation activity compared to troglitazone, a PPARγ full agonist; this result was similar to that of a PPARγ binding assay (Figure 4D). PPARγ partial agonists can upregulate adiponectin production and also improve glucose homeostasis.46 The adiponectin-promoting activity of 1a and related A3 AR ligands was partially contributed by PPARγ partial agonism. To elucidate a functional consequence for PPARδ, we performed a TR-FRET PPARδ coactivator assay, which measures the level of interaction between the PPARδ ligand-binding domain and a fluorescein-labeled coactivator peptide. A selective PPARδ agonist 22 increased the signal associated with the activated PPARδ in a concentration-dependent manner (Figure 5B). Like a PPARδ antagonist 3-[[[2-methoxy-4-(phenylamino)phenyl]-amino]sulfonyl]-2-thiophenecarboxylic acid methyl ester (23, GSK0660),47 1a, 1c, and 3a had no effect on the recruitment of the labeled coactivator peptide, suggesting that 1a and related A3 AR ligands are PPARδ antagonists (Figure 5B).
To confirm the PPARδ antagonism, we evaluated whether 1a, 1c, and 3a competitively inhibited the 22-induced coactivator recruitment to PPARδ (Figure 6A). Compound 3a (1 µM) significantly decreased the effect of 22 on PPARδ coactivator recruitment by 57%. At 1 µM concentration, both 1a and 1c tended to inhibit the effect of 22 by 14% and 22%, respectively, although the inhibition was not statistically significant. PPARδ agonists interfere with the interaction between PPARδ and its corepressors such as silencing mediator of retinoid and thyroid hormone receptors (SMRT).48 Therefore, we analyzed whether compound 3a affected the interaction between PPARδ and the labeled corepressor peptide, which was derived from the interaction domain 2 (ID2) of SMRT (Figure 6B). As expected, 22 decreased the interaction between PPARδ and the corepressor peptide (Figure 6B). The PPARδ antagonists, 23 and 4-chloro-N-[2-[[5-(trifluoromethyl)-2-pyridinyl]sulfonyl]ethyl]benzamide (24, GSK3787)49 significantly promoted the recruitment of the corepressor peptide by PPARδ, an effect that was antagonized by 22. Importantly, similar to the effects of PPARδ antagonists 23 and 24, the promotion of the interaction between PPARδ and corepressor peptide by 3a, an A3 AR antagonist, was concentration-dependent (Figure 6C). The A3 AR agonist 1c also enhanced the interaction of PPARδ with the corepressor peptide (Figure 6C). In mammalian cells, PPARδ agonists increase the gene transcription of angiopoietin-like 4 (ANGPLT4) and pyruvate dehydrogenase kinase 4 (PDK4).48,50 To confirm that compound 3a is a PPARδ antagonist, we measured ANGPLT4 and PDK4 mRNA levels during adipogenesis in hBM-MSCs (Figure 6D and Figure 6E). Consistent with previous reports, the PPARδ agonist 22 significantly increased ANGPTL4 and PDK4 gene transcription, as measured on the 3rd day after the induction of adipogenesis in hBM-MSCs (Figure 6D and Figure 6E). Similar to the other PPARδ antagonists 23 and 24, compound 3a did not affect ANGPTL4 and PDK4 mRNA levels in hBM-MSCs. Recently, it has been reported that the overexpression or transcriptional activation of PPARδ inhibits PPARγ activity, suggesting the regulatory role of PPARδ in PPARγ functions.50 Therefore, the adiponectin-promoting activity of 1a and related A3 AR ligands during adipogenesis in hBM-MSCs is associated with both PPARγ partial agonism and PPARδ antagonism.
Figure 6.

Validation of 1a and related A3 AR ligands as PPARδ antagonists. (A) The effects of 1a, 1c, 3a, and 23 (1 µM) on the 22 (0.03 µM) induced interaction between fluorescein-C33 coactivator peptides and PPARδ LBD in a TR-FRET PPARδ coactivator assay were evaluated. (B) In a TR-FRET PPARδ corepressor assay with SMRT-ID2 peptides, the activities of 23, 24, and 3a (1 µM) were assessed in the condition that 22 (0.1 µM) existed or not. (C) 23, 24, 3a, and 1c were evaluated in a TR-FRET PPARδ corepressor assay with SMRT-ID2 peptides at various concentrations. For functional validation assay for PPARδ antagonism, hBM-MSCs were differentiated in the IDX condition and co-treated with 23, 24, or 3a in the medium. On the 3rd day in culture, total RNA was extracted and Q-RT-PCR was performed for ANGPTL4 (D) and PDK4 (E). GAPDH was used as an internal control for Q-RT-PCR standardization. Values represent the mean ± SD (n = 3, three independent experiments): (*) p ≤ 0.05 and (**) p ≤ 0.01.
Effects of 1a and Related A3 AR Ligands on Insulin Sensitivity in Streptozotocin (STZ)-Induced Diabetic Mice
Antidiabetic drugs like PPARγ agonists and sulfonylureas increase adiponectin production during adipogenesis in hBM-MSCs, which correlates with improved insulin sensitivity.12,13 Compound 1a and related A3 AR ligands have a polypharmacophore to bind A3 AR, PPARγ, and PPARδ. However, whether 1a and related A3 AR ligands promote insulin sensitivity has not been experimentally tested under in vivo conditions. Therefore, we evaluated the insulin-sensitizing effect of these compounds in the STZ-induced diabetes model in C57BL/6J mice (Figure 7A).
Figure 7.

Effects of 1a, 1c, and 3a on STZ-induced diabetic mice. STZ-induced diabetic male C57BL/6J mice were orally administered potential antidiabetic drugs at 20 mg/kg for 5 days. Eight mice were used in each group. Glibenclamide was used as a positive control. Serum glucose (A), triglyceride (B), and lactate (C) levels were measured. Results are the mean ± SD of three independent experiments. Statistical analyses were performed using one-way ANOVA followed by Tukey’s post-test. (D) Serum glucose concentrations were measured just before drug administration (0 h) and at 1 and 4 h after drug administration. Each symbol represents the mean ± SD of the difference in the time spent after the drug or vehicle treatments (8 animals per each group): (*) p ≤ 0.05 and (**) p ≤ 0.01, significantly different from the vehicle-treated STZ-induced diabetic mouse group.
Compounds 1c and 3a significantly decreased serum glucose levels in the STZ-induced diabetic mice, suggesting insulin-sensitizing activity. Compound 1a tended to decrease serum glucose levels, although the effect was not statistically significant. These compounds also downregulated serum triglyceride levels in the mouse model (Figure 7B). Compound 3a tended to decrease the serum lactate levels in this model, but the effect was not significant (Figure 7C). The glucose-lowering effect of 3a in diabetic mice was dose-dependent (Figure 7D). Thus, a compound with a polypharmacological profile of an A3 AR modulator, a PPARγ partial agonist, and a PPARδ antagonist has insulin-sensitizing activity.
It is known that partial PPARγ agonists improve pathologic parameters in various human metabolic diseases.46,51 The insulin-sensitizing effects of PPARδ antagonists are still controversial.52 PPARδ itself may not affect insulin sensitivity but can competitively inhibit the transactivation of cellular PPARγ.50 Recent reports showed that the overexpression or transcriptional activation of PPARδ inhibits PPARγ activity, suggesting a regulatory role for PPARδ in PPARγ functions.50 Therefore, the PPARδ antagonist activity of 1a and related A3 AR ligands may improve insulin sensitivity by blocking the inhibitory effect of PPARδ on PPARγ transactivation. Notably, the adiponectin-promoting activity of 1a and its related A3 AR ligands correlated more significantly with their binding affinity to PPARδ than that to PPARγ (Figure 4). The polypharmacological outcome of 1a and related A3 AR ligands for adiponectin production during adipogenesis in hBM-MSCs is primarily dependent on the binding activity to the nuclear transcription factors PPARγ and PPARδ. Regardless of being A3 AR agonists or antagonists, 1a and the related A3 AR ligand 3a showed significant glucose-lowering effects in STZ-induced diabetic C57BL6/J mice. The antidiabetic potential of 1a and related A3 AR ligands may be due to their effect on both PPARγ and PPARδ.
Implications of Polypharmacology in the Pleiotropic Activities of 1a on Cancer and Inflammatory Diseases
The polypharmacology profile of 1a and its related A3 AR ligands is consistent with their pleiotropic activities in clinical trials to treat cancer and autoimmune inflammatory diseases. The effects of 1a and 1b in their clinical development have been evaluated in many human diseases such as cancer, psoriasis, rheumatoid arthritis, and dry eye syndrome.17,53 However, the A3 AR-mediated pharmacology alone may not fully explain these diverse clinical activities of 1a and 1b against various human diseases. Compounds 1a and 1b have been studied for their anticancer effects on various human cancerous tumors such as melanoma, lymphoma, colon carcinoma, and hepatocellular carcinoma.54 In fact, A3 AR has been reported to be upregulated in different tumor cell types, generating interest in A3 AR as an anticancer drug target.55 Compound 1a inhibits colon carcinoma growth in syngeneic and xenograft murine models.56 Compound 1b has been shown to suppress the growth of hepatocellular carcinoma (HCC) with upregulated A3 AR expression.57 However, the results of studies with various cancer cell lines indicated that 1a and 1b have A3 AR-independent anticancer mechanisms.58 Interaction of 1a with estrogen receptor (ER) α in a breast cancer cell line was reported as a potential A3 AR-independent mechanism for its anticancer activity.59
The polypharmacologlical profile of 1a demonstrated in this study suggests the existence of an alternative A3 AR-independent anticancer mechanism for 1a and 1b. Interaction with both PPARγ and PPARδ can account for the anticancer activity of 1a and 1b. Recently, it was reported that the ligand-induced PPARγ activation could lead to apoptosis of cancer cells, suggesting that PPARγ agonists have anticancer potential in some cancer subtypes.59 In this regard, the PPARγ partial agonist activity of 1a and 1b may contribute to their diverse anticancer activity. PPARδ is upregulated in colorectal cancers and associated with the direct promotion of colorectal tumorigenesis.60 In addition, a PPARδ antagonist inhibits the cell growth of human carcinoma lines61 whereas the PPARδ agonist 22 promotes the tumorigenesis of some cancer cell lines in animal models.61 The tumor-promoting effects of 22 were significantly attenuated in intestinal PPARδ-deleted Apc(±) mice.62 The PPARδ antagonist activity of 1a and 1b may be beneficial to prevent tumorigenesis in human cancers associated with PPARδ dysregulation. Therefore, the anticancer activity of 1a and 1b must be explained with their polypharmacological characteristics of an A3 AR agonist, a PPARγ partial agonist, and a PPARδ antagonist. The molecular characterization of human cancers in the polypharmacological context of A3 AR, PPARγ, and PPARδ may provide therapeutic benefits in the future clinical development of 1a and related A3 AR ligands as anticancer drugs.
In addition to their anticancer potential, 1a and 1b have been clinically evaluated for the treatment of human inflammatory diseases such as rheumatoid arthritis and psoriasis.17,36,53 The polypharmacology profile of 1a and related A3 AR ligands provides a good mechanistic explanation for their anti-inflammatory activity. PPARγ agonists inhibit proinflammatory cytokine production from monocytes or macrophages, whose PPARγ expression is correlated with the disease severity in rheumatoid arthritis.63 In a randomized clinical trial, the PPARγ agonist pioglitazone mildly improved the pathological symptoms of rheumatoid arthritis.64 Therefore, the anti-inflammatory activity of 1a and related A3 AR ligands can be explained by both A3 AR- and PPARγ-mediated mechanisms in various clinical conditions. Recently, inhibition of PPARδ by selective antagonists such as 23 and 24 was reported to improve inflammatory psoriatic conditions in animal models.65 Because 1a and related A3 AR ligands have PPARδ antagonist activity, it is necessary to address the possible involvement of PPARδ-mediated pathways in their in vivo anti-inflammatory outcomes under various clinical conditions.
CONCLUSION
This study demonstrated that the antidiabetic potential of 1a and related A3 AR ligands is associated with previously undetected interactions, i.e., both PPARγ partial agonism and PPARδ antagonism. In order to develop these compounds to treat human metabolic diseases, further studies will be necessary because clinical outcomes associated with efficacy or toxicity have not yet been clearly addressed depending on their A3 AR agonist or A3 AR antagonist activity. In addition, when 1a and related A3 AR ligands are clinically developed as A3 AR modulators to treat A3 AR-associated clinical conditions, the adverse effects or clinical benefits associated with PPARγ partial agonism and PPARδ antagonism should be considered. So far, the molecular targets of polypharmacology case studies have been limited to a structurally similar protein family, such as an inhibitor against multiple tyrosine kinases.3,4 In this study, 1a provides a good case study for a single drug molecule simultaneously targeting different protein families: the GPCR and NR families. Defining polypharmacology in the context of the accurate prediction of clinical efficacy and safety is one of the technical goals of systems pharmacology and future medicinal chemistry. To accomplish this, more evidence on a single drug molecule simultaneously targeting multiple targets that belong to different protein families must be presented. In this regard, the polypharmacological feature of 1a and related A3 AR ligands may provide therapeutic insight into their multipotent efficacy against various human diseases such as cancers, rheumatoid arthritis, psoriasis, and dry eye syndrome.
EXPERIMENTAL SECTION
General Methods
1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were measured in CDCl3, CD3OD, or DMSO-d6, and chemical shifts are reported as parts per million (δ) relative to the solvent peak. Coupling constants (J) are reported in hertz (Hz). Melting points are uncorrected. Elemental analyses (C, H, and N) were used to determine the purity of all synthesized compounds, and the results were within ±0.4% of the calculated values, confirming ≥95% purity. Flash column chromatography was performed on silica gel 60 (230–400 mesh). Unless otherwise noted, materials were obtained from commercial suppliers and were used without purification. All solvents were purified and dried by standard techniques just before use.
General Procedure for the Preparation of 2a–d.27a
Oxidation
To a solution of 9 (1 mmol) in anhydrous DMF (0.05 M) was added PDC (10.0 equiv) in one portion at room temperature under N2. After being stirred at the same temperature for 20 h, the reaction mixture was quenched with H2O (50 mL) and stirred additional 1 h. The precipitate was filtered and the filter cake was washed with H2O many times and dried under high vacuum to give acid intermediate as a brownish solid, which was used in the next step without further purification.
Amide Coupling
To a solution of above-generated acid (1 mmol) in CH2Cl2 (0.05 M) were added EDC (1.5 equiv), HOBt (1.5 equiv), ethylamine·HCl (1.5 equiv), and N,N-diisopropylethylamine (3.0 equiv) at room temperature under N2, and the reaction mixture was stirred at the same temperature for 15 h and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/EtOAc = 10:1 to 5:1) to give the corresponding silyl amide intermediate as white foam.
TBS Deprotection
To a solution of silyl amide intermediate (1 mmol) in anhydrous THF (0.2 M) was dropwise added tetra-n-butylammonium fluoride (1 M solution in THF, 2.5 equiv) at room temperature under N2, and the reaction mixture was stirred at the same temperature for 1 h and concentrated in vacuo. The resulting residue was purified by silica gel column chromatography (CH2Cl2/MeOH = 10:1) to give 2a–d.
(2S,3S,4R,5R)-N-Ethyl-3,4-dihydroxy-5-(6-((3-iodobenzyl)-amino)-9H-purin-9-yl)tetrahydrothiophene-2-carboxamide (2a).27a
Yield = 66%, white solid; [α]D20 −45.6° (c 0.15, MeOH); UV (MeOH) λmax 273.0 nm (pH 7); 1H NMR (400 MHz, DMSO-d6) δ 1.09 (t, J = 7.0 Hz, 3 H), 3.19 (q, J = 6.0 Hz, 2 H), 3.82 (d, J = 4.0 Hz, 1 H), 4.38 (d, J = 4.5 Hz, 1 H), 4.59 (d, J = 3.6 Hz, 1 H), 4.67 (d, J = 4.5 Hz, 2 H), 5.60 (d, J = 5.0 Hz, exchangeable with D2O, OH, 1 H), 5.77 (d, J = 4.8 Hz, exchangeable with D2O, OH, 1 H), 5.88 (d, J = 5.0 Hz, 1 H), 7.12 (t, J = 8.0 Hz, 1 H), 7.38 (d, J = 7.6 Hz, 1 H), 7.60 (d, J = 7.6 Hz, 1 H), 7.73 (s, 1 H), 8.25 (s, 1 H), 8.50 (br s, exchangeable with D2O, NH, 1 H), 8.55 (s, 1 H); 13C NMR (CD3OD) δ 15.4, 35.3, 44.5, 54.4, 68.0, 79.1, 80.1, 94.5, 120.3, 125.8, 128.5, 130.5, 135.3, 140.4, 142.7, 151.4, 155.5, 172.3, 174.5; FAB-MS m/z 541(M+ + 1). Anal. Calcd for C19H21IN6O3S: C, 42.23; H, 3.92; N, 15.55; S, 5.93. Found: C, 42.51; H, 3.95; N, 15.73; S, 5.95.
(2S,3S,4R,5R)-N-Cyclopropyl-3,4-dihydroxy-5-(6-((3-iodobenzyl)amino)-9H-purin-9-yl)tetrahydrothiophene-2-carboxamide (2b).27a
Yield = 66%; white solid; [α]D20 −35.8 (c 0.15, MeOH); UV (MeOH) λmax 272.0 nm (pH 7); 1H NMR (400 MHz, DMSO-d6) δ 0.48(br s, 2 H), 0.72 (m, 2 H), 2.54 (m, 1 H), 3.80 (d, J = 4.3 Hz, 1 H), 4.18 (dd, J = 4.0, 8.5 Hz, 1 H), 4.42 (m, 1 H), 4.70(br s, 2 H), 5.63 (d, J = 5.5 Hz, exchangeable with D2O, 1 H), 5.83 (d, J = 5.0 Hz, exchangeable with D2O, 1 H), 5.90 (d, J = 5.4 Hz, 1 H), 7.13 (t, J = 7.8 Hz, 1 H), 7.35 (d, J = 7.6 Hz, 1 H), 7.60 (d, J = 7.8 Hz, 1 H), 7.73 (s, 1 H), 8.27 (br s, 1 H), 8.58 (s, 1 H), 8.59 (br s, exchangeable with D2O, 1 H); 13C NMR (DMSO-d6) δ 8.4, 10.5, 24.5, 50.5, 64.3, 66.8, 77.0, 80.4, 116.5, 127.4, 133.5, 135.4, 135.9, 140.3, 141.5, 148.5, 150.3, 152.9, 153.5, 171.4 ; FAB-MS m/z 553 (M+ + 1). Anal. Calcd for C20H21IN6O3S: C, 43.49; H, 3.83; N, 15.21; S, 5.80. Found: C, 43.54; H, 3.92; N, 15.28; S, 5.85.
(2S,3S,4R,5R)-N-Cyclopropyl-3,4-dihydroxy-5-(6-((3-iodobenzyl)amino)-9H-purin-9-yl)-N-methyltetrahydrothiophene-2-carboxamide (2c).27a
Yield 61%; white solid; [α]D20 −14.8 (c 0.15, MeOH); UV (MeOH) λmax 274.0 nm (pH 7); 1H NMR (400 MHz, DMSO-d6) δ 0.18 (m, 2 H), 0.23 (m, 2 H), 0.75 (m, 1 H), 2.54 (m, 1 H), 2.87 (t, J = 3.8 Hz, 2 H), 3.67 (d, J = 4.2 Hz, 1 H), 4.19 (dd, J = 4.0, 8.7 Hz, 1 H), 4.42 (m, 1 H), 4.47 (br s, 2 H), 5.41 (d, J = 5.0 Hz, 1 H), 5.59 (d, J = 4.8 Hz, exchangeable with D2O, OH, 1 H), 5.70 (d, J = 5.3 Hz, 1 H), 6.92 (t, J = 7.6 Hz, 1 H), 7.18 (d, J = 7.6 Hz, 1 H), 7.40 (d, J = 7.8 Hz, 1 H), 7.53 (s, 1 H), 8.05 (br s, 1 H), 8.33 (s, 1 H), 8.43 (br s, 1 H, exchangeable with D2O, NH); 13C NMR (DMSO-d6) δ 2.3, 3.2, 13.4, 23.8, 54.3, 63.5, 67.0, 77.4, 81.0, 116.4, 126.3, 132.1, 133.4, 135.2, 139.8, 140.2, 147.5, 150.1, 151.9, 153.4, 171.8; FAB-MS m/z 567 (M+ + 1). Anal. Calcd for C21H23IN6O3S: C, 44.53; H, 4.09; N, 14.84; S, 5.66. Found: C, 44.60; H, 4.12; N, 14.95; S, 5.62.
(2S,3S,4R,5R)-N-Cyclobutyl-3,4-dihydroxy-5-(6-((3-iodobenzyl)amino)-9H-purin-9-yl)tetrahydrothiophene-2-carboxamide (2d).27a
Yield = 53%; white solid; [α]D20 −15.3 (c 0.10, MeOH); UV (MeOH) λmax 270 nm (pH 7); 1H NMR (400 MHz, DMSO-d6) δ 1.71 (m, 2 H), 1.96 (m, 2 H), 2.25 (m, 2 H), 3.84 (d, J = 4.0 Hz, 1 H), 4.30 (m, 1 H), 4.41 (dd, J = 4.5, 8.7 Hz, 1 H), 4.61 (m, 1 H), 4.70 (br s, 2 H), 5.63 (d, J = 5.5 Hz, exchangeable with D2O, OH, 1 H), 5.83 (d, J = 5.0 Hz, exchangeable with D2O, OH, 1 H), 5.90 (d, J = 5.4 Hz, 1 H), 7.15 (t, J = 8.0 Hz, 1 H), 7.41 (d, J = 7.6 Hz, 1 H), 7.62 (d, J = 7.8 Hz, 1 H), 7.76 (s, 1 H), 8.28 (br s, 1 H), 8.57 (s, 1 H), 8.72 (br s, exchangeable with D2O, NH, 1 H); 13C NMR (DMSO-d6) δ 20.4, 35.3, 37.2, 50.5, 54.8, 64.2, 66.5, 78.0, 79.5, 116.4, 126.5, 132.7, 135.3, 135.8, 141.7, 142.3, 148.4, 151.3, 153.0, 154.5, 172.0; FAB-MS m/z 567 (M+ + 1). Anal. Calcd for C21H23IN6O3S: C, 44.53; H, 4.09; N, 14.84; S, 5.66. Found: C, 44.55; H, 4.12; N, 14.96; S, 5.70.
General Procedure for the Preparation of 3a–d.29
To a solution of 13a or 13b (1 mmol) in EtOH (5 mL) were dropwise added appropriate amines (1.5 equiv) at room temperature under N2. After being stirred at the same temperature for 1–3 d, the reaction mixture was concentrated in vacuo. The residue was purified by silica gel column chromatography (CH2Cl2/MeOH = 20:1) to give 3a–d.
(2R,3R,4S)-2-(2-Chloro-6-((3-iodobenzyl)amino)-9H-purin-9-yl)tetrahydrothiophene-3,4-diol (3a).29
Yield = 84%; mp 198.7–199.9 °C; [α]D25 −78.91 (c 0.13, DMSO); UV (MeOH) λmax 274.0 nm; 1H NMR (DMSO-d6) 1H NMR (400 MHz, DMSO-d6) δ 8.90 (t, J = 6.4 Hz, 1 H), 8.51 (s, 1 H), 7.74 (s, 1 H), 7.60 (d, J = 7.6 Hz, 1 H), 7.35 (d, J = 7.6 Hz, 1 H), 7.13 (t, J = 8.0 Hz, 1 H), 5.82 (d, J = 7.6 Hz, 1 H), 5.56 (d, J = 6.4 Hz, 1 H), 5.37 (d, J = 4.0 Hz, 1 H), 4.60 (d, J = 4.4 Hz, 3 H), 4.34 (brs, 1 H), 3.38 (dd, J = 4.0, 10.8 Hz, 1 H), 2.80 (dd, J = 4.0, 10.8 Hz, 1 H); 13C NMR (DMSO-d6) δ 154.7, 153.0, 150.3, 141.9, 140.6, 136.0, 135.5, 130.5, 126.8, 118.4, 94.7, 78.5, 72.1, 61.5, 42.5, 34.4; FAB-MS m/z 504 [M + H]+. Anal. Calcd for C16H15ClIN5O2S: C, 38.15; H, 3.00; N, 13.90; S, 6.37. Found: C, 38.31; H, 2.96; N, 13.98; S, 6.21.
(2R,3R,4S)-2-(2-Chloro-6-((3-chlorobenzyl)amino)-9H-purin-9-yl)tetrahydrothiophene-3,4-diol (3b).29
Yield = 82%; mp 163.3–165.3 °C; [α]D25 −69.92 (c 0.13, DMSO); UV (MeOH) λmax 274.5 nm; 1H NMR (400 MHz, CD3OD) δ 8.34 (s, 1 H), 7.41 (s, 1 H), 7.24–7.34 (m, 3 H), 5.94 (d, J = 6.4 Hz, 1 H), 4.75 (brs, 2 H), 4.61 (q, J = 3.2 Hz, 1 H), 4.45 (q, J = 4.0 Hz, 1 H), 3.51 (dd, J = 4.8, 11.2 Hz, 1 H), 2.95 (dd, J = 3.6, 10.8 Hz, 1 H); 13C NMR (CD3OD) δ 141.8, 135.5, 131.2, 128.9, 128.5, 127.3, 80.9, 74.4, 64.1, 44.6, 35.3; FAB-MS m/z 411 [M]+. Anal. Calcd for C16H15Cl2N5O2S: C, 46.61; H, 3.67; N, 16.99; S, 7.78. Found: C, 46.65; H, 3.67; N, 16.74; S, 7.39.
(2R,3R,4S)-2-(2-Chloro-6-((2-chlorobenzyl)amino)-9H-purin-9-yl)tetrahydrothiophene-3,4-diol (3c).29
Yield = 81%; mp 198.7–199.7 °C; UV (MeOH) λmax 273.5 nm; 1H NMR (400 MHz, CD3OD) δ 8.35 (brs, 1 H), 7.45–7.47 (m, 1 H), 7.39–7.43 (m, 1 H), 7.25–7.29 (m, 2 H), 5.95 (d, J = 6.4 Hz, 1 H), 4.60–4.63 (m, 1 H), 4.45 (dd, J = 3.6, 8.0 Hz, 1 H), 3.51 (dd, J = 4.8, 10.8 Hz, 1 H), 2.95 (dd, J = 4.0, 10.8 Hz, 1 H); 13C NMR (CD3OD) δ 141.8, 130.8, 130.6, 130.0, 128.2, 80.9, 74.4, 64.1, 43.2, 35.3; [α]D25 −96.21 (c 0.12, DMSO); FAB-MS m/z 412 [M + H]+. Anal. Calcd for C16H15Cl2N5O2S: C, 46.61; H, 3.67; N, 16.99; S, 7.78. Found: C, 46.58; H, 3.77; N, 16.65; S, 7.60.
(2R,3R,4S)-2-(6-((3-Iodobenzyl)amino)-9H-purin-9-yl)-tetrahydrothiophene-3,4-diol (3d).29
Yield = 88%; mp 198.8–199.8 °C; UV (MeOH) λmax 271.5 nm; [α]D23.8 −97.08 (c 0.137, DMSO); 1H NMR (400 MHz, DMSO-d6) δ 8.45 (s, 1 H), 8.43 (br s, 1 H, D2O exchangeable), 8.21 (s, 1 H), 7.72 (s, 1 H), 7.56 (d, J = 7.2 Hz, 1 H), 7.35 (d, J = 7.6 Hz, 1 H), 7.10 (merged dd, J = 7.6 Hz, 1 H), 5.90 (d, J = 7.2 Hz, 1 H), 5.53 (d, J = 6.4 Hz, D2O exchangeable, 1 H), 5. 35 (d, J = 4.4 Hz, D2O exchangeable, 1 H), 4.71–4.66 (m, 2 H), 4.37–4.34 (m, 1 H), 3.41 (dd, J = 2.8, 10.8 Hz, 1 H), 3.15 (d, J = 5.2 Hz, 1 H), 2.79 (dd, J = 2.8, 10.8 Hz, 1 H); 13C NMR (DMSO-d6) δ 154.2, 152.4, 149.2, 142.9, 140.0, 137.0, 135.7, 135.3, 130.4, 126.6, 94.7, 78.3, 72.2, 61.6, 42.2, 34.4; FAB-MS m/z 370 [M + H]+. Anal. Calcd for C16H16IN5O2S: C, 40.95; H, 3.44; N, 14.92; S, 6.83. Found: C, 41.04; H, 3.43; N, 14.82; S, 6.81.
(S)-2-Hydroxy-2-((4S,5R)-5-(hydroxymethyl)-2,2-dimethyl-1,3-dioxolan-4-yl)ethyl Benzoate (6).27a
Benzoylation
To a cooled (0 °C) solution of 5 (6.65 g, 35.35 mmol) in anhydrous CH2Cl2 (25 mL) were dropwise added pyridine (8.6 mL, 100.0 mmol) and benzoyl chloride (6.2 mL, 50.0 mmol) under N2. After being stirred at the room temperature for 12 h, the reaction mixture was quenched with saturated aqueous NH4Cl (30 mL) and diluted with CH2Cl2 (20 mL). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (2 × 50 mL). The combined organic layers were washed successively with 5% aqueous HCl (2 × 100 mL), saturated aqueous NaHCO3 solution (2 × 100 mL), and H2O (2 × 100 mL), dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The crude residue was recrystallized in hot ethanol to give benzoyl protected intermediate (9.0 g, 92%) as white crystalline solid: 1H NMR (400 MHz, CDCl3) δ 8.05 (d, J = 7.3 Hz, 2 H), 7.57 (t, J = 7.4 Hz, 1 H), 7.44 (t, J = 7.7 Hz, 2 H), 4.84–4.92 (m, 2 H), 4.76–4.83 (m, 2 H), 4.52–4.56 (m, 1 H), 1.49 (s, 3 H), 1.40 (s, 3 H).
Reduction
To a cooled (0 °C) solution of 6 (16.0 g, 55.0 mmol) in MeOH (100 mL) was added NaBH4 (6.22 g, 165.0 mmol) in one portion. After being stirred at room temperature for 3 h, the reaction mixture was concentrated in vacuo and diluted with EtOAc (100 mL) and H2O (100 mL). The layers were separated, and the aqueous layer was extracted with EtOAc (3 × 100 mL). The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/EtOAc = 1:1) to give 6 (14.6 g, 90%) as white solid: 1H NMR (400 MHz, CDCl3) δ 8.04 (d, J = 7.8 Hz, 2 H), 7.55 (t, J = 7.5 Hz, 1 H), 7.42 (t, J = 7.6 Hz, 2 H), 4.45–4.50 (m, 1 H), 4.37–4.41 (m, 1 H), 4.28–4.32 (m, 1 H), 4.24–4.26 (m, 1 H), 4.12 (br s, 1 H), 3.83–3.88(m, 2 H), 3.14 (s, 1 H), 2.58 (s, 1 H), 1.51 (s, 3 H), 1.36 (s, 3 H).
((3aS,4R,6aR)-2,2-Dimethyltetrahydrothieno[3,4-d][1,3]-dioxol-4-yl)methyl Benzoate (7).27a
Mesylation
To a cooled (0 °C) solution of 6 (19.5 g, 65.0 mmol) in anhydrous CH2Cl2 (150 mL) was added 4-dimethylaminopyridine (1.61 g, 15.0 mmol) in one portion, followed by dropwise addition of triethylamine (75 mL, 530.0 mmol) and methanesulfonyl chloride (20.9 mL, 265.0 mmol) under N2. After being stirred at the same temperature for 30 min, the mixture was quenched with saturated aqueous NH4Cl (100 mL) and diluted with CH2Cl2 (100 mL). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 50 mL). The combined organic layers were washed successively with 5% aqueous HCl (3 × 50 mL), H2O (100 mL), dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The crude product was used for the next step without further purification.
Cyclization
To a solution of above crude dimesylate in DMF (500 mL) was added sodium sulfide monohydrate (19.0 g, 78.0 mmol) in one portion at room temperature under N2. After being heated at 100 °C (bath temperature) with stirring for 15 h, the reaction mixture was cooled to room temperature. The reaction mixture was quenched with H2O (100 mL) and diluted with E2O (100 mL). The layers were separated, and the aqueous layer was extracted with E2O (3 × 50 mL). The combined organic layers were washed successively with H2O (3 × 100 mL) and saturated brine, dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/EtOAc = 5:1) to give 7 (8.14 g, 42% for 2 steps) as colorless solid: 1H NMR (400 MHz, CDCl3) δ 8.01 (d, J = 7.6 Hz, 2 H), 7.55 (t, J = 7.3 Hz, 1 H), 7.43 (t, J = 7.7 Hz, 2 H), 4.95 (t, J = 4.8 Hz, 1 H), 4.78 (d, J = 5.5 Hz, 1 H), 4.37–4.41 (m, 1 H), 4.27–4.32 (m, 1 H), 3.60–3.63 (m, 1 H), 3.13–3.17 (m, 1 H), 2.94 (d, J = 13.0 Hz, 1 H), 1.51 (s, 3 H), 1.30 (s, 3 H).
((3aS,4R,6R,6aR)-6-(6-((3-Iodobenzyl)amino)-9H-purin-9-yl)-2,2-dimethyltetrahydrothieno[3,4-d][1,3]dioxol-4-yl)methyl Benzoate (8).27a
Oxidation
To a cooled (−78 °C) solution of 7 (1.68 g, 5.71 mmol) in anhydrous CH2Cl2 (30 mL) was dropwise added a solution of mCPBA (77%, 1.92 g, 8.57 mmol) in CH2Cl2 (30 mL) under N2. After being stirred at the same temperature for 1 h, the reaction mixture was quenched with saturated aqueous NaHCO3 (70 mL) and extracted with CH2Cl2 (3 × 100 mL). The combined organic layers were washed successively with saturated brine, dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/EtOAc = 3:2) to give 8 (1.68 g, 5.43 mmol, 95%) which was used immediately for the next step; colorless syrup.
Base Condensation
To a suspension of 6-chloropurine (1.68 g, 10.85 mmol) in anhydrous CH3CN (10 mL) and 1,2-dichloroethane (5 mL) were dropwise added triethylamine (1.5 mL, 10.85 mmol) and TMSOTf (4.82 g, 21.69 mmol) at room temperature under N2, and the resulting mixture was stirred until clear solution was obtained. To a solution of silylated 6-chloropurine was added above-generated sulfoxide (1.68 g, 5.43 mmol) in anhydrous 1,2-dichloroethane (5 mL) in one portion at room temperature. Upon additional dropwise addition of triethylamine (1.5 mL, 10.85 mmol), the reaction mixture were initiated to Pummerer reaction. The resulting reaction mixture was heated at 80 °C for 4 d, during which time the initially formed N7-isomer was converted to N9-isomer. The reaction mixture was quenched with saturated aqueous NaHCO3 (20 mL), diluted with EtOAc (20 mL). The layers were separated, and the aqueous layer was extracted with EtOAc (20 mL × 3). The combined organic layers were washed successively with H2O and saturated brine, dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/EtOAc = 5:1) to give 6-chloropurine condensed product (1.29 g, 53%).
N6 Amination
To a solution of 6-chloropurine condensed product (1.29 g, 2.88 mmol) in anhydrous EtOH (60 mL) were dropwise added successively triethylamine (1.2 mL, 8.63 mmol) and 3-iodobenzylamine (0.45 mL, 3.45 mmol) at room temperature under N2. After being stirred at the same temperature for 24 h, the reaction mixture was concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/EtOAc = 1:1) to give the N6-substituted product 8 (1.63 g, 88%) as white foam: UV (MeOH) λmax 272 nm (pH 7); 1H NMR (400 MHz, CDCl3) δ 1.23 (s, 3 H), 1.37 (s, 3 H), 4.06 (td, J = 2.4, 7.3 Hz, 1 H), 4.46 (dd, J = 6.8, 11.4 Hz, 1 H), 4.53 (dd, J = 2.7, 11.4 Hz, 1 H), 4.73 (d, J = 5.8 Hz, 2 H), 4.89 (dd, J = 2.4, 5.6 Hz, 1 H), 5.02 (dd, J = 2.0, 5.6 Hz, 1 H), 5.96 (s, 1 H), 6.59 (br s, 1 H), 7.11–7.84 (m, 9 H), 8.52 (s, 1 H), 8.58 (s, 1 H); FAB-MS m/z 644 (M+ + 1). Anal. Calcd for C27H26IN5O4S: C, 50.40; H, 4.07; N, 10.88; S, 4.98. Found: C, 50.33; H, 4.21; N, 10.90; S, 4.88.
((2R,3S,4R,5R)-3,4-Bis((tert-butyldimethylsilyl)oxy)-5-(6-((3-iodobenzyl)amino)-9H-purin-9-yl)tetrahydrothiophen-2-yl)-methanol (9).27a
Acetonide Deprotection
A solution of 8 in 80% aqueous AcOH (30 mL) was heated at 70 °C (bath temperature) with stirring under N2. After being stirred at the same temperature for 15 h, the reaction mixture was concentrated in vacuo and neutralized with saturated methanolic ammonia. After concentration in vacuo, the residue was purified by silica gel column chromatography (CH2Cl2/MeOH = 10:1) to give diol intermediate as white foam.
TBS Protection
To a solution of diol intermediate in anhydrous pyridine (0.05 M) was dropwise added TMSOTf (5.0 equiv) at room temperature under N2. The reaction mixture was heated at 50 °C (bath temperature) with stirring for 5 h. The reaction mixture was quenched with H2O and diluted with CH2Cl2. The organic layer was washed with H2O, aqueous saturated NaHCO3, saturated brine, dried over anhydrous MgSO4, and concentrated in vacuo. The crude disilyl ether was used in the next step without further purification.
Benzoyl Deprotection
To a solution of above-generated benzoylated disilyl ether in anhydrous MeOH (0.033 M) was added NaOMe (1.5 equiv) in one portion at room temperature under N2. After being stirred at the same temperature for 4 h, the reaction mixture was neutralized with glacial acetic acid and concentrated in vacuo. The resulting residue was purified by silica gel column chromatography (hexanes/EtOAc = 1:1) to give 9 (82%) as white solid: 1H NMR (400 MHz, CDCl3) δ 0.01 (m, 12 H), 0.62 (s, 9 H), 0.83 (s, 9 H), 3.27 (dd, J = 5.6, 11.7 Hz, 1 H), 3.74 (m, 1 H), 3.86 (m, 1 H), 4.14 (dd, J = 5.7, 11.5 Hz, 1 H), 4.63 (br s, 2 H), 5.20 (dd, J = 6.3, 11.9 Hz, 1 H), 5.60 (d, J = 4.6 Hz, 1 H), 5.93 (br s, 1 H), 6.93 (t, J = 7.7 Hz, 1 H), 7.13 (s, 1 H), 7.58 (d, J = 7.5 Hz, 1 H), 7.60 (d, J = 7.8 Hz, 1 H), 7.67 (s, 1 H), 8.01(s, 1 H), 8.28 (s, 1 H).
(1R)-(2,2-Dimethyl-1,3-dioxolan-4-yl)((4S,5S)-2,2-dimethyl-5-(((methylsulfonyl)oxy)methyl)-1,3-dioxolan-4-yl)methyl Methanesulfonate (10).29
Acetonide Protection
To a cooled (0 °C) suspension of d-mannose (0.87 g, 3.26 mmol) in acetone acetone (30 mL) was dropwise added 2,2-dimethoxypropane (1.23 mL, 9.78 mmol) followed by addition of camphosulfonic acid (0.23 g, 0.98 mmol) in one portion under N2. After being stirred at room temperature for 24 h. The mixture was neutralized with triethylamine and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/EtOAc = 1:1) to give 10 (0.81 g, 95%) as white solid: 1H NMR (400 MHz, CDCl3) δ 5.34 (s, 1 H), 4.76–4.79 (m, 1 H), 4.58 (d, J = 6.0 Hz, 1 H), 4.34–4.39 (m, 1 H), 4.15 (dd, J = 3.6, 7.2 Hz, 1 H), 4.00–4.08 (m, 2 H).
Reduction
To a cooled (0 °C) solution of diacetonide 10 (0.75 g, 2.88 mmol) in EtOH (15 mL) was added NaBH4 (220 mg, 5.77 mmol) cautiously in several portions under N2. After being stirred at room temperature for 2 h, the mixture was neutralized with acetic acid and concentrated in vacuo. The reaction mixture was quenched with H2O (30 mL), diluted with EtOAc (30 mL). The layers were separated, and the aqueous layer was extracted with EtOAc (30 mL × 3). The combined organic layers were washed successively with H2O and saturated brine, dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/EtOAc = 1:1) to give diol intermediate (0.69 g, 92%) as colorless syrup: 1H NMR (400 MHz, CDCl3) δ 4.33 (dd, J = 1.6, 7.2 Hz, 1 H), 4.24–4.28 (m, 1 H), 4.06–4.13 (m, 2 H), 3.92–3.97 (m, 1 H), 3.76–3.85 (m, 2 H), 3.59–3.61 (m, 1 H), 1.48 (s, 3 H), 1.38 (s, 3 H), 1.36 (s, 3 H), 1.33 (s, 3 H).
Mesylation
To a cooled (0 °C) solution of diol intermediate (19.26 g, 73.43 mmol) in anhydrous CH2Cl2 (150 mL) was added 4-dimethylaminopyridine (2.69 g, 22.03 mmol) in one portion, followed by dropwise addition of triethylamine (82 mL, 0.585 mol) and methanesulfonyl chloride (23.80 mL, 293.71 mmol) under N2. After being stirred at room temperature for 1 h, the mixture was quenched with saturated aqueous NH4Cl (100 mL) and diluted with CH2Cl2 (100 mL). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 100 mL). The combined organic layers were washed successively with H2O (100 mL), dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/EtOAc = 5:1) to give a dimesylate 10 (28.92 g, 94%) as colorless syrup: 1H NMR (400 MHz, CDCl3) δ 4.75 (pseudo t, J = 7.4 Hz, 1 H), 4.33–4.45 (m, 4 H), 4.06–4.20 (m, 3 H), 3.12 (s, 3 H), 3.07 (s, 3 H), 1.51 (s, 3 H), 1.43 (s, 3 H), 1.37 (s, 3 H), 1.33 (s, 3 H).
(S)-1-((3aR,4S,6aS)-2,2-Dimethyltetrahydrothieno[3,4-d]-[1,3]dioxol-4-yl)ethane-1,2-diol (11).29
Cyclization
To a solution of 10 (466.9 mg, 1.12 mmol) in anhydrous DMF (50 mL) was added sodium sulfide (174.15 g, 2.23 mmol) in one portion at room temperature under N2. After being heated at 80 °C (bath temperature) with stirring overnight, the reaction mixture was concentrated in vacuo. The reaction mixture was quenched with H2O and diluted with EtOAc (30 mL). The layers were separated, and the aqueous layer was extracted with EtOAc (30 mL × 3). The combined organic layers were washed successively with H2O and saturated brine, dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/EtOAc = 8:1) to give cyclized intermediate (227.0 mg, 78%) as colorless syrup: mp 100.7–101.8 °C; [α]D25 −52.38 (c 0.13, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 4.93 (dt, J = 1.8, 5.6 Hz, 1 H), 4.76 (dd, J = 2.0, 5.6 Hz, 1 H), 3.76–3.80 (m, 1 H), 3.70–3.74 (m, 1 H), 3.64–3.68 (m, 1 H), 3.40 (dd, J = 1.6, 5.2 Hz, 1 H), 3.16 (dd, J = 5.0, 12.8 Hz, 1 H), 2.90 (dd, J = 1.6, 12.8 Hz, 1 H), 1.52 (s, 3 H), 1.33 (s, 3 H); 13C NMR (CDCl3) δ 111.6, 87.3, 84.0, 72.3, 65.3, 57.7, 32.0, 26.9, 24.9; FAB-MS m/z 221 [M + H]+. Anal. Calcd for C9H16O4S: C, 49.07; H, 7.32; S, 14.56. Found: C, 49.47; H, 7.72; S, 14.15.
Deprotection
A solution of cyclized intermediate (10.89 g, 41.83 mmol) in 60% aqueous AcOH (120 mL) was stirred at room temperature under N2. After being stirred at the same temperature for 2 h, the reaction mixture was concentrated in vacuo, and the resulting residue was purified by silica gel column chromatography (hexane/EtOAc = 1:2) to give the diol 11 (7.43 g, 81%) as white solid along with recovered starting material (3.08 g): 1H NMR (400 MHz, CDCl3) δ 4.93 (dt, J = 1.8, 5.6 Hz, 1 H), 4.76 (dd, J = 2.0, 5.6 Hz, 1 H), 3.76–3.80 (m, 1 H), 3.70–3.74 (m, 1 H), 3.64–3.68 (m, 1 H), 3.40 (dd, J = 1.6, 5.2 Hz, 1 H), 3.16 (dd, J = 5.0, 12.8 Hz, 1 H), 2.90 (dd, J = 1.6, 12.8 Hz, 1 H), 1.52 (s, 3 H), 1.33 (s, 3 H).
(3aR,6aS)-2,2-Dimethyltetrahydrothieno[3,4-d][1,3]dioxol-4-yl Acetate (12).29
To a cooled (0 °C) solution of diol 11 (1.49 g, 6.74 mmol) in anhydrous EtOAc (30 mL) was added lead(IV) acetate (15.73 g, 33.71 mmol) in one portion under N2. After being stirred for 15 h at the room temperature, the reaction mixture was filtered using Celite pad and the filtrate was diluted with EtOAc. The combined organic layers were washed with saturated aqueous NaHCO3, dried over anhydrous MgSO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexanes/EtOAc = 8:1) to give acetate 12 (0.88 g, 60%) as colorless syrup: [α]D25 −258.15 (c 0.18, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 5.03 (dd, J = 5.6, 9.6 Hz, 1 H), 4.79 (dd, J = 5.6, 8.8 Hz, 1 H), 3.21–3.27 (m, 2 H), 3.01 (dt, J = 0.8, 12.8 Hz, 2 H), 2.05 (s, 3 H), 1.50 (s, 3 H), 1.31 (s, 3 H); FAB-MS m/z 218 [M]+. Anal. Calcd for C9H14O4S: C, 49.52; H, 6.46; S, 14.69. Found: C, 49.51; H, 6.35; S, 14.65.
(2R,3R,4S)-2-(6-Chloro-9H-purin-9-yl)tetrahydrothiophene-3,4-diol (13a).29
Base Condensation
A suspension of dried 6-chloropurine (0.39 g, 2.53 mmol) and ammonium sulfate (8.4 mg, 0.063 mmol) in anhydrous hexamethyldisilazane (HMDS, 5 mL) was refluxed under N2 for overnight. After being stirred overnight, the reaction mixture was concentrated in vacuo. To a cooled 0 °C suspension of above-generated solid in 1,2-dichloroethane (2 mL) was dropwise added 12 (0.276 g, 1.26 mmol) in 1,2-dichloroethane (2 mL) followed by dropwise addition of TMSOTf (0.5 mL, 2.53 mmol). After being stirred at the same temperature for 30 min, the reaction mixture was warmed to room temperature and stirred for 1 h and heated at 80 °C (bath temperature) with stirring for 2 h. The mixture was cooled to room temperature and quenched with saturated aqueous NaHCO3, diluted with CH2Cl2, and washed with saturated brine. The organic layer was dried with anhydrous MgSO4 and concentrated in vacuo. The residue yellow syrup was purified by silica gel column chromatography (CH2Cl2/MeOH = 50:1) to give base condensed intermediate (0.359 g, 90%) as white foam: 1H NMR (400 MHz, CDCl3) δ 8.67 (s, 1 H), 8.23 (s, 1 H), 5.88 (s, 1 H), 5.25–5.19 (m, 1 H), 3.69 (dd, J = 4.0, 13.2 Hz, 1 H), 3.18 (d, J = 12.8 Hz, 1 H), 1.51 (s, 3 H), 1.28 (s, 3 H).
Deprotection
To a cooled (0 °C) solution of base condensed intermediate (0.259 g, 0.828 mmol) in THF (2 mL) was dropwise added 2 N HCl (2 mL) under N2. After being stirred at room temperature overnight, the mixture was neutralized with 1 N NaOH solution, and the reaction mixture was concentrated in vacuo. The residue was purified by silica gel column chromatography (CH2Cl2/MeOH = 20:1) to give 13a (0.90 g, 79%) as white solid: 1H NMR (400 MHz, DMSO-d6) δ 9.02 (s, 1 H), 8.81 (s, 1 H), 6.02 (d, J = 7.2 Hz, 1 H), 5.62 (d, J = 6.0 Hz, D2O exchangeable, 1 H), 5.43 (d, J = 4.1 Hz, D2O exchangeable, 1 H), 4.74–4.70 (m, 1 H), 4.40–4.36 (m, 1 H), 3.47 (dd, J = 4.0, 11.2 Hz, 1 H), 2.83 (dd, J = 2.8, 11.2 Hz, 1 H).
(2R,3R,4S)-2-(2,6-Dichloro-9H-purin-9-yl)tetrahydrothiophene-3,4-diol (13b).29
Compound 13b was prepared according to similar procedure used in the preparation of 13a.
Base Condensation
Yield = 79%; white foam; 1H NMR (400 MHz, CDCl3) δ 8.17 (s, 1 H), 5.87 (s, 1 H), 5.32 (pseudo t, 1 H, J = 4.8 Hz), 5.21 (d, 1 H, J = 5.6 Hz), 3.79 (dd, 1 H, J = 4.4, 12.8 Hz), 3.26 (d, 1 H, J = 13.2 Hz), 1.59 (s, 3 H), 1.36 (s, 3 H).
Deprotection
Yield = 96%; white solid; UV (CH2Cl2) λmax 275.0 nm; 1H NMR (400 MHz, CD3OD) δ 8.87 (s, 1 H), 6.08 (d, J = 6.8 Hz, 1 H), 4.69 (q, J = 3.2 Hz, 1 H), 4.48 (q, J = 3.6 Hz, 1 H), 3.56 (dd, J = 4.4, 11.2 Hz, 1 H), 2.97 (dd, J = 3.4, 11.2 Hz, 1 H); 13C NMR (CDCl3) δ 153.3, 152.5, 152.4, 145.0, 131.8, 112.2, 89.8, 84,6, 70.6, 41.2, 26.5, 24.7; [α]D25 −42.04 (c 0.16, CH2Cl2); FAB-MS m/z 347 [M + H]+. Anal. Calcd for C12H12Cl2N4O2S: C, 41.51; H, 3.48; N, 16.14; S, 9.24. Found: C, 41.84; H, 3.78; N, 15.99; S, 8.98.
Cell Culture and Adipogenic Differentiation
hBM-MSCs were purchased from Lonza (Walkersville, MD, USA) and cultured in Dulbecco’s modified Eagle’s medium (DMEM; glucose 1 g/L) containing 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin (Invitrogen, Carlsbad, CA, USA). To induce adipocyte differentiation, the growth medium was replaced by DMEM containing 4.5 g/L of glucose and supplemented with 10% FBS, 10 µg/mL insulin, 0.5 µM dexamethasone, and 0.5 mM 3-isobutyl-1-methylxanthine (IBMX) (IDX condition). Dexamethasone, insulin, IBMX, glibenclamide, troglitazone, caffeine, 14, 15, 16, and 22 were purchased from Sigma-Aldrich (St. Louis, MO, USA). Compounds 17, 18, 19, 20, 21, 23, and 24 were purchased from Tocris Bioscience (Bristol, U.K.). In hBM-MSCs, cell culture media were exchanged every 2nd or 3rd day during cell differentiation.
Oil Red O and Hematoxylin Staining
The level of adipocyte differentiation of hBM-MSCs was evaluated using an Oil Red O (ORO, Sigma-Aldrich) staining method to measure intracellular lipid accumulation. Cells were rinsed twice with phosphate buffered saline (PBS) and then fixed with 10% formalin in PBS (pH 7.4) for 30 min. Fixed cells were washed with 60% isopropanol and dried completely. Lipid droplets were stained with 0.2% ORO reagent in 60% isopropanol for 10 min at 25 °C and then washed with running tap water four times. To visualize the nucleus, cells were counterstained with hematoxylin reagent for 2 min and then washed four times with tap water. The differentiated adipocytes were observed and photographed using an Eclipse TS100 inverted microscope (Nikon Co., Tokyo, Japan).
Enzyme-Linked Immunosorbent Assay (ELISA)
For quantitative measurement of adiponectin in cell culture supernatants, a Quantikine immunoassay kit (R&D Systems, Minneapolis, MN, USA) was used. The media treated with 1a and related A3 AR ligands were centrifuged for 5 min at 1000g, and the supernatants were subsequently diluted for use in the quantification reaction for adiponectin by ELISA.
Nuclear Receptor (NR) Assays
The time-resolved fluorescence resonance energy transfer (TR-FRET) based receptor binding assay was performed using Lanthascreen competitive binding assay kits (Invitrogen) to evaluate binding of ligand to NRs, PPARα, PPARγ, PPARδ, and GR. Lanthascreen coactivator assay kits were used as previously described66 to determine the receptor activation of PPARδ, LXRα, and LXRβ. The Lanthascreen PPARδ coactivator and corepressor assay was performed using fluorescein-C33 coactivator peptide (sequence HVEMHPLLMGLLMESQWGA) and SMRT-ID2 peptide (sequence HASTNMGLEAHRKALMGKYDQW), respectively.67 All assay measurements were performed using a CLARIOstar (BMG LABTECH, Ortenberg, Germany). The luciferase reporter gene assay was performed as previously described.45
Kinase Assays
The kinase inhibitor assay with KinaseProfiler was performed (Eurofins Pharma Discovery, Dundee, U.K.). Briefly, human CDK5/p25 and CDK5/p35 were incubated with histone H1 and γ-32P-ATP. The reaction was initiated by the addition of magnesium ATP mixture in KinaseProfiler kit. After incubation for 40 min at room temperature, the reaction was stopped by the addition of 3% phosphoric acid solution. The reaction mixture (10 µL) was spotted onto a P30 filtermat (PerkinElmer, Richmond, CA, USA) and washed three times for 5 min in 75 mM phosphoric acid and once in methanol. The filtermat was dried, and the remaining radioactivity was measured with a scintillation counter (Beckman Coulter, Indianapolis, IN, USA).
Total RNA Isolation and Quantitative Real-Time PCR (Q-RT-PCR)
The total RNA from hBM-MSCs or differentiated cells was extracted with Trizol (Invitrogen), followed by a purification step using the Qiagen RNeasy kit (Qiagen, Valencia, CA). The RNA concentration was determined spectrophotometrically at 260 and 280 nm using an Epoch microplate spectrophotometer (BioTeK, Winooski, VT, USA). An amount of 2 µg of RNA from each sample was reverse transcribed into cDNA using Maxima First Strand cDNA synthesis kit for Q-RT-PCR (Thermo Scientific, Waltham, MA, USA). TaqMan Universal Master Mix II and Q-RT-PCR primer sets (Applied Biosystems, Foster City, CA, USA) were used to determine the transcription levels of ANGPTL4 (Hs01101127_m1, Applied Biosystems) and PDK4 (Hs01037712_m1, Applied Biosystems). Human glyceraldehyde 3-phosphate dehydrogenase (GAPDH, 4333764F) was used to normalize sample variations. Q-RT-PCR was performed with an Applied Biosystems 7500 real-time PCR system (Applied Biosystems). Relative gene expression levels were quantified using equations from a mathematical model developed by Pfaffl.68 Q-RT-PCR results were represented as the mean ± SD of three measurements using hBM-MSCs from three independent donors.
Streptozotocin-Induced Diabetes Mellitus Mouse Experiments
All experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee in Seoul National University in accordance with the Principles of Laboratory Animal Care (NIH) and the Animal Care and Use Guidelines of Sahmyook University. A single dose of 180 mg/kg of STZ was intraperitoneally administered to 5-week-old male C57BL/6J mice. From the 7th day after the STZ treatment, serum glucose concentration was monitored daily for 3 consecutive days after 2 h of fasting. The glucose concentration was measured with a portable glucose meter Accu-Check Active (Boehringer-Mannheim Biochemicals, Indianapolis, IN, USA). Mice with a serum glucose concentration higher than 300 mg/dL were considered diabetic. For each experimental group, eight diabetic mice were randomly selected to evaluate antidiabetic activity. Drugs were formulated in 0.5% carboxymethylcellulose (CMC), and control groups received vehicle. Before administration of drugs, serum glucose concentration was confirmed again at 2 h after fasting and potential antidiabetic drugs were orally administered daily for 5 d. On the 5th day, serum glucose levels were measured just before drug administration (0 h) and 1 and 4 h after drug administration. Blood samples were obtained from the tail vein with heparinized syringes. Serum triglycerides (TGs) were determined with a serum triglyceride determination kit (TR0100, Sigma-Aldrich), and lactate levels were quantified with a lactate assay kit (MAK064, Sigma-Aldrich).
Statistical Analysis
Statistical analysis was conducted using RStudio for Windows (RStudio Inc., Boston, MA, USA). Experimental values are expressed as the mean ± standard deviation (SD) from three or four independent experiments. For multiple comparisons, statistical analysis was performed using one-way analysis of variance (ANOVA) followed by Tukey’s post-tests. The correlation coefficient was calculated by Pearson’s correlation. The threshold of significance was set at (*) P ≤ 0.05 and (**) P ≤ 0.01.
Supplementary Material
Acknowledgments
This study was partly supported by National Research Foundation of Korea (NRF) grants funded by the Ministry of Science, ICT, and Future Planning (Grants 2014M3C9A- 2064603, 2015R1A2A2A01008408, and 370C-20160046), by NIDDK Intramural Research Program (Grant ZIA DK031117), and by Promising-Pioneering Researcher Program through the Seoul National University (SNU) in 2015.
ABBREVIATIONS USED
- IB-MECA
N6-(3-iodobenzyl)adenosine-5′-N-methyluronamide
- PPAR
peroxisome proliferator activated receptor
- AR
adenosine receptor
- hBM-MSC
human bone marrow mesenchymal stem cell
- NSAID
nonsteroidal anti-inflammatory drug
- GPCR
G-protein-coupled receptor
- cAMP
cyclic adenosine monophosphate
- IRS-2
insulin receptor substrate 2
- mCPBA
meta-chloroperbenzoic acid
- TMOOTf
trimethylsilyl trifluoromethanesulfonate
- TBS
tert-butyldimethylsilyl
- PDC
pyridinium dichromate
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- HOBt
1-hydroxybenzotriazole
- NECA
5′-(N-ethylcarboxamido)adenosine
- CCPA
2-chloro-N6-cyclopentyladenosine
- IDX
insulin, dexamethasone, and isobutylmethylxanthine
- SAR
structure–activity relationship
- NR
nuclear receptor
- LXR
liver X receptor
- GR
glucocorticoid receptor
- TR-FRET
|time-resolved fluorescence resonance energy transfer
- CDK5
cyclin-dependent kinase 5
- SMRT
silencing mediator of retinoid and thyroid hormone receptor
- ANGPLT4
angiopoietin-like 4
- PDK4
pyruvate dehydrogenase kinase 4
- STZ
streptozotocin
- HCC
hepatocellular carcinoma
- ER
estrogen receptor
- DMEM
Dulbecco modified Eagle’s medium
- FBS
fetal bovine serum
- ORO
Oil Red O
- PBS
phosphate buffered saline
- ELISA
enzyme-linked immunosorbent assay
- Q-RT-PCR
quantitative real-time polymerase chain reaction
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- CMC
carboxymethylcellulose
- TG
triglyceride
- ANOVA
one-way analysis of variance
Footnotes
ASSOCIATED CONTENT
The authors declare no competing financial interest.
References
- 1.Hopkins AL. Network pharmacology: the next paradigm in drug discovery. Nat. Chem. Biol. 2008;4:682–690. doi: 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]
- 2.Anighoro A, Bajorath J, Rastelli G. Polypharmacology: challenges and opportunities in drug discovery. J. Med. Chem. 2014;57:7874–7887. doi: 10.1021/jm5006463. [DOI] [PubMed] [Google Scholar]
- 3.Dar AC, Das TK, Shokat KM, Cagan RL. Chemical genetic discovery of targets and anti-targets for cancer polypharmacology. Nature. 2012;486:80–84. doi: 10.1038/nature11127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gujral TS, Peshkin L, Kirschner MW. Exploiting polypharmacology for drug target deconvolution. Proc. Natl. Acad. Sci. U. S. A. 2014;111:5048–5053. doi: 10.1073/pnas.1403080111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moffat JG, Rudolph J, Bailey D. Phenotypic screening in cancer drug discovery - past, present and future. Nat. Rev. Drug Discovery. 2014;13:588–602. doi: 10.1038/nrd4366. [DOI] [PubMed] [Google Scholar]
- 6.Byun Y, Park J, Hong SH, Han MH, Park S, Jung HI, Noh M. The opposite effect of isotype-selective monoamine oxidase inhibitors on adipogenesis in human bone marrow mesenchymal stem cells. Bioorg. Med. Chem. Lett. 2013;23:3273–3276. doi: 10.1016/j.bmcl.2013.03.117. [DOI] [PubMed] [Google Scholar]
- 7.Noh M. Interleukin-17A increases leptin production in human bone marrow mesenchymal stem cells. Biochem. Pharmacol. 2012;83:661–670. doi: 10.1016/j.bcp.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 8.Rho HS, Hong SH, Park J, Jung HI, Park YH, Lee JH, Shin SS, Noh M. Kojyl cinnamate ester derivatives promote adiponectin production during adipogenesis in human adipose tissue-derived mesenchymal stem cells. Bioorg. Med. Chem. Lett. 2014;24:2141–2145. doi: 10.1016/j.bmcl.2014.03.034. [DOI] [PubMed] [Google Scholar]
- 9.Shin JH, Shin DW, Noh M. Interleukin-17A inhibits adipocyte differentiation in human mesenchymal stem cells and regulates pro-inflammatory responses in adipocytes. Biochem. Pharmacol. 2009;77:1835–1844. doi: 10.1016/j.bcp.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 10.Finucane FM, Luan J, Wareham NJ, Sharp SJ, O’Rahilly S, Balkau B, Flyvbjerg A, Walker M, Hojlund K, Nolan JJ, Savage DB. Correlation of the leptin:adiponectin ratio with measures of insulin resistance in non-diabetic individuals. Diabetologia. 2009;52:2345–2349. doi: 10.1007/s00125-009-1508-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oda N, Imamura S, Fujita T, Uchida Y, Inagaki K, Kakizawa H, Hayakawa N, Suzuki A, Takeda J, Horikawa Y, Itoh M. The ratio of leptin to adiponectin can be used as an index of insulin resistance. Metab., Clin. Exp. 2008;57:268–273. doi: 10.1016/j.metabol.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 12.Shetty S, Kusminski CM, Scherer PE. Adiponectin in health and disease: evaluation of adiponectin-targeted drug development strategies. Trends Pharmacol. Sci. 2009;30:234–239. doi: 10.1016/j.tips.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 13.Fukuen S, Iwaki M, Yasui A, Makishima M, Matsuda M, Shimomura I. Sulfonylurea agents exhibit peroxisome proliferator-activated receptor gamma agonistic activity. J. Biol. Chem. 2005;280:23653–23659. doi: 10.1074/jbc.M412113200. [DOI] [PubMed] [Google Scholar]
- 14.Riera-Guardia N, Rothenbacher D. The effect of thiazolidinediones on adiponectin serum level: a meta-analysis. Diabetes, Obes. Metab. 2008;10:367–375. doi: 10.1111/j.1463-1326.2007.00755.x. [DOI] [PubMed] [Google Scholar]
- 15.Kellinsalmi M, Parikka V, Risteli J, Hentunen T, Leskela HV, Lehtonen S, Selander K, Vaananen K, Lehenkari P. Inhibition of cyclooxygenase-2 down-regulates osteoclast and osteoblast differentiation and favours adipocyte formation in vitro. Eur. J. Pharmacol. 2007;572:102–110. doi: 10.1016/j.ejphar.2007.06.030. [DOI] [PubMed] [Google Scholar]
- 16.Lehmann JM, Lenhard JM, Oliver BB, Ringold GM, Kliewer SA. Peroxisome proliferator-activated receptors alpha and gamma are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J. Biol. Chem. 1997;272:3406–3410. doi: 10.1074/jbc.272.6.3406. [DOI] [PubMed] [Google Scholar]
- 17.Jacobson KA, Gao ZG. Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discovery. 2006;5:247–264. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DiMarco JP, Sellers TD, Berne RM, West GA, Belardinelli L. Adenosine: electrophysiologic effects and therapeutic use for terminating paroxysmal supraventricular tachycardia. Circulation. 1983;68:1254–1263. doi: 10.1161/01.cir.68.6.1254. [DOI] [PubMed] [Google Scholar]
- 19.Wilbur SL, Marchlinski FE. Adenosine as an antiarrhythmic agent. Am. J. Cardiol. 1997;79:30–37. doi: 10.1016/s0002-9149(97)00261-0. [DOI] [PubMed] [Google Scholar]
- 20.Biaggioni I, Paul S, Puckett A, Arzubiaga C. Caffeine and theophylline as adenosine receptor antagonists in humans. J. Pharmacol. Exp. Ther. 1991;258:588–593. [PubMed] [Google Scholar]
- 21.Su SH, Shyu HW, Yeh YT, Chen KM, Yeh H, Su SJ. Caffeine inhibits adipogenic differentiation of primary adipose-derived stem cells and bone marrow stromal cells. Toxicol. In Vitro. 2013;27:1830–1837. doi: 10.1016/j.tiv.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 22.Gharibi B, Abraham AA, Ham J, Evans BA. Contrasting effects of A1 and A2B adenosine receptors on adipogenesis. Int. J. Obes. 2012;36:397–406. doi: 10.1038/ijo.2011.129. [DOI] [PubMed] [Google Scholar]
- 23.He W, Mazumder A, Wilder T, Cronstein BN. Adenosine regulates bone metabolism via A1, A2A, and A2B receptors in bone marrow cells from normal humans and patients with multiple myeloma. FASEB J. 2013;27:3446–3454. doi: 10.1096/fj.13-231233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johansson SM, Lindgren E, Yang JN, Herling AW, Fredholm BB. Adenosine A1 receptors regulate lipolysis and lipogenesis in mouse adipose tissue-interactions with insulin. Eur. J. Pharmacol. 2008;597:92–101. doi: 10.1016/j.ejphar.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 25.Johnston-Cox H, Koupenova M, Yang D, Corkey B, Gokce N, Farb MG, LeBrasseur N, Ravid K. The A2B adenosine receptor modulates glucose homeostasis and obesity. PLoS One. 2012;7:e40584. doi: 10.1371/journal.pone.0040584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gallo-Rodriguez C, Ji XD, Melman N, Siegman BD, Sanders LH, Orlina J, Fischer B, Pu Q, Olah ME, van Galen PJ, Stiles GL, Jacobson KA. Structure-activity relationships of N6-benzyladenosine-5′-uronamides as A3-selective adenosine agonists. J. Med. Chem. 1994;37:636–646. doi: 10.1021/jm00031a014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.(a) Choi WJ, Lee HW, Kim HO, Chinn M, Gao ZG, Patel A, Jacobson KA, Moon HR, Jung YH, Jeong LS. Design and synthesis of N6-substituted-4′-thioadenosine-5′-uronamides as potent and selective human A3 adenosine receptor agonists. Bioorg. Med. Chem. 2009;17:8003–8011. doi: 10.1016/j.bmc.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Qu S, Mulamoottil VA, Nayak A, Ryu S, Hou X, Song J, Yu J, Sahu PK, Zhao LX, Choi S, Lee SK, Jeong LS. Design, synthesis and anticancer activity of C8-substituted-4′-thionucleosides as potential HSP90 inhibitors. Bioorg. Med. Chem. 2016;24:3418–3428. doi: 10.1016/j.bmc.2016.05.041. [DOI] [PubMed] [Google Scholar]
- 28.Batra H, Moriarty RM, Penmasta R, Sharma V, Stanciuc G, Staszewski JP, Tuladhar SM, Walsh DA, Datla S, Krishnaswamy S. A concise, efficient and production-scale synthesis of a protected L-lyxonolactone derivative: an important aldonolactone core. Org. Process Res. Dev. 2006;10:484–486. [Google Scholar]
- 29.(a) Jeong LS, Choe SA, Gunaga P, Kim HO, Lee HW, Lee SK, Tosh DK, Patel A, Palaniappan KK, Gao ZG, Jacobson KA, Moon HR. Discovery of a new nucleoside template for human A3 adenosine receptor ligands: D-4′-thioadenosine derivatives without 4′-hydroxymethyl group as highly potent and selective antagonists. J. Med. Chem. 2007;50:3159–3162. doi: 10.1021/jm070259t. [DOI] [PubMed] [Google Scholar]; (b) Jeong LS, Pal S, Choe SA, Choi WJ, Jacobson KA, Gao ZG, Klutz AM, Hou X, Kim HO, Lee HW, Lee SK, Tosh DK, Moon HR. Structure-activity relationships of truncated D- and L-4′-thioadenosine derivatives as species-independent A3 adenosine receptor antagonists. J. Med. Chem. 2008;51:6609–6613. doi: 10.1021/jm8008647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lohse MJ, Klotz KN, Schwabe U, Cristalli G, Vittori S, Grifantini M. 2-Chloro-N6-cyclopentyladenosine: a highly selective agonist at A1 adenosine receptors. Naunyn-Schmiedeberg's Arch. Pharmacol. 1988;337:687–689. doi: 10.1007/BF00175797. [DOI] [PubMed] [Google Scholar]
- 31.Cusack NJ, Hourani SM. 5′-N-ethylcarboxamidoadenosine: a potent inhibitor of human platelet aggregation. Br. J. Pharmacol. 1981;72:443–447. doi: 10.1111/j.1476-5381.1981.tb10995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nekooeian AA, Tabrizchi R. Effects of CGS 21680, a selective A2A adenosine receptor agonist, on cardiac output and vascular resistance in acute heart failure in the anaesthetized rat. Br. J. Pharmacol. 1998;123:1666–1672. doi: 10.1038/sj.bjp.0701764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.(a) Vassaux G, Gaillard D, Mari B, Ailhaud G, Negrel R. Differential expression of adenosine A1 and A2 receptors in preadipocytes and adipocytes. Biochem. Biophys. Res. Commun. 1993;193:1123–1130. doi: 10.1006/bbrc.1993.1742. [DOI] [PubMed] [Google Scholar]; (b) Borglum JD, Vassaux G, Richelsen B, Gaillard D, Darimont C, Ailhaud G, Negrel R. Changes in adenosine A1- and A2-receptor expression during adipose cell differentiation. Mol. Cell. Endocrinol. 1996;117:17–25. doi: 10.1016/0303-7207(95)03728-4. [DOI] [PubMed] [Google Scholar]
- 34.(a) Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]; (b) Janderova L, McNeil M, Murrell AN, Mynatt RL, Smith SR. Human mesenchymal stem cells as an in vitro model for human adipogenesis. Obes. Res. 2003;11:65–74. doi: 10.1038/oby.2003.11. [DOI] [PubMed] [Google Scholar]
- 35.Kim HO, Ji XD, Siddiqi SM, Olah ME, Stiles GL, Jacobson K. A.2-Substitution of N6-benzyladenosine-5′-uronamides enhances selectivity for A3 adenosine receptors. J. Med. Chem. 1994;37:3614–3621. doi: 10.1021/jm00047a018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.(a) Silverman MH, Strand V, Markovits D, Nahir M, Reitblat T, Molad Y, Rosner I, Rozenbaum M, Mader R, Adawi M, Caspi D, Tishler M, Langevitz P, Rubinow A, Friedman J, Green L, Tanay A, Ochaion A, Cohen S, Kerns WD, Cohn I, Fishman-Furman S, Farbstein M, Yehuda SB, Fishman P. Clinical evidence for utilization of the A3 adenosine receptor as a target to treat rheumatoid arthritis: data from a phase II clinical trial. J. Rheumatol. 2008;35:41–48. [PubMed] [Google Scholar]; (b) Wan TC, Ge ZD, Tampo A, Mio Y, Bienengraeber MW, Tracey WR, Gross GJ, Kwok WM, Auchampach JA. The A3 adenosine receptor agonist CP-532,903 [N6-(2,5-dichlorobenzyl)-3′-aminoadenosine-5′-N-methylcarboxamide] protects against myocardial ischemia/reperfusion injury via the sarcolemmal ATP-sensitive potassium channel. J. Pharmacol. Exp. Ther. 2008;324:234–243. doi: 10.1124/jpet.107.127480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tosh DK, Deflorian F, Phan K, Gao ZG, Wan TC, Gizewski E, Auchampach JA, Jacobson KA. Structure-guided design of A(3) adenosine receptor-selective nucleosides: combination of 2-arylethynyl and bicyclo[3.1.0]hexane substitutions. J. Med. Chem. 2012;55:4847–4860. doi: 10.1021/jm300396n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baraldi PG, Cacciari B, Romagnoli R, Spalluto G, Klotz KN, Leung E, Varani K, Gessi S, Merighi S, Borea PA. Pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine derivatives as highly potent and selective human A(3) adenosine receptor antagonists. J. Med. Chem. 1999;42:4473–4478. doi: 10.1021/jm991114s. [DOI] [PubMed] [Google Scholar]
- 39.Rosen ED, MacDougald OA. Adipocyte differentiation from the inside out. Nat. Rev. Mol. Cell Biol. 2006;7:885–896. doi: 10.1038/nrm2066. [DOI] [PubMed] [Google Scholar]
- 40.Brown PJ, Stuart LW, Hurley KP, Lewis MC, Winegar DA, Wilson JG, Wilkison WO, Ittoop OR, Willson TM. Identification of a subtype selective human PPARα agonist through parallel-array synthesis. Bioorg. Med. Chem. Lett. 2001;11:1225. doi: 10.1016/s0960-894x(01)00188-3. [DOI] [PubMed] [Google Scholar]
- 41.Koldamova RP, Lefterov IM, Staufenbiel M, Wolfe D, Huang S, Glorioso JC, Walter M, Roth MG, Lazo JS. The liver X receptor ligand T0901317 decreases amyloid beta production in vitro and in a mouse model of Alzheimer’s disease. J. Biol. Chem. 2005;280:4079–4088. doi: 10.1074/jbc.M411420200. [DOI] [PubMed] [Google Scholar]
- 42.Choi JH, Banks AS, Estall JL, Kajimura S, Bostrom P, Laznik D, Ruas JL, Chalmers MJ, Kamenecka TM, Bluher M, Griffin PR, Spiegelman BM. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature. 2010;466:451–456. doi: 10.1038/nature09291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brown KK, Henke BR, Blanchard SG, Cobb JE, Mook R, Kaldor I, Kliewer SA, Lehmann JM, Lenhard JM, Harrington WW, Novak PJ, Faison W, Binz JG, Hashim MA, Oliver WO, Brown HR, Parks DJ, Plunket KD, Tong WQ, Menius JA, Adkison K, Noble SA, Willson TM. A novel N-aryl tyrosine activator of peroxisome proliferator-activated receptor-gamma reverses the diabetic phenotype of the Zucker diabetic fatty rat. Diabetes. 1999;48:1415–1424. doi: 10.2337/diabetes.48.7.1415. [DOI] [PubMed] [Google Scholar]
- 44.Oliver WR, Jr, Shenk JL, Snaith MR, Russell CS, Plunket KD, Bodkin NL, Lewis MC, Winegar DA, Sznaidman ML, Lambert MH, Xu HE, Sternbach DD, Kliewer SA, Hansen BC, Willson TM. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc. Natl. Acad. Sci. U. S. A. 2001;98:5306–5311. doi: 10.1073/pnas.091021198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shin DW, Kim SN, Lee SM, Lee W, Song MJ, Park SM, Lee TR, Baik JH, Kim HK, Hong JH, Noh M. (−)-Catechin promotes adipocyte differentiation in human bone marrow mesenchymal stem cells through PPARγ transactivation. Biochem. Pharmacol. 2009;77:125–133. doi: 10.1016/j.bcp.2008.09.033. [DOI] [PubMed] [Google Scholar]
- 46.(a) Kroker AJ, Bruning JB. Review of the structural and dynamic mechanisms of PPARγ partial agonism. PPAR Res. 2015;2015:816856. doi: 10.1155/2015/816856. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Frkic RL, He Y, Rodriguez BB, Chang MR, Kuruvilla D, Ciesla A, Abell AD, Kamenecka TM, Griffin PR, Bruning JB. Structure-activity relationship of 2,4-dichloro-N-(3,5-dichloro-4-(quinolin-3-yloxy)phenyl)benzenesulfonamide (INT131) analogs for PPARγ-targeted antidiabetics. J. Med. Chem. 2017;60:4584–4593. doi: 10.1021/acs.jmedchem.6b01727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shearer BG, Steger DJ, Way JM, Stanley TB, Lobe DC, Grillot DA, Iannone MA, Lazar MA, Willson TM, Billin AN. Identification and characterization of a selective peroxisome proliferator-activated receptor beta/delta (NR1C2) antagonist. Mol. Endocrinol. 2008;22:523–529. doi: 10.1210/me.2007-0190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu S, Reddy JK. Transcription coactivators for peroxisome proliferator-activated receptors. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids. 2007;1771:936–951. doi: 10.1016/j.bbalip.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 49.Shearer BG, Wiethe RW, Ashe A, Billin AN, Way JM, Stanley TB, Wagner CD, Xu RX, Leesnitzer LM, Merrihew RV, Shearer TW, Jeune MR, Ulrich JC, Willson TM. Identification and characterization of 4-chloro-N-(2-{[5-trifluoromethyl)-2-pyridyl]sulfonyl}ethyl)benzamide (GSK3787), a selective and irreversible peroxisome proliferator-activated receptor delta (PPARdelta) antagonist. J. Med. Chem. 2010;53:1857–1861. doi: 10.1021/jm900464j. [DOI] [PubMed] [Google Scholar]
- 50.(a) Shi Y, Hon M, Evans RM. The peroxisome proliferator-activated receptor delta, an integrator of transcriptional repression and nuclear receptor signaling. Proc. Natl. Acad. Sci. U. S. A. 2002;99:2613–2618. doi: 10.1073/pnas.052707099. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zuo X, Wu Y, Morris JS, Stimmel JB, Leesnitzer LM, Fischer SM, Lippman SM, Shureiqi I. Oxidative metabolism of linoleic acid modulates PPAR-beta/delta suppression of PPAR-gamma activity. Oncogene. 2006;25:1225–1241. doi: 10.1038/sj.onc.1209160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burgermeister E, Schnoebelen A, Flament A, Benz J, Stihle M, Gsell B, Rufer A, Ruf A, Kuhn B, Marki HP, Mizrahi J, Sebokova E, Niesor E, Meyer M. A novel partial agonist of peroxisome proliferator-activated receptor-gamma (PPARgamma) recruits PPARgamma-coactivator-1alpha, prevents triglyceride accumulation, and potentiates insulin signaling in vitro. Mol. Endocrinol. 2006;20:809–830. doi: 10.1210/me.2005-0171. [DOI] [PubMed] [Google Scholar]
- 52.Tanaka T, Yamamoto J, Iwasaki S, Asaba H, Hamura H, Ikeda Y, Watanabe M, Magoori K, Ioka RX, Tachibana K, Watanabe Y, Uchiyama Y, Sumi K, Iguchi H, Ito S, Doi T, Hamakubo T, Naito M, Auwerx J, Yanagisawa M, Kodama T, Sakai J. Activation of peroxisome proliferator-activated receptor delta induces fatty acid beta-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc. Natl. Acad. Sci. U. S. A. 2003;100:15924–15929. doi: 10.1073/pnas.0306981100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fishman P, Bar-Yehuda S, Liang BT, Jacobson KA. Pharmacological and therapeutic effects of A3 adenosine receptor agonists. Drug Discovery Today. 2012;17:359–366. doi: 10.1016/j.drudis.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.(a) Madi L, Bar-Yehuda S, Barer F, Ardon E, Ochaion A, Fishman P. A3 adenosine receptor activation in melanoma cells: association between receptor fate and tumor growth inhibition. J. Biol. Chem. 2003;278:42121–42130. doi: 10.1074/jbc.M301243200. [DOI] [PubMed] [Google Scholar]; (b) Lu J, Pierron A, Ravid K. An adenosine analogue, IB-MECA, down-regulates estrogen receptor alpha and suppresses human breast cancer cell proliferation. Cancer Res. 2003;63:6413–6423. [PubMed] [Google Scholar]; (c) Bar-Yehuda S, Madi L, Silberman D, Gery S, Shkapenuk M, Fishman P. CF101, an agonist to the A3 adenosine receptor, enhances the chemotherapeutic effect of 5-fluorouracil in a colon carcinoma murine model. Neoplasia. 2005;7:85–90. doi: 10.1593/neo.04364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Madi L, Ochaion A, Rath-Wolfson L, Bar-Yehuda S, Erlanger A, Ohana G, Harish A, Merimski O, Barer F, Fishman P. The A3 adenosine receptor is highly expressed in tumor versus normal cells: potential target for tumor growth inhibition. Clin. Cancer Res. 2004;10:4472–4479. doi: 10.1158/1078-0432.CCR-03-0651. [DOI] [PubMed] [Google Scholar]
- 56.Fishman P, Bar-Yehuda S, Ohana G, Barer F, Ochaion A, Erlanger A, Madi L. An agonist to the A3 adenosine receptor inhibits colon carcinoma growth in mice via modulation of GSK-3 beta and NF-kappa B. Oncogene. 2004;23:2465–2471. doi: 10.1038/sj.onc.1207355. [DOI] [PubMed] [Google Scholar]
- 57.Cohen S, Stemmer SM, Zozulya G, Ochaion A, Patoka R, Barer F, Bar-Yehuda S, Rath-Wolfson L, Jacobson KA, Fishman P. CF102 an A3 adenosine receptor agonist mediates antitumor and anti-inflammatory effects in the liver. J. Cell. Physiol. 2011;226:2438–2447. doi: 10.1002/jcp.22593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.(a) Kim SG, Ravi G, Hoffmann C, Jung YJ, Kim M, Chen A, Jacobson KA. p53-Independent induction of Fas and apoptosis in leukemic cells by an adenosine derivative, Cl-IB-MECA. Biochem. Pharmacol. 2002;63:871–880. doi: 10.1016/s0006-2952(02)00839-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Morello S, Petrella A, Festa M, Popolo A, Monaco M, Vuttariello E, Chiappetta G, Parente L, Pinto A. Cl-IB-MECA inhibits human thyroid cancer cell proliferation independently of A3 adenosine receptor activation. Cancer Biol. Ther. 2008;7:278–284. doi: 10.4161/cbt.7.2.5301. [DOI] [PubMed] [Google Scholar]
- 59.(a) Youssef J, Badr M. Peroxisome proliferator-activated receptors and cancer: challenges and opportunities. Br. J. Pharmacol. 2011;164:68–82. doi: 10.1111/j.1476-5381.2011.01383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Piemontese L, Cerchia C, Laghezza A, Ziccardi P, Sblano S, Tortorella P, Iacobazzi V, Infantino V, Convertini P, Dal Piaz F, Lupo A, Colantuoni V, Lavecchia A, Loiodice F. New diphenylmethane derivatives as peroxisome proliferator-activated alpha/gamma dual agonists endowed with antiproliferative effects and mitochondrial activity. Eur. J. Med. Chem. 2017;127:379–397. doi: 10.1016/j.ejmech.2016.12.047. [DOI] [PubMed] [Google Scholar]
- 60.(a) Wang D, Wang H, Guo Y, Ning W, Katkuri S, Wahli W, Desvergne B, Dey SK, DuBois RN. Crosstalk between peroxisome proliferator-activated receptor delta and VEGF stimulates cancer progression. Proc. Natl. Acad. Sci. U. S. A. 2006;103:19069–19074. doi: 10.1073/pnas.0607948103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xu M, Zuo X, Shureiqi I. Targeting peroxisome proliferator-activated receptor-beta/delta in colon cancer: how to aim? Biochem. Pharmacol. 2013;85:607–611. doi: 10.1016/j.bcp.2012.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zaveri NT, Sato BG, Jiang F, Calaoagan J, Laderoute KR, Murphy BJ. A novel peroxisome proliferator-activated receptor delta antagonist, SR13904, has anti-proliferative activity in human cancer cells. Cancer Biol. Ther. 2009;8:1252–1261. doi: 10.4161/cbt.8.13.8691. [DOI] [PubMed] [Google Scholar]
- 62.Gupta RA, Wang D, Katkuri S, Wang H, Dey SK, DuBois RN. Activation of nuclear hormone receptor peroxisome proliferator-activated receptor-delta accelerates intestinal adenoma growth. Nat. Med. 2004;10:245–247. doi: 10.1038/nm993. [DOI] [PubMed] [Google Scholar]
- 63.Palma A, Sainaghi PP, Amoruso A, Fresu LG, Avanzi G, Pirisi M, Brunelleschi S. Peroxisome proliferator-activated receptor-gamma expression in monocytes/macrophages from rheumatoid arthritis patients: relation to disease activity and therapy efficacy-a pilot study. Rheumatology. 2012;51:1942–1952. doi: 10.1093/rheumatology/kes177. [DOI] [PubMed] [Google Scholar]
- 64.Ormseth MJ, Oeser AM, Cunningham A, Bian A, Shintani A, Solus J, Tanner S, Stein CM. Peroxisome proliferator-activated receptor gamma agonist effect on rheumatoid arthritis: a randomized controlled trial. Arthritis Res. Ther. 2013;15:R110. doi: 10.1186/ar4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hack K, Reilly L, Palmer C, Read KD, Norval S, Kime R, Booth K, Foerster J. Skin-targeted inhibition of PPAR beta/delta by selective antagonists to treat PPAR beta/delta-mediated psoriasis-like skin disease in vivo. PLoS One. 2012;7:e37097. doi: 10.1371/journal.pone.0037097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stafslien DK, Vedvik KL, De Rosier T, Ozers MS. Analysis of ligand-dependent recruitment of coactivator peptides to RXRbeta in a time-resolved fluorescence resonance energy transfer assay. Mol. Cell. Endocrinol. 2007;264:82–89. doi: 10.1016/j.mce.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 67.(a) Chen JD, Evans RM. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature. 1995;377:454–457. doi: 10.1038/377454a0. [DOI] [PubMed] [Google Scholar]; (b) Chang C, Norris JD, Gron H, Paige LA, Hamilton PT, Kenan DJ, Fowlkes D, McDonnell DP. Dissection of the LXXLL nuclear receptor-coactivator interaction motif using combinatorial peptide libraries: discovery of peptide antagonists of estrogen receptors alpha and beta. Mol. Cell. Biol. 1999;19:8226–8239. doi: 10.1128/mcb.19.12.8226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.