

Abstract
Interleukin-4 (IL-4) mediates important pro-inflammatory functions in asthma including induction of the IgE isotype switch, expression of vascular cell adhesion molecule-1 (VCAM-1), promotion of eosinophil transmigration across endothelium, mucus secretion, and differentiation of T helper type 2 lymphocytes leading to cytokine release. Asthma is a complex genetic disorder that has been linked to polymorphisms in the IL-4 gene promoter and proteins involved in IL-4 signaling. Soluble recombinant IL-4 receptor lacks transmembrane and cytoplasmic activating domains and can therefore sequester IL-4 without mediating cellular activation. We report the results of initial clinical trials, which demonstrate clinical efficacy of this naturally occurring IL-4 antagonist as a therapeutic agent in asthma.
Keywords: asthma, genetics, soluble recombinant human interleukin-4 receptor, T helper lymphocytes
Introduction
Interleukin (IL)-4 is a key cytokine in the development of allergic inflammation. It is associated with induction of the ε isotype switch and secretion of IgE by B lymphocytes [1]. IgE-mediated immune responses are further enhanced by IL-4 through its ability to upregulate IgE receptors on the cell surface: the low-affinity IgE receptor (FcεRII; CD23) on B lymphocytes and mononuclear phagocytic cells and the high-affinity IgE receptor (FcεRI) on mast cells and basophils [2]. IgE-dependent mast cell activation induced by IL-4 has a pivotal role in the development of immediate allergic reactions. An additional mechanism by which IL-4 contributes to airway obstruction in asthma is through the induction of mucin gene expression and the hypersecretion of mucus [3]. IL-4 increases the expression of eotaxin and other inflammatory cytokines from fibroblasts that might contribute to inflammation and lung remodelling in chronic asthma [4].
An important activity of IL-4 in promoting cellular inflammation in the asthmatic lung is the induction of vascular cell adhesion molecule (VCAM)-1 on vascular endothelium [5]. Through the interaction of VCAM-1, IL-4 is able to direct the migration of T lymphocytes, monocytes, basophils, and eosinophils to inflammatory loci. In addition, IL-4 inhibits eosinophil apoptosis and promotes eosinophilic inflammation by inducing eosinophil chemotaxis and activation through the increased expression of eotaxin [6].
An essential biological activity of IL-4 in the development of allergic inflammation is the ability to drive the differentiation of naive T helper type 0 (TH0) lymphocytes into TH2 lymphocytes [7,8]. These TH2 cells are able to secrete IL-4, IL-5, IL-9 and IL-13 but lose the ability to produce interferon-γ [9]. Using human cells, administration of IL-4 generates TH2-like lymphocyte clones, whereas incubation with anti-IL-4 blocks this differentiation. The induction of TH2-like lymphocytes is a unique biological activity of IL-4 because IL-4 receptors and not IL-13 receptors are expressed on T cells [10]. IgE production and the induction of VCAM-1 are activities shared with the related cytokine IL-13.
In addition to driving the differentiation of TH0 lymphocytes into the TH2 phenotype, IL-4 is important in allergic immune responses owing to its ability to prevent apoptosis of T lymphocytes. Activation of these cells results in rapid proliferation and secretion of cytokines. In the absence of an appropriate signal, activated T helper lymphocytes rapidly become apoptotic and are eliminated. Several cytokines, including IL-2, IL-4, IL-7, and IL-15, are effective in preventing the death of activated T cells. Of these, IL-4 and IL-15 are the most effective [11]. Inhibition of apoptosis by IL-4 might be mediated partly by the ability of this cytokine to maintain levels of the survival-promoting protein Bcl-2 in T cells [11]. Apoptosis of T lymphocytes can be induced through signals mediated by Fas ligand through the Fas (CD95) receptor expressed on these cells. T cells from asthmatic lungs show defective expression of Fas. Co-culturing of T cells with IL-4 downregulates Fas expression on the cell surface. The expression of IL-4 in the asthmatic lung and the secondary lack of Fas expression might explain the persistence of inflammatory cellular infiltrates in allergic asthma. Apoptosis of TH2 lymphocytes (and possibly eosinophils) could represent a pathway by which IL-4 blockade produces rapid clinical benefits in asthma. Corticosteroids normally cause apoptosis in mature T helper cell lines. Induction of cell death is prevented by IL-2 in TH1 cells and by IL-4 in TH2 cells [12]. IL-4 and IL-2 synergize to render lymphocytes refractory to the anti-inflammatory influences of corticosteroids. This is a result of alternative splicing of the glucocorticoid receptor (GCR) mRNA, generating GCR-β. GCR-β is unable to bind glucocorticoids but can bind and antagonize the transactivating activity of the classic GCR-α. Through these mechanisms, the autocrine production of IL-4 by TH2 cells in the asthmatic lung might render these cells refractory to the beneficial influences of corticosteroids [13]. Synergistic benefits of interleukin-4 receptor (IL-4R) with inhaled corticosteroids can be expected in asthmatic patients.
Clinical observations of IL-4 in allergic disease
IL-4 is increased in the serum and bronchoalveolar lavage of allergic individuals [14,15], and peripheral blood mononuclear cells from atopic asthmatics increase IL-4 production in response to dust mite antigen [16]. Nebulized administration of IL-4 to patients with mild asthma resulted in a significant increase in airway hyper-responsiveness that was associated with the elevation of sputum eosinophil numbers [17]. A variety of findings suggest that atopic individuals have altered regulation in their IL-4 production. In response to bacterial antigens, CD4+ T cell clones from atopics produce IL-4 and IL-5, whereas non-atopic CD4+ T cell clones produce TH1 cytokines [18]. Atopic subjects have a higher frequency of IL-4-producing T cells than do normal subjects [19], and T cell clones generated from cord blood lymphocytes of newborns with atopic parents produce higher IL-4 concentrations than neonatal lymphocytes of newborns with non-atopic parents [20].
IL-4 genetics and asthma
Asthma has been linked to chromosome 5q31-33 through genome searches and screening of candidate genes [21,22]. This locus includes the genes for the TH2 cytokines IL-4, IL-5, IL-9, and IL-13. Aberrant production of IL-4 or hyperresponsiveness to this cytokine owing to inherited defects might further contribute to the pathophysiology of asthma. Optimal signaling by IL-4 (reviewed in [23]) involves its interaction with receptors consisting of a heterodimer of high-affinity IL-4Rα and either the common γ chain or the IL-13 receptor α chain. Binding of IL-4 results in the tyrosine phosphorylation of signal transduction molecules including motifs similar to those involved in insulin signaling, the insulin receptor substrate (IRS)-1, IRS2 and signal transducer and activator of transcription (Stat)-6 [23]. IRS1 and IRS2 regulate proliferation and protection from apoptosis. Stat-6 is necessary for IL-4-dependent expression of CD23, MHC class II, ε heavy chain, and IL-4Rα [23] and, as noted, is essential for the differentiation of TH2 lymphocytes. BCL6 is a protein that counteracts the stimulatory effects of Stat-6 by binding to Stat-6 responsive promoter elements and repressing transcription. Through this mechanism, BCL6 has been shown to suppress IL-4-induced IgE production [24]. Asthma has been genetically linked to polymorphisms in the genes encoding IL-4Rα, IL-13Rα, Stat-6, and BCL6. A summary of the naturally occurring polymorphisms that are linked to atopy and asthma and might influence IL-4 production or IL-4 signaling is given in Table 1. Individuals genetically programmed to be hyper-responsive to IL-4 represent a cohort of subjects who might be therapeutically responsive to an IL-4 antagonist.
Table 1.
Genes associated with IL-4 and IL-4 signaling linked to asthma and allergies
| Gene | Polymorphism | Location | Reference |
| Interleukin-4 promoter | A→G | -81 | [32] |
| C→T | -285 | [32] | |
| C→T | -590 | [33] | |
| Interleukin-4 receptor, α chain | Ile→Val | +50 | [34] |
| Arg→Gln | +551 | [35] | |
| Ser→Pro | +503 | [36] | |
| Interleukin-13 receptor, α chain | A→G | +1398 | [37] |
| Stat-6 | G→A | +2964 (3' UTR) | [38] |
| BCL6 | HindIII | 5' untranslated region | [39] |
Animal studies demonstrating the role of IL-4 in asthma
Neutralizing IL-4 with anti-IL-4 antibodies in mice inhibits the development of allergen-specific IgE [25], and reduces eosinophilic inflammation [25] and airway reactivity [26]. These results have been confirmed by using IL-4 knockout mice. In addition to these effects, recombinant soluble IL-4 receptor, which acts as a decoy for IL-4 binding and neutralizes IL-4 activity, has been shown in murine models to block allergen-induced airway hyperreactivity [27] and to inhibit VCAM-1 expression, eosinophil influx, and excessive production of mucus [28]. By inhibiting TH2-like lymphocyte differentiation and promoting the apoptosis of established TH2-like cells, IL-4 blockade inhibits the biological activities of IL-4 and, equally important, reduces the production of IL-5. Nonetheless, mice deficient in IL-4 (IL-4 knockouts) maintain residual TH2 responses, which might explain the persistent low-level expression of IL-5, eosinophilia, and airway hyperreactivity observed in some of these murine studies [10]. Although there is always a danger in over-interpreting data derived from animal models, these observations support the role of IL-4 in asthma and allergic disorders.
Soluble cytokine receptors as therapeutic agents
The IL-4R is a cell-surface, heterodimeric complex consisting of a specific, high-affinity α chain (IL-4Rα) and a second chain that can be either the common γ chain or the α chain of the IL-13 receptor (IL-13Rα) [23]. The common γ chain is found in multiple cytokine receptors. Although both chains of the heterodimer are required to mediate cellular activation, only IL-4Rα is necessary for binding IL-4. Secreted forms of IL-4Rα occur naturally and are expressed in allergic inflammation [29]. Soluble IL-4R is capable of interacting with IL-4 even though it lacks the transmembrane and cytoplasmic domains. Because it does not induce cellular activation but instead binds and sequesters IL-4, soluble IL-4R serves as an anti-inflammatory mechanism that can counter the effects of IL-4 (Fig. 1) and might represent an endogenous autoregulatory or homeostatic mechanism. Acting as a decoy to bind and neutralize circulating cytokine, coupled with the high specificity and high affinity of binding for the cytokine, makes the soluble receptor ideal as a cytokine antagonist. Soluble recombinant human IL-4 receptor (rhuIL-4R; Nuvance™; Immunex) is the extracellular portion of human IL-4Rα, the gene encoding which has been cloned and its product expressed in a mammalian expression system. Because the amino acid and carbohydrate or glycosylation sequences are identical to those of human IL-4R, soluble receptors are relatively nonimmunogenic. This is in contrast to chimerized or humanized monoclonal antibodies, which retain some murine sequences, or IL-4 muteins that are not authentic. The soluble tumor necrosis factor receptor, Enbrel®, has been demonstrated to be safe and highly effective for the long-term treatment of rheumatoid arthritis in adults and children.
Figure 1.
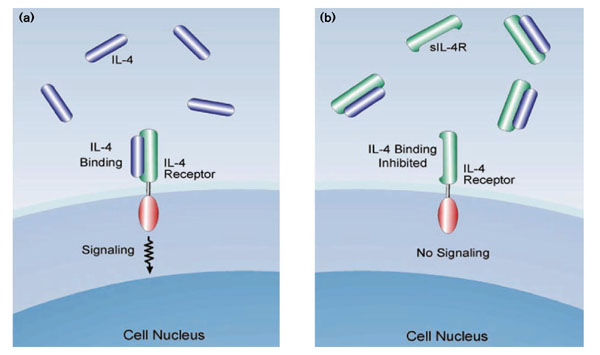
IL-4 binding to cellular IL-4R and its inhibition by soluble IL-4R (sIL-4R). (a) IL-4 binds to cellular IL-4R, mediating cellular activation and important pro-inflammatory functions in asthma. For clarity, only the IL-4Rα chain of the heterodimer is shown, represented by three domains: extracellular (green), transmembrane (white) and cytoplasmic (red). It should be noted, however, that both chains of the heterodimer are required to initiate intracellular signaling. (b) The sIL-4R consists of the extracellular portion of IL-4Rα. It retains the ability to bind IL-4 with high affinity and high specificity and thereby functions as a decoy receptor which can sequester naturally occurring IL-4 and prevent it from activating the cell. sIL-4R cannot activate cellular signaling; thus, it serves as an anti-inflammatory agent that can counter the effects of IL-4 in asthma.
Clinical studies with rhuIL-4R (NuvanceTM)
The promising results in preclinical studies led to preliminary investigations in which rhuIL-4R proved safe and effective in the treatment of patients with asthma [30,31]. In the phase I study, subjects with mild or moderate persistent asthma were withdrawn from their inhaled corticosteroids and randomly assigned to receive placebo or rhuIL-4R at 0.5 or 1.5 mg by nebulizer [30]. There were no significant adverse events related to the study drug and no patients developed antibodies against rhuIL-4R. Pharmacokinetic analysis demonstrated a prolonged serum half-life of about 5 days, suggesting that weekly therapy would be effective. After acute discontinuation of inhaled corticosteroids, no subject in the rhuIL-4R 1.5 mg group withdrew for asthma exacerbations, compared with three of eight in the rhuIL-4R 0.5 mg group and two of eight in the placebo group. Treatment with 1.5 mg of rhuIL-4R was associated with significantly better forced expiratory volume in 1 s (FEV1) at 2 h after treatment and on days 2, 4, and 15 (P < 0.05). Statistically significant differences in asthma symptom score (P < 0.05) and β2-agonist use (P < 0.05) were associated with treatment with rhuIL-4R. Scores on the third section of the AQLQ (Asthma Quality of Life Questionnaire, showing a patient's perception of general health and physical functioning) worsened in the placebo group and improved in the rhuIL-4R 1.5 mg group (P < 0.05). Methacholine testing showed decreased sensitivity in six out of eight patients tested in the 1.5 mg group. Exhaled nitric oxide scores were significantly improved in patients receiving rhuIL-4R (P < 0.05), which is consistent with an anti-inflammatory effect.
In the phase I/II double-blind, placebo-controlled study, 62 moderate persistent asthmatic patients were randomized to 12 weekly nebulizations of 0.75, 1.5, or 3.0 mg of rhuIL-4R (Nuvance™) or placebo [31]. Before the study, patients documented their dependence on inhaled corticosteroids by an exacerbation in asthma induced by one or two 50% reductions in inhaled corticosteroid dose at 2-week intervals. After being restabilized on inhaled corticosteroids for 2 weeks, the inhaled corticosteroids were discontinued at the time that study medication was begun. IL-4R was safe and well tolerated. Antibodies against rhuIL-4R occurred in one patient that were non-neutralizing and resulted in no symptoms. Efficacy was demonstrated by a significant decline in FEV1 observed in the placebo group (-0.35 l; -13% predicted) which did not occur in the 3.0 mg treatment group (-0.09 l; -2% predicted; P = 0.053 over the three-month treatment period). Daily patient-measured morning FEV1 also demonstrated a significant decline in the placebo group (-0.5 l; -18% predicted) that did not occur in the 3.0 mg treatment group (-0.1 l; P = 0.02 over the three-month treatment period; -4% predicted). The afternoon FEV1 also improved at the highest dose and was 19% better than placebo. The efficacy of rhuIL-4R was further confirmed by the absence of an increase in asthma symptom score (a change of 0.1) in the 3.0 mg treatment group in comparison with the placebo group (a change of 1.4 over onemonth; P = 0.075).
Conclusions
These studies demonstrate that IL-4R is a potentially safe and effective new treatment for asthma without the use of corticosteroids. Dosing once a week, with an inhaled medication targeting the lungs, will probably improve patient compliance, which is one of the greatest challenges to the effective treatment of asthma. Inhibiting inflammation at a key regulatory point, IL-4R might affect the long-term disease progression in asthma. IL-4R should also be effective in patients with non-allergic forms of asthma. Although these patients do not demonstrate allergen-specific IgE, the presence of eosinophilic inflammation and elevated total IgE suggests the differentiation of TH2-like lymphocytes, which are responsible for the production of IL-5 and other cytokines that promote the development of eosinophilia. On the basis of our current knowledge of the differentiation of IL-5-producing TH2-like lymphocytes, this process is IL-4-dependent and should be susceptible to suppression by IL-4R therapy. Other atopic disorders such as allergic rhinitis and atopic dermatitis are thought to be mediated by IL-4 and might also respond to IL-4 blockade with IL-4R therapy. Soluble IL-4 receptor studies are continuing and this class of drugs represents the next generation of asthma therapy.
Abbreviations
FEV1 = forced expiratory volume in 1 second; GCR = glucocorticoid receptor; IL = interleukin; IL-4R = interleukin-4 receptor; IRS = insulin receptor substrate; rhuIL-4R = soluble recombinant human interleukin-4 receptor; Stat = signal transducer and activator of transcription; VCAM = vascular cell adhesion molecule.
References
- Coffman RL, Ohara J, Bond MW, Carty J, Zlotnik A, Paul WE. B cell stimulatory factor-1 enhances the IgE response of lipopolysaccharide-activated B cells. J Immunol. 1986;136:4538–4541. [PubMed] [Google Scholar]
- Pawankar R, Okuda M, Yssel H, Okumura K, Ra C. Nasal mast cells in perennial allergic rhinitis exhibit increased expression of the Fc epsilonRI, CD40L, IL-4, and IL-13, and can induce IgE synthesis in B cells. J Clin Invest. 1997;99:1492–1499. doi: 10.1172/JCI119311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabbagh K, Takeyama K, Lee HM, Ueki IF, Lausier JA, Nadel JA. IL-4 induces mucin gene expression and goblet cell metaplasia in vitro and in vivo. J Immunol. 1999;162:6233–6237. [PubMed] [Google Scholar]
- Doucet C, Brouty-Boye D, Pottin-Clemenceau C, Jasmin C, Canonica GW, Azzarone B. IL-4 and IL-13 specifically increase adhesion molecule and inflammatory cytokine expression in human lung fibroblasts. Int Immunol. 1998;10:1421–1433. doi: 10.1093/intimm/10.10.1421. [DOI] [PubMed] [Google Scholar]
- Moser R, Fehr J, Bruijnzeel PL. IL-4 controls the selective endothelium-driven transmigration of eosinophils from allergic individuals. J Immunol. 1992;149:1432–1438. [PubMed] [Google Scholar]
- Hoontrakoon R, Kailey J, Bratton D. IL-4 and TNF-α synergize to enhance eosinophil survival [abstract]. J Allergy Clin Immunol. 1999;103:A239. [Google Scholar]
- Hsieh CS, Heimberger AB, Gold JS, O'Garra A, Murphy KM. Differential regulation of T helper phenotype development by interleukins 4 and 10 in an αβ T-cell-receptor transgenic system. Proc Natl Acad Sci USA. 1992;89:6065–6069. doi: 10.1073/pnas.89.13.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seder RA, Paul WE, Davis MM, Fazekas de St Groth B. The presence of interleukin 4 during in vitro priming determines the lymphokine-producing potential of CD4+ T cells from T cell receptor transgenic mice. J Exp Med. 1992;176:1091–1098. doi: 10.1084/jem.176.4.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutel M, Pichler WJ, Skrbic D, Urwyler A, Dahinden C, Muller UR. Bee venom immunotherapy results in decrease of IL-4 and IL-5 and increase of IFN-γ secretion in specific allergen-stimulated T cell cultures. J Immunol. 1995;154:4187–4194. [PubMed] [Google Scholar]
- Kopf M, Le Gros G, Bachmann M, Lamers MC, Bluethmann H, Kohler G. Disruption of the murine IL-4 gene blocks Th2 cytokine responses. Nature. 1993;362:245–248. doi: 10.1038/362245a0. [DOI] [PubMed] [Google Scholar]
- Vella A, Teague TK, Ihle J, Kappler J, Marrack P. Interleukin 4 (IL-4) or IL-7 prevents death of resting T cells: Stat-6 is probably not required for the effect of IL-4. J Exp Med. 1997;186:325–330. doi: 10.1084/jem.186.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Seward RJ, Huber BT. Cytokine rescue from glucocorticoid induced apoptosis in T cells is mediated through inhibition of Iκ-Bα. Mol Immunol. 1997;34:987–994. doi: 10.1016/S0161-5890(97)00128-4. [DOI] [PubMed] [Google Scholar]
- Leung DYM, Hamid Q, Vottero A, Szefler SJ, Surs W, Minshall E, Chrousos GP, Klemm DJ. Association of glucocorticoid insensitivity with increased expression of glucocorticoid receptor β. J Exp Med. 1997;186:1567–1574. doi: 10.1084/jem.186.9.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daher S, Santos LM, Sole D, De Lima MG, Naspitz CK, Musatti CC. Interleukin-4 and soluble CD23 serum levels in asthmatic atopic children. J Invest Allergol Clin Immunol. 1995;5:251–254. [PubMed] [Google Scholar]
- Walker C, Bauer W, Braun RK, Menz G, Braun P, Schwarz F, Hansel TT, Villiger B. Activated T cells and cytokines in bron-choalveolar lavages from patients with various lung diseases associated with eosinophilia. Am J Respir Crit Care Med. 1994;150:1038–1048. doi: 10.1164/ajrccm.150.4.7921434. [DOI] [PubMed] [Google Scholar]
- Leonard C, Tormey V, Burke C, Poulter LW. Allergen-induced cytokine production in atopic disease and its relationship to disease severity. Am J Respir Cell Mol Biol. 1997;17:368–375. doi: 10.1165/ajrcmb.17.3.2797. [DOI] [PubMed] [Google Scholar]
- Shi HZ, Deng JM, Xu H, Nong ZX, Xiao CQ, Liu ZM, Qin SM, Jiang HX, Liu GN, Chen YQ. Effect of inhaled interleukin-4 on airway hyperreactivity in asthmatics. Am J Respir Crit Care Med. 1998;157:1818–1821. doi: 10.1164/ajrccm.157.6.9710023. [DOI] [PubMed] [Google Scholar]
- Parronchi P, De Carli M, Manetti R, Simonelli C, Piccinni MP, Macchia D, Maggi E, Del Prete G, Ricci M, Romagnani S. Aberrant interleukin (IL)-4 and IL-5 production in vitro by CD4+ helper T cells from atopic subjects. Eur J Immunol. 1992;22:1615–1620. doi: 10.1002/eji.1830220640. [DOI] [PubMed] [Google Scholar]
- Chan SC, Brown MA, Willcox TM, Li SH, Stevens SR, Tara D, Hanifin JM. Abnormal IL-4 gene expression by atopic dermatitis T lymphocytes is reflected in altered nuclear protein interactions with IL-4 transcriptional regulatory element. J Invest Dermatol. 1996;106:1131–1136. doi: 10.1111/1523-1747.ep12340181. [DOI] [PubMed] [Google Scholar]
- Piccinni MP, Beloni L, Giannarini L, Livi C, Scarselli G, Romagnani S, Maggi E. Abnormal production of T helper 2 cytokines interleukin-4 and interleukin-5 by T cells from newborns with atopic parents. Eur J Immunol. 1996;26:2293–2298. doi: 10.1002/eji.1830261004. [DOI] [PubMed] [Google Scholar]
- Marsh DG, Neely JD, Breazeale DR, Ghosh B, Freidhoff LR, Ehrlich-Kautzky E, Schou C, Krishnaswamy G, Beaty TH. Linkage analysis of IL4 and other chromosome 5q31.1 markers and total serum immunoglobulin E concentrations. Science. 1994;264:1152–1156. doi: 10.1126/science.8178175. [DOI] [PubMed] [Google Scholar]
- Rosenwasser LJ, Borish L. Genetics of atopy and asthma: the rationale behind promoter-based candidate gene studies (IL-4 and IL-10). Am J Respir Crit Care Med. 1997;156:S152–155. doi: 10.1164/ajrccm.156.4.12tac-14. [DOI] [PubMed] [Google Scholar]
- Jiang H, Harris MB, Rothman P. IL-4/IL-13 signaling beyond JAK/STAT. J Allergy Clin Immunol. 2000;105:1063–1070. doi: 10.1067/mai.2000.107604. [DOI] [PubMed] [Google Scholar]
- Harris M, Chang CC, Berton M, Danial N, Zhang J, Kuehner D, Ye BH, Cattoretti GM, Kvatyuk GM, Pandolfi PP, Dalla-Favera R, Rothman P. Transcriptional repression of Stat-6-dependent IL-4 induced genes by BCL6: specific regulation of Iε transcript expression. Mol Cell Biol. 1999;19:7264–7275. doi: 10.1128/mcb.19.10.7264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle AJ, Le Gros G, Bertrand C, Tsuyuki S, Heusser CH, Kopf M, Anderson GP. Interleukin-4 is required for the induction of lung Th2 mucosal immunity. Am J Respir Cell Mol Biol. 1995;13:54–59. doi: 10.1165/ajrcmb.13.1.7598937. [DOI] [PubMed] [Google Scholar]
- Corry DB, Folkesson HG, Warnock ML, Erle DJ, Matthay MA, Wiener-Kronish JP, Locksley RM. Interleukin 4, but not interleukin 5 or eosinophils, is required in a murine model of acute airway hyperreactivity. J Exp Med. 1996;183:109–117. doi: 10.1084/jem.183.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renz H, Bradley K, Enssle K, Loader JE, Larsen GL, Gelfand EW. Prevention of the development of immediate hypersensitivity and airway hyperresponsiveness following in vivo treatment with soluble IL-4 receptor. Int Arch Allergy Immunol. 1996;109:167–176. doi: 10.1159/000237216. [DOI] [PubMed] [Google Scholar]
- Henderson W, Chi E, Maliszweski C. Soluble interleukin-4 receptor inhibits airway inflammation following allergen challenge in a mouse model of asthma. J Immunol. 2000;164:1086–1095. doi: 10.4049/jimmunol.164.2.1086. [DOI] [PubMed] [Google Scholar]
- Benson M, Strannegard IL, Wennergren G, Strannegard O. Cytokines in nasal fluids from school children with seasonal allergic rhinitis. Pediatric Allergy Immunol. 1997;8:143–149. doi: 10.1111/j.1399-3038.1997.tb00168.x. [DOI] [PubMed] [Google Scholar]
- Borish L, Nelson H, Lanz M, Claussen L, Whitmore J, Agosti J, Garrison L. Recombinant human interleukin-4 receptor in moderate astopic asthma: a randomized double-blind, placebo-controlled pilot study. Am J Respir Crit Care Med. 1999;160:1816–1823. doi: 10.1164/ajrccm.160.6.9808146. [DOI] [PubMed] [Google Scholar]
- Borish L, Nelson H, Corren J, Bensch G, Busse W, Whitmore J, Agosti J. Phase I/II study of recombinant interleukin-4 receptor (IL-4R) in adult patients with moderate asthma [abstract]. J Allergy Clin Immunol. 2000;105:A828. [Google Scholar]
- Song Z, Casolaro V, Chen R, Georas SN, Monos D, Ono SJ. Polymorphic nucleotides within the human IL-4 promoter that mediate overexpression of the gene. J Immunol. 1996;156:424–429. [PubMed] [Google Scholar]
- Burchard EG, Silverman EK, Rosenwasser LJ, Borish L, Yandava C, Pillari A, Weiss ST, Hasday J, Lilly CM, Ford JG, Drazen JM. Association between a sequence variant in the IL-4 gene promoter and FEV1 in asthma. Am J Respir Crit Care Med. 1999;160:919–922. doi: 10.1164/ajrccm.160.3.9812024. [DOI] [PubMed] [Google Scholar]
- Mitsuyasu H, Izuhara K, Mao XQ, Gao PS, Arinobu Y, Enomoto T, Kawai M, Sasaki S, Dake Y, Hamasaki N, Shirakawa T, Hopkin JM. Ile50Val variant of IL4Rα upregulates IgE synthesis and associates with atopic asthma. Nat Genet. 1998;19:119–120. doi: 10.1038/472. [DOI] [PubMed] [Google Scholar]
- Hershey GKK, Friedrich MF, Esswein LA, Thomas ML, Chatila TA. The association of atopy with a gain-of-function mutation in the α subunit of the interleukin-4 receptor. New Engl J Med. 1997;337:1720–1725. doi: 10.1056/NEJM199712113372403. [DOI] [PubMed] [Google Scholar]
- Kruse S, Japha T, Tedner M, Sparholt SH, Forster J, Kuehr J, Deichmann KA. The polymorphisms S503P and Q576R in the interleukin-4 receptor α gene are associated with atopy and influence signal transduction. Immunology. 1999;96:365–371. doi: 10.1046/j.1365-2567.1999.00705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzmann A, Mao XQ, Akaiwa M, Kreomer RT, Gao PS, Ohshima K, Umeshita R, Abe Y, Braun S, Yamashita T, Roberts MH, Sugimoto R, Arima K, Arinobu Y, Yu B, Kruse S, Enomoto T, Dake Y, Kawai M, Shimazu S, Sasaki S, Adra CN, Kitaichi M, Inoue H, Yamauchi K, Tomichi N, Kurimoto F, Hamasaki N, Hopkin JM, Izuhara K, Shirakawa T, Deichmann KA. Genetic variants of IL-13 signaling and human asthma and atopy. Hum Mol Genet. 2000;9:549–559. doi: 10.1093/hmg/9.4.549. [DOI] [PubMed] [Google Scholar]
- Gao PS, Mao XQ, Roberts MH, Arinobu Y, Akaiwa M, Enomoto T, Dake Y, Kawai M, Sasaki S, Hamasaki N, Izubara K, Shirakawa T, Hopkin JM. Variants of STAT-6 (signal transducer and activator of transcription 6) in atopic asthma. J Med Genet. 2000;66:380–382. doi: 10.1136/jmg.37.5.380a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron BW, Billstrand C, Madronero L, McKeithan TW. Hin dIII polymorphism in the BCL6 gene. Hum Mol Genet. 1993;9:1513. doi: 10.1093/hmg/2.9.1513. [DOI] [PubMed] [Google Scholar]