

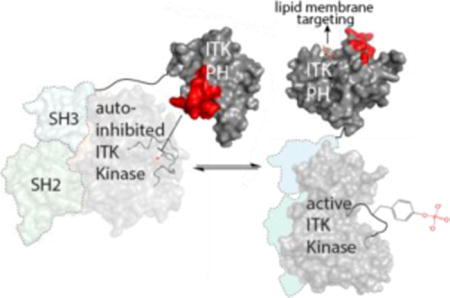
Abstract
Pleckstrin homology (PH) domains are well known as phospholipid binding modules yet evidence is mounting that PH domain function extends beyond lipid recognition. In the current work, we characterize a protein binding function for the PH domain of Interleukin-2-inducible tyrosine kinase (ITK), an immune cell specific signaling protein that belongs to the TEC family of non-receptor tyrosine kinases. Its N-terminal Pleckstrin homology (PH) domain is a well characterized lipid-binding module that localizes ITK to the membrane via phosphatidylinositol (3,4,5)-trisphosphate (PIP3) binding. Using a combination of NMR spectroscopy and mutagenesis, we have mapped an autoregulatory protein interaction site on the ITK PH domain that makes direct contact with the catalytic kinase domain of ITK inhibiting the phospho-transfer reaction. Moreover, we have elucidated an important interplay between lipid binding by the ITK PH domain and the stability of the autoinhibitory complex formed by full length ITK. The ITK activation loop in the kinase domain becomes accessible to phosphorylation to the exogenous kinase LCK upon binding of the ITK PH domain to PIP3. By clarifying the allosteric role of the ITK PH domain in controlling ITK function we have expanded the functional repertoire of the PH domain generally and opened the door to alternative strategies to target this specific kinase in the context of immune cell signaling.
Graphical abstract
Introduction
Protein kinase activity is tightly regulated before, during and after cellular signaling events1. The molecular mechanisms responsible for controlling kinase catalytic activity are varied and can depend on the nature of the non-catalytic domains within a particular kinase family. For example, biochemical data and high resolution crystal structures revealed the regulatory role of the SRC Homology 2 and SRC Homology 3 (SH2 and SH3) domains within the SRC kinases (the largest family of non-receptor tyrosine kinases)2. In the autoinhibited SRC kinase, these regulatory modules pack against the kinase domain in a manner that stabilizes the inactive conformation of the catalytic domain3. Mutations, small molecules or peptides that displace the SH2 and/or SH3 domains from the kinase domain allow the catalytic domain to sample the active conformation leading to efficient phosphorylation of substrates.
The TEC kinases (ITK, BTK, TEC, BMX and TXK) are the second largest family of non-receptor tyrosine kinases and serve important signaling roles in hematopoietic cells during development as well as during activation of mature lymphocytes4, 5. Dysregulation of TEC family members are associated with immunodeficiencies and malignancies6, 7 and so a better understanding of the regulatory machinery of these kinases will aid therapeutic developments. A simple extrapolation of the SRC regulatory mechanism to the TEC family has not been possible, in part due to the presence of a Pleckstrin Homology (PH) domain. PH domains are well known lipid-binding modules that exhibit specific binding to a range of phosphatidylinositol molecules8-12. The TEC family PH domains bind specifically to phosphatidylinositol (3,4,5)-trisphosphate (PIP3) that is transiently produced at the plasma membrane upon antigen receptor activation13. PH domain mediated lipid binding co-localizes the TEC family kinases with the requisite adaptor proteins and substrate.
The PH domain of Interleukin-2 inducible tyrosine kinase (ITK) also appears to play an important regulatory role in controlling ITK catalytic activity. We have previously reported biochemical data that show a significant increase in turnover number upon deletion of the N-terminal PH domain of ITK14. More recently, crystal structures of a tethered fragment of Bruton’s tyrosine kinase (BTK) reveal contacts between the BTK PH and kinase domains15. For the non-TEC family kinase, AKT, the regulatory role of the PH domain has also become clear through crystallography16.
The full length T cell specific TEC kinase ITK has so far eluded crystallization and so we set out to characterize the regulatory role of the ITK PH domain in detail using a range of biochemical approaches and solution NMR spectroscopy. Our findings suggest that the ITK PH domain contacts the ITK kinase domain directly and exerts an inhibitory effect on ITK catalytic activity. We identify a specific surface of the ITK PH domain that mediates contacts with the ITK kinase domain and show that mutation of this region of the N-terminal PH domain activates ITK kinase activity. The soluble head group of PIP3, inositol (1,3,4,5)-tetrakisphosphate (IP4), competes with the regulatory interaction between ITK PH and kinase domains consistent with the long standing observation that PIP3 production contributes to ITK activation in the activated T cell. Moreover, we find that PIP3 containing liposomes activate full length ITK but not an ITK fragment that lacks the PH domain. Finally, we show that phospholipid engagement of the ITK PH domain exposes the activation loop tyrosine in the ITK kinase domain making it more accessible to phosphorylation by the SRC family kinase LCK. The negative regulatory interface we have identified on the ITK PH domain overlaps with the previously reported Ca2+/Calmodulin (CaM) binding site on ITK, consistent with the observation that Ca2+/CaM amplifies ITK signaling17. The findings reported here provide further insight into the expanding repertoire of the PH domain beyond lipid recognition and sets the stage for targeting this newly described allosteric mechanism in developing therapeutic approaches that complement more traditional ATP inhibitors that bind to the kinase active site.
Materials and Methods
Constructs
Baculoviral constructs encoding FLAG-tagged full length ITK (WT and K390R) and SH3-SH2-kinase (K390R) fragment (P171-L619) of ITK has been described elsewhere14. Similarly, bacterially expressed construct of His6-GB1 tagged ITK PHTH (C96E/T110I) domain has been described previously18. The baculoviral construct of ITK kinase domain was produced using the Gateway cloning system (Invitrogen). Briefly, His6 tagged mouse ITK kinase domain (residues 356-619) was PCR amplified and cloned into pENTR/D-TOPO vector (Invitrogen). The pENTR/D-TOPO vector was then recombined with BaculoDirect C-Term Linear DNA (Invitrogen) using LR Clonase II (Invitrogen). The recombined linear DNA was then transfected into Sf9 cells using Cellfectin II reagent (Invitrogen). Baculovirus was selected and amplified for three rounds. All the mutations in the ITK PHTH domain and full length ITK were introduced by site directed mutagenesis (QuikChange Lightning Site-Directed mutagenesis kit, Agilent). N-terminal His6-GB1-tagged BTK PHTHSH3 domain (mouse, aa 1-270), or BTK PHTH domain (aa 1-176) were cloned into the pET20b vector (EMD-Millipore). C-terminally His6-tagged BTK kinase domain (mouse, aa 396-659) and linker-kinase domain (mouse, aa 382-659) were cloned into the pET28a vector (EMD-Millipore). In addition, both the BTK kinase domain and the BTK linker kinase domain constructs had a Y617P mutation which enabled soluble expression in bacteria. The mouse LCK kinase domain (aa Q230-P509) construct with an N-terminal 6His-tag in pET-28a vector was a kind gift from Dr. John Kuriyan. All constructs were verified by sequencing at the Iowa State University DNA Synthesis and Sequencing Facility.
Protein purification
Baculovirus production, expression and purification of all constructs of FLAG tagged full length ITK (WT and K390R), and FLAG tagged SH3-SH2-Kinase (K390R) fragment of ITK were carried out as described previously14 with the exception of use of suspension High Five™ cells (Invitrogen) rather than adherent Sf9 cells for protein production. For His6 tagged ITK kinase domain production, High Five™ cells were infected with baculovirus and harvested 48 hours post infection. The cell pellet was resuspended in lysis buffer containing 20 mM Tris pH 8, 150 mM NaCl and 10 mM Imidazole. The cells were lysed by dounce homogenization and centrifuged at 16,000 rpm at 4°C for 1 hour. Glycerol was added to supernatant to a final volume of 10% and incubated with Ni-NTA resin overnight at 4°C on a rotary mixer. The resin was washed 5× with wash buffer containing 20 mM Tris pH 8, 150 mM NaCl, 20 mM Imidazole and 10% glycerol. Protein was eluted with elution buffer containing 20 mM Tris pH 8, 150 mM NaCl, 250 mM Imidazole and 10% glycerol. Eluted protein was passed through HiLoad 26/60 Superdex 75 prep grade column (GE Healthcare Life Sciences) pre-equilibrated with buffer 20 mM Tris pH 8, 150 mM NaCl and 10% glycerol. The pure protein containing fractions were pooled, concentrated and snap frozen and stored at −80°C. His6-GB1 tagged ITK PHTH domain, BTK PHTH domain, BTK PHTHSH3 domain, His6-tagged BTK kinase domain or His6-tagged BTK linker kinase domain proteins were expressed in Escherichia coli BL21 (DE3) cells and purified as described previously with some modifications14, 18. After elution of protein from Ni-NTA column the protein was concentrated to 5 ml and injected onto a HiLoad 26/60 Superdex 75 prep grade column equilibrated with buffer composed of 50 mM KH2PO4 pH 7.5, 150 mM NaCl. The eluted protein was then concentrated and subjected to His6-GB1 tag cleavage with Factor Xa (Novagen) for 16 hours at room temperature. The cleavage reaction was stopped with 1 mM PMSF and loaded onto a Ni NTA column to remove His6-GB1 and any uncleaved fusion protein. The flow-through was again passed through HiLoad 26/60 Superdex 75 prep grade column pre equilibrated with buffer containing Tris pH 8, 150 mM NaCl and 10% glycerol. His6 tagged LCK kinase domain was expressed and purified from ArcticExpress cells as described previously19. The LCK protein was eluted with elution buffer containing 20 mM Tris pH 8, 150mM NaCl, 250mM Imidazole and 10% glycerol. The protein was flash frozen and stored at −80°C.
PHTH/Kinase domain interaction assay
Purified ITK His6-GB1 PHTH domain (2 μM or 4 μM) was immobilized on 20 μL (50% slurry) IgG Sepharose beads (GE Healthcare) and was incubated with purified His6-tagged ITK kinase domain (4 μM) in buffer containing 20 mM Tris pH 8, 150 mM NaCl and 10% glycerol at 4°C overnight. The samples were washed 5 times in the same buffer, suspended in 2X SDS loading buffer and boiled. Samples were run on a 12% SDS-PAGE gel for 120 minutes at 180 volts and transferred onto a PVDF membrane and western blotted with anti His6 antibody (Millipore). For pull down assays involving various soluble inositol phosphates (Echelon Biosciences and Cayman chemicals), purified ITK His6-GB1 PHTH (2 μM) was incubated with specified inositol phosphates for 20 minutes before adding His6-tagged ITK kinase domain.
NMR spectroscopy
NMR spectra for ITK PHTH domain in NMR buffer (50mM HEPES pH 8, 150mM NaCl, 10% glycerol) was obtained on a Bruker AVII 700 spectrometer with a 5 mm HCN z-gradient cryoprobe operating at 1H frequency 700.13 MHz with sample temperature of 298K. NMR titration was carried out by collecting 1H-15N HSQC spectra of 200 μM 15N labeled ITK PHTH domain with increasing concentration of unlabeled ITK kinase domain (0, 26, and 50 μM). Buffer with 10% glycerol was necessary to ensure the stability of ITK kinase domain. NMRviewJ20 was used for NMR spectra visualization and analysis.
In vitro phosphorylation assay
150 nM full length ITK wild type and mutants (K48D/R49D, V28E/F30Y) and 1μM PLCγ1 SH2C-linker substrate21 were incubated in an in vitro kinase assay buffer composed of 50 mM HEPES pH 7, 10 mM MgCl2, 1 mM DTT, 1 mg/ml BSA, 1 mM Pefabloc, and 200 μM ATP for various time points (0, 30, 60 and 120 minutes) at room temperature. The samples were boiled for 5 minutes, separated by SDS-PAGE and transferred into a PVDF membrane. The membranes were blotted with anti-BTK pY551 (BD Biosciences) to detect pY511 levels in ITK. Anti-BTK pY511 recognizes the Y511 phosphorylated form of ITK. Anti-ITK (2F12, Thermo Scientific) and PLCγ1 pY783 (EMD Millipore) monoclonal antibody were used to detect total enzyme levels and PLCγ1 pY783 levels, respectively. Total PLCγ1 level was probed by Coomassie staining. In kinase assays to test the effect of inositol lipids, ITK enzyme was pre incubated with soluble inositol phosphates or PIP3 liposomes for ten minutes before starting the reaction.
For the ITK loop accessibility kinase assay using active LCK kinase domain, 1 μM full length ITK (K390R) or ITK SH3-SH2-kinase (K390R) fragment were incubated with 100 μM liposomes for 10 minutes prior to the addition of 200 nM LCK kinase domain to start the reaction. The reaction was carried out for 5 minutes at room temperature. The pY511 level was detected as described above. Total ITK and ITK SH3-SH2-Kinase level was detected with an anti-FLAG antibody (Sigma) and total LCK level was detected with anti His6 antibody (Millipore).
Liposome preparation
1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dioleol-sn-glycero-3-[phospho-L-serine] (DOPS) and 1-stearoyl-2-arachidonoyl-sn-glycero-3[phosphoinsositol-3,4,5-triphosphate] (PIP3) (Avanti polar lipids) were separately dissolved in chloroform. For liposomes containing PIP3, all three lipids were mixed in 75:10:5 molar ratio (DOPC:DOPS:PIP3). For control liposome (without PIP3) DOPC and DOPS were mixed in 79:21 molar ratio (DOPC:DOPS). After mixing, chloroform was evaporated by gentle flow of nitrogen into the test tube containing the lipid mixture. The lipid mixture was then dried overnight at room temperature in a vacuum desiccator. The dried lipid films were hydrated for one hour at room temperature with buffer containing 50 mM HEPES (pH 7) such that the final liposome concentration is 12 mM. The hydrated lipid films were then subjected to five freeze thaw cycles using liquid nitrogen and a 37°C water bath. Unilamellar liposomes were created using the mini extrusion set (Avanti polar lipids) by passing through a 100 nM filter (Whatman). Liposomes were then stored at 4°C and used within 48 hours.
CD spectroscopy
CD spectra of 5 μM ITK PHTH wild type and mutants were acquired on a Jasco J-710 in the far UV region (190-240 nm). The samples were prepared in buffer containing 10 mM NaH2PO4 (pH 7.4). CD spectra were obtained at a scanning rate of 100 nm/min with a spectral bandwidth of 2 nm and response time of 1 s. All CD spectra are the average of two scans and were converted from mdegree into molar residue ellipticity (deg cm2 dmol−1).
Results
Full length ITK kinase consists of three regulatory domains: PHTH, SH3, and SH2 that are positioned N-terminal to the catalytic kinase domain (Fig. 1a). It should be noted here that the TH (or TEC Homology) region of the TEC family kinases is considered an extension of the PH fold and so throughout this work we are using the larger ITK fragment that contains both PH and TH regions (ITK PHTH). Based on previous biochemical findings that suggest a role for the PHTH domain in controlling kinase activity14, we set out to better understand how the ITK PHTH region exerts control over ITK mediated phosphorylation. We first expressed and purified His6-GB1 fused ITK PHTH domain and a separate His6 tagged ITK kinase domain (Fig. 1a).
Figure 1. The ITK PHTH domain interacts with the ITK kinase domain in trans.
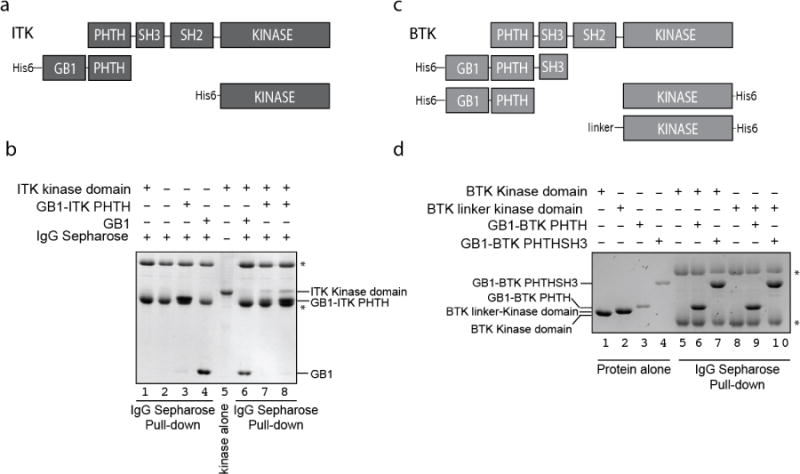
(a) Domain constructs of ITK used in this study. Full length ITK is shown above the His6-GB1-tagged PHTH domain fragment of ITK (residues 1-154) and the His6-tagged Kinase domain fragment (residues 356-619). (b) Coomassie gel showing ITK kinase domain (4 μM) associates with ITK PHTH domain (2 and 4 μM in lanes 7 and 8, respectively) immobilized on IgG sepharose beads. Lanes 1 shows the ITK kinase domain alone does not associate with IgG sepharose, lanes 2, 3 and 4 are loading controls. Lane 5 shows the ITK kinase domain and asterisks show the IgG heavy and light chains. Lane 6 shows that the ITK kinase domain does not associate with IgG sepharose bound His6-GB1 that lacks ITK PHTH. (c) Domain constructs of BTK used in this study. Full length BTK is shown above the His6-GB1-tagged PHTHSH3 (residues 1-270), the His6-GB1-tagged PHTH (residues 1-176) and the His6-tagged Kinase domain fragments of BTK (residues 396-659 (Kinase domain) and residues 382-659 (Linker-Kinase domain)). (d) Coomassie gel showing that neither the BTK kinase domain (1 μM) or the BTK linker-kinase domain (1 μM) associates in this assay with either the BTK PHTH domain (2 μM) or the larger BTK PHTHSH3 construct (2 μM) immobilized on IgG sepharose beads (lanes 6,7,9 and 10). Protein alone loading controls are shown on the left side of the gel. As in (b) asterisks indicate IgG heavy and light chains.
The purified ITK N-terminal fragment, His6-GB1-PHTH (Fig. 1a) was immobilized using IgG sepharose and incubated with the ITK kinase domain. Following extensive washing of the sepharose beads, we find that the ITK kinase domain binds to the ITK PHTH fragment in a concentration dependent manner (Fig. 1b, lanes 7,8). Interestingly, the analogous interaction is not detected in similar experiments with the BTK PHTH and kinase domains (Fig. 1c,d, lanes 6,9). A large loop insertion in the BTK PHTH domain makes it similar in size to the BTK kinase domain and so we assessed the possibility of a direct interaction using the larger BTK His6-GB1-PHTH-SH3 fragment and the BTK His6-Kinase as well as the BTK linker-kinase domain (Fig. 1c,d, lanes 7,10). No interaction is detected between these N-terminal and C-terminal domains of BTK (Fig. 1d) under the same conditions used to detect the ITK PHTH/kinase interaction (Fig. 1b). These data therefore suggest that ITK PHTH interacts in trans with the ITK kinase domain and a corresponding in trans interaction between the BTK PHTH and kinase domains is not detected in the assay.
To further characterize the ITK PHTH/kinase interaction we expressed and purified 15N-labeled ITK PHTH for NMR spectroscopy. Using previously published assignments for an ITK PHTH variant harboring two mutations ((C96E/T110I) that improve expression and solubility17, 18, we titrated unlabeled ITK kinase domain into the labeled ITK PHTH domain and acquired 1H-15N HSQC spectra at each point of the titration (Fig. 2a-c). Many of the PHTH amide NH resonances in the HSQC spectrum do not change throughout the course of the titration (Fig. 2b,c). A subset of ITK PHTH resonances, however, show selective disappearance as the concentration of ITK kinase domain is increased (Fig. 2b,c). Mapping these spectral changes onto a structural model of ITK PHTH shows the residues that are affected by binding of ITK kinase domain are primarily located in two regions: across β-strands 2-4 of the PH domain and one side of the long α-helix of the PH domain fold (Fig. 2d).
Figure 2. NMR mapping of the ITK PHTH regulatory interface.
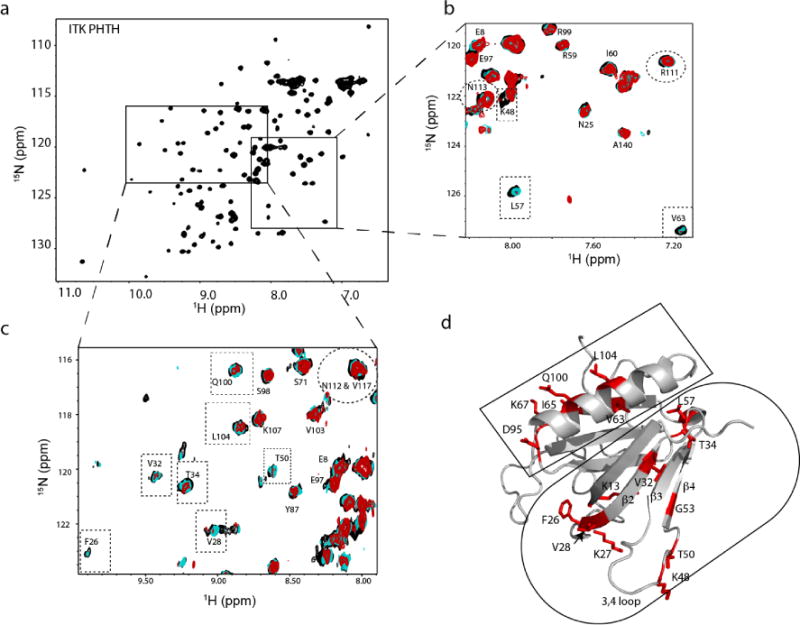
(a) [15N,1H]-HSQC spectrum of uniformly 15N-labeled ITK PHTH domain. (b,c) Addition of unlabeled ITK kinase domain to uniformly 15N-labeled ITK PHTH domain results in significant broadening and in some cases chemical shift changes of selected peaks (boxed) in the overlay of [15N, 1H] HSQC spectra acquired during the titration. A large subset of ITK PHTH resonances do not change over the course of the titration. ITK R111, N112 and N113 (corresponding to R133, Y134 and N135 in BTK), do not change during the titration (circled). Black spectrum corresponds to ITK PHTH alone (200 μM), cyan spectrum acquired after addition of 26 μM unlabeled ITK kinase domain and red spectrum acquired after addition of 50 μM unlabeled ITK kinase domain. (d) Computational model of ITK PHTH based on crystal structures of the corresponding BTK domain fragment (PDB IDs: 1BTK and 1B55); threading was performed using I-TASSER34. ITK PHTH residues for which spectral changes were observed in the NMR data shown in (a-c) are shown in red and labeled. The residues located on β-strands 2-4 are circled and those on the α-helix are boxed. All structural figures were made using PyMol8.
NMR spectral changes are sensitive to small changes in local environment and often map a surface or region that is larger than the actual interface between two proteins in a complex. We therefore used mutagenesis to refine the binding site on the ITK PHTH domain that mediates the interaction with the ITK kinase domain. Based on NMR spectral changes, we first mutated residues within the beta strand region (β2-β4) as well as residues on the β3-β4 loop. Mutations were chosen to directly probe residues for which NMR spectral changes were observed (Fig. 2d) and to assess other residues in this beta-strand region that have surface exposed sidechains but lacked an observable spectral change either due to intramolecular hydrogen bonding, adverse conformational dynamics and/or lack of assignment. Specific amino acid mutations were based on either the corresponding residue in BTK, a charge reversal or mutation to alanine. The extent of interaction of wild type ITK PHTH and each PHTH mutant with the ITK kinase domain was assessed using the same pull down assay described in Figure 1 (Fig. 3a,b). A greater than 50% decrease in binding of the ITK kinase domain to ITK PHTH was observed upon mutation of K48/R49 or V28/F30 (Fig. 3b). Separate mutation of two additional residues, L51D and E56A, causes a 50% decrease in binding between the ITK kinase and PHTH domains (Fig. 3b). To further probe the ITK PHTH surface, we mutated additional surface residues across the entire ITK PHTH domain (including surface exposed side chains on the α-helix) and find only small changes in ITK kinase/PHTH binding (Fig. 3c,d) suggesting that these additional sidechains do not substantially contribute to the interaction between the two domains.
Figure 3. NMR based mutagenesis refines the surface of the ITK PHTH domain that contacts the ITK kinase domain.
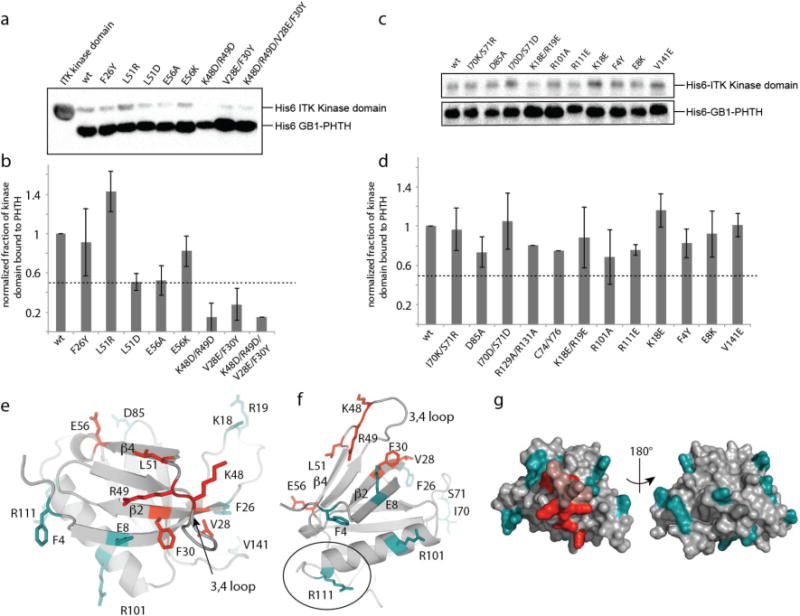
(a,c) Anti-His6 blots showing ITK kinase domain (4μM) associates to different extents with wild type and mutant ITK PHTH domain (2μM) that is immobilized on IgG sepharose beads. (b,d) The fraction of ITK kinase domain bound to wild type and mutant ITK PHTH domains; bound kinase is normalized to His6-GB1 PHTH level in each lane and then compared to wild type (wt). The dotted line shows a decrease in ITK PHTH/kinase domain interaction of 50%. Data shown in (a) and (c) are representative of three independent experiments. (e,f) The binding surface determined from the data in (a) and (c) is mapped onto the ITK PHTH domain computational model, labeled and shown in red. The interface residues are located on the β4 and β2 strands as well as the β3-β4 loop. Those residues that, upon mutation, do not disrupt the ITK PHTH/kinase domain interaction are labeled and shown in cyan. In (f) R111 is circled as it corresponds to the BTK residue R133 that is at the interface of the tethered BTK PHTH and kinase domains for which a crystal structure was recently solved15. (g) Two views of the ITK PHTH surface residues shown in (e) and (f) where red indicates involvement in the ITK PHTH/kinase interaction and cyan corresponds to those residues for which mutation does not affect the ITK PHTH/kinase interaction. The β3-β4 loop residues that are within the binding site identified here are colored pink but were not probed directly by mutagenesis.
Mapping the mutagenesis results onto a model of the ITK PHTH domain shows that the residues that mediate binding of the ITK kinase domain to ITK PHTH cluster in one region of the PHTH domain and those residues that do not contribute to the interaction based on mutagenesis data are outside of this newly defined binding surface (Fig. 3e-g). K48 and R49 are located at the center of the binding site and the surrounding secondary structural elements that mediate the PHTH/kinase interaction include the beta strands β2 and β4 and the β3-β4 loop (Fig. 3e,f).
Our previous biochemical work assessing the activity of ITK deletions14 suggested that the ITK PHTH region inhibits ITK kinase domain activity. Having mapped the surface on the ITK PHTH domain responsible for the interaction with the ITK kinase domain, we next introduced specific PHTH domain mutations into full length ITK and measured both ITK activation loop autophosphorylation (pY511) and phosphorylation of the ITK substrate PLCγ1 (pY783) to determine the extent to which specific PHTH domain residues affect ITK catalytic activity. Mutation of two pairs of PHTH domain residues, K48/R49 and V28/F30, caused the largest decrease in the ITK PHTH/kinase domain interaction (Fig. 3a,b) and so these residues were mutated in full length ITK. Using CD spectroscopy, we first established that these mutations do not alter the overall fold of the PHTH domain but retain the secondary structure of the wild type sequence (Fig. 4a). The kinase activity of full length wild type ITK was then compared to the full length double mutants, ITK K48D/R49D and ITK V28E/F30Y, ensuring that each enzyme is not phosphorylated on the activation loop tyrosine (Y511) at the beginning of the assay (Fig. 4b-d). Both sets of mutations in the PHTH domain affect ITK catalytic activity; activation loop phosphorylation levels (pY511) and phosphorylation of exogenous PLCγ1 are increased compared to wild type ITK (Fig. 4b-d). While both sets of mutations increase ITK activity, mutation of K48/R49, at the center of the newly defined binding surface on the ITK PHTH domain (Fig. 3e), has a greater effect on the kinase activity of full length ITK than does the more peripheral V28E/F30Y mutation. Thus, mutations in the ITK PHTH domain that disrupt the interaction with the kinase domain appear to reduce autoinhibition and increase catalytic function of the ITK kinase domain.
Figure 4. Mutation of the ITK PHTH interface residues in full length ITK increases ITK catalytic activity.
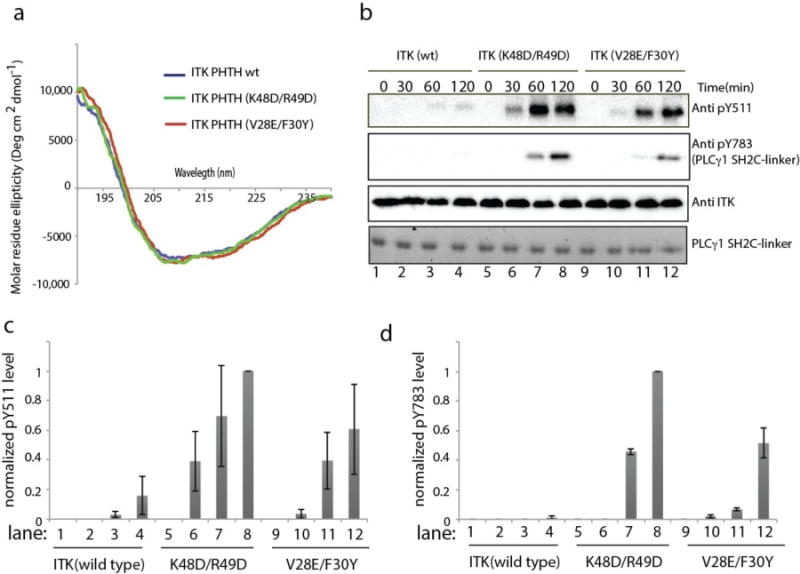
(a) Circular dichroism spectra for wild type ITK PHTH (blue), ITK PHTH (K48D/R49D) (green) and ITK PHTH (V28E/F30Y) (red). (b) Kinase assay spanning 0-120 minutes showing ITK activation loop autophosphorylation (pY511) and phosphorylation of exogenous PLCγ1 (pY783) for full length, wild type ITK (150 nM) in lanes 1-4, full length ITK mutant K48D/R49D (150 nM) in lanes 5-8, and full length ITK mutant V28E/F30Y (150 nM) in lanes 9-12. Phosphorylation of ITK Y511 is detected using anti-BTK pY551 (labeled Anti pY511 for clarity throughout), PLCγ1 pY783 levels are detected using anti-pY783 antibody and total enzyme levels are detected using anti-ITK antibody. Total PLCγ1 substrate levels (SH2C-linker) are detected using Coomassie stain. (c,d) Histogram representation of normalized pY511 (c) and pY783 (d) levels from the ITK kinase assays shown in (b). The intensities of the band corresponding to pY511 and pY783 were divided by the total ITK enzyme level in each lane. The value of these ratios for ITK (K48D/R49D) at time point 120 minutes was set to 1. All other intensity ratios are shown relative to this value. Experiments were run in triplicate.
In the context of cell signaling, the PHTH domain of ITK binds to phosphatidylinositol (3,4,5)-trisphosphate (PIP3) at the membrane via a conserved binding pocket. The lipid binding pocket on ITK PHTH is adjacent to the cluster of amino acids we have identified here that mediate an autoinhibtory interaction with the ITK kinase domain (Fig. 5a). In fact, R29, which is located between V28 and F30 in the ITK PHTH domain, is the conserved arginine that mediates binding to the negatively charged lipid head group. Given the juxtaposition of this lipid binding residue between two residues that mediate the autoinhibitory PHTH/kinase interaction, we decided to avoid mutagenesis and instead test whether binding of the soluble head group of PIP3, inositol (1,3,4,5)-tetrakisphosphate (IP(1,3,4,5)P4), affects the interaction between ITK PHTH and ITK kinase domain. In this assay we compare the interaction between ITK PHTH and ITK kinase domain in the presence of increasing concentration of IP(1,3,4,5)P4 and a control inositol (1,3,4,6)-tetrakisphosphate (IP(1,3,4,6)P4) (Fig. 5b,c). The amount of ITK kinase domain that associates with ITK PHTH decreases in the presence of increasing inositol 1,3,4,5 tetrakisphosphate ligand concentration but not the inositol 1,3,4,6 tetrakisphosphate regioisomer (Fig. 5b,c). To further test specificity we examined a panel of inositol phosphates and find that the inositol tetrakisphosphate (IP4), specifically, inositol (1,3,4,5)-tetrakisphosphate is the only soluble lipid among eleven tested that disrupts the ITK PHTH/kinase domain interaction to greater than 50% (Fig. 5d,e (lane 8)). This suggests that the ITK PHTH/kinase domain interaction is mutually exclusive with lipid binding to the ITK PHTH domain and is specific to the soluble lipid head group of PIP3.
Figure 5. The ITK PHTH/Kinase interaction is selectively disrupted by binding of soluble inositol 1,3,4,5-tetrakisphosphate (IP(1,3,4,5)P4).
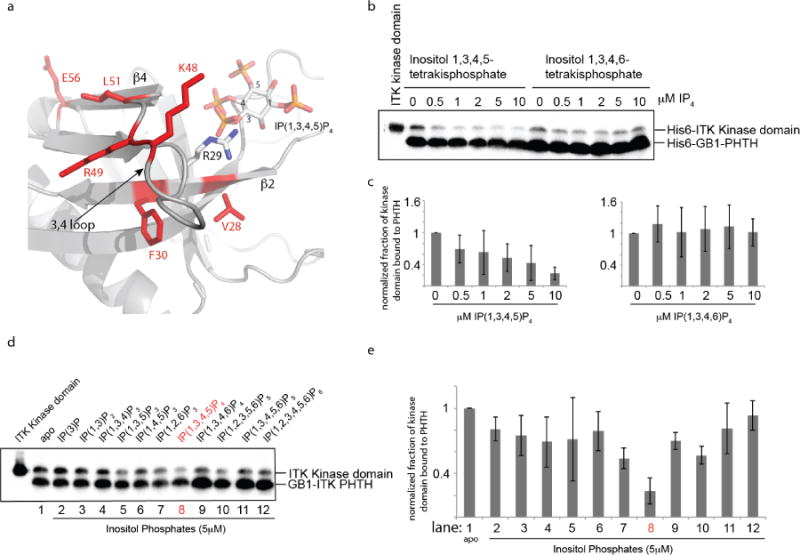
(a) Close up view of the ITK PHTH interface residues that mediate contact to the ITK kinase domain (labeled and in red) and the adjacent lipid binding site. The computational model of the ITK PHTH domain was aligned with the crystal structure of BTK PHTH domain bound to IP(1,3,4,5)P4 (PDB ID: 1B55). BTK PHTH domain is not shown for clarity. The IP4 head group of PIP3 is shown bound to the PHTH domain and positions 1,3,4, and 5 are labeled. The position of the conserved arginine that binds IP4 is indicated to show that it sits between two residues that mediate contacts to the ITK kinase domain (V28 and F30). (b) Anti-His6 blots showing ITK kinase domain (4μM) binding to the ITK PHTH domain (2μM) immobilized on IgG sepharose beads in the presence of soluble inositol 1,3,4,5-tetrakisphosphate (IP(1,3,4,5)P4) and the regioisomeric IP4, inositol 1,3,4,6-tetrakisphosphate (IP(1,3,4,6)P4). The first lane shows total ITK kinase domain input followed by increasing concentration of IP4 compounds (0-10 μM). (c) Histogram representation of the fraction of ITK kinase domain bound to the ITK PHTH domain at 0, 0.5, 1, 2, 5 and 10 μM IP4 (data for inositol 1,3,4,5-tetrakisphosphate is on the left and inositol 1,3,4,6-tetrakisphosphate is on the right). Bound kinase is normalized to His6-GB1 PHTH level in each lane and then compared to the amount of bound kinase in the absence of IP4 (0 μM). (d,e) The same experiment described for panels (b) and (c) is carried out for a panel of inositol phosphates. Lane 1 (apo) contains no added inositol phosphate and the specific compound used in lanes 2-12 is indicated above the blot in (d). The concentration of all inositol phosphates is 5 μM. The IP4 compound that is the soluble head group of the PIP3 target of ITK in T cells (inositol 1,3,4,5-tetrakisphosphate) is labeled in red.
Since inositol (1,3,4,5)-tetrakisphosphate specifically interferes with the ITK PH/kinase interaction we next examined the effect of IP4 on ITK kinase activity. ITK activation loop phosphorylation decreases with increasing IP4, whether the isomer corresponding to the soluble head group of the physiologically relevant PIP3 (IP(1,3,4,5)P4) or the regioisomer IP(1,3,4,6)P4 is used in the assay (data not shown). Previous findings showed no change in ITK mediated in vitro phosphorylation of a non-physiological peptide substrate22 and so together these results suggest that either IP4 by itself does not directly activate ITK in spite of interfering with the regulatory PHTH/Kinase interaction, or at the concentrations required to disrupt the PHTH/kinase interaction, IP4 is incompatible with the in vitro kinase assay causing non-specific inhibition of ITK activity. The observation that the regioisomeric IP4 compound decreases ITK activation loop phosphorylation to the same extent as the physiologically relevant IP4 (IP(1,3,4,5)P4) but does not disrupt the ITK PHTH/kinase interaction (Fig. 5c) supports the conclusion that the in vitro ITK kinase assay is not a reliable measure of the effect of IP4 on ITK function.
In an effort to circumvent the non-specific kinase inhibition caused by soluble IP4, we expanded the ITK in vitro kinase assay to examine the effect of the ITK PHTH domain target, PIP3; the plasma membrane bound phospholipid produced by phosphoinositide 3-kinase following T cell receptor activation23. ITK activation loop phosphorylation levels (pY511) were monitored in the presence of PIP3 containing liposomes as well as control liposomes that lack PIP3. Unlike the equivocal effects of IP4 on ITK activity, PIP3 activates ITK as measured by phosphorylation on the activation loop tyrosine Y511 (Fig. 6a, lane 2, 6b) whereas control liposomes do not enhance activation loop phosphorylation (Fig. 6a, lane 3, 6b). This result is consistent with binding of the PIP3 headgroup in a manner that competes with an autoregulatory interaction mediated by the ITK PHTH domain (Fig. 5a).
Figure 6. PIP3 activates ITK and the activation loop in the ITK kinase domain is sterically blocked by the PHTH domain in autoinhibted ITK.
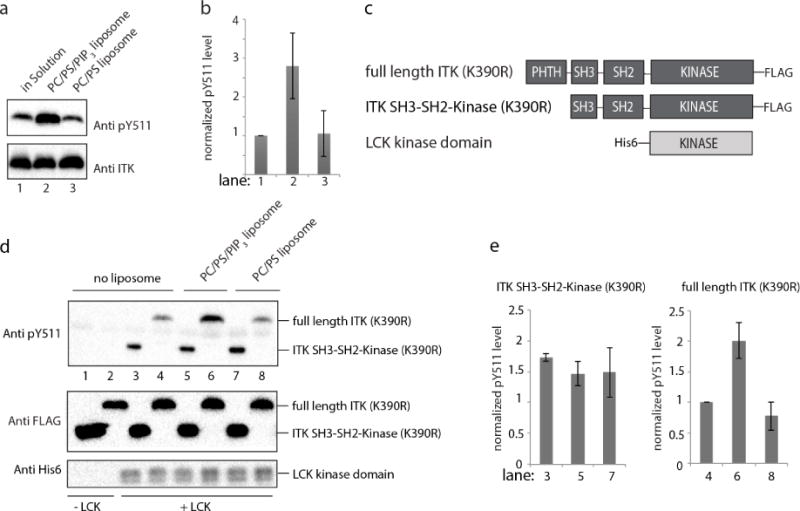
(a) In vitro ITK kinase assay (500 nM ITK) in solution (lane 1), in the presence of PIP3 containing liposomes (lane 2) and control liposomes that lack PIP3 (lane 3). PC indicates 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and PS is 1,2-dioleoyl-sn-glycero-3-[phospho-L-serine] (DOPS). ITK activation loop autophosphorylation (pY511) is detected at 30 minutes using anti-BTK pY551 and total enzyme levels are detected using anti-ITK antibody. (b) Histogram representation of normalized pY511 levels from the ITK kinase assays shown in (a). Normalization was done as in Figure 4(c and d), except that the value for ITK in the absence of any phosphoinositol compound was set to 1. Experiments were run in triplicate. (c) Domain constructs of ITK and LCK. Full length ITK and the fragment lacking the PHTH domain (ITK SH3-SH2-Kinase) are FLAG-tagged at the C-termini and the LCK kinase domain carries a His6 tag at the N-terminus. Full length ITK and the ITK SH3-SH2-kinase fragment contain the K390R mutation rendering both inactive24. (d) In vitro LCK kinase assay in solution (lanes 3 and 4), in the presence of PIP3 containing liposomes (lanes 5 and 6) and control liposomes that lack PIP3 (lanes 7 and 8). ITK SH3-SH2-Kinase (K390R) (1 μM, lanes 3,5,7) and full length ITK (K390R) (1 μM, lanes 4,6,8) serve as substrates for LCK (200 nM). Lanes 1 and 2 are no enzyme (LCK) controls. Phosphorylation of ITK Y511 is detected using anti-BTK pY551 and total substrate and enzyme levels are detected using anti-FLAG and anti-His6 antibodies, respectively. (e) Histogram representation of normalized pY511 levels from the LCK kinase assays shown in (d). Normalization as in panel (b) with the value for full length ITK (K390R) in the absence of liposomes set to 1. Data for the ITK SH3-SH2-Kinase (K390R) substrate is on the left and the full length ITK (K390R) substrate is on the right. Experiments were run in triplicate.
To gain additional insight into precisely how PIP3 leads to increased phosphorylation on the ITK activation loop, we next carried out a combined kinase assay that makes use of kinase inactive full length ITK (K390R), the inactive ITK fragment lacking the PHTH domain (ITK SH3-SH2-Kinase (K390R)) and active LCK kinase domain, the SRC family kinase that is responsible for phosphorylation of ITK Y511 in activated T cells24 (Fig. 6c). The catalytically inactive full length ITK (K390R) and ITK SH3-SH2-Kinase (K390R) are used in this assay to ensure that phosphorylation on ITK Y511 is solely due to LCK activity and not due to intrinsic ITK activity. Thus, the extent to which ITK Y511 is phosphorylated by exogenous LCK reveals the accessibility of the ITK activation loop under different conditions25. Both full length ITK (K390R) and the ITK SH3-SH2-Kinase (K390R) fragment are subjected to phosphorylation by active LCK in the presence and absence of PIP3. In the absence of PIP3 (either no liposomes or control liposomes (PC/PS) that do not contain PIP3) we find the ITK SH3-SH2-Kinase (K390R) fragment is phosphorylated on Y511 to a greater extent than full length ITK (K390R) (Fig. 6d,e lanes 3 & 4 and 7 & 8) suggesting that the PHTH domain reduces accessibility of the ITK activation loop to LCK. For full length ITK (K390R) in the presence of PIP3 containing liposomes (PC/PS/PIP3), LCK dependent phosphorylation of the ITK activation loop tyrosine (pY511) is higher than full length ITK (K390R) in the absence of liposomes or in the presence of control liposomes (Fig. 6d,e lane 6 versus 2, 4 & 8). In contrast, LCK mediated phosphorylation of the activation loop tyrosine Y511 in the ITK SH3-SH2-Kinase fragment that lacks PHTH is unaffected by the presence or absence of PIP3 (Fig. 6d,e lanes 3,5,7). These observations suggest that binding of PIP3 to the ITK PHTH domain ‘activates’ ITK by unmasking of the ITK activation loop making it more accessible to phosphorylation by LCK.
Discussion
The findings reported here support an autoregulatory role for the PHTH domain in controlling the catalytic activity of ITK. The role of the PHTH domain in ITK regulation has been hinted at in previous deletion studies14 but not previously dissected at the molecular level. In the context of the full length ITK enzyme, a specific surface on the PHTH domain encompassing the β3-β4 loop of the PH fold mediates a direct interaction with the ITK kinase domain resulting in diminished ITK catalytic activity. Mutation of ITK PHTH residues at this interface leads to activation of the remote catalytic domain in the full length ITK enzyme and deletion of the PHTH domain or binding to phospholipid leads to increased accessibility of the ITK activation loop for phosphorylation by LCK.
Binding of the soluble head group of the PIP3 ligand, IP(1,3,4,5)P4, selectively disrupts the ITK PHTH/kinase interaction (Fig. 5). This finding may be related to earlier observations that soluble IP(1,3,4,5)P4 promotes activation of ITK in T lymphocytes22 and so IP(1,3,4,5)P4 might, at least in part, pry open the autoinhibited form of ITK. However, the results of in vitro assays in the presence of inositol tetrakisphosphates suggest difficulty in directly measuring the effect of IP4 on ITK activity. It should also be noted that the autoinhibitory region of the ITK PH domain that we have identified here overlaps with a peripheral binding site identified on the BTK PH domain that binds soluble IP615. IP6 binding to the BTK PH domain may stabilize transient dimerization and subsequent BTK autophosphorylation15. Yet, as the authors in that work note, the peripheral BTK IP6 binding site is not conserved in ITK and so a specific IP6 regulatory role for ITK may not exist.
A structural model of the autoinhibited form of BTK has recently been reported15. Consistent with our observation that the BTK PHTH and BTK kinase domains do not interact in trans (Fig. 1d), crystallographic analysis of the BTK PHTH/kinase interaction required a tethered BTK PHTH-Kinase construct, which yielded a structure defining a PHTH/kinase interface centered on the base of the PH domain α-helix (Fig. 7a). R133 and Y134 in the BTK PH domain mediate primary sidechain contacts between BTK PH and kinase domains in the tethered BTK crystal structure15 yet NMR resonances for the corresponding ITK residues, R111 and N112, as well as that of the neighboring residue N113, are unaffected by addition of unlabeled ITK kinase domain (Fig. 2b,c). Moreover, mutation of ITK R111 to glutamate does not affect the ITK PHTH/kinase interaction (Fig. 3d). These findings support the conclusion that the PHTH/kinase interface observed in the crystal structure of the tethered BTK PHTH-kinase construct is not the interface that mediates the autoregulatory ITK PHTH/kinase contact. Instead, ITK residues in the β3-β4 loop of the PHTH domain are primarily responsible for mediating the ITK PHTH/kinase autoinhibitory interaction (Fig. 7a).
Figure 7. Autoinhibition and allosteric activation of the TEC kinases.
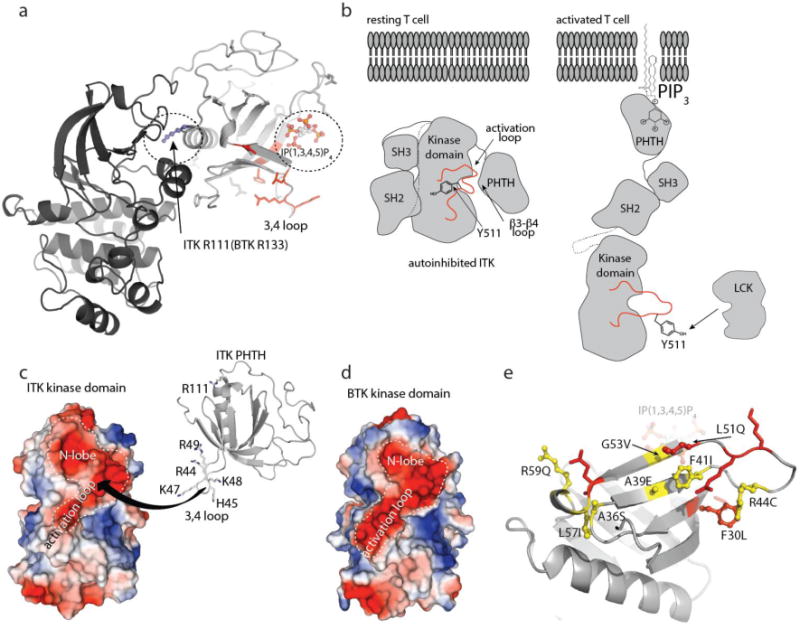
(a) The structure of tethered BTK PHTH-Kinase domain (PDB ID: 4Y93) where the ITK PHTH domain has been superimposed with the BTK PHTH domain (BTK PHTH not shown for clarity). R133 in the BTK PHTH domain makes contact with the BTK kinase domain. R111 is the corresponding residue in the ITK PHTH domain but mutation of R111 does not affect the ITK PHTH/Kinase interaction. The ITK PHTH interface residues located in and around the β3-β4 loop (colored red) are not located at the interface with the kinase domain defined in the crystal structure for the tethered BTK domains but are close to the IP(1,3,4,5)P4 binding site (circled). (b) left, Model of closed, autoinhibited ITK in a resting T cell (arrangement of the SH3 and SH2 domains is based on the BTK crystal structure of the SH3-SH2-Kinase fragment (4XI2)). Right, model of an open form of ITK (based on SAXS structure of full length BTK35 in the presence of membrane anchored PIP3 revealing Y511 for phosphorylation by LCK. (c) The ITK kinase domain presents a large negatively charged surface that spans the N-lobe and activation loop and may serve as a binding site for the positively charged region of ITK PHTH defined in this study. The highly basic stretch of residues on the β3-β4 loop are shown and labeled. (d) The BTK kinase domain shares the acidic patch across the N-lobe and activation loop. (e) Autoinhibitory ITK PHTH surface defined in this study. Residues shown in red are those side chains mutated in the current study that have an effect on the inhibitory interaction with the ITK kinase domain, those in yellow, ball and stick are ITK residues listed in the COSMIC database. F30 is shown in red with ball and stick to indicate that it both affects the PHTH/Kinase interface and is mutated in the database of somatic mutations in cancer.
Typically protein kinases belonging to the same family are regulated by related mechanisms. In fact, mutation of the BTK β3-β4 loop residues that correspond to the ITK PHTH domain surface we have defined here (BTK R49E and K52E (K48 and L51 in ITK)) activate BTK15. Since the β3-β4 loop residues of the BTK PHTH domain are not located at the PHTH/kinase interface in the BTK structural model (Fig. 7a), the authors of that work suggested that mutations in the BTK β3-β4 loop might alleviate electrostatic repulsion facilitating transient dimerization (and hence autophosphorylation). Our new insights into ITK regulation add the possibility that the TEC kinases share structural features of the fully suppressed state wherein the β3-β4 loop serves as a latch that stabilizes and the autoinhibited conformation. In this light, the contact between PHTH and kinase domain in the crystallographically determined model of BTK15 (Fig. 7a) might represent an intermediate state between the fully autoinhibited and active kinase.
Differences persist within the TEC kinase family since we find the ITK PHTH/kinase interaction is readily detected intermolecularly while the BTK PHTH/kinase interaction is not (Fig. 1). This observation suggests that the intramolecular regulatory interaction between PHTH and kinase domains is stronger in ITK than BTK, an idea that is consistent with earlier work suggesting that B cell proteins such as BTK and SYK are more easily activated than their T cell counterparts, ITK and ZAP-7025, 26. The reason for differences in regulation of ITK and BTK may be related to the notion that, compared to B cells, T cell activation needs to be under stricter control given the deleterious effects of over production of inflammatory cytokines25.
Crystal contacts in the tethered BTK PH-kinase structure15 reveal that this construct forms the “Saraste Dimer”27 mediated by the PHTH β3-β4 loop as well as the large insert between the β5 and β6 strands. The extent to which the ITK PHTH domain forms a similar dimer is unclear and ITK lacks the large β5-β6 loop of BTK that forms much of the “Saraste Dimer” interface. The ITK SH3-SH2 fragment have been shown to form oligomers28 and so the full length ITK protein may access higher order states albeit by alternative domain contacts. This is important in considering the activating effect we observe for ITK in the presence of PIP3 (Fig. 6a). While the data support a role for specific lipid binding in destabilizing the autoinhibitory contact between PHTH and kinase domains (Fig. 5), it is also possible that PIP3 mediated increase in ITK activity arises in part from increased local concentration of the kinase favoring dimerization/oligomerization that promotes trans autophosphorylation. Nevertheless, the observations that targeted mutations such as K48D/R49D in the ITK PHTH β3-β4 loop activate the kinase toward various substrates in the absence of lipid and that deletion of the PHTH domain increases the accessibility of the kinase activation loop both support a regulatory role for this region that is independent of aggregation at a lipid surface.
The effect of the ITK PHTH domain on activation loop accessibility is intriguing and the possibility that the PHTH domain masks this important region of the kinase domain is further buttressed by the presence of a large acidic patch extending from the N-lobe over the activation loop (Fig. 7c). The acidic nature of this region of the kinase domain might provide a complementary binding surface for the positively charged amino acids in the ITK PHTH β3-β4 loop (Fig. 7c) and it is noteworthy that the BTK kinase domain shares the large negatively charged surface (Fig. 7d). Further structural work will definitively determine whether this acidic surface serves as the binding interface with the PHTH domain in the fully suppressed, autoinhibitory conformation of the TEC kinases. Precedence exists for this mode of regulation as demonstrated by the crystal structure of the autoinhibited serine/threonine kinase, AKT, showing PH domain occlusion of the kinase active site16. AKT is also allosterically activated by PIP329 extending the similarities between these PH domain containing kinases.
The ITK PHTH β3-β4 loop has also been previously implicated in binding the calcium binding protein, Calmodulin17. Binding of Ca2+/Calmodulin to the ITK PHTH domain (via β3-β4 loop residues) was shown to result in sustained ITK activation and potentiated calcium signaling in T cells. Based on the negative regulatory role we have now demonstrated for this region of ITK PHTH, it is possible that one of the consequences of Ca2+/Calmodulin binding to ITK is to maintain an open, active state and prevent formation of the autoinhibitory state. Along these lines, it is interesting to note that the COSMIC (catalogue of somatic mutations in cancer) database lists several mutations in ITK that span the PHTH interface responsible for binding to the ITK kinase domain (Fig. 7e). It is possible that these mutations activate ITK kinase activity in much the same way that the targeted mutations studied here abolish the autoregulatory PHTH/kinase interaction (Fig. 3) and increase ITK mediated phosphorylation of PLCγ1 (Fig. 4). Similar oncogenic activation via PH domain mutations has been reported for AKT30.
It is becoming increasingly clear that Pleckstrin Homology domains target non-lipid binding partners in addition to membrane-bound phospholipids. A recent review by Huang and co-workers31 highlights the varied roles of the PH domain in lymphocyte signaling as well as the regulatory role of the PH domain in non-lymphocytic proteins generally. In one example, the DH-PH domain module of Dbs, a Cdc42 specific Guanine Exchange Factor, contacts Cdc42 via the β3-β4 loop of the Dbs PH domain32. In another example, the guanine nucleotide exchange factor P-Rex2 recognizes its substrate G protein via the β3-β4 loop region of the PH domain33. With our observations of a critical role for the same β3-β4 loop of ITK PHTH in binding the ITK kinase domain as well as Ca2+/Calmodulin17, it is possible that this region of the PH fold may serve as a general protein recognition motif that is sensitive to phospholipid binding. The growing examples of PH domain mediated regulation also suggest that the specific protein interactions mediated by the PH fold are potential targets for allosteric regulation of signaling molecules via the activity of small molecules. Indeed, targeting AKT in this manner has already been demonstrated providing proof of principle for other systems such as the TEC family kinases.
Acknowledgments
The authors would like to thank Thamotharan Subbiah for making the baculoviral ITK kinase domain construct.
Funding Information: This work was supported by a grant from the NIH, National Institute of Allergy and Infectious Diseases, AI043957.
Abbreviations
- Interleukin-2-inducible tyrosine kinase
(ITK)
- Bruton’s tyrosine kinase
(BTK)
- Pleckstrin homology
(PH)
- phosphatidylinositol (34,5)-trisphosphate
(PIP3)
- inositol 3-phosphate
(IP(3)P)
- inositol 13-diphosphate
(IP(1,3)P2)
- inositol 13,4-trisphosphate
(IP(1,3,4)P3)
- inositol 13,5-trisphosphate
(IP(1,3,5)P3)
- inositol 14,5-trisphosphate
(IP(1,4,5)P3)
- inositol 12,6-trisphosphate
(IP(1,2,6)P3)
- inositol 13,4,5-tetrakisphosphate
(IP(1,3,4,5)P4)
- inositol 13,4,6-tetrakisphosphate
(IP(1,3,4,6)P4)
- inositol 12,3,5,6-pentakisphosphate
(IP(1,2,3,5,6)P5)
- inositol 13,4,5,6-pentakisphosphate
(IP(1,3,4,5,6)P5)
- inositol 12,3,4,5,6-hexakisphosphate
(IP(1,2,3,4,5,6)P6)
- SRC Homology 2 and SRC Homology 3
(SH2 and SH3)
- Calmodulin
(CaM)
- Heteronuclear single quantum coherence
(HSQC)
- Nuclear Magnetic Resonance
(NMR)
- hexa-histidine tag
(His6)
References
- 1.Lee MJ, Yaffe MB. Protein Regulation in Signal Transduction. Cold Spring Harb Perspect Biol. 2016;8 doi: 10.1101/cshperspect.a005918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu W, Harrison SC, Eck MJ. Three-dimensional structure of the tyrosine kinase c-Src. Nature. 1997;385:595–602. doi: 10.1038/385595a0. [DOI] [PubMed] [Google Scholar]
- 3.Xu W, Doshi A, Lei M, Eck MJ, Harrison SC. Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol Cell. 1999;3:629–638. doi: 10.1016/s1097-2765(00)80356-1. [DOI] [PubMed] [Google Scholar]
- 4.Andreotti AH, Schwartzberg PL, Joseph RE, Berg LJ. T-cell signaling regulated by the Tec family kinase, Itk. Cold Spring Harb Perspect Biol. 2010;2:a002287. doi: 10.1101/cshperspect.a002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bradshaw JM. The Src, Syk, and Tec family kinases: distinct types of molecular switches. Cell Signal. 2010;22:1175–1184. doi: 10.1016/j.cellsig.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Tsukada S, Saffran DC, Rawlings DJ, Parolini O, Allen RC, Klisak I, Sparkes RS, Kubagawa H, Mohandas T, Quan S, et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell. 1993;72:279–290. doi: 10.1016/0092-8674(93)90667-f. [DOI] [PubMed] [Google Scholar]
- 7.Huck K, Feyen O, Niehues T, Ruschendorf F, Hubner N, Laws HJ, Telieps T, Knapp S, Wacker HH, Meindl A, Jumaa H, Borkhardt A. Girls homozygous for an IL-2-inducible T cell kinase mutation that leads to protein deficiency develop fatal EBV-associated lymphoproliferation. J Clin Invest. 2009;119:1350–1358. doi: 10.1172/JCI37901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lemmon MA. Pleckstrin homology (PH) domains and phosphoinositides. Biochem Soc Symp. 2007:81–93. doi: 10.1042/BSS0740081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moravcevic K, Oxley CL, Lemmon MA. Conditional peripheral membrane proteins: facing up to limited specificity. Structure. 2012;20:15–27. doi: 10.1016/j.str.2011.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harlan JE, Hajduk PJ, Yoon HS, Fesik SW. Pleckstrin homology domains bind to phosphatidylinositol-4,5-bisphosphate. Nature. 1994;371:168–170. doi: 10.1038/371168a0. [DOI] [PubMed] [Google Scholar]
- 11.Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol. 2012;13:195–203. doi: 10.1038/nrm3290. [DOI] [PubMed] [Google Scholar]
- 12.Cantrell DA. Phosphoinositide 3-kinase signalling pathways. J Cell Sci. 2001;114:1439–1445. doi: 10.1242/jcs.114.8.1439. [DOI] [PubMed] [Google Scholar]
- 13.Okoh MP, Vihinen M. Pleckstrin homology domains of tec family protein kinases. Biochem Biophys Res Commun. 1999;265:151–157. doi: 10.1006/bbrc.1999.1407. [DOI] [PubMed] [Google Scholar]
- 14.Joseph RE, Min L, Andreotti AH. The linker between SH2 and kinase domains positively regulates catalysis of the Tec family kinases. Biochemistry-Us. 2007;46:5455–5462. doi: 10.1021/bi602512e. [DOI] [PubMed] [Google Scholar]
- 15.Wang Q, Vogan EM, Nocka LM, Rosen CE, Zorn JA, Harrison SC, Kuriyan J. Autoinhibition of Bruton’s tyrosine kinase (Btk) and activation by soluble inositol hexakisphosphate. Elife. 2015;4 doi: 10.7554/eLife.06074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu WI, Voegtli WC, Sturgis HL, Dizon FP, Vigers GP, Brandhuber BJ. Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS One. 2010;5:e12913. doi: 10.1371/journal.pone.0012913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, Boyken SE, Hu J, Xu X, Rimer RP, Shea MA, Shaw AS, Andreotti AH, Huang YH. Calmodulin and PI(3,4,5)P(3) cooperatively bind to the Itk pleckstrin homology domain to promote efficient calcium signaling and IL-17A production. Sci Signal. 2014;7:ra74. doi: 10.1126/scisignal.2005147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boyken SE, Fulton DB, Andreotti AH. Rescue of the aggregation prone Itk Pleckstrin Homology domain by two mutations derived from the related kinases, Btk and Tec. Protein Sci. 2012;21:1288–1297. doi: 10.1002/pro.2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joseph RE, Andreotti AH. Bacterial expression and purification of interleukin-2 tyrosine kinase: single step separation of the chaperonin impurity. Protein Expr Purif. 2008;60:194–197. doi: 10.1016/j.pep.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson BA, Blevins RA. NMR View: A computer program for the visualization and analysis of NMR data. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 21.Joseph RE, Min L, Xu R, Musselman ED, Andreotti AH. A remote substrate docking mechanism for the tec family tyrosine kinases. Biochemistry. 2007;46:5595–5603. doi: 10.1021/bi700127c. [DOI] [PubMed] [Google Scholar]
- 22.Huang YH, Grasis JA, Miller AT, Xu R, Soonthornvacharin S, Andreotti AH, Tsoukas CD, Cooke MP, Sauer K. Positive regulation of Itk PH domain function by soluble IP4. Science. 2007;316:886–889. doi: 10.1126/science.1138684. [DOI] [PubMed] [Google Scholar]
- 23.August A, Sadra A, Dupont B, Hanafusa H. Src-induced activation of inducible T cell kinase (ITK) requires phosphatidylinositol 3-kinase activity and the Pleckstrin homology domain of inducible T cell kinase. Proc Natl Acad Sci U S A. 1997;94:11227–11232. doi: 10.1073/pnas.94.21.11227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heyeck SD, Wilcox HM, Bunnell SC, Berg LJ. Lck phosphorylates the activation loop tyrosine of the Itk kinase domain and activates Itk kinase activity. J Biol Chem. 1997;272:25401–25408. doi: 10.1074/jbc.272.40.25401. [DOI] [PubMed] [Google Scholar]
- 25.Joseph RE, Kleino I, Wales TE, Xie Q, Fulton DB, Engen JR, Berg LJ, Andreotti AH. Activation loop dynamics determine the different catalytic efficiencies of B cell- and T cell-specific tec kinases. 2013;6:ra76. doi: 10.1126/scisignal.2004298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Latour S, Chow LM, Veillette A. Differential intrinsic enzymatic activity of Syk and Zap-70 protein-tyrosine kinases. J Biol Chem. 1996;271:22782–22790. doi: 10.1074/jbc.271.37.22782. [DOI] [PubMed] [Google Scholar]
- 27.Hyvonen M, Saraste M. Structure of the PH domain and Btk motif from Bruton’s tyrosine kinase: molecular explanations for X-linked agammaglobulinaemia. Embo J. 1997;16:3396–3404. doi: 10.1093/emboj/16.12.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brazin KN, Fulton DB, Andreotti AH. A specific intermolecular association between the regulatory domains of a Tec family kinase. J Mol Biol. 2000;302:607–623. doi: 10.1006/jmbi.2000.4091. [DOI] [PubMed] [Google Scholar]
- 29.Ebner M, Lucic I, Leonard TA, Yudushkin I. PI(3,4,5)P3 Engagement Restricts Akt Activity to Cellular Membranes. Mol Cell. 2017;65:416–431 e416. doi: 10.1016/j.molcel.2016.12.028. [DOI] [PubMed] [Google Scholar]
- 30.Parikh C, Janakiraman V, Wu WI, Foo CK, Kljavin NM, Chaudhuri S, Stawiski E, Lee B, Lin J, Li H, Lorenzo MN, Yuan W, Guillory J, Jackson M, Rondon J, Franke Y, Bowman KK, Sagolla M, Stinson J, Wu TD, Wu J, Stokoe D, Stern HM, Brandhuber BJ, Lin K, Skelton NJ, Seshagiri S. Disruption of PH-kinase domain interactions leads to oncogenic activation of AKT in human cancers. Proc Natl Acad Sci U S A. 2012;109:19368–19373. doi: 10.1073/pnas.1204384109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Hills LB, Huang YH. Lipid and Protein Co-Regulation of PI3K Effectors Akt and Itk in Lymphocytes. Front Immunol. 2015;6:117. doi: 10.3389/fimmu.2015.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rossman KL, Worthylake DK, Snyder JT, Siderovski DP, Campbell SL, Sondek J. A crystallographic view of interactions between Dbs and Cdc42: PH domain-assisted guanine nucleotide exchange. Embo J. 2002;21:1315–1326. doi: 10.1093/emboj/21.6.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Joseph RE, Norris FA. Substrate specificity and recognition is conferred by the pleckstrin homology domain of the Dbl family guanine nucleotide exchange factor P-Rex2. J Biol Chem. 2005;280:27508–27512. doi: 10.1074/jbc.M412495200. [DOI] [PubMed] [Google Scholar]
- 34.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marquez JA, Smith CI, Petoukhov MV, Lo Surdo P, Mattsson PT, Knekt M, Westlund A, Scheffzek K, Saraste M, Svergun DI. Conformation of full-length Bruton tyrosine kinase (Btk) from synchrotron X-ray solution scattering. Embo J. 2003;22:4616–4624. doi: 10.1093/emboj/cdg448. [DOI] [PMC free article] [PubMed] [Google Scholar]