

Abstract
Background
Long non-coding RNAs (lncRNAs) have a role in physiological and pathological processes, including cancer. The aim of this study was to investigate the expression of the long intergenic non-protein coding RNA 665 (LINC00665) gene and the cell cycle in hepatocellular carcinoma (HCC) using database analysis including The Cancer Genome Atlas (TCGA), the Gene Expression Omnibus (GEO), and quantitative real-time polymerase chain reaction (qPCR).
Material/Methods
Expression levels of LINC00665 were compared between human tissue samples of HCC and adjacent normal liver, clinicopathological correlations were made using TCGA and the GEO, and qPCR was performed to validate the findings. Other public databases were searched for other genes associated with LINC00665 expression, including The Atlas of Noncoding RNAs in Cancer (TANRIC), the Multi Experiment Matrix (MEM), Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) and protein-protein interaction (PPI) networks.
Results
Overexpression of LINC00665 in patients with HCC was significantly associated with gender, tumor grade, stage, and tumor cell type. Overexpression of LINC00665 in patients with HCC was significantly associated with overall survival (OS) (HR=1.47795%; CI: 1.046–2.086). Bioinformatics analysis identified 469 related genes and further analysis supported a hypothesis that LINC00665 regulates pathways in the cell cycle to facilitate the development and progression of HCC through ten identified core genes: CDK1, BUB1B, BUB1, PLK1, CCNB2, CCNB1, CDC20, ESPL1, MAD2L1, and CCNA2.
Conclusions
Overexpression of the lncRNA, LINC00665 may be involved in the regulation of cell cycle pathways in HCC through ten identified hub genes.
MeSH Keywords: Carcinoma, Hepatocellular; Real-Time Polymerase Chain Reaction; RNA, Long Noncoding
Background
Non-coding RNA (ncRNA) is a general term for RNAs that lacks protein-coding functionality. With recent developments in high-throughput sequencing techniques, increasing numbers of ncRNAs have been discovered, and are now categorized into two classes, depending on the transcript length. One ncRNA class is up to 200 nucleotides in length and includes microRNAs (miRNAs), small interfering RNAs (siRNAs), and small nuclear RNAs (snRNAs) [1,2]. Another ncRNA class is composed of long non-coding RNAs (lncRNAs) that contain 200 or more nucleotides in length [3]. Previously, lncRNAs were unknown entities within the genome [4,5]. Recently, although only a few lncRNAs have been discovered, they are now thought to play critical regulatory roles in physiological and pathological processes, including in cancer [6,7]. Recently, increasing numbers of studies on the roles of lncRNA have shown that changes in expression levels of lncRNAs have been found in several types of malignant tumors, indicating a role for lncRNAs in the development and progression of malignancy. Therefore, studies of lncRNA expression levels might have a future role as diagnostic or prognostic biomarkers in human cancer [8,9]. The lncRNAs that have been most commonly associated with cancer-related pathways have been studied in human tumors and have been found to function as regulators via several molecular mechanisms [10]. However, these studies on the role of lncRNAs in are still at an early stage, and many of the potential mechanisms and clinical significance of lncRNAs remain unclear and require further studies.
Worldwide, hepatocellular carcinoma (HCC) is one of the most common malignant tumors [11]. Recently published studies have shown that lncRNAs play a regulatory function in the onset and development of HCC, providing a new direction for the clinical diagnosis and treatment of this cancer [12,13]. Currently, there are several lncRNAs that have been found to be abnormally expressed in HCC. These lncRNAs influence the onset, development, invasion, metastasis, and recurrence of HCC by participating in multiple tumor-related pathways [14]. Considering the important molecular mechanisms of lncRNA in the onset and progression of HCC, the potential role of lncRNA in clinical practice has gained research interest. Recently, Qu et al. undertook a meta-analysis that included data from 27 published and found that the upregulation of lncRNA was associated with a reduced overall survival in HCC [15]. Although some studies have reported that lncRNAs play critical functions as oncogenes or tumor suppressor genes, the lncRNAs studied thus far represent only a small fraction of the lncRNAs defined in Gene Expression Profiling Interactive Analysis (GEPIA) [16]. Therefore, the investigation of new lncRNAs in HCC is an important area of research.
In this study, lncRNAs that were abnormally expressed in HCC were identified from liver hepatocellular carcinoma (LIHC) data in The Cancer Genome Atlas (TCGA) database (http://cancergenome.nih.gov/). The lncRNA, LINC0065 was selected as it had not been previously studied in HCC. The aim of this study was to investigate the expression of the lncRNA, LINC0065 gene and the cell cycle in HCC using database analysis including The Cancer Genome Atlas (TCGA), the Gene Expression Omnibus (GEO), and quantitative real-time polymerase chain reaction (qPCR), and its relationship to clinical parameters, and its prognostic value from cases at our hospital. Genes related to LINC00665 were determined through the Atlas of Noncoding RNAs in Cancer (TANRIC), and Multi Experiment Matrix (MEM) databases and the relationship between LINC00665 and cell cycle pathways were investigated using bioinformatics methodology.
Material and Methods
Data extraction from The Cancer Genome Atlas (TCGA) database and analysis of differentially expressed long non-coding RNAs (lncRNAs)
From the differentially expressed long non-coding RNAs (lncRNAs) found in The Cancer Genome Atlas (TCGA), the long intergenic non-protein coding RNA 665 (LINC00665) gene was selected for this study. The log2 transcripts per million (TPM+1) algorithm for TCGA data developed by Gene Expression Profiling Interactive Analysis (GEPIA) (http://gepia.cancer-pku.cn/) was applied to study the expression of LINC00665 in hepatocellular carcinoma (HCC) tissue and in normal adjacent liver tissue [16].
Expression of LINC00665 using other open databases
The relevant lncRNA chip or sequencing data for HCC from Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/), ArrayExpress (http://www.ebi.ac.uk/arrayexpress/), and Oncomine (https://www.oncomine.org/resource/login.html) databases were collected. The search terms used were: malignant OR cancer OR tumor OR neoplasia OR carcinoma AND hepatocellular OR liver OR hepatic OR HCC. All relevant expression data and clinical parameters related to LINC00665 were extracted.
Data from quantitative real-time polymerase chain reaction (qPCR) examination of clinical samples
HCC and corresponding adjacent normal liver tissue samples from 39 cases receiving hepatectomies at the First Affiliated Hospital of Guangxi Medical University, between January 2012 to August 2013 were selected. Patients were excluded from the study if they were receiving adjuvant chemotherapy or radiotherapy. The HCC tumors and adjacent normal liver tissue were diagnosed independently by two pathologists (Yu-yan Pang and Gang Chen). Control adjacent normal liver specimens were collected from sites at least 2 cm from the surgical margin; no tumor cells were found in the control specimens on histological examination. All tissue samples were fixed in 10% formaldehyde and subjected to routine paraffin embedding, and tissue sectioning. This study was approved by the local Ethics Committee of the First Affiliated Hospital of Guangxi Medical University. All patients signed informed consents to participate in the study.
RNA extraction and quantitative real-time polymerase chain reaction (qPCR)
Total RNA was extracted from 39 tissue samples of HCC, and paired adjacent normal liver tissues, using the Qiagen RNeasy FFPE Kit (Qiagen, Netherlands) to purify total RNA from formalin-fixed, paraffin-embedded tissue sections, according to the manufacturer’s protocol [17,18]. Quantification of LINC00665 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was performed using the PCR7900 (Applied Biosystems, Amsterdam, Netherlands).
The sequences of the LINC00665 primers used were as follows:
5′-AGCACCCCTAGTGTCAGTCA-3′ (forward),
5′-TGGTCTCTAGGGAGGCAGAA-3′ (reverse).
For GAPDH (internal control), the primers were as follows:
5′-AGTGGCAAAGTGGAGATT-3′ (forward),
5′-GTGGAGTCATACTGGAACA-3′ (reverse).
Expression levels of LINC00665 were evaluated using the 2−ΔCt method.
Statistical analysis
Data were analyzed using SPSS version 24.0 (Chicago, IL, USA). GraphPad Prism 7 software (GraphPad Software, San Diego, CA, USA) was used to plot data. Data were expressed as the mean ± standard deviation (SD). Independent sample t-tests were used to analyze the expressions of LINC00665 and related genes in HCC and adjacent normal liver tissues. Spearman’s correlation test was used to analyze the relationship between LINC00665 and the clinicopathological features of patients with HCC.
The expression of LINC00665 and related genes were demonstrated by scatter plots and box plots. One-way analysis of variance (ANOVA) was used to process and demonstrate the differences in data from three or more groups. The total standardized mean difference (SMD) with 95% confidence interval (CI) and summary receiver operating characteristic (ROC) curves were calculated with STATA version 12.0 (StataCorp, College Station, TX, USA). When SMD was >0 and the 95% CI did not include 0, the expression of LINC00665 in tumors was considered to be overexpressed when compared with that in normal adjacent liver tissue.
To test the performance of LINC00665 expression in the differentiation between HCC and normal liver tissues, SPSS was used to construct the ROC curve for each dataset and to calculate the area under the ROC curve (AUC). Meta-Disc1.4 software was used to evaluate the combined effect, sensitivity (SEN), specificity (SPE), positive likelihood ratio (PLR), negative likelihood ratio (NLR), diagnostic score (DS), and diagnostic odds ratio (DOR). The I2 test was applied to examine the heterogeneity between studies; when the P value was <0.05 or when the I2 value was >50%, then heterogeneity is considered to exist between studies. A random-effects model was selected to combine data; otherwise, a fixed-effects model was implemented. The correlation between the expressions of LINC00665 and related hub genes was analyzed using Pearson’s correlation analysis. For all statistical analysis, two-tailed analysis of differences was considered to be statistically significant with a P-value <0.05.
Potential genes that correlated to LINC00665 in HCC
Potential target genes of LINC00665 were predicted through The Atlas of Noncoding RNAs in Cancer (TANRIC) and MEM databases [19,20]. To obtain a more reliable pathway analysis and a correlated gene network, genes predicted by both TANRIC and the Multi Experiment Matrix (MEM) databases were identified.
Analysis of associated gene-enriched pathways and their functions
To study the potential molecular mechanisms of LINC00665-associated genes, the Web-based Gene Set Analysis Toolkit (WebGestalt) was used (http://www.webgestalt.org/option.php) for Gene Ontology (GO) analysis, the Kyoto Encyclopedia of Genes and Genomes (KEGG), Protein Analysis Through Evolutionary Relationships (PANTHER), and the Reactome pathway analysis databases were used [21]. The results were considered to be statistically significant when the false discovery rate (FDR) was <0.05. GOview (http://www.webgestalt.org/GOView/) was used to visualize Gene Ontology (GO) pathways [22]. R3.4.1 with the ggplot2 package was used to visualize pathway annotations from KEGG, PANTHER, and Reactome.
Construction of a protein-protein interaction (PPI) network and confirmation of hub genes
To identify the connections and interactions between overlapping genes, version 10.5 of STRING (http://www.string-db.org) was applied to construct the Protein-Protein Interaction (PPI) network for overlapping correlating genes [23]. Genes with combined scores >0.9 were obtained. Those genes that intersected with the gene enriched in the first KEGG pathway were identified as the hub genes of LINC00665.
Results
Expression of the long intergenic non-protein coding RNA 665 (LINC00665) gene in hepatocellular carcinoma (HCC) based on database analysis
Prior to this study, using The Cancer Genome Atlas (TCGA), RNA-Seq-based transcriptome data was obtained for the LIHC gene and analyzed using the DESeq package with the R-language. The long non-coding RNAs (lncRNAs) differentially expressed in hepatocellular carcinoma (HCC) were obtained. The long intergenic non-protein coding RNA 665 (LINC00665) gene, which had not previously appeared in the published literature, was selected as the subject of this study (Figure 1).
Figure 1.

Flow diagram of the study design to investigate the expression of the long intergenic non-protein coding RNA 665 (LINC00665) gene and the cell cycle in hepatocellular carcinoma (HCC) using database analysis.
Data on LINC00665 from TCGA that could be used for expression analysis were extracted. The results of data analysis indicated that among the 370 identified cases of HCC the expression of LINC00665 was significantly increased in HCC tumor tissues compared with the paired normal liver tissues (7.2643±2.2990 vs. 5.7526±0.9214) (FC=1.2628; P<0.0001) (Table 1) (Figure 2A). Receiver operating characteristic (ROC) curve analysis showed an area under the curve (AUC) of 0.673 (P<0.0001; 95% CI: 0.626–0.717) (Figure 2B) for LINC00665. Subsequently, an analysis of the transcripts per million (TPM) data from Gene Expression Profiling Interactive Analysis (GEPIA) showed that the expression of LINC00665 in 369 cases of HCC was increased in HCC tumor tissues compared with the paired normal liver tissues (Figure 3A), with numerical values of 2.16 and 0.48, respectively (Figure 3B). From several online databases, including Gene Expression Omnibus (GEO), ArrayExpress, and Oncomine, one chip was obtained from the GEO database that was expressed in both HCC and matched normal liver tissues, which was the GSE54236 chip from the GEO database (Agilent-014850 Whole Human Genome Microarray). However, in the GSE54236 chip, the expression levels of LINC00665 in HCC and normal adjacent liver tissues were not significantly different (5.7859±0.9652 vs. 5.8175±1.0390; FC=0.9946) (P=0.842) (Figure 2C). ROC curve analysis showed an AUC of 0.519 (P=0.682, 95% CI: 0.439–0.598) (Figure 2D).
Table 1.
Relationship between the expression of the long intergenic non-protein coding RNA 665 (LINC00665) gene and clinical parameters in The Cancer Genome Atlas (TCGA).
| Clinicopathological feature | n | LINC00665 expression in TCGA database | |||
|---|---|---|---|---|---|
| M ±SD | t | P | |||
| Tissue | Adjacent | 50 | 5.7526±0.9214 | −8.550a | <0.0001 |
| HCC | 370 | 7.2643±2.2990 | |||
| Gender | Male | 255 | 6.7842±2.5317 | −4.178 | <0.0001 |
| Female | 122 | 7.8508±2.2100 | |||
| Neoplasm histology grade 1 | I | 55 | 6.8179±1.7326 | F=4.045b | 0.008 |
| II | 177 | 7.0568±2.2893 | |||
| III | 122 | 7.6324±2.4559 | |||
| IV | 12 | 8.7823±2.2915 | |||
| Neoplasm histology grade 2 | I–II | 232 | 7.0001±2.1691 | −2.878 | 0.004 |
| III–IV | 134 | 7.7354±2.4556 | |||
| Pathologic stage I | I | 170 | 7.0134±2.2042 | F=2.318 | 0.075 |
| II | 86 | 7.2462±2.4777 | |||
| III | 82 | 7.7017±2.3357 | |||
| IV | 5 | 8.7059±1.5359 | |||
| Pathologic stage II | I–II | 156 | 7.0916±2.2975 | −2.341 | 0.020 |
| III–IV | 87 | 7.7594±2.3029 | |||
| Pathologic tumor I | TI | 180 | 7.0226±2.1824 | F=1.845 | 0.139 |
| TII | 93 | 7.3853±2.4490 | |||
| TIII | 80 | 7.7032±2.3744 | |||
| TIV | 13 | 6.9036±1.9319 | |||
| Pathologic tumor II | I–II | 273 | 7.1462±2.2787 | −1.619 | 0.106 |
| III–IV | 93 | 7.5914±2.3250 | |||
| Pathologic lymph node | − | 252 | 7.3046±2.3385 | −2.900 | 0.057 |
| + | 4 | 9.5284±1.5048 | |||
| Metastasis | − | 266 | 7.4298±2.3195 | −1.857 | 0.155 |
| + | 4 | 8.9711±1.6359 | |||
| Person neoplasm cancer status | − | 202 | 7.3287±2.1887 | 0.510 | 0.611 |
| + | 150 | 7.2021±2.3876 | |||
| Radiation therapy | − | 338 | 7.2844±2.2843 | 1.312 | 0.190 |
| + | 9 | 6.2725±2.2347 | |||
| Relative family cancer history | − | 207 | 7.3254±2.3148 | 0.167 | 0.867 |
| + | 112 | 7.2810±2.1503 | |||
| Vascular invasion I | None | 205 | 6.9705±2.2014 | F=4.728 | 0.009 |
| Micro | 93 | 7.4040±2.4094 | |||
| Macro | 17 | 8.6168±2.3080 | |||
| Vascular invasion II | − | 205 | 6.9705±2.2014 | −2.237 | 0.026 |
| + | 110 | 7.5914±2.4240 | |||
Student’s two-sample independent t-test was performed;
One-way analysis of variance (ANOVA) test was performed.
Figure 2.

Relative expression levels of the long intergenic non-protein coding RNA 665 (LINC00665) gene in hepatocellular carcinoma (HCC) tissue and adjacent normal liver tissue. (A) The expression of LINC00665 in HCC tissues and matched adjacent normal liver tissue, based on The Cancer Genome Atlas (TCGA) database. (B) Receiver operating characteristic (ROC) curve analysis of LINC00665 in HCC from TCGA. (C) The expression of LINC00665 in HCC tissues and adjacent normal liver tissue from the GSE54236 chip from the Gene Expression Omnibus (GEO) database. (D) ROC curve analysis of LINC00665 in HCC from GSE54236. (E) Quantitative real-time polymerase chain reaction (qPCR) was used to measure LINC00665 expression in HCC tissues and matched adjacent normal liver tissue. (F) Diagnostic value of LINC00665 in HCC with the ROC curve analysis by qPCR analysis.
Figure 3.

The median expression levels of the long intergenic non-protein coding RNA 665 (LINC00665) gene in normal and hepatocellular carcinoma (HCC) tissues in the body map by Gene Expression Profiling Interactive Analysis (GEPIA). Green represents normal tissues, and red represents the HCC tissues.
The quantitative real-time polymerase chain reaction (qPCR) was used to evaluate the expression of LINC00665 in 39 pairs of HCC tissues and normal adjacent liver tissues from our hospital. The results showed that the expression of LINC00665 in HCC samples was significantly increased in HCC tumor tissues compared with the paired normal liver tissues (3.1972±1.4164 vs. 2.1518±0.8978; FC=1.4858) (P<0.0001) (Table 2) (Figure 2E). ROC curve analysis showed an AUC of 0.709 (P<0.001; 95% CI: 0.596–0.807) (Figure 2F). To obtain results from a comprehensive analysis of LINC00665 expression representing different databases and study methods, data were combined, and meta-analysis of data from TCGA, GEO, and in-house qPCR was undertaken.
Table 2.
Relationship between expression of the long intergenic non-protein coding RNA 665 (LINC00665) gene, the clinical parameters and real-time quantitative polymerase chain reaction (qPCR).
| Clinicopathological features | n | LINC00665 expression in qPCR | |||
|---|---|---|---|---|---|
| M ±SD | t | P | |||
| Tissue | Adjacent non-tumor tissues | 39 | 2.1518±0.8978 | 3.893a | <0.0001 |
| HCC | 39 | 3.1972±1.4164 | |||
| Gender | Male | 29 | 3.2714±1.3679 | 0.552 | 0.584 |
| Female | 10 | 2.9820±1.6063 | |||
| Age | <50 y | 18 | 3.1428±1.4865 | 0.219 | 0.828 |
| ≥50 y | 21 | 3.2438±1.3888 | |||
| Pathologic tumor | Stage I–II | 25 | 3.1376±1.5405 | −0.347 | 0.731 |
| Stage III–IV | 14 | 3.3036±1.2100 | |||
| Nodes | Single | 34 | 3.2776±1.4548 | 0.923 | 0.362 |
| Multi | 5 | 2.6500±1.0813 | |||
| Metastasis | No | 36 | 3.1592±1.4230 | −0.575 | 0.568 |
| Yes | 3 | 3.6533±1.5312 | |||
| Embolus | No | 38 | 2.2200±1.4281 | 0.615 | 0.542 |
| Yes | 1 | 2.3300 | |||
| Status | Alive | 17 | 3.1424±1.2660 | 1.090 | 0.287 |
| Death | 7 | 3.7843±1.4241 | |||
| Diameter | <5 cm | 11 | 3.1782±1.6146 | −0.052 | 0.959 |
| ≥5 cm | 28 | 3.2046±1.3630 | |||
| Vascular invasion | No | 33 | 3.2276±1.4398 | 0.311 | 0.758 |
| Yes | 6 | 3.0300±1.3926 | |||
| Tumor capsular infiltration | Infiltration or no capsule | 14 | 3.1314±1.3340 | 0.214 | 0.832 |
| With complete capsule | 25 | 3.2340±1.4862 | |||
| Differentiation | Low | 10 | 3.0270±1.0942 | F=0.688b | 0.509 |
| Moderate | 25 | 3.1404±1.4919 | |||
| High | 4 | 3.9775±1.7545 | |||
| AFP | No | 21 | 3.5405±1.6405 | −1.490 | 0.146 |
| Yes | 12 | 2.8575±0.9916 | |||
| Cirrhosis | No | 25 | 3.2744±1.3854 | −0.450 | 0.655 |
| Yes | 14 | 3.0593±1.5129 | |||
Student’s two-sample independent t-test was performed;
One-way analysis of variance (ANOVA) test was performed.
AFP – alpha-feto protein.
Using the random-effects model, the pooled total standardized mean difference (SMD) of LINC00665 was 0.50 (95% CI: −0.06–1.05; P=0.078; I2=86.8%; P=0.001) (Figure 4A). The results of a meta-analysis of diagnostic tests showed that in the summary receiver operating characteristic (SROC) curves, the AUC was 0.614 (95% CI: 0.525–0.702) (Figure 4B). The pooled sensitivity (SEN), specificity (SPE), positive likelihood ratio (PLR), negative likelihood ratio (NLR), diagnostic score (DS), and diagnostic odds ratio (DOR) of LINC00665 in these studies were 0.55 (95% CI: 0.51–0.60), 0.53 (95% CI: 0.45–0.61), 2.57 (95% CI: 0.32–20.48), 0.85 (95% CI: 0.37–1.97), and 17.84 (95% CI: 5.46–58.34), respectively (Figure 5A–5E). Due to heterogeneity, GSE54236 data were eliminated. The results based on other combined data indicated that LINC00665 was highly expressed in HCC (SMD=0.75; 95% CI: 0.50–1.00; P<0.001; I2=0, P=0.503) (Figure 4C). In conclusion, LINC00665 was highly expressed in HCC tissues, but this finding was obtained from only two studies that included a total of 498 cases.
Figure 4.

The expression levels of the long intergenic non-protein coding RNA 665 (LINC00665) gene in hepatocellular carcinoma (HCC). (A) Forest plot of all eligible datasets to compare LINC00665 expression between HCC and matched adjacent normal liver tissue. (B) The summary receiver operating characteristic (SROC) curves of LINC00665 expression to compare HCC with matched adjacent normal liver tissue based on The Cancer Genome Atlas (TCGA), the independent microarray dataset, GSE54236, derived from HCC patients in the Gene Expression Omnibus (GEO) database, and quantitative real-time polymerase chain reaction (qPCR) datasets. (C) Forest plot of TCGA and in-house qPCR datasets evaluating LINC00665 expression between HCC and matched adjacent normal liver tissue.
Figure 5.

Forest plots show the diagnostic performance of the long intergenic non-protein coding RNA 665 (LINC00665) gene in hepatocellular carcinoma (HCC). (A) The pooled sensitivity. (B) The pooled specificity. (C) The pooled positive likelihood ratio. (D) The pooled negative likelihood ratio (E) The pooled diagnostic odds ratio. (F) Receiver operating characteristic (ROC) curve plots based on the eligible datasets.
Clinical significance of LINC00665 expression in the development and prognosis of HCC
Considering its possible carcinogenic function in HCC, LINC00665 may facilitate the development of HCC. For this reason, the expression of LINC00665 against various parameters in clinical cases was analyzed, as well as the correlation of LINC00665 expression with clinical progress. The results of analysis based on the TCGA database indicated that LINC00665 was correlated to gender, histological grade of HCC, tumor stage, and the presence of vascular invasion (Table 1). For gender, tumor stage, tumor grade, and the presence of vascular invasion, the results of independent sample t-tests and Spearman’s correlation tests showed that the expression of LINC00665 was significantly greater in women compared with men (7.8508±2.2100 vs. 6.7842±2.5317; P<0.0001; r=−0.1978) (Figure 6A, 6B), and significantly greater in HCC stage III–IV compared with stage I–II (7.7594±2.3029 vs. 7.0916±2.2975; P=0.020; r=0.1227) (Figure 6C, 6D), and significantly greater in HCC grade 3–4 compared with grade 1–2 (7.7354±2.4556 vs. 7.0001±2.1691; P=0.004; r=−0.1523) (Figure 7A, 7B), and significantly greater when there was the presence of vascular invasion compared with no vascular invasion (7.5914±2.4240 vs. 6.9705±2.2014; P=0.026) (Figure 7D, 7E) (Table 1). LINC00665 expression was significantly associated with histological grade (F=4.045, P=0.008) and the presence of vascular invasion (F=4.728, P=0.009) (Figure 7C, 7F). The GEPIA site used the log2 (TPM + 1) method to analyze clinical parameters in the TCGA database. LINC00665 expression was significantly associated with differences in stages I–IV of HCC (F=2.71; P=0.045) (Figure 6E).
Figure 6.

The relationship between the long intergenic non-protein coding RNA 665 (LINC00665) gene and the progression of hepatocellular carcinoma (HCC) Scatter plots (A: Gender, C: Stage). Receiver operating characteristic (ROC) curves to evaluate the status of tumor progression (B: Gender, D: Stage). (E) The expression of LINC00665 in the stages (I–IV) of HCC by Gene Expression Profiling Interactive Analysis (GEPIA).
Figure 7.

The relationship between the long intergenic non-protein coding RNA 665 (LINC00665) gene and clinical parameters of hepatocellular carcinoma (HCC). Scatter plots (A: Major grade; C: Sub-grade; D: Major tumor cell type; F: Minor tumor cell type. Receiver operating characteristic (ROC) curves to forecast the status of progression (B: Gender; E: Tumor cell type).
Because other databases did not provide usable data on clinical parameters in HCC, qPCR was also used to study the expression of LINC00665 in 39 patients with HCC in our hospital, with different clinical subgroups. Differential expression of LINC00665 in subgroups was not detected (Table 2); a possible reason for this finding was the small number of cases in the study.
To investigate the effect of the increased expression of LINC00665 on the prognosis and survival of patients with HCC, the cases of HCC from TCGA were split into high-expression and low-expression groups, based on the median level of LINC00665 expression. In this study, the Kaplan-Meier method was used for statistical analysis. The results of survival curve analysis indicated that the median overall survival (OS) for the high-expression group was 1,397 days; the median OS for the low-expression group was 2,116 days. The hazard ratio (HR) of OS was 1.477 (95% CI: 1.046–2.086). These results showed that the OS of patients with HCC with LINC00665 overexpression were shorter than those of patients with LINC00665 expressed at low levels (log-rank, P=0.027) (Figure 8A). However, there was no statistically significant alteration in the disease-free survival (DFS) of patients with high versus low expression levels of LINC00665 (log-rank P=0.364; HR=1.129; 95% CI: 0.8687–1.468) (Figure 8B). Therefore, the ability of LINC00665 to predict DFS was limited.
Figure 8.

The correlation between the long intergenic non-protein coding RNA 665 (LINC00665) gene expression and survival in patients with hepatocellular carcinoma (HCC). (A) Kaplan-Meier survival analysis of overall survival (OS) in 364 patients via GEPIA database. (B) Kaplan-Meier survival analysis of disease-free survival (DFS) in 364 patients from the Gene Expression Profiling Interactive Analysis (GEPIA) database.
LINC00665-correlated genes and analysis of gene-enrichment pathways
A total of 1,277 genes that correlated with LINC00665 were obtained from The Atlas of Noncoding RNAs in Cancer (TANRIC) website. Additionally, 4,296 correlated genes were obtained from the Multi Experiment Matrix (MEM) database. Based on genes obtained from the above databases, 469 overlapped genes were obtained (Figure 9A). The WebGestalt (WEB-based GEne SeT AnaLysis Toolkit) analysis was used to confirm Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), Protein Analysis Through Evolutionary Relationships (PANTHER), and Reactome pathways. GO: 0051301-Cell division (P=2.28, E-23) (Table 3) (Figure 9B), GO: 0005654-nucleoplasm (P=3.47, E-29) (Table 3) (Figure 9C), and GO: 0003676-nucleic acid binding (P=6.39, E-19) (Table 3) (Figure 9D) were the first GO annotated pathways for biological process (BP), cellular component (CC), and molecular function (MF).
Figure 9.

The Gene Ontology (GO) enrichment analyses categories of the potentially related genes of the long intergenic non-protein coding RNA 665 (LINC00665) gene in hepatocellular carcinoma (HCC). (A) Venn diagram shows the areas of overlap between the number of predicted related genes of LINC00665 using the Multi Experiment Matrix (MEM) and The Atlas of Noncoding RNAs in Cancer (TANRIC) databases. (B) The Biological Process (BP) network analysis with the predicted genes related to LINC00665. (C) The Cellular Component (CC) network analysis with the predicted genes related to LINC00665. (D) The Molecular Function (MF) network analysis for the predicted genes related to LINC00665.
Table 3.
Gene Ontology (GO) analysis of overlapping correlated genes of the long intergenic non-protein coding RNA 665 (LINC00665) gene.
| Category | Term | Count | P-value |
|---|---|---|---|
| GOTERM_BP | GO: 0051301~cell division | 51 | 2.28E-23 |
| GOTERM_BP | GO: 0007067~mitotic nuclear division | 40 | 6.18E-20 |
| GOTERM_BP | GO: 0007062~sister chromatid cohesion | 25 | 9.50E-17 |
| GOTERM_BP | GO: 0006355~regulation of transcription, DNA-templated | 91 | 1.61E-14 |
| GOTERM_BP | GO: 0006260~DNA replication | 25 | 1.54E-12 |
| GOTERM_BP | GO: 0006351~transcription, DNA-templated | 98 | 9.01E-11 |
| GOTERM_BP | GO: 0000070~mitotic sister chromatid segregation | 11 | 2.65E-10 |
| GOTERM_BP | GO: 0007059~chromosome segregation | 14 | 1.57E-08 |
| GOTERM_BP | GO: 0007052~mitotic spindle organization | 10 | 4.02E-08 |
| GOTERM_BP | GO: 0008283~cell proliferation | 28 | 9.94E-07 |
| GOTERM_CC | GO: 0005654~nucleoplasm | 163 | 3.47E-29 |
| GOTERM_CC | GO: 0005634~nucleus | 235 | 3.26E-25 |
| GOTERM_CC | GO: 0000775~chromosome, centromeric region | 16 | 4.06E-12 |
| GOTERM_CC | GO: 0000777~condensed chromosome kinetochore | 18 | 2.67E-11 |
| GOTERM_CC | GO: 0000776~kinetochore | 16 | 8.80E-10 |
| GOTERM_CC | GO: 0005819~spindle | 17 | 3.80E-08 |
| GOTERM_CC | GO: 0005876~spindle microtubule | 11 | 7.55E-08 |
| GOTERM_CC | GO: 0005622~intracellular | 64 | 2.37E-07 |
| GOTERM_CC | GO: 0035098~ESC/E(Z) complex | 7 | 1.28E-06 |
| GOTERM_CC | GO: 0000922~spindle pole | 14 | 2.38E-06 |
| GOTERM_MF | GO: 0003676~nucleic acid binding | 79 | 6.39E-19 |
| GOTERM_MF | GO: 0003677~DNA binding | 106 | 5.62E-18 |
| GOTERM_MF | GO: 0046872~metal ion binding | 97 | 1.22E-08 |
| GOTERM_MF | GO: 0005515~protein binding | 285 | 1.35E-07 |
| GOTERM_MF | GO: 0005524~ATP binding | 74 | 1.42E-07 |
| GOTERM_MF | GO: 0044822~poly(A) RNA binding | 60 | 2.95E-07 |
| GOTERM_MF | GO: 0000166~nucleotide binding | 28 | 5.47E-07 |
| GOTERM_MF | GO: 0008017~microtubule binding | 20 | 2.55E-06 |
| GOTERM_MF | GO: 0003682~chromatin binding | 28 | 5.05E-06 |
| GOTERM_MF | GO: 0003723~RNA binding | 34 | 8.46E-06 |
Only the top ten pathways were listed for each category (BP – biological process; CC – cellular component; MF – molecular function).
The first pathways in the PANTHER and Reactome databases were P00022: General transcription by RNA polymerase I (P=3.48, E-01) (Table 4) (Figure 10B) and R-has-1640170: the Cell Cycle pathway (P=0.00, E+00) (Table 4) (Figure 10C). KEGG pathway analysis showed a significant association between LINC0066-correlated genes and the Cell Cycle pathway (P=4.86, E-15) (Table 4) (Figure 10A). The cell cycle pathway included 24 genes: CDC7, E2F2, CDK1, E2F3, ANAPC5, CDC14A, DBF4, TTK, PRKDC, CDC20, ESPL1, PTTG1, MCM3, MCM4, CDC25A, CCNB1, MAD2L1, MCM7, HDAC2, CCNB2, PLK1, BUB1, BUB1B and CCNA2 (Figure 11).
Table 4.
Kyoto Encyclopedia of Genes and Genomes (KEGG), Protein ANalysis THrough Evolutionary Relationships (PANTHER) and Reactome Pathway of overlapping correlated genes of the long intergenic non-protein coding RNA 665 (LINC00665) gene.
| Category | Term | Count | P-value |
|---|---|---|---|
| KEGG_PATHWAY | hsa04110: Cell cycle | 24 | 2.69E-13 |
| KEGG_PATHWAY | hsa04914: Progesterone-mediated oocyte maturation | 12 | 5.22E-04 |
| KEGG_PATHWAY | hsa03460: Fanconi anemia pathway | 9 | 5.44E-04 |
| KEGG_PATHWAY | hsa04114: Oocyte meiosis | 12 | 2.98E-03 |
| KEGG_PATHWAY | hsa00400: Phenylalanine, tyrosine and tryptophan biosynthesis | 3 | 8.01E-03 |
| KEGG_PATHWAY | hsa03030: DNA replication | 6 | 9.61E-03 |
| KEGG_PATHWAY | hsa03440: Homologous recombination | 6 | 1.72E-02 |
| KEGG_PATHWAY | hsa03013: RNA transport | 11 | 8.06E-02 |
| KEGG_PATHWAY | hsa04068: FoxO signaling pathway | 9 | 1.64E-01 |
| KEGG_PATHWAY | hsa04120: Ubiquitin mediated proteolysis | 9 | 1.71E-01 |
| PANTHER_PATHWAY | P00022: General transcription by RNA polymerase I | 3 | 3.48E-01 |
| PANTHER_PATHWAY | P00017: DNA replication | 3 | 3.48E-01 |
| PANTHER_PATHWAY | P04385: Histamine H1 receptor mediated signaling pathway | 4 | 3.48E-01 |
| PANTHER_PATHWAY | P00020: FAS signaling pathway | 3 | 5.69E-01 |
| PANTHER_PATHWAY | P00042: Muscarinic acetylcholine receptor 1 and 3 signaling pathway | 4 | 5.69E-01 |
| PANTHER_PATHWAY | P05911: Angiotensin II-stimulated signaling through G proteins and beta-arrestin | 3 | 6.40E-01 |
| PANTHER_PATHWAY | P00060: Ubiquitin proteasome pathway | 3 | 6.59E-01 |
| PANTHER_PATHWAY | P00055: Transcription regulation by bZIP transcription factor | 3 | 6.59E-01 |
| PANTHER_PATHWAY | P00059: p53 pathway | 4 | 6.59E-01 |
| PANTHER_PATHWAY | P05730: Endogenous cannabinoid signaling | 2 | 6.59E-01 |
| Reactome_PATHWAY | R-HSA-1640170: Cell cycle | 84 | 0.00E+00 |
| Reactome_PATHWAY | R-HSA-2500257: Resolution of sister chromatid cohesion | 27 | 0.00E+00 |
| Reactome_PATHWAY | R-HSA-68877: Mitotic prometaphase | 30 | 0.00E+00 |
| Reactome_PATHWAY | R-HSA-69278: Cell cycle, Mitotic | 71 | 0.00E+00 |
| Reactome_PATHWAY | R-HSA-74160: Gene expression | 110 | 2.69E-12 |
| Reactome_PATHWAY | R-HSA-212436: Generic transcription pathway | 69 | 3.78E-12 |
| Reactome_PATHWAY | R-HSA-68886: M phase | 40 | 4.05E-12 |
| Reactome_PATHWAY | R-HSA-68882: Mitotic anaphase | 29 | 1.44E-10 |
| Reactome_PATHWAY | R-HSA-5663220: RHO GTPases activate formins | 24 | 1.44E-10 |
| Reactome_PATHWAY | R-HSA-2555396: Mitotic metaphase and anaphase | 29 | 1.44E-10 |
Only the top ten pathways were listed for each category.
Figure 10.

(A–C) Bar chart for the enriched annotation of the Kyoto Encyclopedia of Genes and Genomes (KEGG), Protein ANalysis THrough Evolutionary Relationships (PANTHER) and Reactome pathway analysis of the predicted genes related to the long intergenic non-protein coding RNA 665 (LINC00665) gene in hepatocellular carcinoma (HCC).
Figure 11.

The function of the hub genes in the cell cycle signaling pathway visualized in the Kyoto Encyclopedia of Genes and Genomes (KEGG) map obtained by the Database for Annotation, Visualization and Integrated Discovery (DAVID).
Construction of the protein-protein interaction (PPI) network and the hub genes
Using version 10.5 of the STRING database, a functional protein network containing the above 469 genes was analyzed. Through the constructed protein-protein interaction (PPI) network, the 30 genes with the most connections were obtained (Figure 12). Genes with combined scores greater than 0.999 are illustrated in Figure 13, using Cytoscape version 3.5.1. The cell cycle pathways described above play critical functions in the growth of tumor cells. Therefore, the overlap between the top 30 genes connected within the PPI network and 24 genes in the cell cycle pathway were reviewed, and ten overlapping genes were identified, which were, in our opinion, the core or hub genes for the facilitation of LINC00665 in tumor growth and development. These ten core or hub genes were CDK1, BUB1B, BUB1, PLK1, CCNB2, CCNB1, CDC20, ESPL1, MAD2L1, and CCNA2.
Figure 12.
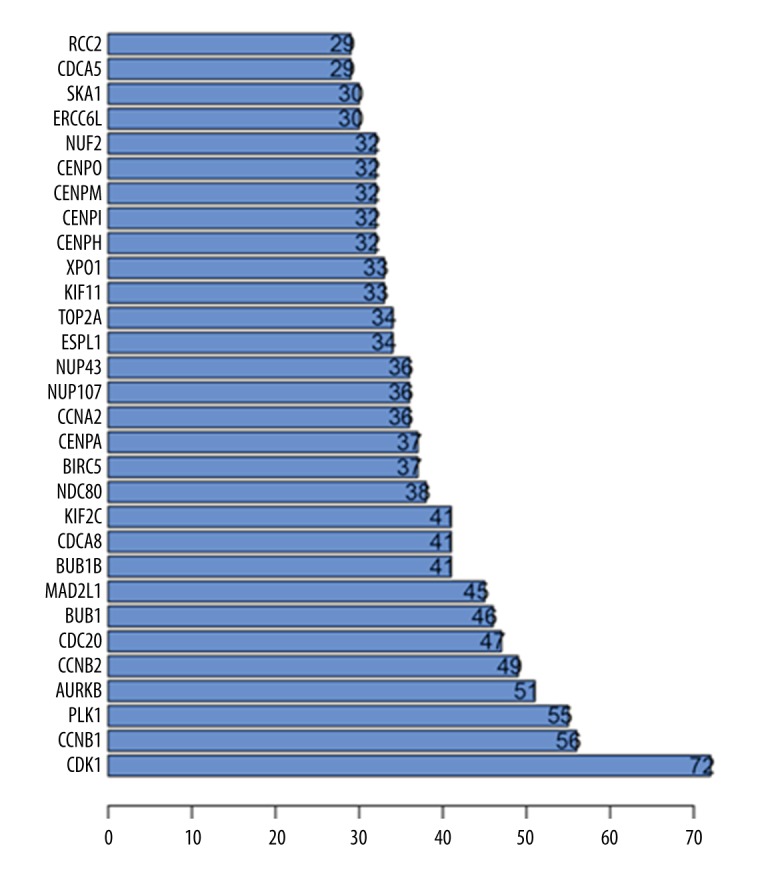
Bar chart shows the top 30 related genes of the long intergenic non-protein coding RNA 665 (LINC00665) gene in hepatocellular carcinoma (HCC).
Figure 13.

Genes that correlated with the long intergenic non-protein coding RNA 665 (LINC00665) gene, with combined scores >0.999 visualized by Cytoscape version 3.5.1
Verification of the relationship between LINC00665 and the ten core or hub genes
Using TCGA database, verification was performed on the relationship between LINC00665 and the expression of the ten identified hub genes in HCC tissues. Based on independent sample t-tests, the expression of the ten hub genes, CDK1, BUB1B, BUB1, PLK1, CCNB2, CCNB1, CDC20, ESPL1, MAD2L1, and CCNA2, were significantly increased in HCC tissues (P<0.0001) when compared with the normal adjacent liver tissue; this finding is similar to the expression of LINC00665. ROC curve analysis showed the following AUCs data: CDK1 (0.965; P<0.0001), BUB1B (0.948; P<0.0001), BUB1 (0.958; P<0.0001), PLK1 (0.960; P<0.0001), CCNB2 (0.962; P<0.0001), CCNB1 (0.967; P<0.0001), CDC20 (0.968; P<0.0001), ESPL1 (0.789; P<0.0001), MAD2L1 (0.923; P<0.0001), and CCNA2 (0.954; P<0.0001).
By Pearson’s correlation analysis, in the TCGA database, LINC00665 expression was found to be positively correlated with the expression of the ten hub genes with the following findings: CDK1 (r=0.3541; P<0.0001), BUB1B (r=0.3461; P<0.0001), BUB1 (r=0.3457; P<0.0001), PLK1 (r=0.3231; P<0.0001), CCNB2 (r=0.3187; P<0.0001), CCNB1 (r=0.3164; P<0.0001), CDC20 (r=0.3085; P<0.0001), ESPL1 (r=0.2960; P<0.0001), MAD2L1 (r=0.2943; P<0.0001) and CCNA2 (r=0.2728; P<0.0001) (Figures 14, 15). A principal component analysis (PCA) was performed on these ten hub genes using GEPIA. A three-dimensional (3-D) plot confirmed that these ten genes had the greatest potentential as biomarkers for HCC (Figure 16).
Figure 14.

Expression levels of the ten hub genes of the long intergenic non-protein coding RNA 665 (LINC00665) gene in hepatocellular carcinoma (HCC) and matched adjacent normal liver tissue, based on The Cancer Genome Atlas (TCGA) database. (A) CDK1 (D) BUB1B (G) BUB1 (J) PLK1 (M) CCNB2. Diagnostic performance of hub genes of LINC00665 in HCC with receiver operating characteristic (ROC) curve analysis. (B) CDK1 (E) BUB1B (H) BUB1 (K) PLK1 (N) CCNB2. Correlation analysis between LINC00665 and hub genes using Pearson’s correlation. (C) CDK1 (F) BUB1B (I) BUB1 (L) PLK1 (O) CCNB2.
Figure 15.

The expression levels of ten hub genes of the long intergenic non-protein coding RNA 665 (LINC00665) gene in hepatocellular carcinoma (HCC) and matched adjacent normal liver tissue, based on The Cancer Genome Atlas (TCGA) database. (A) CCNB1 (D) CDC20 (G) ESPL1 (J) MAD2L1 (M) CCNA2. Diagnostic validity of hub genes of LINC00665 in HCC using receiver operating characteristic (ROC) curve analysis. (B) CCNB1 (E) CDC20 (H) ESPL1 (K) MAD2L1 (N) CCNA2. Correlation analysis between LINC00665 and hub genes using Pearson’s correlation. (C) CCNB1 (F) CDC20 (I) ESPL1 (L) MAD2L1 (O) CCNA2.
Figure 16.

Correlation between the hub genes of the long intergenic non-protein coding RNA 665 (LINC00665) gene in hepatocellular carcinoma (HCC) using principal component analysis (PCA) by Gene Expression Profiling Interactive Analysis (GEPIA).
Discussion
In this study, differential investigation of long non-coding RNA (lncRNA) was based on sequencing data from patients with hepatocellular carcinoma (HCC) provided by the open database, The Cancer Genome Atlas (TCGA). This study is the first to report that the expression of long intergenic non-protein coding RNA 665 (LINC00665) gene was significantly increased in HCC tissues compared with adjacent normal tissues. Confirmation of the findings was performed with the use of other online databases and using the findings from quantitative real-time polymerase chain reaction (qPCR) results in cases from our hospital.
In this study, expression of LINC00665 was significantly upregulated in HCC tumor tissue from patients with HCC and was correlated with certain clinical and pathological features, including poor tumor differentiation (grade), a high TNM score (stage), and the presence of vascular invasion, which is a requirement for tumor metastasis. As these clinical parameters can reflect the clinical prognosis in HCC, LINC00665 may function as an oncogene in the initiation and progression of HCC. This view is supported by the finding from this study that patients with HCC who had highly upregulated LINC00665 had a worse overall survival (OS).
In this study, the association between malignant biological behavior of HCC and upregulation of LINC00665 were studied in terms of facilitating the onset and development of HCC. Through using bioinformatics analysis, increased expression of LINC00665 was shown to facilitate the development of HCC by regulating a series of genes that trigger changes in the cell cycle pathways. The results of this study indicated that upregulation of LINC00665 might be important in the onset and development of HCC. Therefore, measurement of LINC00665 expression may have important future clinical applications.
The primary aim of this study was to explore the expression of LINC00665 in HCC, by performing an analysis of TCGA data and the study findings showed that the LINC00665 was significantly overexpressed in HCC tissues when compared with the normal adjacent liver tissues. Other open databases such as Gene Expression Omnibus (GEO), Arrayexpres, and Oncomine, as well as quantitative real-time polymerase chain reaction (qPCR) data from cases at our hospital, were used for confirmation. Comprehensive results were obtained via meta-analysis. The overall total standardized mean difference (SMD) was 0.75 (95% CI: 0.50–1.00). These results indicated that there was a trend of increasing LINC00665 expression as HCC progressed. Although these findings require further examination through an independent cohort study and other methods, LINC00665 might also participate in the onset of HCC as a tumor-promoting factor.
The most important finding in this study was the close correlation between the increased expression of LINC00665 and the presence of tumor cell vascular invasion, which also supports the possibility that LINC00665 can also be a potential predictive biomarker for the progress of HCC. For histological grades 3–4, clinical stages III–IV, and for patients who had HCC with the presence of vascular invasion, LINC00665 expression was significantly greater than in patients with histological grades 1–2, stages I–II, and patients without the presence of vascular invasion. This study found that the OS of HCC patients with high expression levels of LINC00665 was less than that of patients with low expression levels of LINC00665 (HR=1.4, P=0.07). However, this trend requires further confirmation with a larger number of study participants. The findings from this study indicate that expression of LINC00665 is closely correlated to the development of HCC, especially vascular invasion. However, due to the lack of reports on LINC00665 in the literature, the molecular mechanism of LINC00665 in the onset and development of HCC remains unknown.
Therefore, in this study, an attempt was made to predict the functional mechanism of LINC00665 in HCC using bioinformatics. We first obtained 469 genes that were correlated to LINC00665 through The Atlas of Noncoding RNAs in Cancer (TANRIC) and Multi Experiment Matrix (MEM) databases. Subsequently, we performed Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), Protein Analysis Through Evolutionary Relationships (PANTHER), Reactome, and protein-protein interaction (PPI) analysis on these 469 genes. The results indicated that these genes function in multiple biological processes and have multiple molecular functions, among which cell cycle pathways had the most significant gene enrichment, as indicated by KEGG analysis. The cell cycle is controlled by multiple mechanisms to ensure the accurate division of cells [24,25]. A disturbance in cell cycle pathways could lead to cell cycle arrest and could be closely correlated to the development and prognosis of tumors [26,27]. Therefore, it may be argued that LINC00665 could facilitate the generation and development of HCC cells by influencing the cell cycle. To further confirm the target molecule of LINC00665 in the HCC cell cycle, ten genes were identified that overlapped with the genes obtained from PPI analysis and the 24 genes enriched in cell cycle pathways; these ten genes were CDK1, CDC20, ESPL1, CCNB1, MAD2L1, CCNB2, PLK1, BUB1, BUB1B, and CCNA2. Also, these ten genes showed significant overexpression in HCC and were positively correlated with LINC00665. Therefore, it is likely that LINC00665 regulates cell cycle pathways through these ten genes and thereby facilitates the onset and development of HCC.
There have been some previously published reports on the functions of these ten hub genes in HCC. CDK1, a serine/threonine kinase, is a vital regulator of the cell cycle [28,29]. A study on the knockdown of CDK1 showed that this significantly reduced apoptin-induced HCC cell apoptosis [30]. Li et al. reported a close correlation between overexpression of CDC20 and the development of HCC, as the proliferation of HCC cells was inhibited by interference in the expression of CDC20 [31]. ESPL1 is thought to be an oncogene that is highly expressed in multiple tumors [32,33]. However, the clinical significance and functional mechanism of ESPL1 have not yet been officially reported in HCC. CCNB1, CCNB2, and CCNA2 belong to the cyclin (CCN) gene family [34,35]. Cyclins primarily regulate the cell cycle by activating CDK kinase [36]. Previous studies have shown that MAD2L1 is highly expressed in HCC tissues and cells [37]. In HCC, expression of MAD2L1 has also been shown to be closely correlated with the size, stage, and grade of the tumor [38]. PLK1 is a gene that has multiple roles in the cell cycle [39,40]. The inhibition of PLK1 expression can inhibit the proliferation of HCC cells [41]. Increased expression of the BUB1 gene triggers the onset of several tumors through the pathway of an altered mitotic spindle assembly checkpoint [42].
Therefore, the expression and upregulation of LINC00665, and the ten associated hub genes, result in changes and functions that may be involved in the development and progression of HCC, especially in the regulation of the cell cycle in HCC.
This study had several limitations. First, the number of cases included that had data on prognosis based on LINC00665 was small, as only 370 cases were provided in the TCGA database. Based on the information provided, the patients with HCC who had increased levels of expression of LINC00665 had a poorer clinical prognosis. However, the potential value of upregulation of LINC00665 gene expression as a potential prognostic biomarker in HCC requires confirmation by larger, multicenter clinical studies. Second, the mechanism of action of LINC00665 in HCC requires confirmation and further evaluation by in vitro and in vivo studies.
Conclusions
This study comprehensively analyzed the expression of the long intergenic non-protein coding RNA 665 (LINC00665) gene in patients with hepatocellular carcinoma (HCC) and evaluated the potential clinical value of LINC00665 expression using data from multiple sources and methods. Bioinformatics analysis was performed to determine the possible functional mechanism of LINC00665 expression in facilitating the onset and development of HCC by regulating cell cycle pathways.
Acknowledgements
The authors thank The Cancer Genome Atlas (TCGA) and the Gene Expression Omnibus (GEO) databases for providing the data.
Footnotes
Source of support: This study was supported by the Fund of the National Natural Science Foundation of China (Fund Numbers: NSFC81560489, NSFC81060202, NSFC81260222)
References
- 1.Gu L, Li H, Chen L, et al. MicroRNAs as prognostic molecular signatures in renal cell carcinoma: A systematic review and meta-analysis. Oncotarget. 2015;6:32545–60. doi: 10.18632/oncotarget.5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cao MX, Jiang YP, Tang YL, Liang XH. The crosstalk between lncRNA and microRNA in cancer metastasis: Orchestrating the epithelial-mesenchymal plasticity. Oncotarget. 2017;8:12472–83. doi: 10.18632/oncotarget.13957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505:344–52. doi: 10.1038/nature12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagano T, Fraser P. No-nonsense functions for long noncoding RNAs. Cell. 2011;145:178–81. doi: 10.1016/j.cell.2011.03.014. [DOI] [PubMed] [Google Scholar]
- 5.Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;17:47–62. doi: 10.1038/nrg.2015.10. [DOI] [PubMed] [Google Scholar]
- 6.Sailer V, Holmes EE, Gevensleben H, et al. PITX2 and PANCR DNA methylation predicts overall survival in patients with head and neck squamous cell carcinoma. Oncotarget. 2016;7:75827–38. doi: 10.18632/oncotarget.12417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Batista PJ, Chang HY. Long noncoding RNAs: Cellular address codes in development and disease. Cell. 2013;152:1298–307. doi: 10.1016/j.cell.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bolha L, Ravnik-Glavac M, Glavac D. Long noncoding RNAs as biomarkers in cancer. Dis Markers. 2017;2017:7243968. doi: 10.1155/2017/7243968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kwok ZH, Tay Y. Long noncoding RNAs: Lincs between human health and disease. Biochem Soc Trans. 2017;45:805–12. doi: 10.1042/BST20160376. [DOI] [PubMed] [Google Scholar]
- 10.Schmitt AM, Chang HY. Long noncoding RNAs in cancer pathways. Cancer Cell. 2016;29:452–63. doi: 10.1016/j.ccell.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 12.Niu J, Lin Y, Liu P, et al. Microarray analysis on the lncRNA expression profile in male hepatocelluar carcinoma patients with chronic hepatitis B virus infection. Oncotarget. 2016;7:76169–80. doi: 10.18632/oncotarget.12732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiong D, Sheng Y, Ding S, et al. LINC00052 regulates the expression of NTRK3 by miR-128 and miR-485-3p to strengthen HCC cells invasion and migration. Oncotarget. 2016;7:47593–608. doi: 10.18632/oncotarget.10250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu YR, Tang RX, Huang WT, et al. Long noncoding RNAs in hepatocellular carcinoma: Novel insights into their mechanism. World J Hepatol. 2015;7:2781–91. doi: 10.4254/wjh.v7.i28.2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qu Z, Yuan CH, Yin CQ, et al. Meta-analysis of the prognostic value of abnormally expressed lncRNAs in hepatocellular carcinoma. Onco Targets Ther. 2016;9:5143–52. doi: 10.2147/OTT.S108599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang Z, Li C, Kang B, et al. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017 doi: 10.1093/nar/gkx247. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo S, Chen W, Luo Y, et al. Clinical implication of long non-coding RNA NEAT1 expression in hepatocellular carcinoma patients. Int J Clin Exp Pathol. 2015;8:5395–402. [PMC free article] [PubMed] [Google Scholar]
- 18.Pan LJ, Zhong TF, Tang RX, et al. Upregulation and clinicopathological significance of long non-coding NEAT1 RNA in NSCLC tissues. Asian Pac J Cancer Prev. 2015;16:2851–55. doi: 10.7314/apjcp.2015.16.7.2851. [DOI] [PubMed] [Google Scholar]
- 19.Li J, Han L, Roebuck P, et al. TANRIC: An interactive open platform to explore the function of lncRNAs in cancer. Cancer Res. 2015;75:3728–37. doi: 10.1158/0008-5472.CAN-15-0273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kolde R, Laur S, Adler P, Vilo J. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics. 2012;28:573–80. doi: 10.1093/bioinformatics/btr709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J, Duncan D, Shi Z, Zhang B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): Update 2013. Nucleic Acids Res. 2013;41:W77–83. doi: 10.1093/nar/gkt439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang J, Vasaikar S, Shi Z, et al. WebGestalt 2017: A more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017 doi: 10.1093/nar/gkx356. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Szklarczyk D, Morris JH, Cook H, et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45:D362–68. doi: 10.1093/nar/gkw937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang JD, Levin PA. Metabolism, cell growth and the bacterial cell cycle. Nat Rev Microbiol. 2009;7:822–27. doi: 10.1038/nrmicro2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hong SH, Eun JW, Choi SK, et al. Epigenetic reader BRD4 inhibition as a therapeutic strategy to suppress E2F2-cell cycle regulation circuit in liver cancer. Oncotarget. 2016;7:32628–40. doi: 10.18632/oncotarget.8701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bretones G, Delgado MD, Leon J. Myc and cell cycle control. Biochim Biophys Acta. 2015;1849:506–16. doi: 10.1016/j.bbagrm.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 27.Kohrman AQ, Matus DQ. Divide or conquer: Cell cycle regulation of invasive behavior. Trends Cell Biol. 2017;27:12–25. doi: 10.1016/j.tcb.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sakurikar N, Eastman A. Critical reanalysis of the methods that discriminate the activity of CDK2 from CDK1. Cell Cycle. 2016;15:1184–88. doi: 10.1080/15384101.2016.1160983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Banyai G, Baidi F, Coudreuse D, Szilagyi Z. Cdk1 activity acts as a quantitative platform for coordinating cell cycle progression with periodic transcription. Nat Commun. 2016;7:11161. doi: 10.1038/ncomms11161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao J, Han SX, Ma JL, et al. The role of CDK1 in apoptin-induced apoptosis in hepatocellular carcinoma cells. Oncol Rep. 2013;30:253–59. doi: 10.3892/or.2013.2426. [DOI] [PubMed] [Google Scholar]
- 31.Li J, Gao JZ, Du JL, et al. Increased CDC20 expression is associated with development and progression of hepatocellular carcinoma. Int J Oncol. 2014;45:1547–55. doi: 10.3892/ijo.2014.2559. [DOI] [PubMed] [Google Scholar]
- 32.Prinzhorn W, Stehle M, Kleiner H, et al. c-MYB is a transcriptional regulator of ESPL1/Separase in BCR-ABL-positive chronic myeloid leukemia. Biomark Res. 2016;4:5. doi: 10.1186/s40364-016-0059-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Finetti P, Guille A, Adelaide J, et al. ESPL1 is a candidate oncogene of luminal B breast cancers. Breast Cancer Res Treat. 2014;147:51–59. doi: 10.1007/s10549-014-3070-z. [DOI] [PubMed] [Google Scholar]
- 34.Jia Q, Dong Q, Qin L. CCN: Core regulatory proteins in the microenvironment that affect the metastasis of hepatocellular carcinoma? Oncotarget. 2016;7:1203–14. doi: 10.18632/oncotarget.6209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hung YH, Huang HL, Chen WC, et al. Argininosuccinate lyase interacts with cyclin A2 in cytoplasm and modulates growth of liver tumor cells. Oncol Rep. 2017;37:969–78. doi: 10.3892/or.2016.5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang P, Chen S, Fang H, et al. miR-214/199a/199a* cluster levels predict poor survival in hepatocellular carcinoma through interference with cell-cycle regulators. Oncotarget. 2016;7:929–45. doi: 10.18632/oncotarget.6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Y, Bai W, Zhang J. MiR-200c-5p suppresses proliferation and metastasis of human hepatocellular carcinoma (HCC) via suppressing MAD2L1. Biomed Pharmacother. 2017;92:1038–44. doi: 10.1016/j.biopha.2017.05.092. [DOI] [PubMed] [Google Scholar]
- 38.Yan H, Li Z, Shen Q, et al. Aberrant expression of cell cycle and material metabolism related genes contributes to hepatocellular carcinoma occurrence. Pathol Res Pract. 2017;213:316–21. doi: 10.1016/j.prp.2017.01.019. [DOI] [PubMed] [Google Scholar]
- 39.Archambault V, Lepine G, Kachaner D. Understanding the Polo Kinase machine. Oncogene. 2015;34:4799–807. doi: 10.1038/onc.2014.451. [DOI] [PubMed] [Google Scholar]
- 40.Kumar S, Sharma AR, Sharma G, et al. PLK-1: Angel or devil for cell cycle progression. Biochim Biophys Acta. 2016;1865:190–203. doi: 10.1016/j.bbcan.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 41.Bouhlal H, Ouled-Haddou H, Debuysscher V, et al. RB/PLK1-dependent induced pathway by SLAMF3 expression inhibits mitosis and control hepatocarcinoma cell proliferation. Oncotarget. 2016;7:9832–43. doi: 10.18632/oncotarget.6954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.King RW. When 2+2=5: The origins and fates of aneuploid and tetraploid cells. Biochim Biophys Acta. 2008;1786:4–14. doi: 10.1016/j.bbcan.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]