

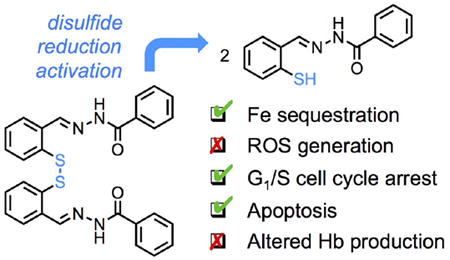
Abstract
The iron metabolism of malignant cells, which is altered to ensure higher acquisition and utilization, motivates the investigation of iron chelation strategies in cancer treatment. In a prochelation approach aimed at increasing intracellular specificity, disulfide reduction/activation switches are incorporated on iron-binding scaffolds resulting in intracellularly activated scavengers. Herein, this strategy is applied to several tridentate donor sets including thiosemicarbazones, aroylhydrazones and semicarbazones. The novel prochelator systems are antiproliferative in breast adenocarcinoma cell lines (MCF-7 and metastatic MDA-MB-231) and do not result in the intracellular generation of oxidative stress. Consistent with iron deprivation, the tested prochelators lead to cell-cycle arrest at the G1/S interface and induction of apoptosis. Notably, although hemoglobin-synthesizing blood cells have the highest iron need in the human body, no significant impact on hemoglobin production was observed in the MEL (murine erythroleukemia) model of differentiating erythroid cells. This study provides new information on the intracellular effects of disulfide-based prochelators and indicates aroylhydrazone (AH1-S)2 as a promising prototype of a new class of antiproliferative prochelator systems.
Keywords: iron, prochelator, anticancer, cell-cycle arrest, hemoglobin production
Graphical abstract
1. Introduction
Rapidly dividing malignant cells are characterized by a reprogrammed iron metabolism, which enhances intracellular iron availability through an altered expression of several key proteins for iron homeostasis [1]. Correspondingly, the expression levels of transferrin receptors, ferroportin and ferritin have been identified as prognostic markers in breast cancer patients [2-4]. At a molecular level, reactivity-based fluorescent probes of intracellular iron have shown recently that the labile iron pool is larger in several cancer cell lines when compared to non-malignant ones [5, 6]. As such, high-affinity scavengers (chelators) can be employed to target the increased iron needs of cancer cells for the development of antineoplastic agents [7]. The scaffolds of chelators studied in this context vary substantially (Fig. 1): from hexadentate hydroxamate-based siderophores (e.g., desferrioxamine, DFO) to bidentate deferiprone (DFP) to tridentate thiosemicarbazones (e.g., Triapine) and aroylhydrazones (e.g., salicyl isonicotinoyl hydrazone, SIH).
Fig. 1.
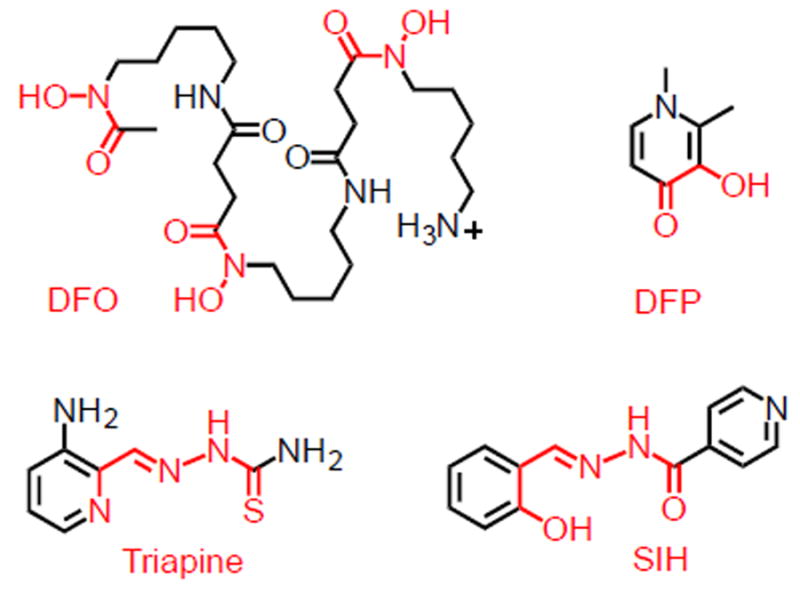
Examples of iron-binding units (in red) in chelators employed for biomedical applications.
As early as the 1950s, thiosemicarbazones were found to have anticancer activity in mice [8, 9]. Since then numerous thiosemicarbazones and hydrazones have been studied for their antineoplastic effects in vitro and in animal models [10]. Furthermore, Triapine has been investigated in several clinical trials [11]. More recently, new classes of thiosemicarbazones were investigated as iron chelators [12], and these studies produced highly potent compounds such as di-2-pyridylketone 4,4-dimethyl-3-thiosemicarbazone (Dp44mT) and di-2-pyridylketone 4-cyclohexyl-4-methyl-3-thiosemicarbazone (DpC), which proved effective in vitro and in vivo through oral and intravenous administration [13, 14]. In particular, these compounds demonstrated the potential to overcome multidrug resistance [15] as well as some of the limitations of Triapine, including methemoglobin formation [16]. DpC has entered clinical trials in 2016 [17].
Within the study of metal-binding pharmaceuticals, prochelation strategies are being pursued to minimize off-target toxicity through inclusion of structural motifs that render the chelator inactive until triggered by disease-specific conditions [18, 19]. Examples of this approach include prochelators that are activated by intracellular enzymes [20, 21], under oxidative stress conditions [22, 23], and by reduction of disulfide bonds [24-26].
The disulfide reduction/activation approach employs disulfide bonds as redox-sensitive switches that are triggered upon cell entry by the high concentrations of cytosolic thiols with respect to the extracellular milieu. Indeed, the intracellular concentrations of reduced glutathione (GSH, 5-11 mM) are orders of magnitude greater than those in the extracellular space and blood plasma (< 5 μM) [27, 28]. As such, disulfide linkages are employed extensively in prodrug and chemosensor design [29, 30], and recent theranostic systems have allowed visualization of selective disulfide-based drug release in vivo [31]. In addition, malignant tissue contains higher levels of GSH when compared to the neighboring normal tissue [28, 32, 33], therefore disulfide switches are particularly attractive for the design of antiproliferative prochelator systems.
We have previously shown that a disulfide bond can be employed to mask a sulfur donor within tridentate thiosemicarbazone chelators (Scheme 1) [24, 25]. The resulting disulfide-based prochelators do not coordinate metal ions in neutral aqueous solutions, whereas the thiolates generated upon intracellular reduction are high-affinity chelators [24]. In the case of prochelator (TC1-S)2 (Scheme 1), intracellular reduction and iron binding lead to the formation of a low-spin ferric complex that is not redox-active [25]. More recently, we have utilized disulfide switches as linkers in glycoconjugate prochelator systems targeting the overexpression of glucose transporters in colon cancer cells [34]. Furthermore, disulfide-based thiosemicarbazone prochelators have the potential to alter iron trafficking and distribution in the tumor microenvironment through their effects on the iron phenotype of tumor-associated macrophages [35].
Scheme 1.
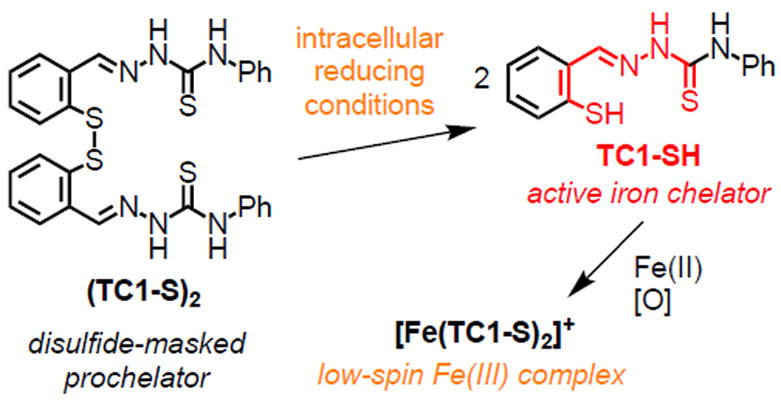
Reduction/activation of a disulfide switch in thiosemicarbazone prochelator (TC1-S)2
In this work, we apply the disulfide-based prochelation design to aroylhydrazone and semicarbazone scaffolds, and we examine the introduction of a methyl substituent at the iminic carbon in the new prototypes and in the original thiosemicarbazone framework. The intracellular effects of the corresponding chelators, which feature both (S, N, S) and (S, N, O) donor sets, are investigated with respect to redox behavior, cell cycle, toxicity and cell death. In addition, the impact of selected chelators on hemoglobin production is studied in a model of differentiating erythroid cells, namely the type of mammalian cells that require the highest amount of iron.
2. Experimental
2.1. Materials and methods
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and propidium iodide (PI) were purchased from VWR and used as received. The fluorescein isothiocyanate conjugate of Annexin V (FITC-AnnV) was purchased from Fisher Scientific and used per manufacturer’s instructions. All other reagents were purchased from common commercial sources and used without further purification. Absorption and fluorescence assays in 96-well plates were recorded on a BioTek Synergy™ 2 microplate reader at the indicated wavelengths. Flow cytometric analyses were performed at the University of Arizona Cytometry Core Facility (Arizona Cancer Center/Arizona Research Laboratories) using a FACSCanto II flow cytometer (BDBiosciences, San Jose, CA) equipped with an air-cooled 15-mW argon ion laser tuned to 488 nm. The emission fluorescence of dichlorofluorescein (DCF), PI, and FITC-AnnV were detected and recorded through a 530/30 bandpass filter in the FL1 channel. List mode data files consisting of 10,000 events gated on FSC (forward scatter) vs SSC (side scatter) were acquired and analyzed using the CellQuest PRO software (BD Biosciences, San Jose, CA). Appropriate electronic compensation was adjusted by acquiring cell populations stained with each dye/fluorophore individually, as well as an unstained control.
2.2. Synthesis of disulfide prochelators
(TC1-S)2, and (TC3-S)2 were synthesized according to reported procedures [24]. The other prochelators were synthesized via condensation of a thiosemicarbazide, semicarbazide, or hydrazide with a 2,2’-dithiodibenzyl dialdehyde or methyldiketone. Detailed procedures and chemical characterization data are reported in the supplementary data.
2.3. Cytotoxicity assays
MTT viability assays were conducted as previously described [24]. Cells were seeded (4000 cells per well for MCF-7, 700 cells per well for MDA-MB-231, and 10,000 cells per well for MRC-5 cells) and allowed to attach for 24 h. Test compounds dissolved in dimethylsulfoxide (DMSO) were diluted in Eagle’s Minimal Essential Medium (EMEM) to the specified concentration with final DMSO concentration limited to 0.1% v/v. Cells were incubated in the presence of the test compounds for 72 h, then the MTT solution (4 mg/mL, 10 μL) was added to each well and incubated for 4 h. Following media removal, DMSO (100 μL) was added to each well to dissolve the purple formazan crystals and the plates were incubated for an additional 30 min. Absorption at 560 nm was recorded and data were analyzed using logarithmic fits (Origin®) to obtain IC50 values. Each experiment was conducted in triplicate, and values are given as mean ± standard deviation of the mean (SDM).
2.4. Detection of intracellular reactive oxygen species (ROS)
Solutions of the fluorescent probe dichlorodihydrofluorescein diacetate (DCFH2-DA) (Invitrogen) were prepared in DMSO, aliquoted in single-use doses and stored at −20 °C. MDA-MB-231 cells were seeded in 6-well plates at a density of 1.0 × 105 cells/mL (2.0 × 105 cells/well) and allowed to incubate overnight. The growth media were then removed and the adherent cells were treated for 2 h with the test compounds in phenol-red-free EMEM (0.1% DMSO was used to solubilize the compounds). The positive control (H2O2) was diluted in phosphate-buffered saline (PBS) and cells were exposed to this solution for 10 min. After incubation, the cells were washed with PBS and then treated with warm PBS containing DCFH2-DA (30 μM) for 20 min. After removal of the probe solution, the cells were washed with PBS (×2) and detached using trypsin/ethylenediaminetetraacetic acid (EDTA) (3 min). The cell suspension was diluted with growth media (2 mL) and centrifuged at 2000 rpm for 10 min. The resulting cell pellet was then suspended in PBS (500 μL), stored on ice, and analyzed by flow cytometry within 1 h.
2.5. Cell cycle analysis
MDA-MB-231 cells were seeded at 0.2 million cells per well (in EMEM supplemented with 0.1 mg/mL human holo-transferrin) in 6-well plates and allowed to adhere overnight. Media were then replaced with fresh media containing the test compounds (final DMSO concentration 0.1% v/v) and cells were incubated for 12 h. Cells were washed with PBS (1 mL) and then detached by addition of 0.25% trypsin-EDTA (0.4 mL) followed by a 3-min incubation. After addition of EMEM (1 mL), the cell suspension was centrifuged at 125g for 15 min. Media were discarded and cells were fixed by addition of ice-cold 70% ethanol (2 mL) and stored in a freezer at -20 °C overnight (and no longer than one week). Cells were spun at 2000 rpm for 20 min, and the resulting pellet was suspended in PBS (0.3 mL) and treated with RNAse and PI (0.5 mg/mL and 40 μg/mL respectively, 30 min), placed on ice and analyzed by flow cytometry within 1 h.
2.6. Apoptosis assays
Jurkat cells were grown to 1.5 million cells/mL, then centrifuged and suspended in fresh Roswell Park Memorial Institute (RPMI) medium at 0.5 million cells/mL. Cells were then treated with either test compounds dissolved in DMSO or DMSO alone for control samples (final DMSO concentration 0.1% v/v). Cells were incubated for the specified time and then aliquots of 1 million cells were centrifuged. The pellets were suspended in binding buffer (10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) with 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, and 1.8 mM CaCl2, pH 7.4) containing 2μg/mL FITC-AnnV (0.3 mL) and solutions were incubated in the dark at room temperature for 10 min. PI was added prior to analysis at a final concentration of 1μg/mL.
2.7. MEL cell culture and spectrophotometric hemoglobin quantitation
Murine erythroleukemia (MEL) strain DS19 is a well-established cell line that can be induced to model differentiation from proerythroblast to orthochromic stages of erythroid development using DMSO [36, 37]. Here, MEL cells were seeded at 2.5×105 cells/mL and maintained at 37°C and 5% CO2 for 72 h in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 25 mM glucose, 1 mM sodium pyruvate, and 4 mM L-glutamine (Cellgro, Corning, NY), supplemented with 10% (v/v) fetal bovine serum (FBS) (Atlanta Biologicals, Atlanta, GA), 1X Pen/Strep (Cellgro), and 1.5% (v/v) DMSO. Media were pretreated with prochelators (0-5 μM) in the presence or absence of 1 mM succinylacetone (SA) (Sigma), a specific inhibitor of heme biosynthesis [38]. All cultures were prepared in triplicate. Hemoglobin levels in MEL cells were quantitated as previously described [39] with modifications. Briefly, whole-cell suspensions washed and resuspended in PBS were scanned in the Olis CLARiTY 1000A spectrophotometer with an integrated RSM-ICAM containing a one-milliliter cuvette. Apparent absorbance values were recorded relative to a PBS baseline at the heme Soret peak of 411 nm. Javorfi correction [40] was carried out to normalize enhanced pathlength values to one centimeter using SpectralWorks software (Olis, Inc.). Soret peak baselines were measured by linear interpolation, and the resulting corrected absorbance intensities were converted to hemoglobin concentrations using an extinction coefficient of 0.291 μM-1 cm-1. The assumptions that all heme measured in the intact MEL cells is bound to hemoglobin and that each hemoglobin tetramer contains four heme molecules were made.
Cell counts were obtained with a Scepter handheld cell counter (Millipore, Billerica, MA) [41] to quantitate cell viability and per-cell hemoglobin concentrations, both of which are reported as mean ± SDM. Statistical significance of these values were determined against untreated (DMSO) controls using two-way ANOVA (without repeated measures), followed by Dunnett’s test, on GraphPad Prism version 7 (GraphPad Software, USA).
3. Results and discussion
3.1 Synthesis
The prochelators examined in this work fall into three classes of metal-binding scaffolds (Fig. 2): the thiosemicarbazones (TC compounds), the aroylhydrazones (AH compounds) and the semicarbazones (SC compounds). The selected compounds allowed comparison of binding groups with (S,N,S) and (S,N,O) donor sets, as well as different functional groups (e.g., aroyl hydrazones vs semicarbazones within the (S,N,O) binding units). In addition, methyl hydrazone analogs were included because studies on SIH and related high-affinity hydrazone chelators indicated that these derivatives are more stable with respect to hydrolytic degradation [42].
Fig. 2.
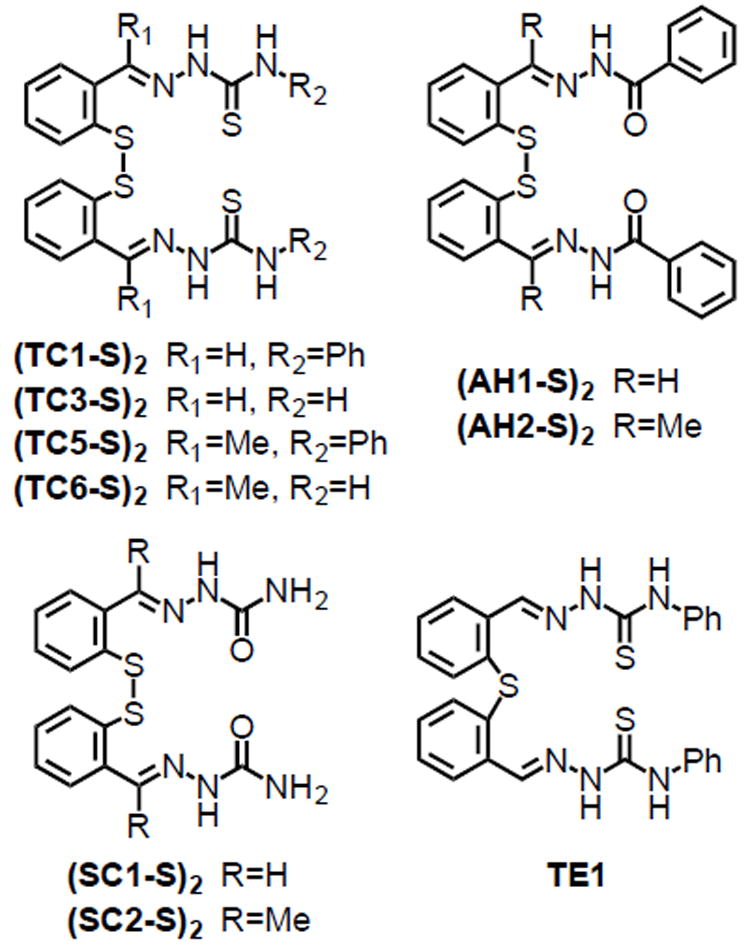
Disulfide-based prochelators and thioether control compound employed in this study.
Thioether TE1 was synthesized as a control analog of prochelator (TC1-S)2 lacking the disulfide bond. TE1 does not bind iron in physiologically relevant conditions (Fig. S1) and cannot be reduced by intracellular reagents to produce an iron-binding species.
The disulfide-based prochelators were synthesized via Schiff-base condensation reactions between a 2,2’-dithiodibenzyl aldehyde or ketone and a thiosemicarbazide, semicarbazide, or aroyhydrazide (Scheme S1). A major advantage in the purification of the compounds in Fig. 2 is the solubility difference between the starting materials and the products of the condensation reactions. Although the starting materials are soluble in refluxing alcohols (e.g., methanol, ethanol, isopropanol), the disulfide prochelators generally precipitate from the reaction mixtures in quantitative yields and high purity. For biological testing, stock solutions of the prochelators were then prepared in DMSO. Compound (SC1-S)2, however, presented limited solubility in DMSO and was not investigated further.
The stability of the disulfide prochelator systems and of the thioether control was assessed in aqueous buffered solutions in the presence of GSH at a physiologically relevant concentration (11 mM). In all cases, no significant decomposition was observed over a period of 10 h (Figs. S2 and S3). A slight increase of the baseline in some cases (TC3 and TC5, Fig. S2, and AH1, Fig. S3) was attributed to slow precipitation, which could be observed by the naked eye upon standing of the solutions over 24 h.
3.2 Antiproliferative activity
Our assessment of the antiproliferative activity of the compounds focused on breast cancer because its association to an altered metabolism of iron is well documented [3, 43]. The prochelators were tested in breast adenocarcinoma cell lines MCF-7 (ATCC® HTB-22™) and MDA-MB-231 (ATCC® HTB-26™) using standard MTT assay protocols (Table 1). For comparison with a normal cell line, the antiproliferative activity was also assessed in lung MRC-5 fibroblasts (ATCC® CCL-171™).
Table 1.
IC50 values for disulfide prochelators in breast cancer cell lines and normal lung fibroblastsa
| IC50 (μM)
|
|||
|---|---|---|---|
| MDA-MB-231 | MCF-7 | MRC-5 | |
| (TC1-S)2 | 4.4 ± 0.7 | 12.3 ± 0.8 | 30 ± 5 |
| (TC3-S)2 | 17.0 ± 0.4 | 48.3 ± 1.2 | 27.3 ± 1.6 |
| (TC5-S)2 | 17 ± 3 | 37 ± 1 | > 300 |
| (TC6-S)2 | 18.1 ± 0.9 | 19.5 ± 1.5 | 29.0 ± 0.6 |
| (AH1-S)2 | 6.7 ± 0.5 | 4.6 ± 0.9 | 20.5 ± 1.0 |
| (AH2-S)2 | 17.1 ± 1.5 | 45.3 ± 2.9 | > 100 |
| (SC2-S)2 | 45 ± 4 | 53 ± 1 | 91 ± 3 |
| TE1 | >200 | >100 | >100 |
aIC50 values were determined using standard MTT assays after 72-h incubations. Values are presented as mean ± SDM.
The antiproliferative activities of the thiosemicarbazone- and hydrazone-based disulfide-masked prochelators in the cancer cell lines fall within a relatively narrow range, with IC50 values mostly ranging from 4 to 50 μM, similar to those previously observed for this type of prochelators [24, 34]. Within the tested panel, we observed that the semicarbazone (SC2-S)2 presented the highest IC50 values. This observation is in line with previous studies showing lower antiproliferative activities of semicarbazones as compared to their thiosemicarbazone analogues [44, 45]. Confirming the requirement for intracellular activation and iron binding, the thioether control TE1 did not affect cell viability across all concentrations tested (up to 200 μM).
In the normal fibroblasts, IC50 values were consistently higher than 20 μM and up to more than 100 μM for (AH2-S)2 and (TC5-S)2. Although selectivity for cancer cells cannot be easily derived from comparisons of toxicity parameters in different cell cultures, these data indicated that the prochelators, including the most active compounds (TC1-S)2 and (AH1-S)2, are generally less toxic to the MRC-5 fibroblasts.
We have previously shown that the exposure of cultured cells to prochelator (TC1-S)2 leads to iron sequestration and intracellular formation of a low-spin Fe(III) complex [25]. This ferric species is not susceptible to intracellular redox cycling and therefore does not elicit catalytic generation of ROS [24, 25]. Indeed, the observed toxicity values for (TC1-S)2 (Table 1) in the micromolar range are expected for a species that interferes with the intracellular availability of labile iron (also micromolar) [46] without causing oxidative stress. Conversely, chelators that form redox-active complexes (of iron and/or copper) typically display higher toxicity parameters [47, 48]. For instance, effective antiproliferative chelators such as Triapine and Dp44mT lead to the oxidation of oxyhemoglobin to methemoglobin (among other effects) [16].
The micromolar IC50 values observed for the series of prochelators examined herein are similar to those of (TC1-S)2 and hence compatible with the notion that these compounds exclude intracellular iron from redox cycling. We sought to examine this possibility through assays of redox activity prior to proceeding to the analysis of the impact of the prochelators on cell cycle and heme biosynthesis.
3.3 Investigation of intracellular ROS generation
Our initial tests of oxidative reactivity of the metal complexes derived from our chelator systems were conducted in vitro using the benzoate hydroxylation assay in the presence of Fe(II) and H2O2 (Fig. S4). These measurements indicated that the compounds did not induce oxidative reactivity, whereas the formation of fluorescent salicylate upon benzoate hydroxylation was clearly observed in the presence of the positive control EDTA.
The ability of the disulfide prochelators to induce oxidative stress intracellularly was then tested using DCFH2-DA as a fluorogenic probe. DCFH2-DA, which is hydrolyzed to DCFH2 and trapped intracellularly upon the action of esterases, reacts with several ROS/RNS species (e.g., hydroxyl radical, peroxynitrite) to produce the fluorescent dichlorofluorescein (DCF) dye [49]. Hydrogen peroxide (used as the positive control in this assay) does not react directly with DCFH2, but its intracellular metal-mediated decomposition produces the detected ROS/RNS species [49].
Treatment of MDA-MB-231 breast adenocarcinoma cells with high-affinity chelator SIH (50 μM, 2 h) results in suppression of inherent ROS as demonstrated by a significant decrease of DCF fluorescence compared to the untreated control (Fig. 3). This behavior is well documented, and in fact SIH can be employed to protect cells against metal-mediated oxidative stress [50]. For a selection of prochelators featuring one compound of each family of binding units (Fig. 3), we found that treatment of MDA-MB-231 cells (50 μM, 2 h) also results in a decrease of ROS/RNS levels compared to the untreated control. Conversely, treatment with thioether TE1, which lacks metal-binding affinity, does not result in suppression of basal ROS/RNS concentrations. The antioxidant behavior of the prochelators can therefore be ascribed to metal sequestration, which excludes Fe(II) ions from Fenton chemistry, rather than to radical/ROS scavenging reactivity by their organic framework.
Fig. 3.
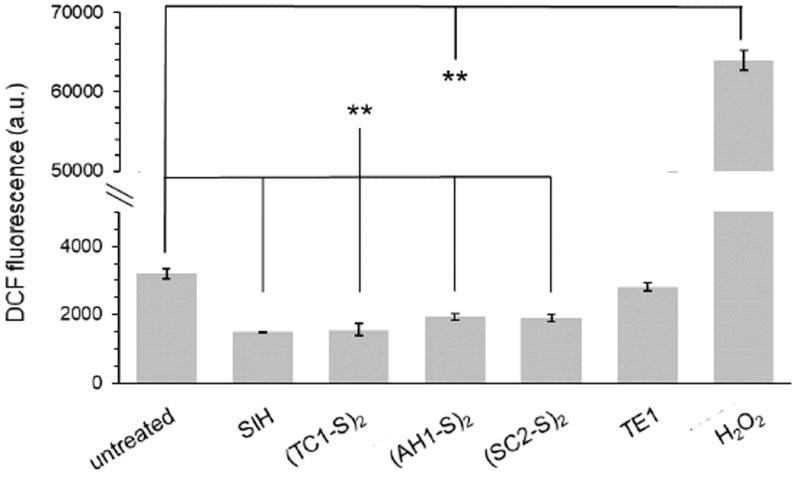
Assessment of intracellular generation of ROS upon incubation with selected prochelators. MDA-MB-231 cells were treated with the indicated compounds (50 μM, 2 h), washed, treated with DCFH2-DA (30 μM, 30 min) in PBS, and then analyzed by flow cytometry. Hydrogen peroxide is used as a positive control and SIH as a negative control. Values are presented as mean ± SDM (n=3), ** p <0.01.
Because the positive control (H2O2) showed a response that is an order of magnitude higher than that of the untreated control [51], we conclude that our prochelators do not result in induction of oxidative stress at the tested concentrations and treatment time. Rather, the disulfide-masked prochelators protect against metal-mediated intracellular oxidative stress as measured by DCFH2-DA.
3.4 Effects on cell cycle progression
Iron chelation often causes cell cycle arrest at the G1/S interface [52, 53], i.e., after the first growth (G1) phase and before the DNA synthesis (S) phase, because cells display lower DNA biosynthetic activity and cannot progress through the cell cycle [54]. Although the decreased availability of iron affects several cellular processes, the G1/S arrest has been attributed, at least in part, to the ability of chelators to decrease the activity of ribonucleotide reductase (RNR), an enzyme that is critical for DNA synthesis [55-58]. We have previously shown that treatment of cultured Jurkat lymphocyte cells with prochelator (TC1-S)2 results in decreased intracellular levels of active RNR as measured by electron paramagnetic resonance (EPR) spectroscopy [25]. Herein, we sought to investigate the effects of our selection of prochelators on cell cycle progression. As in the redox activity assays (vide supra), high-affinity chelator SIH was employed as a positive control [59].
We tested cell cycle distributions in MDA-MB-231 cells after 10 and 50 μM treatment and 12-h incubations (Fig. 4). At 10 μM, SIH resulted in arrest at the G1/S interface, with accumulation of G0/1 compared to the untreated control. At this concentration, treatment with (AH1-S)2 resulted in G1/0 accumulation as significant as that for SIH. This accumulation of cells in the G1/0 phase is accompanied by statistically significant depletion of cells in the S phase, with (AH1-S)2 being more effective than SIH (Fig. 4, top panel). At the higher concentration (50 μM), all the prochelators have similar impact on cell cycle, with G1/0 accumulation and S depletion being statistically significant in all cases (Fig. 4, bottom panel). These data therefore indicate that all the tested prochelators have effects on cell cycle that are consistent with iron sequestration.
Fig. 4.
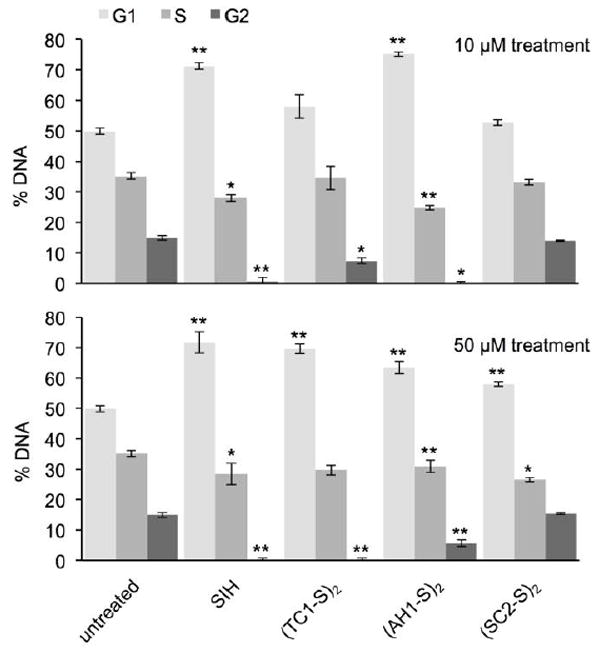
Effect of prochelators on cell cycle in MDA-MB-231 cells. Cells were treated with the compounds (10 or 50 μM, 12 h), harvested, fixed, pelleted and then treated with RNAse and propidium iodide (0.5 mg/mL and 40 μg/mL, respectively, 30 min) prior to analysis by flow cytometry. Values are presented as mean ± SDM (n=3), * p < 0.05 and ** p < 0.01.
3.5 Effects on cell death
Apoptosis is an important form of programmed cell death that is often implicated as the pathway of chelation-induced cytotoxicity [53]. Several iron chelators, such as DFO [60] and thiosemicarbazone Dp44mT [61], initiate the apoptotic pathway of cell death. In the context of this analysis of the biological activity of disulfide-based prochelators, we investigated their effects on cell death using PI and the fluorescein isothiocyanate conjugate of Annexin V (FTIC-AnnV) as probes for apoptotic markers.
Preliminary experiments indicated that MDA-MB-231 cells suffered partial rupture in the conditions of this assay after a 48-h incubation, and a significant portion of cellular debris was detected by flow cytometry. Nevertheless, reliable results (Fig. 5) were obtained using suspended cultures of Jurkat lymphocyte cells, in which the activation of prochelator (TC1-S)2 has been previously documented [25]. In this assay, we used potent antineoplastic agent taxol (paclitaxel) as a positive control known to induce apoptosis [62], and tridentate chelator SIH was included as a comparison leading to iron sequestration with no need for intracellular activation.
Fig. 5.
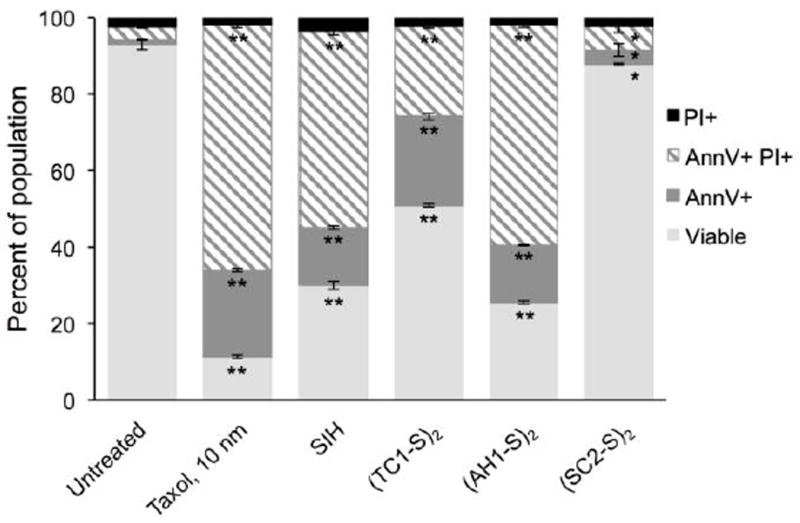
Investigation of cell death in the presence of disulfide-based prochelators. Jurkat cells were incubated with the tested compounds (20 μM, 48 h) or vehicle only (DMSO, untreated control). Following treatment with FTIC-AnnV and propidium iodide (PI), the cells were analyzed by flow cytometry. Values are presented as mean ± SDM (n=3), * p <0.05 and ** p <0.01.
For all tested prochelators, 48-hr treatments resulted in a decrease in viable cells compared to the untreated control. This observation is accompanied by a significant increase in populations that stain positive for AnnV only (termed apoptotic) or AnnV and PI (termed late-apoptotic). Within the prochelator series, (AH1-S)2 was as effective as SIH at inducing apoptosis. The thiosemicarbazone-based prochelator (TC1-S)2 is less effective and the semicarbazone analogue (SC2-S)2 is the least potent of all the compounds tested. These results are consistent with the antiproliferative activity of this series of disulfide-masked prochelators as assessed by colorimetric assays as well as their efficacy in halting cell cycle progression (Section 3.4).
The induction of apoptosis in Jurkat cells by disulfide-masked prochelators is concentration- and time-dependent, and we observe an increase in AnnV+/PI+ populations with longer incubations or higher concentrations (data not shown). None of the treatments resulted in significant populations that stained positive for PI only (less than 4% of the population in all cases), ruling out necrosis as a pathway of cell death. In fact, iron scavengers are known to suppress iron-mediated necrosis that results from Fenton reactions and ROS generation [63]. The lack of a significant necrotic population is indeed in line with the fact that our prochelators suppress basal ROS levels (vide supra).
Collectively, and in agreement with the IC50 values (Table 1), the data from cell cycle and cell death assays indicate that aroylhydrazone (AH1-S)2 and thiosemicarbazone (TC1-S)2 are significantly more effective than semicarbazone (SC2-S)2 as antiproliferative prochelators.
3.6 Effects on hemoglobin production
Rapidly dividing cells, such as malignant cells, require increased iron levels during proliferation; however, the greatest need for this element and the highest expression of transferrin receptors are characteristic of differentiating erythroid cells [64]. These blood cells are responsible for hemoglobin (Hb) biosynthesis during erythropoiesis, a process that consumes 20 mg of iron per day in an average adult [65]. In the context of this initial biological investigation of disulfide-based prochelators, we sought to examine the impact of these iron-binding compounds on hemoglobin-synthesizing cells and hence their potential to interfere with the critical iron-dependent process of red blood cell production. As models of erythroid cells, we employed murine erythroleukemia (MEL) cells, in which differentiation from proerythroblast to orthochromic stages of erythroid development can be induced by DMSO [36, 37].
In this set of experiments, we limited the concentration of prochelators in growth media to 0.5 μM or 5 μM in order to maintain cell viability. The incubations at 5 μM showed some effect on cell counts (Fig S5); however, the hemoglobin content per cell could be quantified reliably in all cases (vide infra). Conversely, incubations at 25 μM led to significantly decreased cell counts (Fig S5) therefore this concentration was deemed unsuitable for this assay of hemoglobin production.
The hemoglobin amount per cell was assessed by established methods from the absorbance of the Soret band of heme at 411 nm [39]. Cells were incubated with the prochelators alone or in the presence of succinylacetone (SA), an inhibitor of δ-aminolevulinic acid dehydratase and hence of heme synthesis [38]. This experimental design was selected to reveal effects on Hb production and additional effects on cells in which the inhibition of heme biosynthesis by SA leads to mitochondrial iron accumulation [66, 67].
Overall, treatment of induced MEL cells with disulfide-based prochelators or with chelator SIH (0.5 μM or 5 μM, 72 h) did not have major effects on hemoglobin production (Fig. 6). For the semicarbazone (SC2-S)2 and the thiosemicarbazone prochelator (TC1-S)2, we found no statistically significant difference in the intracellular Hb concentrations even at 5 μM. A small (less than 20%) but statistically significant decrease in intracellular hemoglobin concentrations was observed in the case of (AH1-S)2 and SIH (Fig. 6, top panel). These data are in line with previous reports showing that MEL cells [68], primary mouse erythroblasts [69], and reticulocytes [70] are not (or only slightly) affected by iron chelators, an effect that has been attributed to distinct mechanisms to regulate and distribute iron in erythroid cells when compared to non-erythroid cells. In particular, this observation is consistent with the notion that iron may be delivered to the mitochondria by direct and transient docking of transferrin-containing endosomes, thus remaining unaffected by cytosolic iron scavengers in erythroid cells [71, 72].
Fig. 6.
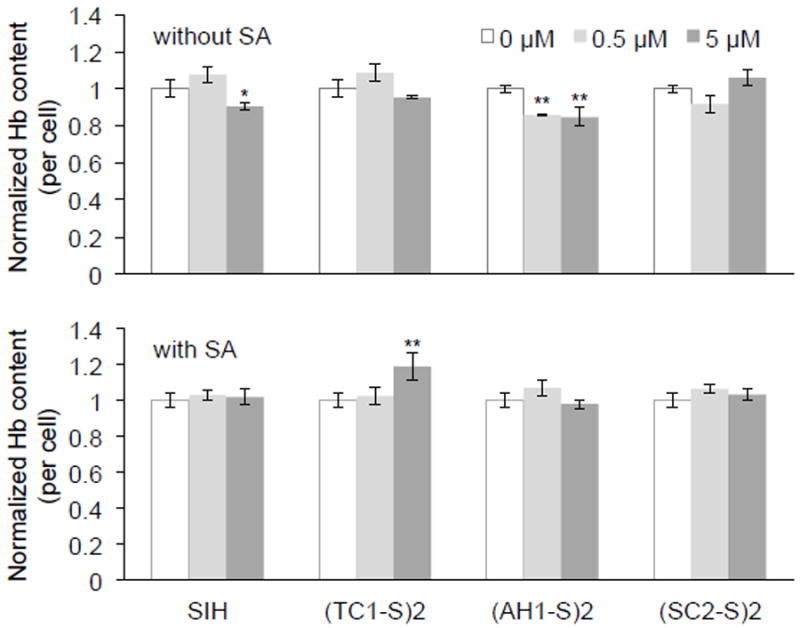
Effect of iron prochelators on intracellular haemoglobin (Hb) concentrations in the MEL model of erythropoiesis. Hb content was assessed after treatment with prochelators alone (top) or along with succinylacetone (SA, 1 mM). Values (mean ± SDM, n=3) are normalized to the untreated control (0 μM), * p <0.05 and ** p <0.01.
Importantly, this established assay for Hb quantification [39] is able to distinguish between Fe(II) and Fe(III) hemoglobin (i.e., methemoglobin) based on Soret peak wavelength differences, and therefore our findings show that the tested prochelators (and the corresponding iron complexes formed intracellularly) do not result in significant oxidation of heme. This observation is thus consistent with our investigation of oxidative stress, which showed no increase of ROS concentrations in cultured cells exposed to the prochelators (Fig. 3).
In the MEL model of erythropoiesis, treatment with heme biosynthesis inhibitor SA results in iron accumulation in the mitochondria [66, 67]. In this experiment, cells were treated with the prochelators in the presence of SA, therefore the overall Hb production was dramatically reduced to 15-25% of the per-cell levels observed in cultures lacking SA. Within normalized data sets (Fig. 6, bottom panel), we found that treatment with SIH, (AH1-S)2 or (SC2-S)2 did not result in statistically significant changes in intracellular Hb concentrations. Treatment with (TC1-S)2 at the higher concentration (5 μM) led to a slightly higher amount of Hb per cell (Fig. 6, bottom panel). This unusual effect could be tentatively ascribed to either a prochelator-associated decrease in ROS [73] or a change in iron distribution (i.e., mitochondrial vs cytosolic) that alters the expression of heme biosynthetic enzymes in competition with SA. For instance, mobilization of iron from mitochondria to the cytosol could alleviate the translational block on δ-aminolevulinic acid synthase 2 (ALAS2), the rate-limiting enzyme in heme synthesis, by iron-responsive protein 2 (IRP2) [74].
In summary, in the MEL model of differentiating erythroid cells, the disulfide-based prochelators examined in this study did not show significant impact on hemoglobin production and/or oxidation to methemoglobin.
4. Conclusions
The chemistry of disulfide bonds in biological settings, and specifically their reduction by abundant intracellular thiols (e.g., glutathione), can be utilized for the design of prochelators that target intracellular iron. This approach was initially tested on thiosemicarbazone scaffolds and was extended herein to aroylhydrazone and semicarbazone binding units. Each prochelator is reductively activated to a tridentate chelator with a (S,N,S) or (S,N,O) donor set. The introduction of methyl hydrazone motifs on all three systems was examined as an additional design feature.
The antiproliferative activity of this cohort of prochelators was assessed in breast adenocarcinoma cell lines (MCF-7 and MDA-MB-231) and IC50 values were determined in the micromolar range, namely at concentrations similar to those of cytosolic iron. None of the prochelator/chelator systems induced intracellular generation of oxidative stress in cell-based assays using DCFH2-DA as fluorogenic probe of ROS. Conversely, we observed that the prochelators result in suppression of inherent ROS, likely because of the exclusion of labile iron from intracellular redox cycling and production of ROS. Both the toxicity assays and the assessment of oxidative damage in cells therefore indicated that the prochelator systems are not leading to the formation of redox-active iron complexes of higher toxicity relative to the parent chelators.
The reduction/activation of the disulfide-masked prochelators is crucial for their antiproliferative action. The thioether compound TE1, which is a close analog of thiosemicarbazone prochelator (TC1-S)2 but lacks the disulfide bond, showed no antiproliferative activity even at 200 μM.
Additional experiments in MDA-MB-231 cells indicated that, similar to other antiproliferative iron chelators, the tested compounds result in G0/1 cell cycle arrest and the extent of accumulation at G0/1 correlated well with the antiproliferative potency of the prochelators. The mechanism of action of these systems likely proceeds through an apoptotic pathway as determined by staining methods through flow cytometry. Finally, we found that the most antiproliferative disulfide-based prochelators of this series do not affect significantly the process of heme biosynthesis in the MEL model of erythropoiesis.
In summary, disulfide/thiol switches afford viable approaches to the development of antiproliferative iron chelators that are activated under reducing intracellular conditions. This strategy is amenable to structural modifications, and the compounds reported herein (featuring (thio)semicarbazones and aroylhydrazones) interfere with cell cycle progression and induce apoptosis, with new aroylhydrazone compound (AH1-S)2 presenting the highest biological activity so far. Relevant to their potential medicinal applications, these prochelators do not lead to oxidative stress or reduced heme production in cultured cells.
Supplementary Material
Synopsis.
Activated to sequester iron under intracellular reducing conditions, disulfide-based prochelators display antiproliferative activity through cell cycle arrest and apoptosis without causing oxidative stress in breast cancer cells or alterations of hemoglobin production in erythroid cells.
Highlights.
Disulfide switches for intracellular activation were introduced in tridentate chelators
(Thio)semicarbazones and aroyl hydrazones were tested as iron-binding units
The prochelators are antiproliferative in cancer cells at micromolar concentrations
G0/1 cell cycle arrest and apoptosis were detected in the absence of oxidative stress
In murine erythroleukemia cells, prochelators did not affect heme biosynthesis
Acknowledgments
This work was supported by the University of Arizona Office for Research, Discovery & Innovation. Support from the National Institute of Health (grant R01 DK096501 to H.A.D.) is also gratefully acknowledged. The UACC/ARL Cytometry Core Facility at the University of Arizona is funded by Cancer Center Support Grant CA 023074.
Abbreviations
- ALAS2
δ-aminolevulinic acid synthase 2
- AnnV
Annexin V
- ANOVA
analysis of variance
- DCF
dichlorofluorescein
- DCFH2-DA
dichlorodihydrofluorescein diacetate
- DFO
desferrioxamine
- DFP
deferiprone
- DMEM
Dulbecco’s Modified Eagle’s Medium
- DMSO
dimethylsulfoxide
- Dp44mT
di-2-pyridylketone 4,4,-dimethyl-3-thiosemicarbazone
- DpC
di-2-pyridylketone 4-cyclohexyl-4-methyl-3-thiosemicarbazone
- EDTA
ethylenediaminetetraacetic acid
- EMEM
Eagle’s Minimal Essential Medium
- EPR
electron paramagnetic resonance
- FBS
fetal bovine serum
- FTIC-AnnV
fluorescein isothiocyanate conjugate of Annexin V
- GSH
reduced glutathione
- Hb
hemoglobin
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- IRP2
iron-responsive protein 2
- MEL
murine erythroleukemia
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PBS
phosphate-buffered saline
- PI
propidium iodide
- RNR
ribonucleotide reductase
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- RPMI
Roswell Park Memorial Institute
- SA
succinylacetone
- SDM
standard deviation of the mean
- SIH
salicyl isonicotinoyl hydrazone
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Torti SV, Torti FM. Nat Rev Cancer. 2013;13:342–355. doi: 10.1038/nrc3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Habashy HO, Powe DG, Staka CM, Rakha EA, Ball G, Green AR, Aleskandarany M, Paish EC, Macmillan RD, Nicholson RI, Ellis IO, Gee JMW. Breast Cancer Res Treat. 2010;119:283–293. doi: 10.1007/s10549-009-0345-x. [DOI] [PubMed] [Google Scholar]
- 3.Pinnix ZK, Miller LD, Wang W, D’Agostino R, Jr, Kute T, Willingham MC, Hatcher H, Tesfay L, Sui G, Di X, Torti SV, Torti FM. Sci Transl Med. 2010;2:43–56. doi: 10.1126/scisignal.3001127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ricolleau G, Charbonnel C, Lode L, Loussouarn D, Joalland MP, Bogumil R, Jourdain S, Minvielle S, Campone M, Deporte-Fety R, Campion L, Jezequel P. Proteomics. 2006;6:1963–1975. doi: 10.1002/pmic.200500283. [DOI] [PubMed] [Google Scholar]
- 5.Spangler B, Morgan CW, Fontaine SD, Vander Wal MN, Chang CJ, Wells JA, Renslo AR. Nat Chem Biol. 2016;12:680–685. doi: 10.1038/nchembio.2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aron AT, Loehr MO, Bogena J, Chang CJ. J Am Chem Soc. 2016;138:14338–14346. doi: 10.1021/jacs.6b08016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Richardson DR, Kalinowski DS, Lau S, Jansson PJ, Lovejoy DB. Biochim Biophys Acta. 2009;1790:702–717. doi: 10.1016/j.bbagen.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 8.Brockman RW, Thomson JR, Bell MJ, Skipper HE. Cancer Res. 1956;16:167–170. [PubMed] [Google Scholar]
- 9.French FA, Freedlander BL, Hoskino A, French J. Cancer Res. 1958;18:1290–1300. [PubMed] [Google Scholar]
- 10.Kalinowski DS, Richardson DR. Pharmacol Rev. 2005;57:547–583. doi: 10.1124/pr.57.4.2. [DOI] [PubMed] [Google Scholar]
- 11.Merlot AM, Kalinowski DS, Richardson DR. Antioxid Redox Sign. 2013;18:973–1006. doi: 10.1089/ars.2012.4540. [DOI] [PubMed] [Google Scholar]
- 12.Yu Y, Kalinowski DS, Kovacevic Z, Siafakas AR, Jansson PJ, Stefani C, Lovejoy DB, Sharpe PC, Bernhardt PV, Richardson DR. J Med Chem. 2009;52:5271–5294. doi: 10.1021/jm900552r. [DOI] [PubMed] [Google Scholar]
- 13.Kovacevic Z, Chikhani S, Lovejoy DB, Richardson DR. Mol Pharmacol. 2011;80:598–609. doi: 10.1124/mol.111.073627. [DOI] [PubMed] [Google Scholar]
- 14.Lovejoy DB, Sharp DM, Seebacher N, Obeidy P, Prichard T, Stefani C, Basha MT, Sharpe PC, Jansson PJ, Kalinowski DS, Bernhardt PV, Richardson DR. J Med Chem. 2012;55:7230–7244. doi: 10.1021/jm300768u. [DOI] [PubMed] [Google Scholar]
- 15.Jansson PJ, Yamagishi T, Arvind A, Seebacher N, Gutierrez E, Stacy A, Maleki S, Sharp D, Sahni S, Richardson DR. J Biol Chem. 2015;290:9588–9603. doi: 10.1074/jbc.M114.631283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quach P, Gutierrez E, Basha MT, Kalinowski DS, Sharpe PC, Lovejoy DB, Bernhardt PV, Jansson PJ, Richardson DR. Mol Pharmacol. 2012;82:105–114. doi: 10.1124/mol.112.078964. [DOI] [PubMed] [Google Scholar]
- 17.Jansson PJ, Kalinowski DS, Lane DJR, Kovacevic Z, Seebacher NA, Fouani L, Sahni S, Merlot AM, Richardson DR. Pharmacol Res. 2015;100:255–260. doi: 10.1016/j.phrs.2015.08.013. [DOI] [PubMed] [Google Scholar]
- 18.Oliveri V, Vecchio G. J Inorg Biochem. 2016;162:31–43. doi: 10.1016/j.jinorgbio.2016.05.012. [DOI] [PubMed] [Google Scholar]
- 19.Wang Q, Franz KJ. Acc Chem Res. 2016;49:2468–2477. doi: 10.1021/acs.accounts.6b00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schugar H, Green DE, Bowen ML, Scott LE, Storr T, Bohmerle K, Thomas F, Allen DD, Lockman PR, Merkel M, Thompson KH, Orvig C. Angew Chem Int Ed. 2007;46:1716–1718. doi: 10.1002/anie.200603866. [DOI] [PubMed] [Google Scholar]
- 21.Zheng HL, Youdim MBH, Fridkin M. ACS Chem Biol. 2010;5:603–610. doi: 10.1021/cb900264w. [DOI] [PubMed] [Google Scholar]
- 22.Charkoudian LK, Pham DM, Franz KJ. J Am Chem Soc. 2006;128:12424–12425. doi: 10.1021/ja064806w. [DOI] [PubMed] [Google Scholar]
- 23.Charkoudian LK, Pham DM, Kwon AM, Vangeloff AD, Franz KJ. Dalton Trans. 2007:5031–5042. doi: 10.1039/b705199a. [DOI] [PubMed] [Google Scholar]
- 24.Chang TM, Tomat E. Dalton Trans. 2013;42:7846–7849. doi: 10.1039/c3dt50824b. [DOI] [PubMed] [Google Scholar]
- 25.Akam EA, Chang TM, Astashkin AV, Tomat E. Metallomics. 2014;6:1905–1912. doi: 10.1039/c4mt00153b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pujol AM, Cuillel M, Jullien AS, Lebrun C, Cassio D, Mintz E, Gateau C, Delangle P. Angew Chem Int Ed. 2012;51:7445–7448. doi: 10.1002/anie.201203255. [DOI] [PubMed] [Google Scholar]
- 27.Schafer FQ, Buettner GR. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 28.Gamcsik MP, Kasibhatla MS, Teeter SD, Colvin OM. Biomarkers. 2012;17:671–691. doi: 10.3109/1354750X.2012.715672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saito G, Swanson JA, Lee KD. Adv Drug Delivery Rev. 2003;55:199–215. doi: 10.1016/s0169-409x(02)00179-5. [DOI] [PubMed] [Google Scholar]
- 30.Lee MH, Yang Z, Lim CW, Lee YH, Dongbang S, Kang C, Kim JS. Chem Rev. 2013;113:5071–5109. doi: 10.1021/cr300358b. [DOI] [PubMed] [Google Scholar]
- 31.Lee MH, Sessler JL, Kim JS. Acc Chem Res. 2015;48:2935–2946. doi: 10.1021/acs.accounts.5b00406. [DOI] [PubMed] [Google Scholar]
- 32.Ballatori N, Krance SM, Notenboom S, Shi SJ, Tieu K, Hammond CL. Biol Chem. 2009;390:191–214. doi: 10.1515/BC.2009.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yeh CC, Hou MF, Wu SH, Tsai SM, Lin SK, Hou LA, Ma H, Tsai LY. Cell Biochem Funct. 2006;24:555–559. doi: 10.1002/cbf.1275. [DOI] [PubMed] [Google Scholar]
- 34.Akam EA, Tomat E. Bioconjugate Chem. 2016;27:1807–1812. doi: 10.1021/acs.bioconjchem.6b00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mertens C, Akam EA, Rehwald C, Brune B, Tomat E, Jung M. PLoS One. 2016;11:e0166164. doi: 10.1371/journal.pone.0166164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Friend C, Scher W, Holland JG, Sato T. Proc Natl Acad Sci U S A. 1971;68:378–382. doi: 10.1073/pnas.68.2.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohta Y, Tanaka M, Terada M, Miller OJ, Bank A, Marks PA, Rifkind RA. Proc Natl Acad Sci U S A. 1976;73:1232–1236. doi: 10.1073/pnas.73.4.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ebert PS, Hess RA, Frykholm BC, Tschudy DP. Biochem Biophys Res Commun. 1979;88:1382–1390. doi: 10.1016/0006-291x(79)91133-1. [DOI] [PubMed] [Google Scholar]
- 39.Marcero JR, Piel RB, Burch JS, Dailey HA. BioTechniques. 2016;61:83–91. doi: 10.2144/000114444. [DOI] [PubMed] [Google Scholar]
- 40.Javorfi T, Erostyak J, Gal J, Buzady A, Menczel L, Garab G, Naqvi KR. J Photochem Photobiol B. 2006;82:127–131. doi: 10.1016/j.jphotobiol.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 41.Ongena K, Das C, Smith JL, Gil S, Johnston G. J Vis Exp. 2010:e2204. doi: 10.3791/2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Potuckova E, Hruskova K, Bures J, Kovarikova P, Spirkova IA, Pravdikova K, Kolbabova L, Hergeselova T, Haskova P, Jansova H, Machacek M, Jirkovska A, Richardson V, Lane DJR, Kalinowski DS, Richardson DR, Vavrova K, Simunek T. PLoS One. 2014;9:e112059. doi: 10.1371/journal.pone.0112059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marques O, da Silva BM, Porto G, Lopes C. Cancer Lett. 2014;347:1–14. doi: 10.1016/j.canlet.2014.01.029. [DOI] [PubMed] [Google Scholar]
- 44.Soares ROA, Echevarria A, Bellieny MSS, Pinho RT, de Leo RMM, Seguins WS, Machado GM, Canto-Cavalheiro MM, Leon LL. Exp Parasitol. 2011;129:381–387. doi: 10.1016/j.exppara.2011.08.019. [DOI] [PubMed] [Google Scholar]
- 45.Potuckova E, Roh J, Machacek M, Sahni S, Stariat J, Sestak V, Jansova H, Haskova P, Jirkovska A, Vavrova K, Kovarikova P, Kalinowski DS, Richardson DR, Simunek T. PLoS One. 2015;10:e0139929. doi: 10.1371/journal.pone.0139929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hider RC, Kong XL. Dalton Trans. 2013;42:3220–3229. doi: 10.1039/c2dt32149a. [DOI] [PubMed] [Google Scholar]
- 47.Jansson PJ, Hawkins CL, Lovejoy DB, Richardson DR. J Inorg Biochem. 2010;104:1224–1228. doi: 10.1016/j.jinorgbio.2010.07.012. [DOI] [PubMed] [Google Scholar]
- 48.Lovejoy DB, Jansson PJ, Brunk UT, Wong J, Ponka P, Richardson DR. Cancer Res. 2011;71:5871–5880. doi: 10.1158/0008-5472.CAN-11-1218. [DOI] [PubMed] [Google Scholar]
- 49.Kalyanaraman B, Darley-Usmar V, Davies KJA, Dennery PA, Forman HJ, Grisham MB, Mann GE, Moore K, Roberts LJ, Ischiropoulos H. Free Radic Biol Med. 2012;52:1–6. doi: 10.1016/j.freeradbiomed.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Charkoudian LK, Dentchev T, Lukinova N, Wolkow N, Dunaief JL, Franz KJ. J Inorg Biochem. 2008;102:2130–2135. doi: 10.1016/j.jinorgbio.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Karlsson M, Kurz T, Brunk UT, Nilsson SE, Frennesson CI. Biochem J. 2010;428:183–190. doi: 10.1042/BJ20100208. [DOI] [PubMed] [Google Scholar]
- 52.Blatt J, Taylor SR, Stitely S. J Lab Clin Med. 1988;112:433–436. [PubMed] [Google Scholar]
- 53.Le NT, Richardson DR. Biochim Biophys Acta. 2002;1603:31–46. doi: 10.1016/s0304-419x(02)00068-9. [DOI] [PubMed] [Google Scholar]
- 54.Eriksson S, Graslund A, Skog S, Thelander L, Tribukait B. J Biol Chem. 1984;259:1695–1700. [PubMed] [Google Scholar]
- 55.Cooper CE, Lynagh GR, Hoyes KP, Hider RC, Cammack R, Porter JB. J Biol Chem. 1996;271:20291–20299. doi: 10.1074/jbc.271.34.20291. [DOI] [PubMed] [Google Scholar]
- 56.Eberhard Y, McDermott SP, Wang XM, Gronda M, Venugopal A, Wood TE, Hurren R, Datti A, Batey RA, Wrana J, Antholine WE, Dick J, Schimmer AD. Blood. 2009;114:3064–3073. doi: 10.1182/blood-2009-03-209965. [DOI] [PubMed] [Google Scholar]
- 57.Aye Y, Long MJC, Stubbe J. J Biol Chem. 2012;287:35768–35778. doi: 10.1074/jbc.M112.396911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Popovic-Bijelic A, Kowol CR, Lind MES, Luo JH, Himo F, Enyedy EA, Anion VB, Graslund A. J Inorg Biochem. 2011;105:1422–1431. doi: 10.1016/j.jinorgbio.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mackova E, Hruskova K, Bendova P, Vavrova A, Jansova H, Haskova P, Kovarikova P, Vavrova K, Simunek T. Chem Biol Interact. 2012;197:69–79. doi: 10.1016/j.cbi.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 60.Pan YJ, Hopkins RG, Loo G. BBA Mol Cell Res. 2004;1691:41–50. doi: 10.1016/j.bbamcr.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 61.Noulsri E, Richardson DR, Lerdwana S, Fucharoen S, Yamagishi T, Kalinowski DS, Pattanapanyasat K. Am J Hematol. 2009;84:170–176. doi: 10.1002/ajh.21350. [DOI] [PubMed] [Google Scholar]
- 62.Wahl AF, Donaldson KL, Fairchild C, Lee FYF, Foster SA, Demers GW, Galloway DA. Nat Med. 1996;2:72–79. doi: 10.1038/nm0196-72. [DOI] [PubMed] [Google Scholar]
- 63.Terman A, Kurz T. Antioxid Redox Sign. 2013;18:888–898. doi: 10.1089/ars.2012.4885. [DOI] [PubMed] [Google Scholar]
- 64.Ponka P. Blood. 1997;89:1–25. [PubMed] [Google Scholar]
- 65.Muckenthaler MU, Rivella S, Hentze MW, Galy B. Cell. 2017;168:344–361. doi: 10.1016/j.cell.2016.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ponka P, Wilczynska A, Schulman HM. Biochim Biophys Acta. 1982;720:96–105. doi: 10.1016/0167-4889(82)90043-x. [DOI] [PubMed] [Google Scholar]
- 67.Adams ML, Ostapiuk I, Grasso JA. Biochim Biophys Acta. 1989;1012:243–253. doi: 10.1016/0167-4889(89)90104-3. [DOI] [PubMed] [Google Scholar]
- 68.Chan RY, Seiser C, Schulman HM, Kuhn LC, Ponka P. Eur J Biochem. 1994;220:683–692. doi: 10.1111/j.1432-1033.1994.tb18669.x. [DOI] [PubMed] [Google Scholar]
- 69.Schranzhofer M, Schifrer M, Cabrera JA, Kopp S, Chiba P, Beug H, Mullner EW. Blood. 2006;107:4159–4167. doi: 10.1182/blood-2005-05-1809. [DOI] [PubMed] [Google Scholar]
- 70.Sheftel AD, Zhang AS, Brown C, Shirihai OS, Ponka P. Blood. 2007;110:125–132. doi: 10.1182/blood-2007-01-068148. [DOI] [PubMed] [Google Scholar]
- 71.Hamdi A, Roshan TM, Kahawita TM, Mason AB, Sheftel AD, Ponka P. Biochim Biophys Acta. 2016;1863:2859–2867. doi: 10.1016/j.bbamcr.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 72.Schultz IJ, Chen CY, Paw BH, Hamza I. J Biol Chem. 2010;285:26753–26759. doi: 10.1074/jbc.R110.119503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vreugdenhil G, Smeets M, Feelders RA, van Eijk HG. Acta Haematol. 1993;89:57–60. doi: 10.1159/000204488. [DOI] [PubMed] [Google Scholar]
- 74.Camaschella C, Campanella A, De Falco L, Boschetto L, Merlini R, Silvestri L, Levi S, Iolascon A. Blood. 2007;110:1353–1358. doi: 10.1182/blood-2007-02-072520. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.