

Abstract
Cerebellins are synaptic organizer molecules that bind to presynaptic neurexins and postsynaptic receptors. They are well studied in the cerebellum, but three of the four cerebellins (Cbln1, Cbln2, and Cbln4) are also broadly expressed outside of the cerebellum, suggesting that they perform general functions throughout the brain. Here, we generated male and female constitutive single (KO), double KO (dKO), and triple KO (tKO) mice of Cbln1, Cbln2, and Cbln4. We found that all constitutive cerebellin-deficient mice were viable and fertile, suggesting that cerebellins are not essential for survival. Cbln1/2 dKO mice exhibited salience-induced seizures that were aggravated in Cbln1/2/4 tKO mice, suggesting that all cerebellins contribute to brain function. As described previously, Cbln1 KO mice displayed major motor impairments that were aggravated by additional KO of Cbln2. Strikingly, the Cbln1/2 dKO did not cause alterations in synapse density in the hippocampus of young adult (1- and 2-month-old) mice, but produced a selective ∼50% decrease in hippocampal synapse density in the stratum lacunosum moleculare of the CA1 region and in the dentate gyrus of aging, 6-month-old mice. A similar decrease in excitatory synapse density was observed in the striatum and retrosplenial cortex. Behaviorally, the Cbln1 KO produced dramatic changes in motor behaviors that were partly aggravated by additional deletion of Cbln2 and/or Cbln4. Our results show that cerebellins are not essential for survival and do not contribute to initial synapse formation, but perform multiple functions throughout the brain; as a consequence, their ablation results in a delayed loss of synapses and in behavioral impairments.
SIGNIFICANCE STATEMENT Cerebellins (Cbln1-4) are trans-synaptic cell adhesion molecules. In the cerebellum, Cbln1 functions as a bidirectional organizer of parallel fiber–Purkinje cell synapses by binding to presynaptic neurexins and postsynaptic GluRδ2. Little is known about the function of cerebellins outside of the cerebellum; therefore, the present study used single, double, and triple constitutive KO mice of Cbln1, Cbln2, and Cbln4 to analyze the overall function of cerebellins. We show that cerebellins act as important synaptic organizers in specific subsets of neurons and likely contribute to many different brain functions. We also show that cerebellins are not initially required for synapse formation, but rather for specification and long-term synapse maintenance and demonstrate that all cerebellins, not just Cbln1, contribute to brain function.
Keywords: cerebellins, neurexins, synapse formation, synapse specification, synaptic organizers
Introduction
Precise control of synapse formation and specification is required for the normal development and functioning of neural networks and, consequently, for cognition, emotion, and behavior. Trans-synaptic cell adhesion molecules are understood to play an essential role in the formation, specification, and functioning of synapses. Cerebellins are a family of secreted proteins (Cbln1-4) that act as trans-synaptic cell adhesion molecules by binding to presynaptic neurexins and postsynaptic GluRδs (Cbln1 and 2) (Ito-Ishida et al., 2008; Uemura et al., 2010; Lee et al., 2012; Cheng et al., 2016; Zhong et al., 2017) or to DCC (“deleted-in-colorectal-cancer”) and neogenin-1 (Cbln4) (Wei et al., 2012; Haddick et al., 2014). Cbln1, Cbln2, and Cbln4 are abundantly expressed in subsets of neurons throughout the brain (Miura et al., 2006; Wei et al., 2007; Otsuka et al., 2016; Seigneur and Südhof, 2017) and function as autonomous homohexamers. In contrast, Cbln3 is restricted to the cerebellum, where it can only function in a hetero-hexameric complex with Cbln1 (Pang et al., 2000; Bao et al., 2006).
Neurexins are presynaptic receptors for cerebellins and other trans-synaptic ligands and are encoded by three genes (Nrxn1-3) that encode both shorter β- and longer α-neurexins (Ushkaryov et al., 1992, 1994; Ushkaryov and Südhof, 1993). Neurexins are extensively alternatively spliced at six canonical sites (Ullrich et al., 1995; Schreiner et al., 2014; Treutlein et al., 2014). Among these sites, alternative splicing at SS#4 is particularly important because it regulates neurexin binding to multiple ligands, including cerebellins that bind exclusively to α- and β-neurexins containing an insert at SS#4 (Uemura et al., 2010; Matsuda and Yuzaki, 2011; for review, see Südhof, 2017).
In the cerebellum, Cbln1 functions as a bidirectional synaptic organizer of parallel fiber-Purkinje cell synapses by binding to presynaptic neurexins and postsynaptic GluRδ2. Mice lacking Cbln1 gene exhibit ataxia, motor learning deficits, an ∼50% decrease in parallel fiber–Purkinje cell synapses and a complete loss of LTD (Hirai et al., 2005; Rong et al., 2012). Single Cbln2 and Cbln4 KO mice have also been reported and found not to exhibit any overt motor deficits or phenotypes on their own (Rong et al., 2012; Wei et al., 2012; Haddick et al., 2014), but no Cbln1/2 double KO (dKO) mice have been reported to test this question.
Cerebellins are expressed broadly throughout the brain in specific, often nonoverlapping patterns that produce relatively high expression levels in specific nuclei and cell types (Miura et al., 2006; Wei et al., 2007; Otsuka et al., 2016; Seigneur and Südhof, 2017). Specifically, Cbln1, Cbln2, and Cbln4 are differentially expressed in areas such as the olfactory bulb and the entorhinal cortex, but coexpressed in all neurons of the parafascicular nucleus of the thalamus, which provides an excitatory input to the striatum (Kusnoor et al., 2010; Otsuka et al., 2016; Seigneur and Südhof, 2017). Cbln2 is generally highly expressed in motor-related areas of the brain, including the motor cortex and the motor thalamus (ventral lateral and ventral anterior nuclei). Given these expression patterns, we hypothesized that, although cerebellins may not interact directly with each other, they may play differential functions in common pathways. If so, then loss of two or all three cerebellins would result in unique phenotypes that are not observed in single cerebellin KO mice.
In the present study, we generated and analyzed constitutive Cbln1/2 dKO and Cbln1/2/4 triple KO (tKO) mice, the latter of which have deletion of all cerebellin expression in the brain because Cbln3 requires Cbln1 for secretion. We show that, although cerebellin deletions cause seizures and major behavioral and morphological impairments, even Cbln1/2/4 KO are viable and fertile, demonstrating that cerebellins are functionally important but that, unlike other neurexin ligands, they are not essential for survival. Strikingly, we observed a secondary loss of synapses in multiple areas throughout the brain, suggesting that cerebellins contribute to synaptic function in many distinct pathways outside of the cerebellum. Our results establish a general important but restricted role for cerebellins as synaptic organizers.
Materials and Methods
Mouse handling and husbandry.
All experiments were performed with male and female adult, C57BL/6/SV129 hybrid mice maintained on a 12 h light/dark schedule and given food and water ad libitum. All animal procedures conformed to National Institutes of Health's Guidelines for the Care and Use of Laboratory Animals and were approved by the Stanford University Administrative Panel on Laboratory Animal Care.
Generation of constitutive KO mice.
Generation of Cbln1flox/flox, Cbln2flox/flox, and Cbln4flox/flox mice was described previously (Seigneur and Südhof, 2017). Homozygous Cbln1flox/flox;Cbln2flox/flox mice were crossed with transgenic mice expressing Cre-recombinase under control of the nestin promoter (The Jackson Laboratory) to produce the following groups: Cbln1+/−;Cbln2+/− (control), Cbln1−/−;Cbln2+/− (Cbln1 KO), Cbln1+/−;Cbln2−/− (Cbln2 KO), and Cbln1−/−;Cbln2−/− (Cbln1/2 dKO).
In the second set of experiments, Cbln4flox/flox mice were crossed with the nestin-cre mice. These mice were then crossed with Cbln1;Cbln2 transgenic mice to generate the following groups: Cbln1+/−;Cbln2+/−;Cbln4+/− (control), Cbln1−/−;Cbln2−/−;Cbln4+/− (Cbln1/2 dKO), and Cbln1−/−;Cbln2−/−;Cbln4−/− (tKO).
Genotyping was performed by PCR using the following primers: Cbln1 forward: 5′-TAGGGTGGACAGAGAAAAGG-3′, reverse: 5′-CTTCTAATCTGTCCTGACCACA-3′, reverse: 5′-TCTAAAGGCAGGAAGGAATCTA-3′ (expected products, WT = 342 bp, floxed = 437 bp, and KO = 544 bp); Cbln2 forward: 5′-TAAAAGACAGTCCAGAGTTTTAGTC-3′, reverse: 5′-TGTGCTTACTCCTCTCTATTTGA-3′, reverse: 5′-CTATTGGAGTCC TTCAAGGAAA-3′ (expected products, WT = 225 bp, floxed = 320 bp, and KO = 420 bp); Cbln4 forward: 5′-CACAGATCTGTATTTCAAGGCA-3′, reverse: 5′-TGGATTTATTTCTT GGTGAGACATG-3′, reverse: 5′-AGTCCTTCAAGGAAACAAACTT-3′ (expected products, WT = 234 bp, floxed = 315 bp, and KO = 459 bp).
Mouse behavior.
In the first set of experiments, behavior was investigated using 2- to 6-month-old male littermate mice, as noted. For all behavior assays, numbers of animals were as follows: control, n = 10; Cbln1 KO, n = 8; Cbln2 KO, n = 9; and Cbln1/2 dKO, n = 13 except where noted. In the second set of experiments, behavior was investigated using 1- to 12-month-old male and female littermate mice, as noted. Because we observed no gender differences on any of the behavioral assays, data from male and female mice were pooled (except for body weight). For this second set of behavior assays, numbers of animals were as follows: control, n = 16 (8 females, 8 males); Cbln1/2 dKO, n = 14 (8 females, 6 males); and tKO, n = 11 (5 females, 6 males) except where noted.
All experiments were performed between 7:00 A.M. and 7:00 P.M.. Mice were moved from the holding facility to the testing room at least 1 h before testing began. All behavior assays were conducted and analyzed by researchers blinded to genotype.
Force-plate actometer.
Mice were placed in the center of a 28 × 28 cm force plate actometer and allowed to explore freely for 15 min. Changes in the center of force (movement) of the mouse was monitored and analyzed using an in-house program as described previously (Fowler et al., 2001). Area measure was adapted from human postural sway analysis and calculated by first measuring the area around the center of force at three successive locations (triangle areas) and then summing these areas together. Increased postural sway reflects deficits in balance and stability. In addition to the automatic analysis, each 15 min session was video recorded and lateral falls (defined as all 4 paws off the ground with the mouse lying on its left or right side) during the first 5 min were counted by hand.
Footprint analysis.
Various gait parameters were quantified using footprint analysis as described previously (Carter et al., 2001). A piece of white paper was placed along the floor of an open-top runway measuring 50 cm long, 10 cm wide, and 10 cm high. Nontoxic paint was used to paint the forepaws (red) and hindpaws (blue) of the mice; the mice then were placed at one end of the runway and allowed to run to a covered goal box at the other end, leaving painted footprints on the sheet of paper along the way. Each mouse was given three training sessions without painted feet and then three trial sessions with painted feet. The following gait parameters were then analyzed (1) stride length, the average distance between successive footprints on each side, that is, the distance between the first left forepaw and second left forepaw, etc.; (2) forepaw and hindpaw base width, the average distance between the left and right forepaw or hindpaw prints, respectively; (3) overlap between forepaw and hindpaw placement, the average distance between the center of the forepaw and hindpaw prints on each side, which measures foot placement accuracy and step pattern uniformity; and (4) step frequency, the average number of steps divided by distance, which measures stride fluidity and length.
Balance beam.
A wooden beam measuring 95 cm long and 2 or 1 cm in diameter was suspended 50 cm in the air. A dark goal box was positioned at one end of the beam (end) and a bright light was positioned above the other end (start). A starting line was marked ∼10 cm from the end so that the total distance between the starting line and the goal box was 80 cm. Mice were given three trials with 60 s between each trial and their best trial was used for analysis.
Accelerating rotarod.
Mice were placed on a rotating rod that accelerated from four to 40 RPMs. The mice were given 3 trials/d for 4 d with 1 h between each trial. The speed in RPMs at which the mouse fell off the rod was recorded.
Acoustic startle and prepulse inhibition (PPI).
The acoustic startle response and PPI were measured using the Kinder Scientific Startle Monitor and analyzed using Startle Monitor II software. The mice were trapped in a small compartment atop a force plate inside a soundproof chamber. The startle response, defined as the change in amplitude of force in response to an unexpected acoustic stimulus, was measured. In the acoustic startle response test, each of the following trial type was presented 10 times in pseudorandom order: no stimulus, 75 dB pulse, 85 dB pulse, 95 dB pulse, 105 dB pulse, and 115 dB pulse. The mean startle amplitude was calculated for each stimulus intensity.
In the PPI test, the baseline startle response was first determined by presenting 10 consecutive 115 dB pulse trials. The following trials were then presented 10 times each in pseudorandom order: pulse alone (115 dB), 68 dB prepulse, 71 dB prepulse, and 77 dB prepulse. A 115 dB pulse followed each prepulse at a 100 ms onset-to-onset interval. The percentage inhibition of the baseline startle amplitude was then calculated for each prepulse/pulse pair.
Immunoblotting.
Six-month-old littermate mice were deeply anesthetized with isoflurane and motor cortex, caudate putamen, thalamus, and cerebellum were dissected and lysed in fresh, prechilled RIPA buffer (10 mm Tris-Cl, pH 8.0, 1 mm EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 140 mm NaCl) with 1 mm PMSF protease inhibitor mixture (Roche Applied Science) for 1 h at 4°C with agitation. Samples were then centrifuged at 20,000 × g for 30 min at 4°C and the supernatants were transferred to prechilled 1.5 ml Eppendorf tubes. Protein concentrations were determined using the Pierce BCA Protein Assay kit. Then, 10 μg of protein was loaded onto a Criterion TGX 4–20% Tris-Glycine precast gel (Bio-Rad) and separated via SDS-PAGE at 120 V for 1.5 h and then transferred onto a nitrocellulose membrane using the Trans-Blot Turbo transfer system (Bio-Rad) according to the manufacturer's instructions. Membranes were then blocked with 5% nonfat dry milk in TBS containing 0.1% Tween 20 (TBST) for 1 h at room temperature (RT) with agitation and incubated in primary antibody overnight at 4°C with agitation. Membranes were washed 3× with TBST for 10 min/each at RT and then incubated in fluorescently labeled secondary antibodies (donkey anti-rabbit IR dye 680CW, 1:5000; donkey anti-mouse IR dye 800CW, 1:5000; and donkey anti-guinea pig IR dye 680RD, 1:5000; LI-COR Bioscience). Membranes were scanned using an Odyssey Infrared Imager and analyzed with Odyssey software (LI-COR Biosciences). The intensity value for each protein of interest was first normalized to actin and then normalized to the control sample. Each protein/sample/group was tested in triplicate and the normalized mean ± SE is depicted in the graphs.
The following antibodies were used in this study: monoclonal mouse anti-β-actin (1:5000; A1978 Sigma-Aldrich; RRID:AB_476692), polyclonal rabbit anti-GluA1 (1:500; PC246 Millipore; RRID:AB_564636), monoclonal mouse anti-GluA2 (1:1000; MAB397 Millipore; RRID:AB_2113875), polyclonal rabbit anti-GluA4 (1:1000; AB1508 Millipore; RRID:AB_90711), polyclonal rabbit anti-Grid2 (1:500; AB1514 Millipore), monoclonal mouse anti-GluN1 (1:1000; 114011 Synaptic Systems; RRID:AB_887750), polyclonal rabbit anti-GluN2 A/B (1:500; AB1548 Millipore; RRID:AB_90759), polyclonal rabbit anti-PSD95 (1:500; L667 Südhof laboratory), polyclonal rabbit anti-Synapsin (1:1000; E028 Südhof laboratory), polyclonal rabbit anti-vGAT (1:1000; 131003 Synaptic Systems; RRID:AB_887869), polyclonal guinea pig anti-vGluT1 (1:1000; AB5905 Millipore; RRID:AB_2301751), and polyclonal guinea pig anti-vGluT2 (1:1000; AB2251 Millipore; RRID:AB_1587626).
Immunohistochemistry.
Six-month-old control, Cbln1/2 dKO, and Cbln1/2/4 tKO littermate mice were anesthetized with isoflurane and their brains were fixed with transcranial perfusion of 4% paraformaldehyde (PFA). After removal from the skull, brains were further incubated in 4% PFA for 2 h at RT with rocking. Brains were then cryoprotected in 30% sucrose (in 1× PBS) at 4°C for 48 h with rotation. The samples were then frozen in OCT compound (TissueTek) on dry ice and 25 μm coronal sections were made with a cryostat (CM 3050S; Leica Biosystems). Sections were carefully transferred to a 1.5 ml Eppendorf tube and blocked with 10% goat serum, 1% BSA, and 0.3% Triton X-100 in 1× PBS for 1 h at RT with rocking. Sections were incubated in primary antibody for 48 h at 4°C with rocking. Sections were washed 3× for 20 min/each with 1× PBS containing 0.3% Triton X-100 and then incubated in species-specific secondary antibodies coupled to Alexa Fluor 488, 546, or 633 (Invitrogen; 1:400 dilution) for 2 h at RT. Sections were then washed and mounted on slides with Vectashield containing the nuclei marker DAPI (Vector Laboratories).
The following antibodies were used in this study: monoclonal mouse anti-calbindin (1:200; C9848 Sigma-Aldrich; RRID:AB_476894), polyclonal chicken anti-GFP (1:1000; GFP-1020 Aves Laboratories; RRID:AB_10000240), polyclonal chicken anti-MAP2 (1:500; CPCA-MAP2, EnCor Biotechnology; RRID:AB_2138173), polyclonal rabbit anti-MAP2 (1:500; AB5622 Millipore; RRID:AB_11213363), polyclonal rabbit anti-vGAT (1:500; 131003 Synaptic Systems; RRID:AB_887869), polyclonal guinea pig anti-vGluT1 (1:500; AB5905 Millipore; RRID:AB_2301751), and polyclonal guinea pig anti-vGluT2 (1:500; AB2251 Millipore; RRID:AB_1587626).
Images were collected using a Nikon A1R confocal with a 60× objective and analyzed using NIS-Elements Advanced Research software (Nikon). For each synaptic marker and brain area, data were collected from three mice/genotype, three sections/mouse, and two regions of interest/section. The fluorescent signal intensity for each synaptic marker was first normalized to the area of MAP2-labeled or calbindin-labeled (for cerebellum sections only) tissue and then to the levels observed in the controls.
Statistics.
All analysis was done using GraphPad Prism 6 software. Graphs depict mean ± SEM. All behavior was analyzed by either one-way or two-way Kruskal–Wallis ANOVA for group differences and significant differences are reported as p-values. Differences between control and individual KO groups or between Cbln1KO and Cbln1/2 dKO were then determined using Tukey's post hoc test, correcting for multiple comparisons, and are reported as adjusted p-values. Immunoblots were analyzed with one-way ANOVA for group differences and significant differences are reported as p-values. Significant differences between control and individual KO groups were determined using Dunnett's post hoc test, correcting for multiple comparisons, and are reported as adjusted p-values. Differences in vGluT1, vGluT2, or vGaT signal intensity were analyzed by one-way ANOVA and are reported as p-values.
Results
Cerebellin-deficient mice exhibit a normal lifespan but suffer from weight loss
We generated constitutive Cbln1 KO, Cbln2 KO, Cbln1/2 dKO, and Cbln1/2/4 tKO mice and analyzed their weight and survival. Note that because Cbln3 requires Cbln1 for surface transport (Bao et al., 2006), Cbln1/2/4 tKO mice are subject to a complete ablation of cerebellins. At 2 months of age, all of these mice were indistinguishable from control littermates (Fig. 1A–C). Starting at 4 months of age, Cbln1 KO, Cbln1/2 dKO, and Cbln1/2/4 tKO mice weighed significantly less than Cbln2 KO mice and control littermate mice; this difference further increased at later ages (at 4 months, F(3,36) = 7.530, p = 0.0005; at 5-months F(3,36) = 8.987, p = 0.0001; and at 6-months, F(3,36) = 27.34, p = 0.0001, ANOVA; Fig. 1C). This phenotype applied to both male and female Cbln1/2 dKO and Cbln1/2/4 tKO mice, with the weight difference manifesting earlier in male (by 3 months of age) than in female mice (by 5 months of age). At 6 months of age, Cbln1/2 dKO mice weighed ∼12% less than Cbln1 KO mice, which was statistically significant when comparing Cbln1 KO with Cbln1/2 dKO mice (t(19) = 2.526, p = 0.0206, Student's t test), but not after correction for multiple comparisons of all groups after one-way ANOVA (adjusted p = 0.1599, Tukey's post hoc test). At 12 months, Cbln1/2 dKO mice weighed half as much as control littermates (Fig. 1A,C). Moreover, apparent muscle degeneration resulted in a convex curvature of the spine in older Cbln1/2 dKO and Cbln1/2/4 tKO mice, which was not observed in control or in single Cbln1 or Cbln2 KO mice (Fig. 1A, right).
Figure 1.

Single, double, and triple Cbln1, Cbln2, and Cbln4 KOs do not impair survival, but Cbln1 KOs cause weight loss and Cbln1/2 dKOs induce seizures. A, Images illustrating the body size of littermate control (Ctl), Cbln1 KO, Cbln2 KO, and Cbln1/2 dKO mice at 2 and 12 months of age. Twelve-month-old dKO mice were significantly smaller than age- and sex-matched Ctl, Cbln1KO, and Cbln2KO mice and exhibited spinal changes (arrow in right), which were absent in Ctl littermates. B, Comparison of the weights of single versus double Cbln1/2 KO mice. Cbln1 KO (n = 8) and Cbln1/2 dKO mice (n = 13) weighed significantly less than Ctl (n = 10) and Cbln2 KO mice (n = 9) at 4, 5, and 6 months of age (****p < 0.0001, two-way ANOVA). dKO mice also weighed significantly less than Cbln1 KO mice at 6 months (p = 0.0206, two-tailed t test); however, this difference lost significance after correcting for multiple comparisons of all groups (adjusted p = 0.0934, Tukey's post hoc test). Data are shown as means ± SEM. C, Comparison of the weights of Cbln1, Cbln2, and Cbln4 dKO versus tKO mice. Male Cbln1/2 dKO (n = 6) and Cbln1/2/4 tKO (n = 6) mice weighed significantly less than Ctl (n = 8) littermate mice beginning at 3 months of age; female dKO (n = 8) and tKO (n = 5) mice weighed less than Ctl (n = 8) littermates beginning at 5 months of age, but tKO and dKO mice of either gender did not differ from each other (****p < 0.0001, two-way ANOVA). Data are shown as means ± SEM. D, Cerebellin deletions do not impair mouse survival despite severe motor dysfunction and weight loss. Cbln1/2 dKO (n = 14 at 1 month; n = 13 at 8–12 months) and Cbln1/2/4 tKO mice (n = 11) have normal lifespans compared with Ctl mice (n = 16 at 1 month; n = 15 at 8 months; n = 14 9–12 months). E, Cbln1/2 dKO mice, but not single Cbln1 or Cbln2 KO mice, exhibit a high incidence of seizures. Data show the number of dKO mice that exhibited seizures during two behavioral challenges, whereas no seizures were ever observed in Ctl, Cbln1KO, or Cbln2 KO mice (*p = 0.0368; **p = 0.0015; ***p < 0.0001; Fisher's exact test). F, Cbln1/2/4 tKO mice exhibit a higher seizure propensity than Cbln1/2 dKO mice and seizures emerge earlier in tKO mice than in dKO mice. Data depict the percentage of dKO (n = 14) and tKO (n = 11) mice that exhibited epileptogenic activity during a 3 min tail suspension assay at the indicated ages (p = 0.0001, Mantel–Cox). No seizures were ever observed in Ctl mice (n = 16).
Previous studies reported that Cbln1 KO and Cbln2 KO mice reproduce normally and have normal lifespans (Hirai et al., 2005; Rong et al., 2012). Here, we found that, despite severe motor deficits, Cbln1/2 dKO and Cbln1/2/4 tKO mice also reproduced normally and had normal lifespans compared with control mice (Fig. 1D). Therefore, unlike neurexins and neuroligins, the other major neurexin ligands (Missler et al., 2003; Varoqueaux et al., 2006), deletion of all cerebellins is not lethal.
Cbln1/2 double and Cbln1/2/4 tKO mice develop epilepsy
During behavioral testing, we observed that ∼92% of Cbln1/2 dKO mice (n = 13) but not Clbn1 or Cbln2 single KO mice exhibited stress-induced seizures (χ(3)2 = 27.5, p < 0.0001, Fisher's exact test). Broken down by assay, ∼62% had a seizure during the tail suspension test (χ(3)2 = 15.37, p = 0.0015, Fisher's exact test), and ∼38% during the forced swim test (χ(3)2 = 8.493, p = 0.037, Fisher's exact test). Seizures were never observed in littermate control mice or in single Cbln1 or Cbln2 KO mice (Fig. 1E).
Because seizures were present in Cbln1/2 dKO mice, but not in single Cbln1 KO or Cbln2 KO mice, we investigated whether the additional deletion of Cbln4 would aggravate the seizure phenotype of Cbln1/2 dKO mice. To address this question, we measured the incidence of seizures in 1-, 2-, and 3-month-old mice during a 3 min tail suspension test. Compared with Cbln1/2 dKO mice, Cbln1/2/4 tKO mice were significantly more susceptible to stress-induced seizures. At 1 month of age, no mice exhibited seizures; at 2 months, 15% of Cbln1/2 dKO and 90% of Cbln1/2/4 tKO mice exhibited seizures; and at 3 months, 46% of Cbln1/2 dKO and 100% of Cbln1/2/4 tKO mice developed seizures (χ(2)2 = 36.28, p = 0.0001, Mantel–Cox). No seizures were observed in control mice of any age (Fig. 1F). Therefore, deletions of multiple, but not of individual, cerebellins cause late-onset seizures, suggesting that all three autonomous cerebellins (Cbln1, Cbln2, and Cbln4) contribute to brain function.
Cbln1 and Cbln2 single and dKOs induce diverse changes in synaptic protein composition
Given the well documented function of Cbln1 in cerebellar synapses (Hirai et al., 2005; Ito-Ishida et al., 2008; Uemura et al., 2010), we next investigated whether cerebellin deletions alter the synaptic proteome in mice (Fig. 2). Using quantitative immunoblotting, we determined the relative levels of synaptic and nonsynaptic proteins in the motor cortex, caudate putamen, thalamus, and cerebellum of constitutive Cbln1 KO, Cbln2 KO, and Cbln1/2 dKO mice compared with littermate controls.
Figure 2.
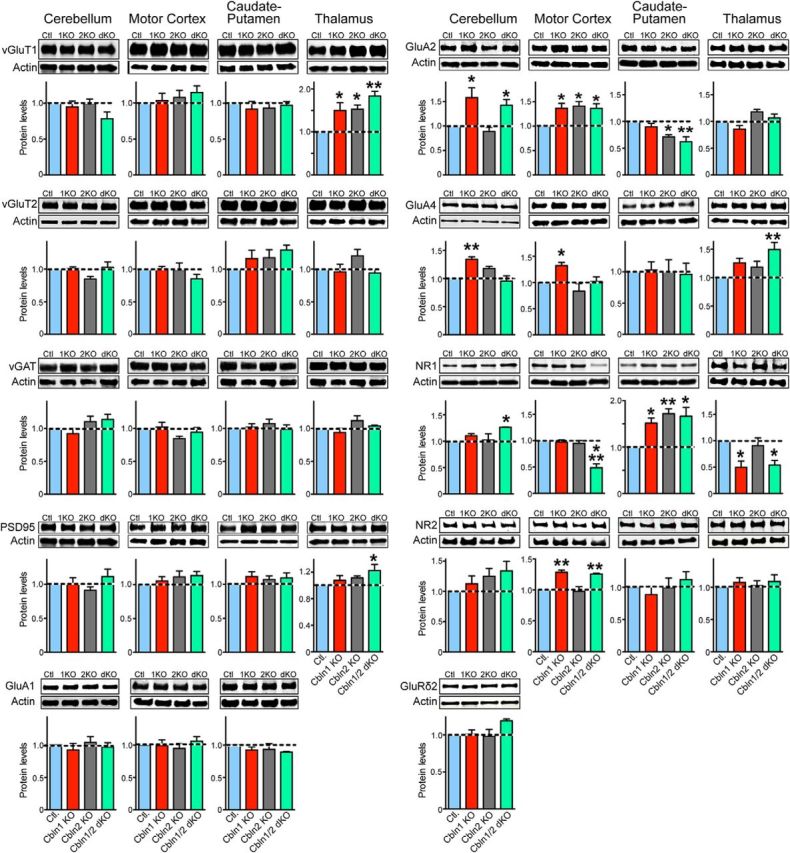
Cbln1 and Cbln2 single KOs and Cbln1/2 dKOs cause region-specific increases or decreases in synaptic proteins as assessed by quantitative immunoblotting. Representative blots of the indicated proteins (top) and summary graphs of their levels (bottom) measured by quantitative immunoblotting in lysates from cerebellum, motor cortex, caudate putamen, and thalamus. Lysates were obtained from littermate control, Cbln1, Cbln2, and Cbln1/2 dKO mice (age = 6 months; n = 3 mice). All data are shown as means ± SEM and were normalized first to the levels of β-actin as an internal loading control and then to the levels observed in the controls. Asterisks denote statistically significant differences between mutant and control samples as assessed by one-way ANOVA followed by Dunnet's multiple-comparisons correction. *adjusted p ≤ 0.05; **adjusted p ≤ 0.01.
The levels of the excitatory presynaptic protein vGluT1 were increased significantly in the thalamus of Cbln1 KO mice (adjusted p = 0.043, Dunnett's post hoc test), Cbln2 KO mice (adjusted p = 0.033, Dunnett's post hoc test), and Cbln1/2 dKO mice (adjusted p = 0.0028, Dunnett's post hoc test) compared with controls (F(3,8) = 8.504, p = 0.0072, ANOVA), but were unchanged in other brain regions tested. Similarly, the excitatory presynaptic protein vGluT2 trended toward an increase in the caudate of Cbln1 or Cbln2 KO mice or Cbln1/2 dKO mice compared with littermate control mice, but this trend did not reach statistical significance as measured by ANOVA (F(3,8) = 1.525, p = 0.2809) and vGluT2 was unchanged in all other brain regions tested. In the thalamus, expression of the excitatory postsynaptic protein PSD95 was also increased in Cbln1/2 dKO mice (adjusted p = 0.0413, Dunnett's post hoc test), but not in Cbln1 KO and Cbln2 KO mice compared with controls (F(3,20) = 2.402, p = 0.0979, ANOVA). In all other brain regions, PSD95 levels were unchanged. In contrast to vGluT1, vGluT2, and PSD95, the inhibitory presynaptic protein vGAT was unchanged in all brain regions tested in various cerebellin KO mice (Fig. 2).
Next, we examined the levels of the AMPA receptor subunits GluA1, GluA2, and GluA4. No changes in GluA1 were detected in any of the brain regions tested in various cerebellin KO mice (note that GluA1 is very weakly expressed in the thalamus (Montgomery et al., 2009) and was therefore excluded from testing). In contrast, GluA2 expression was increased in the motor cortex of Cbln1 KO (adjusted p = 0.036, Dunnett's post hoc test), Cbln2 KO (adjusted p = 0.021, Dunnett's post hoc test), and Cbln1/2 dKO mice (adjusted p = 0.037, Dunnett's post hoc test) compared with controls (F(3,8) = 5.246, p = 0.027, ANOVA). GluA2 levels were likewise increased in the thalamus of Cbln2 KO mice (F(3,8) = 5.665, p = 0.022, ANOVA) and in the cerebellum of Cbln1 KO (adjusted p = 0.011, Dunnett's post hoc test) and Cbln1/2 dKO mice (adjusted p = 0.0429, Dunnett's post hoc test; F(3,8) = 7.102, p = 0.012, ANOVA). Conversely, GluA2 levels were significantly decreased in the caudate putamen of Cbln2 KO (adjusted p = 0.025, Dunnett's post hoc test) and Cbln1/2 dKO mice (adjusted p = 0.0058, Dunnett's post hoc test) compared with controls (F(3,8) = 8.034, p = 0.0085, ANOVA). Moreover, GluA4 levels were significantly increased in Cbln1 KO mice compared with control mice in the motor cortex (F(3,8) = 5.045, p = 0.030, ANOVA; adjusted p = 0.0358, Dunnett's post hoc test) and cerebellum (F(3,8) = 10.83, p = 0.003, ANOVA; adjusted p = 0.0053, Dunnett's post hoc test). GluA4 levels were also significantly increased in the thalamus of Cbln1 KO, Cbln2 KO, and Cbln1/2 dKO mice as measured by ANOVA (F(3,8) = 5.164, p = 0.028), but after correcting for multiple comparisons, this increase only reached significance in Cbln1/2 dKO mice (adjusted p = 0.0047, Dunnett's post hoc test). GluA4 expression was unchanged in caudate putamen of any of the KO mice.
We also studied the NMDA-receptor subunits NR1 and NR2 in the same samples. NR1 expression was unchanged in motor cortex of Cbln1 KO and Cbln2 KO mice, but dramatically reduced in Cbln1/2 dKO mice compared with controls (F(3,8) = 23.21, p = 0.0003, ANOVA; adjusted p = 0.0003, Dunnett's post hoc test). NR1 expression was also reduced in the thalamus of Cbln1 KO (adjusted p = 0.023, Dunnett's post hoc test) and Cbln1/2 dKO mice (adjusted p = 0.035, Dunnett's post hoc test), but not in Cbln2 KO mice (adjusted p = 0.8565, Dunnett's post hoc test) compared with controls (F(3,8) = 5.950, p = 0.020, ANOVA). Conversely, NR1 expression was increased in the caudate putamen of Cbln1 KO (F(3,8) = 7.267, p = 0.011, ANOVA; adjusted p = 0.0417, Dunnett's post hoc test), Cbln2 KO (adjusted p = 0.0079, Dunnett's post hoc test), and Cbln1/2 dKO mice (adjusted p = 0.012, Dunnett's post hoc test) and in the cerebellum of Cbln1/2 dKO mice (F(3,8) = 3.489, p = 0.070, ANOVA; adjusted p = 0.048, Dunnett's post hoc test). NR2 expression was increased in the motor cortex of Cbln1 KO (F(3,8) = 14.90, p = 0.0012, ANOVA; adjusted p = 0.0038, Dunnett's post hoc test) and Cbln1/2 dKO mice (adjusted p = 0.0072, Dunnett's post hoc test), but unchanged in Cbln2 KO mice compared with controls. NR2 expression was unchanged in the caudate putamen, thalamus, and cerebellum in all KO mice. Finally, expression of the postsynaptic Cbln-binding protein GluRδ2 was unchanged in the cerebellum in Cbln1 KO, Cbln2 KO, and Cbln1/2 dKO mice compared with controls (Fig. 2).
Combined, these results are rather surprising: They show that individual or combined deletions of Cbln1 and Cbln2 produce no consistent change in the levels of any synaptic protein; in fact, increases in synaptic protein levels were observed on balance as often as decreases. We found different patterns of changes in different brain regions, consistent with the notion that Cbln1 and Cbln2 do not simply act as synapse assembly proteins and perform partly nonoverlapping functions in the brain regions examined.
Loss of Cbln1 and Cbln2 causes a delayed, input-specific loss of synapses in the hippocampus
In the cerebellum, deletions of Cbln1 were shown to cause a major but incomplete loss of parallel fiber synapses (Hirai et al., 2005). Specifically, parallel fiber synaptic contacts develop initially normally in Cbln1 KO mice, but are then eliminated preferentially in distal domains of the Purkinje cell dendritic arbors (Hirai et al., 2005; Ito-Ishida et al., 2008; Uemura et al., 2010). However, little is known about the effects of the deletions of Cbln2 or Cbln4 on synapses in the cerebellum and of Cbln1, Cbln2, or Cbln4 on synapses in brain outside of the cerebellum. To investigate this, we used immunohistochemistry and quantified the intensity of excitatory synapses identified by labeling with antibodies to vGluT1 and/or vGluT2 and of inhibitory synapses identified by labeling with antibodies to vGAT. Using this approach, we examined the effect of the Cbln1/2 dKO and Cbln1/2/4 tKO on the intensity of excitatory synapses in the hippocampus at 3 different ages: 1, 2, and 6 months. We sampled excitatory synapse intensity, as monitored by vGluT1 staining, in three regions of the hippocampus of littermate control and Cbln1/2 dKO mice:the molecular layer of the dentate gyrus and the stratum radiatum and stratum lacunosum moleculare of the CA1 region.
In the hippocampal formation, layer 2 neurons of the entorhinal cortex form synapses in the molecular layer of the dentate gyrus and the stratum lacunosum moleculare of the CA3 region, whereas layer 3 neurons project to the stratum lacunosum moleculare of the CA1 region (Chrobak et al., 2000). Although expression of the cerebellins is extremely low in the hippocampus proper, Cbln2 is highly expressed in the subiculum. In addition, Cbln1 and Cbln4 are highly expressed in the entorhinal cortex and Cbln1+ or Cbln4+ projections from these neurons are observed in the molecular layer of the dentate gyrus and the stratum lacunosum moleculare of the CA1 region (Otsuka et al., 2016; Seigneur and Südhof, 2017; Fig. 3A). Furthermore, we investigated three different ages of mice because, in the cerebellum, a loss of synapses is only observed after a delay (Kurihara et al., 1997; Hirai et al., 2005), prompting us to explore whether a delayed loss of synapses may also be evident in other brain regions. Indeed, we detected no change in synapse intensity in 1- and 2-month-old Cbln1/2 dKO mice, suggesting that the deletion of Cbln1 and Cbln2 does not impair initial synapse formation (Fig. 3B,D). Strikingly, at 6 months of age, we observed a dramatic loss of synapse intensity in both the molecular layer of the dentate gyrus (F(2,53) = 32.74, p < 0.0001, ANOVA) and the stratum lacunosum moleculare of the CA1 region (F(2,54) = 47.70, p < 0.0001, ANOVA; Fig. 3C,D). To establish that the decrease in vGluT1 signal intensity reflects a decrease in synapses and not merely a decrease in vGluT1 protein expression, we also quantified the signal intensity of synapsin in the 6-month-old mice. Again, we observed a significant decrease in synapsin intensity in both the molecular layer of the dentate gyrus (F(2,40) = 6.576, p = 0.0034, ANOVA) and the CA1 region stratum lacunosum moleculare (F(2,41) = 15.47, p < 0.0001, ANOVA; Fig. 3E,G).
Figure 3.
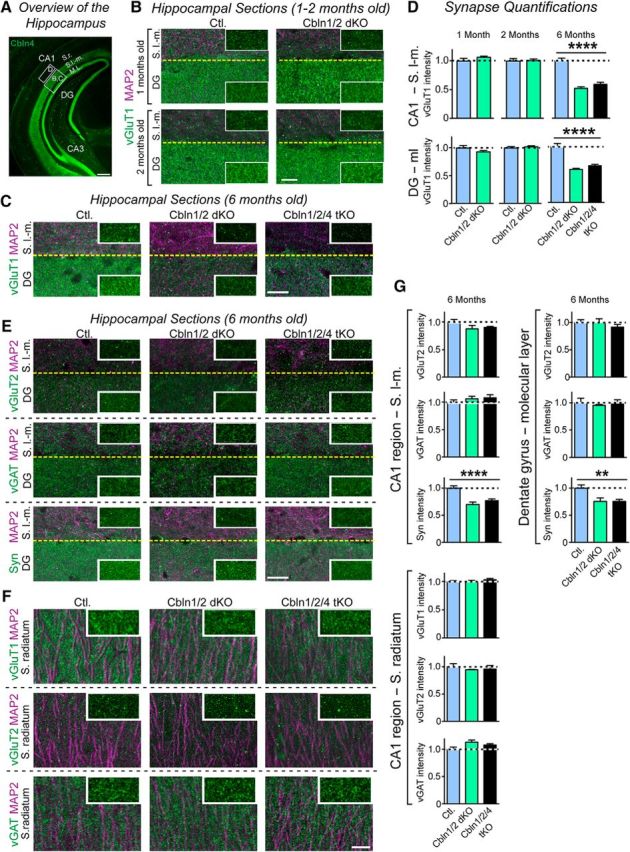
Cbln1/2 dKOs have no effect on hippocampal synapse intensity in 1- and 2-month-old mice, but decrease excitatory synapse intensity in the statum lacunosum moleculare of the CA1 region and the molecular layer of the dentate gyrus in 6-month-old mice. A, Illustration of the hippocampal projection patterns of Cbln4-positive neurons in the entorhinal cortex and of the areas sampled in the synapse intensity quantifications. Images display representative coronal sections from a Cbln4-mVenus reporter mouse in which mVenus was visualized by immunofluorescence labeling for GFP (Seigneur and Südhof, 2017). Boxed area “B,C” outlines the stratum lacunosum moleculare (SLM) in the CA1 region and the molecular layer (ML) of the dentate gyrus (DG); boxed area “D” marks the stratum radiatum (SR) in the CA1 region. Scale bar, 100 μm. B, Representative images of sections from the CA1 region stratum lacunosum moleculare and dentate gyrus molecular layer in 1-month-old (top) and 2-month-old (bottom) mice. Sections in each panel were from littermate control (left) and Cbln1/2 dKO (right) mice and were labeled by immunofluorescence for vGluT1 (green) and MAP2 (magenta). Inserts show a high-magnification example of the synaptic marker labeling without MAP2. Scale bar, 20 μm. C, Representative images of sections from the CA1 region stratum lacunosum moleculare in 6-month-old mice. Sections in each panel were from littermate control (left), Cbln1/2 dKO (middle), and Cbln1/2/4 tKO mice (right) and were colabeled by immunofluorescence for MAP2 (magenta) and vGluT1 (green). Insets show a high-magnification example of the synaptic marker labeling without MAP2. Scale bar, 20 μm. D, Cerebellin deletions selectively decrease the intensity of vGluT1-positive synapses in the CA1 region stratum lacunosum moleculare and the molecular layer of the dentate gyrus in 6-month-old, but not in 1- or 2-month-old mice. Summary graphs depict the intensity of vGluT1-postive puncta in these two regions, imaged as a function of age and cerebellin deletions as described in B and C. E, Representative images of sections from the CA1 region stratum lacunosum moleculare and dentate gyrus molecular layer in 6-month-old mice. Sections in each panel were from littermate control (left), Cbln1/2 dKO (middle), and Cbln1/2/4 tKO mice (right) and were colabeled by immunofluorescence for MAP2 (magenta) and vGluT2 (green/top), vGAT (green/middle), or synapsin (green/bottom). Insets show a high-magnification example of the synaptic marker labeling without MAP2. Scale bar, 20 μm. F, Same as in E but sections are from the CA1 region stratum radiatum and were colabeled by immunofluorescence for MAP2 (magenta) and vGluT1 (green/top), vGluT2 (green/middle), or vGAT (green/bottom). G, Summary graphs displaying the intensity of vGluT1-, vGluT2-, vGAT-, or synapsin-positive puncta in the three regions sampled imaged as a function of cerebellin deletions as described in E and F. All data in D and G are shown as means ± SEM. Synaptic puncta intensity was normalized to the area of MAP2-labeled tissue and to the levels observed in the controls. Statistical analysis was performed by one-way ANOVA (n = 3 mice for each genotype). ****p < 0.0001; **p = 0.0034.
We detected no changes in the stratum radiatum of the CA1 region, which receives inputs, not from the entorhinal cortex, but from the CA3 region. The synapse loss in aging Cbln1/2 dKO mice was specific for vGluT1-expressing excitatory synapses because we did not observe changes in the intensity of synapses, as quantified by vGluT2 staining (CA1, F(2,56) = 1.580, p = 0.22; dentate gyrus, F(2,55) = 0.6862, p = 0.51, ANOVA) or vGAT staining (CA1, F(2,51) = 0.9358, p = 0.40; F(2,51) = 0.2590, p = 0.77, ANOVA). Moreover, deletion of Cbln4 in addition to Cbln1 and Cbln2 did not enhance the synapse loss, suggesting that Cbln4 makes little contribution to the phenotype (Fig. 3C–G). These results, along with the delayed onset of parallel fiber synapse loss and only partial loss of parallel fiber synapses in Cbln1 KO mice (Hirai et al., 2005; Ito-Ishida et al., 2008; Uemura et al., 2010), suggests that cerebellins are not essential for synapse formation and that the synapse loss in Cbln1 KO and Cbln1/2 dKO mice may be secondary to a functional deficit (Südhof, 2017).
Cerebellin deletions induce synapse loss in diverse brain regions
We next investigated whether the hippocampal synapse loss observed in older Cbln1/2 dKO mice is also evident in other brain regions. To test this question, we sampled the dorsal striatum, the retrosplenial cortex, and the cerebellum. We selected to analyze the dorsal striatum because the Cbln1 KO was reported to lead to an increase instead of a decrease in dendritic spines in the striatum (Kusnoor et al., 2010), the retrosplenial cortex because of its unusual, segregated expression patterns of Cbln1, Cbln2, and Cbln4 (Otsuka et al., 2016; Seigneur and Südhof, 2017), and the cerebellum because we aimed to test whether the double deletion of both Cbln1 and Cbln2 may aggravate the only partial loss of parallel fiber synapses in Cbln1 KO mice (Hirai et al., 2005; Ito-Ishida et al., 2008; Uemura et al., 2010).
Dorsal striatum
Cbln2 is highly expressed in corticostriatal projecting neurons and Cbln1, Cbln2, and Cbln4 are coexpressed in thalamostriatal projecting neurons in the parafascicular nucleus (Seigneur and Südhof, 2017), leading us to quantify changes in synapses in the dorsal striatum of 6-month-old littermate control, Cbln1/2 dKO, and Cbln1/2/4 tKO mice (Fig. 4). We observed a significant decrease in vGluT1 signal intensity (F(2,60) = 10.47, p = 0.0001, ANOVA), but an increase in vGluT2 signal intensity (F(2,60) = 5.243, p = 0.0080, ANOVA) in Cbln1/2 double and Cbln1/2/4 tKO mice compared with control. Because the striatum receives vGluT1-positive inputs from the cortex and vGluT2-positive inputs from the thalamus (Doig et al., 2010), these data suggest that loss of cerebellins results in a loss of excitatory corticostriatal inputs and gain of excitatory thalamostriatal inputs, analogous to the situation observed in the cerebellum (see below). There was no change in vGAT staining intensity (F(2,51) = 0.5273, p = 0.59, ANOVA) and, for all markers, Cbln1/2 double and Cbln1/2/4 tKO mice did not differ from each other.
Figure 4.

Cerebellin deletions decrease the intensity of vGluT1-positive synapses, but increase the intensity of vGluT2-positive synapses in the dorsal striatum in 6-month-old mice. A, Representative immunofluorescence images of sections from the dorsolateral subdivision of the caudate putamen of control (left), Cbln1/2 dKO (middle), and Cbln1/2/4 tKO (right) adult mice at 6 months of age. Sections were colabeled for MAP2 and vGluT1 (top), vGluT2 (middle), or vGAT (bottom). Insets show a high-magnification example of the synaptic marker labeling without MAP2. Scale bar, 20 μm. B, Cerebellin deletions cause opposite effects on the intensity of vGluT1- and vGluT2-positive synapses in the dorsolateral striatum. Summary graphs display the intensity of vGluT1-, vGluT2-, or vGAT-positive puncta in striatum imaged as a function of cerebellin deletions as described in A. All data are shown as means ± SEM. Synaptic puncta intensity was normalized to the area of MAP2-labeled tissue and to the levels observed in the controls. Statistical analysis was performed by one-way ANOVA (n = 3 mice). **p = 0.0080; ***p = 0.0001.
Retrosplenial cortex
The retrosplenial cortex is divided into granular (Rg) and dysgranular (Rdg) sections that are organized into a layered structure similar to other cortical regions (Wyss and van Groen, 1992). In the retrosplenial cortex, Cbln1, Cbln2, and Cbln4 are expressed in distinct cell populations, such that Cbln1 is expressed in layer 4 of only the Rg (Otsuka et al., 2016), Cbln2 is expressed in layers 2/3 and 5 of both the Rg and Rdg (Fig. 5A) and Cbln4 is expressed in layers 2/3 of only the Rdg (Fig. 5F).
Figure 5.
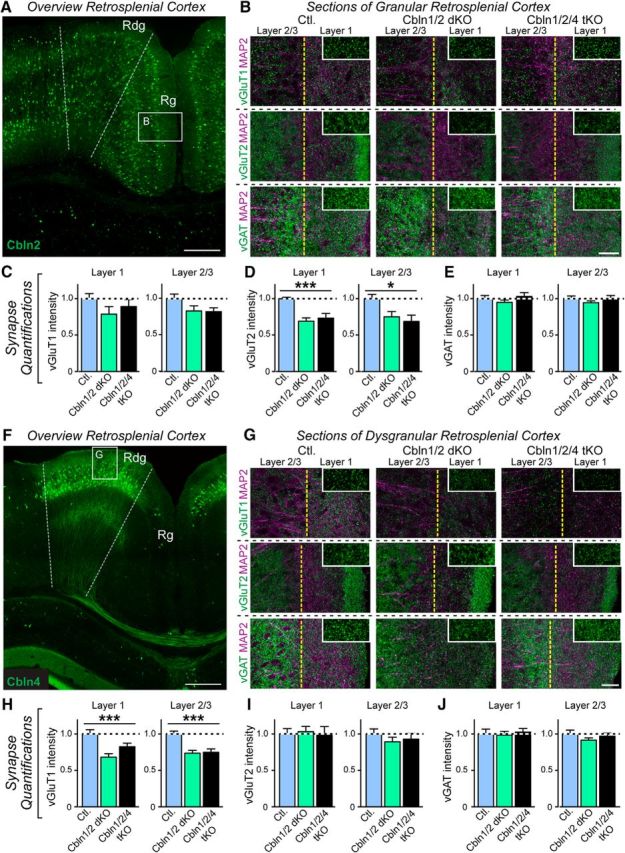
Cerebellin deletions selectively decrease vGluT2-positive synapses in the superficial layers of the granular retrosplenial cortex and vGluT1-positive synapses in the superficial layers of the dysgranular retrosplenial cortex in 6-month-old mice. A, Illustration of Cbln2 expressing neurons in the Rg and Rdg retrosplenial cortex and of the granular cortex area sampled in the synapse intensity quantifications. Image shows a coronal section from a representative Cbln2-mVenus reporter mouse; Cbln2 expression is visualized by mVenus fluorescence and does not reflect subcellular localization (Seigneur and Südhof, 2017). Scale bar, 100 μm. B, Coimmunolabeling of MAP2 (magenta) along with vGluT1 (top), vGluT2 (middle), or vGAT (bottom) in layers 1 (right half) and 2/3 (left half) of Rg (boxed area in A) in control (left), Cbln1/2 dKO (middle), and Cbln1/2/4 tKO (right) mice. Insets show a high-magnification example of the synaptic marker labeling in layer 1 without MAP2. Scale bar, 20 μm. C–E, Quantification of B. vGluT2 intensity was significantly reduced in dKO and tKO mice compared with control in layer 1 (p = 0.0002, denoted by ***) and layers 2/3 (p = 0.0159, denoted by *). vGluT1 and vGAT labeling was unchanged. F, Illustration of Cbln4 expressing neurons in the Rdg but not Rg retrosplenial cortex and of the dysgranular cortex area sampled in the synapse intensity quantifications. Section is from a representative Cbln4-mVenus reporter mouse; Cbln4 expression is visualized by immunofluorescence using antibodies to GFP and does not reflect subcellular localization (Seigneur and Südhof, 2017). Scale bar, 100 μm. G, Shows coimmolabeling of MAP2 (magenta) with vGluT1 (top), vGluT2 (middle), or vGAT (bottom) in layers 1 (right half) and 2/3 (left half) of Rdg (boxed area in F) in control (left), Cbln1/2 dKO (middle), and Cbln1/2/4 tKO (right) mice. Insets show a high-magnification example of the synaptic marker labeling in layer 1 without MAP2. Scale bar, 20 μm. H–J, Quantification of G. vGluT1 intensity was significantly reduced in dKO and tKO mice compared with control in layer 1 (p = 0.0007, denoted by ***) and layers 2/3 (p = 0.0001, denoted by ***). vGluT2 and vGAT labeling was unchanged. Images were obtained from littermate control, Cbln1/2 dKO, and Cbln1/2/4 tKO mice at 6 months of age (n = 3 mice). All numerical data are shown as means ± SEM and were normalized first to the area of MAP2-labeled tissue and then to the levels observed in the controls. Statistical analysis was done by one-way ANOVA.
In Rg (Fig. 5B,D), the vGluT2 signal intensity was significantly decreased in Cbln1/2 double and Cbln1/2/4 tKO mice in both layer 1 (F(2,24) = 12.92, p = 0.0002, ANOVA) and layer 2/3 (F(2,24) = 4.943, p = 0.016, ANOVA), but vGluT1 (Fig. 5B,C) and vGAT (Fig. 5B,E) were unchanged compared with controls. Conversely, in Rdg (Fig. 5F–J), the vGluT1 signal intensity was decreased in Cbln1/2 double and in Cbln1/2/4 tKO mice in layer 1 (F(2,24) = 9.944, p = 0.0007, ANOVA) and layer 2/3 (F(2,24) = 13.25, p = 0.0001, ANOVA), whereas vGluT2 and vGAT were unchanged compared with controls. In both regions, Cbln1/2 double and Cbln1/2/4 tKO mice did not differ from each other. In the retrosplenial cortex, vGluT1-positive synapses largely arise from corticocortical projections, whereas vGluT2-positive synapses are largely derived from thalamocortical projections (Wyss and van Groen, 1992; Nakamura et al., 2005; Oda et al., 2014). Therefore, our data suggest that loss of cerebellins results in a decrease in thalamocortical synapses to the Rg and in a decrease of corticocortical synapses to the Rdg.
Cerebellum
In the cerebellum, excitatory parallel and climbing fiber synapses can be specifically labeled with antibodies to vGluT1 and vGluT2, respectively. During early development, cerebellar granule cells express high levels of Cbln2, but not Cbln1, whereas later in development, in the course of synapse maturation Cbln2 expression decreases and Cbln1 becomes extremely abundant (Rong et al., 2012; Seigneur and Südhof, 2017). Deletion of Cbln1 causes a partial loss of vGluT1-positive parallel fiber synapses and an increase in vGluT2-positive climbing fiber synapses (Hirai et al., 2005; Ito-Ishida et al., 2008; Uemura et al., 2010). The question now arises whether the remaining parallel fiber synapses observed in Cbln1 KO mice, a not insignificant fraction of the total synapses, are maintained by expression of Cbln2 or if cerebellins are only essential for maintaining a subset of synapses in aging mice.
We found that the vGluT1 signal intensity was decreased by ∼50% in the cerebella of Cbln1/2 dKO and Cbln1/2/4 tKO mice compared with controls (F(2,38) = 41.16, p < 0.0001, ANOVA; Fig. 6A,B, top), whereas the vGluT2 signal intensity doubled (F(2,41) = 6.486, p = 0.0036, ANOVA; Fig. 6A,B, middle). The vGAT signal intensity was also increased by ∼50% in Cbln1/2 dKO and Cbln1/2/4 tKO mice (F(2,43) = 9.671, p = 0.0003, ANOVA; Fig. 6A,B, bottom), possibly due to a reduction in size of the molecular layer. For all three markers, there were no significant differences between Cbln1/2 dKO and Cbln1/2/4 tKO. Therefore, the remaining parallel fiber synapses in Cbln1 KO mice are not sensitive to the additional deletion of Cbln2, suggesting that Cbln2 does not redundantly support parallel fiber synapse numbers in the cerebellum.
Figure 6.

Double Cbln1/2 and triple Cbln1/2/4 deletions cause a similarly large decrease in vGluT1-staining intensity as a marker of parallel fiber synapses and a similarly large increase in vGluT2 and in vGAT staining intensity as a marker of excitatory climbing fiber and inhibitory interneuron synapses, respectively, in 6-month-old mice. A, Representative immunofluorescence images of sections of the cerebellum from littermate control (left), Cbln1/2 dKO (middle), and Cbln1/2/4 tKO mice (right; age = 6 months). Sections were double labeled for calbindin as a Purkinje cell marker and for vGluT1 (top), vGluT2 (middle), or vGAT (bottom). Scale bar, 20 μm. B, Cerebellin deletions cause opposite effects on the intensity of vGluT1-positive parallel fiber versus vGluT2-positive climbing fiber and vGAT-positive inhibitory synapses. Summary graphs display the staining intensity of vGluT1, vGluT2, or vGAT in cerebellar sections imaged as a function of cerebellin deletions as described in A, All Data are shown as means ± SEM. Synaptic puncta intensity was normalized to the area of calbindin-labeled tissue and to the levels observed in the controls. Statistical analysis was performed by one-way ANOVA (n = 3 mice). **p = 0.0036; ***p = 0.0003; ****p < 0.0001.
Viewed together, these data show that deletions of Cbln1 and Cbln2 produce a delayed loss of synapses in all brain regions that we tested that express these genes. The effect sizes of the deletions vary, but the overall result is consistent with a general function of Cbln1 and Cbln2 in many synaptic connections throughout the brain. Conversely, additional deletion of Cbln4 had no detectable effect in any brain region tested, leaving open the question of the function of this interesting isoform.
Loss of Cbln1 and/or Cbln2 produces diverse but discrete behavioral changes
Next, we performed behavioral analyses to assess the functional consequences of the constitutive Cbln1, Cbln2, Cbln1/2 double, or the Cbln1/2/4 triple deletions. As shown in the data described below, these experiments support the hypothesis emerging from the morphological studies that cerebellins perform distinct functions in different behaviorally relevant circuits.
Cbln1 deletions induce well characterized motor deficits, including a wide gait, ataxia, and loss of motor learning on the rotarod (Hirai et al., 2005; Ito-Ishida et al., 2008; Rong et al., 2012). Conversely, Cbln2-null mice appear to have no overt motor deficits (Rong et al., 2012). However, because Cbln2 is expressed in the cerebellum early in development and in other motor-related brain regions throughout life, such as primary and secondary motor cortex and the ventrolateral thalamus (Seigneur and Südhof, 2017), we hypothesized that loss of both Cbln1 and Cbln2 may result in additional motor deficits compared with Cbln1 KO mice.
Indeed, Cbln1/2 dKO mice presented with unique gait abnormalities in addition to the ataxia and gait abnormalities observed in Cbln1 KO mice (Fig. 7). These gait abnormalities (Wolfson et al., 1990; Wolfson, 2001; Sanders and Gillig, 2010) included waddling (exaggerated lateral movement of the trunk), staggering (sudden loss of balance resulting in lateral lurching), and festination (compensation for displacement of center of gravity resulting in fast, short steps). To quantify these differences, we marked the mice's feet with nontoxic paint to produce walking tracks that allow analysis of four gait parameters: stride length, paw base width, forepaw/hindpaw overlap, and stride frequency (Fig. 7A; Carter et al., 2001). Cbln1 KO and Cbln1/2 dKO mice had a significantly shorter forepaw stride (F(3,73) = 42.36, p < 0.0001, ANOVA) and significantly decreased hindpaw stride length (F(3,73) = 50.57, p < 0.0001, ANOVA) compared with control or Cbln2 KO mice; Cbln1 KO and Cbln1/2 dKO did not differ significantly from each other in these parameters (Fig. 7B, top). Cbln1 KO and Cbln1/2 dKO mice also had a wider forepaw (F(3,35) = 37.92, p < 0.0001, ANOVA) and hindpaw (F(3,35) = 54.69, p < 0.0001, ANOVA) base compared with control or Cbln2 KO mice, but, again, they did not differ from each other (Fig. 7B, bottom). A large paw base width indicates the presence of ataxia (Carter et al., 2001).
Figure 7.

The Cbln2 KO has no detectable effect on gait performance, whereas the Cbln1 KO and the Cbln1/2 dKO severely impair gait performance in 2-month-old littermate mice. A, Representative images from footprint analyses comparing the gait of control, Cbln1 KO, Cbln2 KO, and Cbln1/2 dKO mice. B, Cbln1 KO and Cbln1/2 dKO mice exhibit a decreased stride length and an increased paw base compared with control and Cbln2 KO mice. Summary graphs show the stride length (top) and paw base (bottom, the distance between right and left paws during gait) of littermate control (n = 10), Cbln1 KO (n = 7), Cbln2 KO (n = 9), and Cbln1/2 dKO mice (n = 13). C, Representative images from footprint analyses illustrating that Cbln1 KO and Cbln1/2 dKO mice exhibit less accurate and uniform foot placement compared with control and Cbln2 KO mice, as evidenced by an increase in the distance between the placement of the right forepaws and hindpaws and left forepaws and hindpaws (****p < 0.0001, one-way ANOVA). In addition, the distance between paws in Cbln1KO mice is significantly greater than in dKO mice (adjusted p = 0.0254, Tukey's post hoc test). D, dKO mice walk with a longer stride, requiring significantly more steps to cover the same distance than Cbln1KO mice (adjusted p = 0.0350, Tukey's post hoc test); both dKO and Cbln1 KO required more steps than control and Cbln2 KO mice (p < 0.0001, one-way ANOVA). All numerical data are shown as means ± SEM. Statistical significance was analyzed by one-way ANOVA followed by Tukey's post hoc test (D). +p = 0.0254 (left graph in D); +p = 0.0350 (right graph in D); ****p < 0.0001.
During locomotion, wild-type mice walk with a uniform step pattern such that the center of the hindpaw print falls on top of the center of the preceding forepaw print on each side (right/left). An increase in the distance between the center of the hindpaw and forepaw (overlap) is indicative of an arrhythmic stride and inaccurate foot placement (Carter et al., 2001). Both in Cbln1 KO and Cbln1/2 dKO mice, the overlap distance was significantly greater than that observed in control or Cbln2 KO mice (F(3,35) = 72.84, p < 0.0001, ANOVA). Interestingly, the overlap distance was significantly smaller in Cbln1/2 dKO mice than in Cbln1 KO mice (adjusted p = 0.0254, Tukey's post hoc test), perhaps because of the dKO mice's festinating gait (Fig. 7C,D).
Moreover, both Cbln1 KO and Cbln1/2 dKO mice had an increased step frequency (required more steps to cover the same distance) compared with control and Cbln2 KO mice (F(3,35) = 29.68, p < 0.0001, ANOVA), consistent with their shorter strides. The step frequency of Cbln1/2 dKO mice was significantly greater than that of Cbln1 KO mice (adjusted p = 0.035, Tukey's post hoc test), providing a quantitative measure of the short, fast footsteps (festination) characteristic of the Cbln1/2 dKO stride.
We next quantified the differences in balance and coordination by using a force plate actometer to measure postural sway, which is the amount of spontaneous motion while standing still (Fowler et al., 2001; Wolfson, 2001). Cbln1/2 dKO mice had significantly more postural sway than Cbln1 KO mice (adjusted p = 0.042, Tukey's post hoc test) and both groups displayed increased postural sway compared with control and Cbln2 KO mice (F(3,34) = 16.05, p < 0.0001, ANOVA; Fig. 3A, left). There was no difference in postural sway between Cbln1/2 dKO and tKO mice (Fig. 8A, right).
Figure 8.

Cbln1 KO causes a major impairment in motor behaviors that is aggravated by additional deletion of Cbln2 even though the Cbln2 KO alone produces no phenotype. A, Cbln1 KO, Cbln1/2 dKO, and Cbln1/2/4 tKO impair postural sway, measured as the area around the center of force of a mouse monitored on a force plate actometer at three successive locations. Experiments were performed on two separate mouse cohorts as follows: left graph, Cbln1 KO (n = 8), Cbln2 KO (n = 9), Cbln1/2 dKO (n = 13), and control mice (Ctl; n = 10); right graph, Cbln1/2 dKO (n = 14), Cbln1/2/4 tKO (n = 11), and Ctl mice (Ctl.; n = 16). Postural sway impairments in Cbln1 KO mice were enhanced in Cbln1/2 dKO mice (adjusted p = 0.0418, Tukey's post hoc test), whereas Cbln2 KO mice exhibited no phenotype and Cbln1/2/4 tKO mice did not significantly differ from Cbln1/2 dKO mice. +p < 0.05, ****p < 0.0001, one-way ANOVA. B, Cbln1/2 dKO causes a loss of balance in mice, resulting in lateral falls, that is more severe than that observed in Cbln1 KO or Cbln1/2/4 tKO mice. A lateral fall was counted when all 4 paws were off the ground and the mouse was laying on its left or right side; the number of lateral falls during a 5 min open-field test was measured at different time points. Left, Lateral falls were observed in the Cbln1/2 dKO (n = 11) mice beginning at ∼2 months of age and the frequency of falling increased as the mouse aged. Some Cbln1 KO mice (n = 7) exhibited this loss of balance starting at 6 months of age, but the phenotype emerged earlier and was more severe in Cbln1/2 dKO mice (time *p = 0.0458; genotype ****p < 0.0001; interaction *p = 0.0385, two-way ANOVA). No lateral falls were ever observed in Ctl or Cbln2 KO mice. Right, Surprisingly, Cbln1/2/4 tKO mice (n = 11) exhibited significantly fewer lateral falls than Cbln1/2 dKO mice across all time points (n = 13; time ****p < 0.0001, genotype ****p < 0.0001, interaction *p = 0.0161, two-way ANOVA). C, Distance traveled across an 80-cm-long balance beam (width = 2 or 1 cm) by 3-month-old mice of the indicated genotypes. Left, On the 2 cm beam, the performance of Cbln1KO (n = 8) and dKO (n = 13) mice did not differ significantly from each other, but on the 1 cm beam, dKO mice traveled less distance (before falling off the beam) than Cbln1KO mice (adjusted p = 0.0005, Tukey's post hoc test, denoted by ***); both groups traveled significantly less distance compared with Ctl (n = 10) and Cbln2KO (n = 9) mice on the 2 cm (p = 0.0003, one-way ANOVA, denoted by +++) and 1 cm (p < 0.0001, one-way ANOVA, denoted by ****) beams. Right, Cbln1/2 dKO (n = 13) and Cbln1/2/4 tKO mice (n = 11) exhibited decreased performance on the balance beam as a function of age, with a dramatic decrease in the traversed distance compared with Ctl (n = 16) seen at 3 and 4 months. ****p < 0.0001, two-way ANOVA. D, Cbln1 KO, Cbln1/2 dKO, and Cbln1/2/4 tKO equally impair motor coordination and motor learning as monitored on the accelerating rotarod, whereas the Cbln2 KO has no effect. Left, Comparison of Cbln1 KO (n = 7), Cbln2 KO (n = 9), Cbln1/2 dKO (n = 13) and Ctl (n = 10) mice. Right, Cbln1/2 dKO mice (n = 14) and Cbln1/2/4 tKO mice (n = 11) exhibit similar impairments in motor coordination and motor learning compared with Ctl mice (n = 16). ****p < 0.0001, two-way ANOVA.
One of the most striking features of the Cbln1/2 dKO mice was their frequent loss of balance, resulting in the mice falling over on the left or right side of their body. To quantify this, we counted the number of lateral falls during the first 5 min of open-field exploration and tested the mice at 2, 4, and 6 months of age (Fig. 8B, left). The phenotype of increased lateral falls became evident in Cbln1/2 dKO mice by 2 months of age and progressively worsened as the mice grew older (within-group time factor F(2,88) = 3.195, p = 0.046, two-way ANOVA). No lateral falls were observed in Cbln1 KO mice at 2 or 4 months of age and only a small number of falls were observed in 6-month-old mice. No falls were ever observed in control or Cbln2 KO mice (gene factor F(3,88) = 18.98, p < 0.0001; interaction F(6,88) = 2.337, p = 0.039, two-way ANOVA).
In a second set of experiments, we quantified the number of lateral falls in Cbln1/2 dKO and Cbln1/2/4 tKO mice over a 12 month period (Fig. 8B, right). Overall, the average number of falls in Cbln1/2 dKO mice during a 5 min period increased ∼22-fold from 1 to 12 months of age. Unexpectedly, this phenotype was less severe in the Cbln1/2/4 tKO mice compared with Cbln1/2 dKO mice, with significantly fewer lateral falls observed in the Cbln1/2/4 tKO mice at all time points (time factor F(8,198) = 5.854, p < 0.0001; gene factor F(1,198) = 33.48, p < 0.0001; interaction F(8,198) = 2.425 p = 0.0161, two-way ANOVA), although motor deficits also became progressively worse in the tKO mice with age. A recent study demonstrated a possible role for Cbln4 in axon guidance (Haddick et al., 2014). If Cbln4 normally helps to establish the circuit or circuits that are dysfunctional in Cbln1/2 dKO mice, then the loss of this circuit due to a lack of Cbln4 could explain why this particular behavior is less severe in the Cbln1/2/4 tKO mice. Future research will need to explore this further.
Next, we measured the total distance traveled across an 80-cm-long balance beam with a 2 or 1 cm diameter (Fig. 8C, left). On the 2 cm beam, the performance of Cbln1 KO and Cbln1/2 dKO mice did not differ significantly from each other, but on the 1 cm beam, Cbln1/2 dKO mice traveled significantly less distance (before falling off the beam) than Cbln1 KO mice (adjusted p = 0.0005, Tukey's post hoc test). Both groups traveled a significantly shorter distance than control and Cbln2 KO mice on both the 2 cm beam (F(3,35) = 8.145, p = 0.0003, ANOVA) and the 1 cm beam (F(3,36) = 59.49, p < 0.0001, ANOVA).
To test whether balance impairments became more severe with age, we measured the total distance traveled across an 80-cm-long, 2-cm-diameter beam every month in mice at 1 to 4 months of age (Fig. 8C, right). We observed a significant decrease in performance in the Cbln1/2 dKO and Cbln1/2/4 tKO mice compared with control (time F(3,147) = 61.89, p < 0.0001; genotype F(2,147) = 42.84, p < 0.0001; interaction F(6,147) = 17.14, p < 0.0001, two-way ANOVA). At 1 and 2 months of age, Cbln1/2 dKO and Cbln1/2/4 tKO mice traveled the same distance as control mice; by 3 months of age, the performance of the KO mice had significantly decreased (control vs Cbln1/2 dKO adjusted p = 0.0015; control vs tKO adjusted p = 0.0078, Tukey's post hoc test) and a steep decline in performance occurred between 3 and 4 months of age (control vs Cbln1/2 dKO adjusted p < 0.0001; control vs tKO adjusted p < 0.0001, Tukey's post hoc test). There was no difference between Cbln1/2 dKO and Cbln1/2/4 tKO mice at any time point.
To further quantify motor performance, we tested motor learning on the accelerating rotarod. Neither Cbln1 KO nor Cbln1/2 dKO mice were able to learn this task, and both groups performed significantly worse than control and Cbln2 KO mice (time F(4,175) = 40.84, p < 0.0001; genotype F(3,175) = 150.6, p < 0.0001; interaction F(12,175) = 3.958, p < 0.0001, two-way ANOVA; Fig. 8D, left). No impairments were observed in Cbln2 KO mice, consistent with previous reports (Rong et al., 2012). There was no difference in performance among Cbln1 KO, Cbln1/2 dKO, and Cbln1/2/4 tKO mice (Fig. 8D). Together, these results show that loss of Cbln1 and Cbln2 causes increased impairments in balance and coordination, but not motor learning, compared with loss of Cbln1 alone, although the severe balance impairments observed in these mice make it difficult to assess the degree to which motor learning is affected.
Next, to determine whether the KO mice had impairments in sensory processing and sensorimotor gating, we tested their acoustic startle response and PPI. There was no significant effect in acoustic startle response in Cbln1 KO, Cbln2 KO, or Cbln1/2 dKO mice compared with control (stimulus intensity F(4,175) = 81.45, p < 0.0001; genotype F(3,175) = 2.215, p = 0.088; interaction F(12,175) = 0.5699, p = 0.86, two-way ANOVA; Fig. 9A). However, we did observe a significant reduction in the startle response in Cbln1/2/4 tKO mice compared with control (stimulus intensity F(4,190) = 25.37, p < 0.0001; genotype F(2,190) = 16.49, p < 0.0001; interaction F(8,190) = 4.015, p = 0.0002, two-way ANOVA) after a 95 dB (adjusted p = 0.0503, Tukey's post hoc test), 105 dB (adjusted p < 0.0001, Tukey's post hoc test), and 115 dB (adjusted p < 0.0001, Tukey's post hoc test; Fig. 9C) acoustic stimulus. After a 68 dB stimulus, PPI was slightly reduced in Cbln1 KO mice compared with controls; however, this reduction only reached statistical significance in a Student's t test (t(16) = 2.898, p = 0.0105) and was not significant when correcting for multiple comparisons (adjusted p = 0.2522, Tukey's post hoc test). In response to all other stimulus intensities, there was no difference among the control, Cbln1 KO, Cbln2 KO, and Cbln1/2 dKO groups (stimulus intensity F(2,108) = 29.16, p < 0.0001; genotype F(3,108) = 1.739, p = 0.1634; interaction F(6,108) = 0.5284, p = 0.7857, two-way ANOVA; Fig. 4B). PPI was significantly reduced in Cbln1/2/4 tKO mice overall (stimulus intensity F(2,117) = 7.530, p = 0.0008; genotype F(2,117) = 7.183, p = 0.0011; interaction F(4,117) = 1.590, p = 0.1817, two-way ANOVA) and compared with both control (adjusted p = 0.0011, Tukey's post hoc test) and Cbln1/2 dKO (adjusted p = 0.0045, Tukey's post hoc test) mice after a 77 dB prepulse stimulus (Fig. 9D). Cbln4-null mice showed no change in acoustic startle response or PPI compared with control (data not shown), suggesting that these deficits are unique to tKO mice lacking all functional cerebellins.
Figure 9.

Cbln1/2/4 tKO causes a major impairment in acoustic startle responses and PPI, whereas Cbln1 or Cbln2 single KOs produce no phenotype and the Cbln1/2 dKO induces only a modest phenotype. A, Cbln1 KO (n = 8), Cbln2 KO (n = 9), or Cbln1/2 dKO mice (n = 13) exhibit no change in acoustic startle response compared with littermate control mice (Ctl) (n = 10). Summary graphs show startle amplitude as a function of the strength of the acoustic stimulus in decibels (dB). B, Cbln1 KO, Cbln2 KO, or Cbln1/2 dKO mice display no change in PPI compared with littermate Ctl mice. Summary graphs show the percentage by which the startle response to a 115 dB stimulus was inhibited by a prepulse stimulus delivered at the indicated strength (in dB) 100 ms before the startle stimulus (numbers of animals are the same as in A). C, Cbln1/2/4 tKO greatly impairs the acoustic startle response, whereas the Cbln1/2 dKO has only a modest effect. Experiments were as described in A, except that Cbln1/2 dKO (n = 11) and Cbln1/2/4 tKO (n = 14) mice were compared with Ctl mice (n = 16). D, Cbln1/2/4 tKO also impairs PPI of the acoustic startle response, presumably because the startle response itself was suppressed. Same as B, but comparing Cbln1/2 dKO mice (n = 11) and Cbln1/2/4 tKO mice (n = 14) with Ctl mice (n = 16). Data are shown as means ± SEM. Statistical significance was assessed by two-way ANOVA. ****p < 0.0001, ***p = 0.0002 in C and 0.0008 in D; **p = 0.0011. Plus signs denote statistical significance between Ctl and tKO in a post hoc Tukey's multiple-comparisons test (in C: +adjusted p = 0.0503 and ++++adjusted p < 0.0001; in D: ++adjusted p = 0.0011).
Viewed together, these data show that most behavioral impairments are dominated by the well characterized cerebellar deficits of Cbln1 KO mice, but that for selected impairments such as gait abnormalities, the Cbln2 KO enhances the Cbln1 KO phenotype significantly even though the Cbln2 KO on its own does not have a significant phenotype.
Discussion
Cbln1 has been studied extensively because of its central role in cerebellar parallel fiber synapses (Hirai et al., 2005; Ito-Ishida et al., 2008; Uemura et al., 2010). However, the overall role of Cbln1 in brain is incompletely understood, and the function of the other cerebellin isoforms is largely unknown. Here, we set out to analyze the overall function of cerebellins in mice by examining the effect of deleting all cerebellins, thus enabling a systematic initial assessment of cerebellin function. Cerebellins are expressed in mammals from four genes, of which Cbln3 cannot be transported to the surface without coexpression of another cerebellin (Bao et al., 2006); therefore, triple deletions of Cbln1, Cbln2, and Cbln4 effectively ablate expression of all cerebellins. We examined constitutive Cbln1 and Cbln2 single KO mice as well as in Cbln1/2 dKO and Cbln1/2/4 tKO mice using genetic, morphological, biochemical, and behavioral studies. Our results allow four major conclusions.
First, cerebellins are clearly functionally important, but are not essential for survival. Cerebellin KO mice exhibited major behavioral phenotypes that included massive motor dysfunction and severe epilepsy (Figs. 1, 7, 8, 9). The Cbln1 single KO already caused large motor impairments, as described previously in pioneering studies by Mishina, Yuzaki, and others (Hirai et al., 2005; Ito-Ishida et al., 2008; Rong et al., 2012) and replicated here. Some of these phenotypes were aggravated by additional deletion of Cbln2 (Fig. 1). Despite these severe impairments, constitutive Cbln1/2 dKO and Cbln1/2/4 tKO mice exhibited an overall normal lifespan (Fig. 1). Therefore, whereas constitutive deletions of neurexins, the presynaptic binding partners of cerebellins, and of neuroligins, another trans-synaptic neurexin ligand, cause lethality (Missler et al., 2003; Varoqueaux et al., 2006), constitutive deletion of cerebellins does not limit survival.
Second, consistent with their expression throughout the brain (Miura et al., 2006; Wei et al., 2007; Otsuka et al., 2016; Seigneur and Südhof, 2017), cerebellins perform functions in many brain regions. This conclusion is based on the observation that constitutive Cbln1, Cbln2, and Cbln1/2 dKO mice exhibit diverse changes in the levels of synaptic proteins in many brain regions (Fig. 2) and display a decrease in synapse intensity in the hippocampus (Fig. 3), dorsal striatum (Fig. 4), and retrosplenial cortex (Fig. 5), in addition to the cerebellum (Fig. 6). Notably, the loss of synapses in various brain regions is selective for subsets of neurons expressing cerebellins and is not uniformly associated with a decrease in synaptic protein levels, but can also cause an increase in such levels (e.g., NMDA receptor subunit NR1 in the striatum or vGluT1 in the thalamus; see Fig. 2).
Third, the major phenotypes we observed in cerebellin KO mice exhibited a delayed onset. For example, synapse density in the hippocampus is normal in 1- and 2-month-old Cbln1/2 dKO mice, but severely decreased in 6-month-old Cbln1/2 dKO mice (Fig. 3). Similarly, the weight of these mice is normal in the first 2 months of life and Cbln1/2 dKO mice exhibit modest motor impairments until 3 months of age (Fig. 8C,D). The only phenotype we analyzed that develops early on is stress-induced seizures in Cbln1/2/4 tKO mice that appear to be already maximally expressed by 2 months of age (Fig. 1D). These observations suggest that the decrease in synapse numbers and at least some of the behavioral changes may be secondary to a primary synaptic impairment that manifests in the increased seizure incidence. Future experiments will have to address this important question.
Fourth, all three cerebellins, not just Cbln1, contribute to brain function. Although the Cbln2 KO did not produce major phenotypes on its own, only Cbln1/2 dKO mice, but not Cbln1 or Cbln2 single KO mice suffered from seizures, suggesting redundancy (Fig. 1). Moreover, the Cbln1/2 dKO mice exacerbated the Cbln1 single KO phenotype in assessments of postural sway, lateral falls, balance beam performance, and startle responses, further indicating a major role for Cbln2 in brain (Fig. 8). Specifically, Cbln1/2 dKO mice were unable to maintain their center of gravity, causing the mice to perpetually sway from side to side, and to waddle, stagger, and walk with short fast steps (festinate). The Cbln1/2 dKO mice would frequently lose their balance completely and fall over on their side. This falling over behavior was only rarely observed in Cbln1 KO mice and, even then, only in 6-month-old mice (Fig. 8C). In addition, Cbln1/2 dKO mice performed less well than Cbln1 KO mice on the balance beam (Fig. 8D), although both groups performed equally poorly on the rotarod, with neither group showing significant motor learning ability (Fig. 8B). Combined, these results show that loss of Cbln1 and Cbln2 has an additive effect on balance and coordination. Cbln4 additionally contributed to some brain functions. Specifically, the Cbln1/2/4 tKO mice exhibited a more severe phenotype than the Cbln1/2 dKO mice in three assays, the incidence of seizures (Fig. 1E), the startle response (Fig. 9C), and PPI (Fig. 9D), documenting a function for Cbln4 as well. Overall, these results show that although Cbln1 single KO mice exhibit major motor phenotypes because of the dominant function of Cbln1 in the cerebellum, Cbln2 and Cbln4 perform possibly redundant functions with Cbln1 in brain in most parameters tested.
Our experiments did not aim for a mechanistic dissection of cerebellins, which would have exceeded the scope of our studies but could now be approached with the mutant mouse lines that we have developed. Therefore, our conclusions are limited in that we have no information about possible synaptic defects produced by cerebellin KOs in synapses outside of the cerebellum. For example, in the cerebellum the major synaptic phenotypes of Cbln1 KO or GluRδ2 KO mice (which phenocopies Cbln1 KO) that is not attributable to the progressive loss of parallel fiber synapses is a complete absence of LTD (Kashiwabuchi et al., 1995; Hirai et al., 2005) and an increase in AMPA-type glutamate receptors (Yamasaki et al., 2011). It would be extremely interesting to learn whether cerebellin KOs produce similar changes in neurotransmitter receptors and LTP in other brain regions. Similarly, it is well established that in cerebellar parallel fiber synapses, Cbln1 acts by forming tripartite complexes with presynaptic neurexins and postsynaptic GluRδ2 (Uemura et al., 2010; Lee et al., 2012), but little is known about the mechanism of action of Cbln2 and Cbln4 and about cerebellin receptors outside of the brain. It seems likely that all cerebellins interact with presynaptic neurexins as receptors, but the postsynaptic binding partners could be GluRδ isoforms or DCC and neogenin or as yet unidentified proteins (Lee et al., 2012; Wei et al., 2012; Haddick et al., 2014). In addition, the synaptic deficits that we observed in the striatum and hippocampus appear to be due to loss of presynaptic Cblns because the expression of Cblns is very low in these regions. However, it is unclear whether the deficits that we observed in the retrosplenial cortex are due to loss of presynaptic or postsynaptic Cblns or both. Clearly, many important questions remain open about how cerebellins function in the brain.
Viewed together, our data suggest that cerebellins act as important synaptic organizers in specific subsets of neurons in the brain and likely contribute to many different brain functions. The fact that deletions of cerebellins cause a delayed loss of synapses that becomes evident after most synapses have matured in the first 2 months of life of a mouse (Fig. 3) shows that cerebellins are not initially required for synapse formation, but rather for long-term synapse maintenance, possibly because the loss of cerebellins causes a functional synaptic impairment that results in a long-term destabilization of synapses. The selective expression of cerebellins in subsets of neurons and synapses indicates that they may confer particular properties on these synapses that are important for overall brain function. Again, future experiments will have to address these important mechanistic questions.
Footnotes
This work was supported by the National Institutes of Health–National Institute of Mental Health (Grant R37-MH052804). We thank Dr. James Morgan (St. Jude Research Hospital) for provision of the Cerebellin-1 KO mice.
The authors declare no competing financial interests.
References
- Bao D, Pang Z, Morgan MA, Parris J, Rong Y, Li L, Morgan JI (2006) Cbln1 is essential for interaction-dependent secretion of Cbln3. Mol Cell Biol 26:9327–9337. 10.1128/MCB.01161-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter RJ, Morton J, Dunnett SB (2001) Motor coordination and balance in rodents. Cur Protoc Neurosci Chapter 8;Unit 8.12. 10.1002/0471142301.ns0812s15 [DOI] [PubMed] [Google Scholar]
- Cheng S, Seven AB, Wang J, Skiniotis G, Özkan E (2016) Conformational plasticity in the transsynaptic neurexin-cerebellin-glutamate receptor adhesion complex. Structure 24:2163–2173. 10.1016/j.str.2016.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrobak JJ, Lörincz A, Buzsáki G (2000) Physiological patterns in the hippocampo-entorhinal cortex system. Hippocampus 10:457–465. 10.1002/1098-1063(2000)10:4%3C457::AID-HIPO12%3E3.0.CO;-Z [DOI] [PubMed] [Google Scholar]
- Doig NM, Moss J, Bolam JP (2010) Cortical and thalamic innervation of direct and indirect pathway medium-sized spiny neurons in mouse striatum. J Neurosci 30:14610–14618. 10.1523/JNEUROSCI.1623-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler SC, Birkestrand BR, Chen R, Moss SJ, Vorontsova E, Wang G, Zarcone TJ (2001) A force-plate actometer for quantitating rodent behaviors: illustrative data on locomotion, rotation, spatial patterning, stereotypies, and tremor. J Neurosci Methods 107:107–124. 10.1016/S0165-0270(01)00359-4 [DOI] [PubMed] [Google Scholar]
- Haddick PC, Tom I, Luis E, Quiñones G, Wranik BJ, Ramani SR, Stephan JP, Tessier-Lavigne M, Gonzalez LC (2014) Defining the ligand specificity of the deleted in colorectal cancer (DCC) receptor. PLoS One 9:e84823. 10.1371/journal.pone.0084823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H, Pang Z, Bao D, Miyazaki T, Li L, Miura E, Parris J, Rong Y, Watanabe M, Yuzaki M, Morgan JI (2005) Cbln1 is essential for synaptic integrity and plasticity in the cerebellum. Nat Neurosci 8:1534–1541. 10.1038/nn1576 [DOI] [PubMed] [Google Scholar]
- Ito-Ishida A, Miura E, Emi K, Matsuda K, Iijima T, Kondo T, Kohda K, Watanabe M, Yuzaki M (2008) Cbln1 regulates rapid formation and maintenance of excitatory synapses in mature cerebellar purkinje cells in vitro and in vivo. J Neurosci 28:5920–5930. 10.1523/JNEUROSCI.1030-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwabuchi N, Ikeda K, Araki K, Hirano T, Shibuki K, Takayama C, Inoue Y, Kutsuwada T, Yagi T, Kang Y (1995) Impairment of motor coordination, purkinje cell synapse formation, and cerebellar long-term depression in GluR delta 2 mutant mice. Cell 81:245–252. 10.1016/0092-8674(95)90334-8 [DOI] [PubMed] [Google Scholar]
- Kurihara H, Hashimoto K, Kano M, Takayama C, Sakimura K, Mishina M, Inoue Y, Watanabe M (1997) Impaired parallel fiber → purkinje cell synapse stabilization during cerebellar development of mutant mice lacking the glutamate receptor delta 2 subunit. J Neurosci 17:9613–9623. 10.1523/JNEUROSCI.17-24-09613.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusnoor SV, Parris J, Muly EC, Morgan JI, Deuth AY (2010) Extracerebellar role for Cerebellin 1: modulation of dendritic spine density and synapses in striatial medium spiny neurons. J Comp Neurol 518:2525–2537. 10.1002/cne.22350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Uemura T, Yoshida T, Mishina M (2012) GluRδ2 assembles four neurexins into trans-synaptic triad to trigger synapse formation. J Neurosci 32:4688–4701. 10.1523/JNEUROSCI.5584-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda K, Yuzaki M (2011) Cbln family proteins promote synapse formation by regulating distinct neurexin signaling pathways in various brain regions. Eur J Neurosci 33:1447–1461. 10.1111/j.1460-9568.2011.07638.x [DOI] [PubMed] [Google Scholar]
- Missler M, Zhang W, Rohlmann A, Kattenstroth G, Hammer RE, Gottmann K, Südhof TC (2003) α-neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature 423:939–948. 10.1038/nature01755 [DOI] [PubMed] [Google Scholar]
- Miura E, Iijima T, Yuzaki M, Watanabe M (2006) Distinct expression of cbln family mRNAs in developing and adult mouse brains. Eur J Neurosci 24:750–760. 10.1111/j.1460-9568.2006.04950.x [DOI] [PubMed] [Google Scholar]
- Montgomery KE, Kessler M, Arai AC (2009) Modulation of agonist binding to AMPA receptors by 1-(1,4-benzodioxan-6-ylcarbonyl)peperidine (CX546): differential effects across brain regions and GluA1-4/transmembrane AMPA receptor regulatory protein combinations. J Pharmacol Exp Ther 331:965–974. 10.1124/jpet.109.158014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Hioki H, Fujiyama F, Kaneko T (2005) Postnatal changes of vesicular glutamate transporter (VGluT)1 and VGluT2 immunoreactivities and their colocalization in the mouse forebrain. J Comp Neurol 492:263–288. 10.1002/cne.20705 [DOI] [PubMed] [Google Scholar]
- Oda S, Funato H, Sato F, Adachi-Akahane S, Ito M, Takase K, Kuroda M (2014) A subset of thalamocortical projections to the retrosplenial cortex possesses two vesicular glutamate transporter isoforms, vGluT1 and vGluT2, in axon terminals and somata. J Comp Neurol 522:2089–2106. 10.1002/cne.23519 [DOI] [PubMed] [Google Scholar]
- Otsuka S, Konno K, Abe M, Motohashi J, Kohda K, Sakimura K, Watanabe M, Yuzaki M (2016) Roles of Cbln1 in non-motor functions of mice. J Neurosci 36:11801–11816. 10.1523/JNEUROSCI.0322-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang Z, Zuo J, Morgan JI (2000) Cbln3, a novel member of the precerebellin family that binds specifically to Cbln1. J Neurosci 20:6333–6339. 10.1523/JNEUROSCI.20-17-06333.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong Y, Wei P, Parris J, Guo H, Pattarini R, Correia K, Li L, Kusnoor SV, Deutch AY, Morgan JI (2012) Comparison of Cbln1 and Cbln2 functions using transgenic and knockout mice. J Neurochem 120:528–540. 10.1111/j.1471-4159.2011.07604.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders RD, Gillig PM (2010) Gait and its assessment in psychiatry. Psychiatry (Edgmont) 7:38–43. [PMC free article] [PubMed] [Google Scholar]
- Schreiner D, Nguyen TM, Russo G, Heber S, Patrignani A, Ahrné E, Scheiffele P (2014) Targeted combinatorial alternative splicing generates brain region-specific repertoires of neurexins. Neuron 84:386–398. 10.1016/j.neuron.2014.09.011 [DOI] [PubMed] [Google Scholar]
- Seigneur E, Südhof TC (2017) Cerebellins are differentially expressed in selective subsets of neurons throughout the brain. J Comp Neurol 525:3286–3311. 10.1002/cne.24278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. (2017) Synaptic neurexin complexes: a molecular code for the logic of neural circuits. Cell 171:745–769. 10.1016/j.cell.2017.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treutlein B, Gokce O, Quake SR, Südhof TC (2014) Cartography of neurexin alternative splicing mapped by single-molecule long-read mRNA sequencing. Proc Natl Acad Sci U S A 111:E1291–E1299. 10.1073/pnas.1403244111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura T, Lee SJ, Yasumura M, Takeuchi T, Yoshida T, Ra M, Taguchi R, Sakimura K, Mishina M (2010) Trans-synaptic interaction of GluRδ2 and neurexin through Cbln1 mediates synapse formation in the cerebellum. Cell 141:1068–1079. 10.1016/j.cell.2010.04.035 [DOI] [PubMed] [Google Scholar]
- Ullrich B, Ushkaryov YA, Südhof TC (1995) Cartography of neurexins: more than 1000 isoforms generated by alternative splicing and expressed in distinct subsets of neurons. Neuron 14:497–507. 10.1016/0896-6273(95)90306-2 [DOI] [PubMed] [Google Scholar]
- Ushkaryov YA, Südhof TC (1993) Neurexin III α: extensive alternative splicing generates membrane-bound and soluble forms. Proc Natl Acad Sci U S A 90:6410–6414. 10.1073/pnas.90.14.6410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushkaryov YA, Petrenko AG, Geppert M, Südhof TC (1992) Neurexins: synaptic cell surface proteins related to the α-latrotoxin receptor and laminin. Science 257:50–56. 10.1126/science.1621094 [DOI] [PubMed] [Google Scholar]
- Ushkaryov YA, Hata Y, Ichtchenko K, Moomaw C, Afendis S, Slaughter CA, Südhof TC (1994) Conserved domain structure of b-neurexins: unusual cleaved signal sequences in receptor-like neuronal cell-surface proteins. J Biol Chem 269:11987–11992. [PubMed] [Google Scholar]
- Varoqueaux F, Aramuni G, Rawson RL, Mohrmann R, Missler M, Gottmann K, Zhang W, Südhof TC, Brose N (2006) Neuroligins determine synapse maturation and function. Neuron 51:741–754. 10.1016/j.neuron.2006.09.003 [DOI] [PubMed] [Google Scholar]
- Wei P, Smeyne RJ, Bao D, Parris J, Morgan JII (2007) Mapping Cbln1-like immunoreactivity in adult and developing mouse brain and its localization to the endolysosomal compartment of neurons. Eur J Neurosci 26:2962–2978. 10.1111/j.1460-9568.2007.05913.x [DOI] [PubMed] [Google Scholar]
- Wei P, Pattarini R, Rong Y, Guo H, Bansal PK, Kusnoor SV, Deutch AY, Parris J, Morgan JI (2012) The cbln family of proteins interacts with multiple signaling pathways. J Neurochem 121:717–729. 10.1111/j.1471-4159.2012.07648.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfson L. (2001) Gait and balance dysfuntion: a model of the interaction of age and disease. Neuroscientist 7:178–183. 10.1177/107385840100700212 [DOI] [PubMed] [Google Scholar]
- Wolfson L, Whipple R, Amerman P, Tobin JN (1990) Gait assessment in the elderly: a gait abnormality rating scale and its relation to falls. J Gerontol 45:M12–M19. 10.1093/geronj/45.1.M12 [DOI] [PubMed] [Google Scholar]
- Wyss JM, Van Groen T (1992) Connections between the retrosplenial cortex and the hippocampal formation in the rat: a review. Hippocampus 2:1–11. 10.1002/hipo.450020102 [DOI] [PubMed] [Google Scholar]
- Yamasaki M, Miyazaki T, Azechi H, Abe M, Natsume R, Hagiwara T, Aiba A, Mishina M, Sakimura K, Watanabe M (2011) Glutamate receptor δ2 is essential for input pathway-dependent regulation of synaptic AMPAR contents in cerebellar purkinje cells. J Neurosci 31:3362–3374. 10.1523/JNEUROSCI.5601-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong C, Shen J, Zhang H, Li G, Shen S, Wang F, Hu K, Cao L, He Y, Ding J (2017) Cbln1 and Cbln4 are structurally similar but differ in GluD2 binding interactions. Cell Rep 20:2328–2340. 10.1016/j.celrep.2017.08.031 [DOI] [PubMed] [Google Scholar]