

Abstract
Background
Arsenic is naturally prevalent in the earth’s crust and widely distributed in air and water. Chronic low arsenic exposure is associated with several cancers in vivo, including skin cancer, and with transformation in vitro of cell lines including immortalized human keratinocytes (HaCaT). Arsenic also is associated with cell cycle dysregulation at different exposure levels in multiple cell lines. In this work, we analyzed gene expression in HaCaT cells to gain an understanding of gene expression changes contributing to transformation at an early time point.
Methods
HaCaT cells were exposed to 0 or 100 nM NaAsO2 for 7 weeks. Total RNA was purified and analyzed by microarray hybridization. Differential expression with fold change ≥|1.5| and p-value ≤0.05 was determined using Partek Genomic Suite™ and pathway and network analyses using MetaCore™ software (FDR ≤0.05). Cell cycle analysis was performed using flow cytometry.
Results
644 mRNAs were differentially expressed. Cell cycle/cell cycle regulation pathways predominated in the list of dysregulated pathways. Genes involved in replication origin licensing were enriched in the network. Cell cycle assay analysis showed an increase in G2/M compartment in arsenite-exposed cells.
Conclusions
Arsenite exposure induced differential gene expression indicating dysregulation of cell cycle control, which was confirmed by cell cycle analysis. The results suggest that cell cycle dysregulation is an early event in transformation manifested in cells unable to transit G2/M efficiently. Further study at later time points will reveal additional changes in gene expression related to transformation processes.
Keywords: Arsenic-induced skin cancer, mRNA, carcinogenesis, transformation, chronic exposure, cell cycle
Introduction
Arsenic is a naturally prevalent metalloid in air and water. Underground drinking water is the major source of human arsenic exposure. Globally, over 200 million people are exposed to arsenic in drinking water in levels higher than the World Health Organization recommends (10 μg/L) (WHO 2008). Most of which are concentrated in Bangladesh, Cambodia, India, Nepal and Vietnam along with countries in Latin and North America, such as Argentina, Bolivia, Chile and Mexico and the U.S.A. Several epidemiological studies in Taiwan, Bangladesh, Chile, India, Argentina and the U.S.A. demonstrated links between the arsenic exposure exceeding 10 μg/L in drinking water and increased all-cause mortality, linked not only to increased risk of lung, bladder and skin cancers, but also cardiovascular, neurological and respiratory diseases, and skin lesions (Ahsan et al. 2006; Argos et al. 2010; Argos et al. 2011; Argos et al. 2012; Chen et al. 2010; Chen and Ahsan 2004; Chen et al. 2011; Gribble et al. 2012; Haque et al. 2003; James et al. 2015; Parvez et al. 2008; Smith and Steinmaus 2009; Wasserman et al. 2014; Wright et al. 2006; Yuan et al. 2010).
Studies from arsenic-endemic regions of Taiwan showed the association of skin cancer prevalence with increased arsenic levels in drinking-water (Tseng 1977). Long-term ingestion of arsenic is associated with different types of skin cancer such as intraepidermal carcinoma (Bowen’s disease, BD), squamous cell carcinoma (SCC), basal cell carcinoma (BCC), and Merkel cell carcinoma (Centeno et al. 2002; Ho et al. 2005). Immortalized human keratinocytes (HaCaT) are an in vitro model for normal human keratinocytes (Boukamp et al. 1988). Pi et al (2008) showed that continuous exposure of HaCaT cells for 28 weeks to 100 nM sodium arsenite malignantly transformed these cells and resulted in an aggressive SCC phenotype when inoculated into nude mice (Pi et al. 2008). The model has also been evaluated by Sun et. al. as an important model to study the induction of skin cancer by arsenic exposure (Sun et al. 2009). However, the early stages of the transformation process in this model have not been studied yet. Thus, within a longitudinal study using this chronic exposure model, we analyzed gene expression to gain an understanding of gene expression changes contributing to transformation at an early time point. The data reveal differential expression of a wide array of genes responsible for cell cycle regulation suggesting that cell cycle dyregulation plays a role in early events leading to transformation.
Materials and Methods
a. Cell Culture and RNA Isolation
We adopted the HaCaT model of Pi et al. (2008) for these studies. HaCaT cells were the kind gift of Dr. TaiHao Quan, University of Michigan. NaAsO2 (CAS 7784-0698) was obtained from Fisher Scientific, Waltham, MA, USA. HaCaT cells were cultured in alpha modification of minimal essential media supplemented with fetal bovine serum (10%), penicillin (100 units/mL), streptomycin (100 μg/mL) and glutamine (2 mM). Cultures were maintained at 37°C in a humidified 5% CO2 atmosphere. Multiple cultures of cells (4 with and 4 without 100 nM NaAsO2) were maintained separately for 7 weeks (Fig. 1). This NaAsO2 concentration was selected based on blood levels observed in an epidemiological study of a population in China that used tube wells containing high concentrations of arsenic (Pi et al. 2000). The study subjects were diagnosed with chronic arsenic intoxication and arsenic-induced skin lesions and epidermal cancers (Pi et al. 2000). Cells were passaged twice a week and a million cells were plated per 100 mm dish at every passage. Total RNA was purified from the cells (quadruplicate unexposed and exposed cultures) using the mirVana™ RNA Isolation Kit (Thermo Fisher Scientific Inc., Waltham, MA, USA). RNA quality was determined using the Agilent RNA 6000 Pico Kit, Eukaryote, version 2.6 and the Agilent 2100 Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA, USA). All samples used had RIN (RNA integrity number) > 9.
Figure 1.
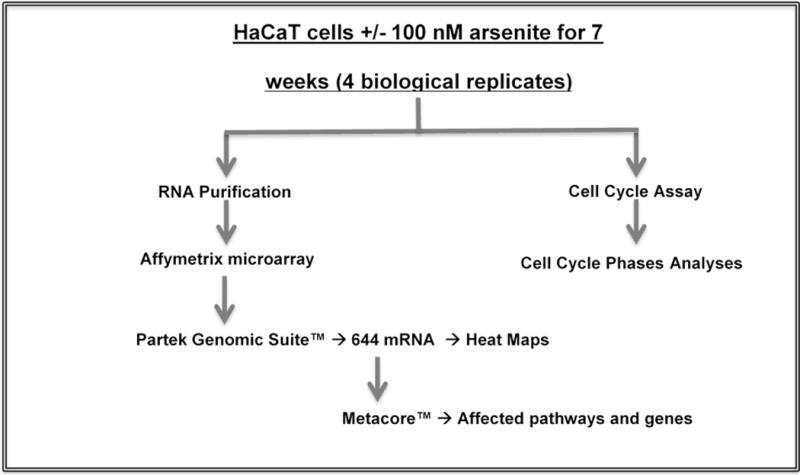
HaCaT cells were exposed to 0 or 100 nM NaAsO2 for 7 weeks. RNA was purified and expression determined on Affymetrix microarrays and analyzed using Metacore software. Cell cycle analyses were performed by flow cytometry.
b. Microarray Analysis
Expression profiles of mRNA were obtained using GeneChip® PrimeView™ Human Gene Expression Affymetrix arrays (Fig. 1). Biotinylated cRNA was prepared according to the standard protocol for Affymetrix 3′ IVT Express Plus Reagent Kit from 250 ng total RNA. Following fragmentation, cRNA was hybridized for 16 h at 45°C to Affymetrix Primeview Human arrays according to the Affymetrix GeneChip 3′ array Hybridization User Manual. GeneChips were scanned using GeneChip Scanner 3000 7G (Affymetrix). The CEL files were imported into Partek software Version 6.6 (Partek Inc) and normalized using Robust Multi-Array (RMA) normalization. Contrasts of interest were analyzed using a 2-way ANOVA considering treatment and time. Using Partek Genomic Suite™ a list of differentially expressed mRNAs (644) (fold change ≥|1.5|) at 7 weeks was obtained (supplementary Table 1). Data have been deposited in the GEO database, accession number GSE97306.
c. Flow cytometry cell cycle assay
HaCaT cells exposed to 0 or 100 nM NaAsO2 for 7 weeks were seeded at density of 1 × 106 cells per 55 cm2 cell culture dish with arsenite exposure maintained. After 48 hours, the cells were trypsinized, washed twice with PBS then fixed in 70% ethanol for 24 hours at 4°C. The cells were then centrifuged at 1500 g for 1 minute, suspended in PBS (400 μl), then RNaseA (10 mg/ml, 50 μl) and propidium iodide (2 mg/ml, 10 μl) were added followed by a 30 minute incubation in the dark at room temperature. Fluorescence was acquired by flow cytometry on a Becton Dickinson FACSCalibur™ (BD Biosciences) (Chowdhury et al. 2010). Cell cycle distribution was determined using FlowJo® v10.2. Student’s T-Test was used for statistical analyses (p-value ≤ 0.05, was considered significant).
Results
a. Arsenite-dependent differential expression of cell cycle involved mRNAs
Analysis of the gene chip data revealed that 644 mRNAs were differentially expressed in HaCaT cells exposed to 100 nM NaAsO2 for seven weeks. More than half of the mRNAs were suppressed with arsenite exposure and the remainder were induced (Fig. 2A). The differentially expressed genes were then loaded into Metacore™ software for pathways analysis. The 15 pathways with FDR values ≤0.05 are listed in Table 1. Cell cycle and cell cycle regulation pathways are highly represented in this list (7 of 15). Other pathways that were dysregulated include epithelial-to-mesenchymal transition (EMT), cytoskeleton remodeling, apoptosis, immune response and gap junction pathways. The mRNAs populating these pathways were analyzed for potential interactions. The interacting genes are shown in a network built by Metacore™ software (Fig. 2B). Three E3 ubiquitin ligase complexes known to regulate cell cycle are present in this network (Siah1/SIP/EBI; CUL1/RBX1; SKP2/TRCP/FBXW) (Frescas and Pagano 2008; Matsuzawa and Reed 2001; Zheng et al. 2002). A subnetwork of genes involved in regulating licensing of the DNA replication origin (CDT1; MCM4/6/7 complex; MCM2) (Nishitani and Lygerou 2002) also is present.
Figure 2.
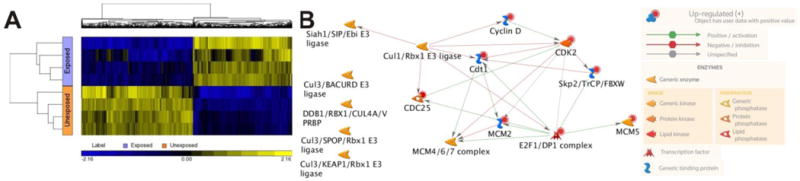
Differential mRNA expression with arsenite exposure. A. Hierarchical clustering of mRNAs differentially expressed in HaCaT cells exposed to 0/100 nM arsenite for 7 weeks. Microarray hybridization data from 4 exposed and 4 unexposed cultures was analyzed using Parteck Genomic Suite to generate the heat map. Yellow indicates mRNAs induced by exposure, blue indicates suppression. B. Network of genes populating multiple dysregulated pathways at 7 weeks. Network of only direct interactions between genes populating multiple pathways at 7 weeks was built by Metacore™ software.
Table 1.
Pathways populated by differentially expressed mRNAs 7 weeks.
| Pathway* | FDR | # of genes in pathway | Induction ↑ Suppression ↓ |
|---|---|---|---|
| Cell cycle Start of DNA replication in early S phase | 4.54E-10 | 13 | ↑ |
| Cell cycle Role of SCF complex in cell cycle regulation | 7.44E-07 | 10 | ↑ |
| Cytoskeleton remodeling Neurofilaments | 0.0006747 | 7 | ↑ |
| Cell cycle Transition and termination of DNA replication | 0.0008922 | 7 | ↑ |
| Immune response Antigen presentation by MHC class I, classical pathway | 0.001548 | 9 | ↑↓ |
| Cell cycle (generic schema) | 0.001548 | 6 | ↑ |
| Immune response IL-4-induced regulators of cell growth, survival, differentiation and metabolism | 0.003904 | 9 | ↑ |
| Development Regulation of epithelial-to-mesenchymal transition (EMT) | 0.003904 | 9 | ↑ |
| Cell cycle Regulation of G1/S transition (part 2) | 0.003904 | 6 | ↑ |
| Cell cycle Regulation of G1/S transition (part 1) | 0.003928 | 7 | ↑ |
| Apoptosis and survival Endoplasmic reticulum stress response pathway | 0.007063 | 8 | ↓ Protein folding |
| Cell cycle ESR1 regulation of G1/S transition | 0.01425 | 6 | ↑ |
| Proteolysis Putative ubiquitin pathway | 0.01568 | 5 | ↑ |
| Transcription Ligand-dependent activation of the ESR1/SP pathway | 0.04928 | 5 | ↑ cell cycle regulation |
| Cell adhesion-Gap junctions | 0.04928 | 5 | ↑ |
Pathway analysis performed by Metacore software with expression criteria ≥|1.5| fold change, FDR ≤0.05.
b. Arsenite-dependent increase in G2/M compartment
The gene expression data indicated dysregulation of cell cycle was occurring in arsenite exposed cells. In order to confirm cell cycle dysregulation, cell cycle analyses were performed on the 4 exposed and 4 unexposed cultures. The results indicate that arsenite-exposed cells are accumulating in the G2/M compartment suggesting a delay in either the G2 to M phase or M to G1 phase transition (Fig. 3; Supp. Fig. 1). These results are consistent with the network results of Figure 2B.
Figure 3.
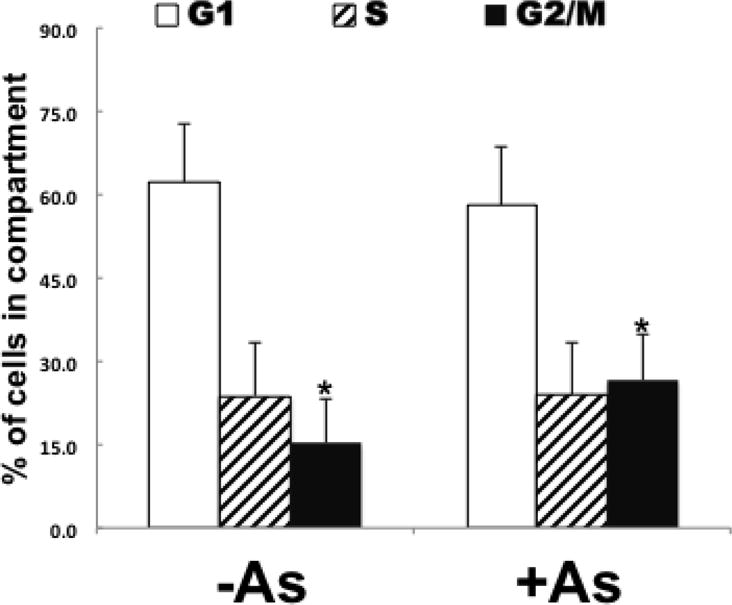
HaCaT cells exposed to arsenite are increased in G2/M compartment. Four cultures each (exposed (+As)/unexposed (−As)) were analyzed by flow cytometry for cell cycle distribution. Fraction in each compartment (G1, S, G2/M) were determined and means ± SD are plotted. Student T-Test was used for statistical analyses. *p-value ≤ 0.05.
Discussion
Arsenic is a known human carcinogen (IARC 2012). Several mechanisms of arsenic carcinogenicity including genomic instability and chromosomal abnormalities have been proposed (Chih-Hung Lee 2010). Arsenic exposure has been associated in several studies with dysregulation of DNA methylation, cell cycle, and DNA repair gene expression (Andrew et al. 2006; Ren et al. 2011; Treas et al. 2013; Zhang et al. 2007). However, there is not yet agreement on the mechanism(s) of arsenic transformation. Chronic low arsenic exposure leads to the malignant transformation of cell lines from several tissues (Achanzar et al. 2002; Barrett et al. 1989; Chang et al. 2010; Pi et al. 2008).
Transformation is likely to be driven by a series of events starting from early times of exposure and extending out to as much as thirty weeks exposure when transformation can be demonstrated to have occurred. Early events in transformation in these model systems have not yet been investigated. Thus, we have examined gene expression changes in the HaCaT model of arsenic-induced skin carcinogenesis after seven weeks’ exposure to gain an understanding of the early events related to transformation.
We (unpublished data) and others (Pi et al. 2008) have observed that HaCaT cells chronically exposed to 100 nM NaAsO2 grow more slowly than parallel cultures of unexposed cells until approximately 19 weeks exposure. Dysregulation of cell cycle control as suggested by the differential expression and pathways data is consistent with these earlier observations on cell growth kinetics. This conclusion is further supported by cell cycle assay analysis on HaCaT cells after seven weeks’ exposure that showed accumulation of cells in the G2/M compartment (Figure 3). This observation is consistent with the network analysis showing induction of genes associated with licensing replication origins (CDT1, MCM2/4/6/7, Figure 2B). These observations are consistent with G2/M delays observed in earlier studies by our laboratory (McNeely et al. 2008; Muenyi et al. 2014; States et al. 2002; Taylor et al. 2006) and by others (Ma et al. 1998; McCollum et al. 2005).
The E3 ubiquitin ligase complexes identified in the network analysis (Figure 3) are all zinc finger RING E3’s that regulate cell cycle transitions other than G2 to M or M to G1. Thus, although the genes involved in replication origin licensing suggest a major impact on M to G1 transition, the induction of these E3 ubiquitin ligase genes suggests that the cells are responding to delays at other cell cycle transitions as well. Impact of arsenite exposure at multiple cell cycle transitions was demonstrated by McCollum et al in U937 cells (McCollum et al. 2005), and by S phase lengthening in MCF7 and H1299 cells (Pozo-Molina et al. 2015). Accumulation in the G0/G1 phase leading to reduction in the S phase also was observed with mouse skin fibroblast cells (m5S), mouse thymocytes and B-cell lymphoma A20 cells (Jutapon Chayapong 2017; Nohara et al. 2008). It was also reported that HL-60; derived from a patient with acute myeloid leukemia (FAB), were arrested at G1 with arsenic exposure (Zhang et al. 1998). Furthermore, Moghaddaskho et al. 2017 showed that arsenic arrested MDA-MB-231 and MDA-MB-468 cells at G2/M phase (Moghaddaskho et al. 2017). Clearly, arsenite exposure can cause cell cycle disruption in all cell cycle phases. However, delays in M phase are likely to contribute to genomic instability via induction of aneuploidy.
Conclusions
Results presented here showed that arsenite exposure induced differential gene expression indicating dysregulation of cell cycle control, consistent with slow growth of arsenite-exposed cells at early times of chronic exposure. Analysis of cell cycle distribution showed a delay at G2/M was occurring in HaCaT cells chronically exposed to 100 nM NaAsO2 for seven weeks. This delay could be related to induction of aneuploidy, known to be caused by arsenic exposure. Further study at later time points will reveal additional changes in gene expression related to transformation processes.
Supplementary Material
Highlights.
Arsenite induces differential expression of 644 mRNAs at early stages of exposure
Arsenite exposure induced differential mRNA expression indicates cell cycle dysregulation
Arsenite delays cells in G2/M compartment
Acknowledgments
This work was supported in part by NIH/NIEHS grant R21ES023627 and by a Competitive Enhancement Grant from the University of Louisville Office of the Executive Vice President for Research and Innovation. Part of this work was performed with assistance of the UofL Genomics Facility, which is supported by NIH grants P20GM103436 (KY IDeA Networks of Biomedical Research Excellence) and P30GM106396. SFJ was supported by NIH/NIEHS grant T35ES014559.
Abbreviations
- BD
Bowen’s disease
- BCC
basal cell carcinoma
- HaCaT cells
immortalized human keratinocytes
- mRNA
messenger RNA
- PBS
phosphate buffered saline
- SCC
squamous cell carcinoma
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare no conflict of interest other than grants as stated in acknowledgements.
References
- Achanzar WE, Brambila EM, Diwan BA, Webber MM, Waalkes MP. Inorganic arsenite-induced malignant transformation of human prostate epithelial cells. Journal of the National Cancer Institute. 2002;94:1888–1891. doi: 10.1093/jnci/94.24.1888. [DOI] [PubMed] [Google Scholar]
- Ahsan H, Chen Y, Parvez F, Argos M, Hussain AI, Momotaj H, et al. Health effects of arsenic longitudinal study (heals): Description of a multidisciplinary epidemiologic investigation. Journal of Exposure Science & Environmental Epidemiology. 2006;16:191–205. doi: 10.1038/sj.jea.7500449. [DOI] [PubMed] [Google Scholar]
- Andrew AS, Burgess JL, Meza MM, Demidenko E, Waugh MG, Hamilton JW, et al. Arsenic exposure is associated with decreased DNA repair in vitro and in individuals exposed to drinking water arsenic. Environmental Health Perspectives. 2006;114:1193–1198. doi: 10.1289/ehp.9008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argos M, Kalra T, Rathouz PJ, Chen Y, Pierce B, Parvez F, et al. Arsenic exposure from drinking water, and all-cause and chronic-disease mortalities in bangladesh (heals): A prospective cohort study. Lancet. 2010;376:252–258. doi: 10.1016/S0140-6736(10)60481-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argos M, Kalra T, Pierce BL, Chen Y, Parvez F, Islam T, et al. A prospective study of arsenic exposure from drinking water and incidence of skin lesions in bangladesh. American Journal of Epidemiology. 2011;174:185–194. doi: 10.1093/aje/kwr062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argos M, Ahsan H, Graziano JH. Arsenic and human health: Epidemiologic progress and public health implications. Reviews on Environmental Health. 2012;27:191–195. doi: 10.1515/reveh-2012-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JC, Lamb PW, Wang TC, Lee TC. Mechanisms of arsenic-induced cell transformation. Biological Trace Element Research. 1989;21:421–429. doi: 10.1007/BF02917284. [DOI] [PubMed] [Google Scholar]
- Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. The Journal of Cell Biology. 1988;106:761–771. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centeno JA, Mullick FG, Martinez L, Page NP, Gibb H, Longfellow D, et al. Pathology related to chronic arsenic exposure. Environmental Health Perspectives. 2002;110(Suppl 5):883–886. doi: 10.1289/ehp.02110s5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q, Pan J, Wang X, Zhang Z, Chen F, Shi X. Reduced reactive oxygen species-generating capacity contributes to the enhanced cell growth of arsenic-transformed epithelial cells. Cancer Research. 2010;70:5127–5135. doi: 10.1158/0008-5472.CAN-10-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CL, Chiou HY, Hsu LI, Hsueh YM, Wu MM, Wang YH, et al. Arsenic in drinking water and risk of urinary tract cancer: A follow-up study from northeastern taiwan. Cancer epidemiology, biomarkers & prevention. 2010;19:101–110. doi: 10.1158/1055-9965.EPI-09-0333. [DOI] [PubMed] [Google Scholar]
- Chen Y, Ahsan H. Cancer burden from arsenic in drinking water in bangladesh. American journal of public health. 2004;94:741–744. doi: 10.2105/ajph.94.5.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Graziano JH, Parvez F, Liu M, Slavkovich V, Kalra T, et al. Arsenic exposure from drinking water and mortality from cardiovascular disease in bangladesh: Prospective cohort study. BMJ. 2011;342:d2431. doi: 10.1136/bmj.d2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chih-Hung Lee W-TL, Hsin-Su Yu. Mechanisms and immune dysregulation in arsenic skin carcinogenesis. Journal of Cancer Therapy. 2010;1:76–86. [Google Scholar]
- Chowdhury R, Chatterjee R, Giri AK, Mandal C, Chaudhuri K. Arsenic-induced cell proliferation is associated with enhanced ros generation, erk signaling and cyclina expression. Toxicology Letters. 2010;198:263–271. doi: 10.1016/j.toxlet.2010.07.006. [DOI] [PubMed] [Google Scholar]
- Frescas D, Pagano M. Deregulated proteolysis by the f-box proteins skp2 and beta-trcp: Tipping the scales of cancer. Nature reviews Cancer. 2008;8:438–449. doi: 10.1038/nrc2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble MO, Howard BV, Umans JG, Shara NM, Francesconi KA, Goessler W, et al. Arsenic exposure, diabetes prevalence, and diabetes control in the strong heart study. American journal of epidemiology. 2012;176:865–874. doi: 10.1093/aje/kws153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque R, Mazumder DN, Samanta S, Ghosh N, Kalman D, Smith MM, et al. Arsenic in drinking water and skin lesions: Dose-response data from west bengal, india. Epidemiology. 2003;14:174–182. doi: 10.1097/01.EDE.0000040361.55051.54. [DOI] [PubMed] [Google Scholar]
- Ho SY, Tsai YC, Lee MC, Guo HR. Merkel cell carcinoma in patients with long-term ingestion of arsenic. Journal of occupational health. 2005;47:188–192. doi: 10.1539/joh.47.188. [DOI] [PubMed] [Google Scholar]
- IARC IWGotEoCRtH. Arsenic, metals, fibres, and dusts. IARC monographs on the evaluation of carcinogenic risks to humans. 2012;100:11–465. [PMC free article] [PubMed] [Google Scholar]
- James KA, Byers T, Hokanson JE, Meliker JR, Zerbe GO, Marshall JA. Association between lifetime exposure to inorganic arsenic in drinking water and coronary heart disease in colorado residents. Environmental health perspectives. 2015;123:128–134. doi: 10.1289/ehp.1307839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutapon Chayapong Hk, Madhyastha Radha, Madhyastha Queen, Intan Nurrahmah, Yuichi Nakajima, Narantsog Choijookhuu, Yoshitaka Hishikawa, Masugi Maruyama. Arsenic trioxide induces ros activity and DNA damage, leading to g0/g1 extension in skin fibroblasts through the atm-atr-associated chk pathway. Environmental Science and Pollution Research. 2017;24:5316–5325. doi: 10.1007/s11356-016-8215-7. [DOI] [PubMed] [Google Scholar]
- Ma DC, Sun YH, Chang KZ, Ma XF, Huang SL, Bai YH, et al. Selective induction of apoptosis of nb4 cells from g2+m phase by sodium arsenite at lower doses. European journal of haematology. 1998;61:27–35. doi: 10.1111/j.1600-0609.1998.tb01057.x. [DOI] [PubMed] [Google Scholar]
- Matsuzawa SI, Reed JC. Siah-1, sip, and ebi collaborate in a novel pathway for beta-catenin degradation linked to p53 responses. Molecular cell. 2001;7:915–926. doi: 10.1016/s1097-2765(01)00242-8. [DOI] [PubMed] [Google Scholar]
- McCollum G, Keng PC, States JC, McCabe MJ., Jr Arsenite delays progression through each cell cycle phase and induces apoptosis following g2/m arrest in u937 myeloid leukemia cells. The Journal of pharmacology and experimental therapeutics. 2005;313:877–887. doi: 10.1124/jpet.104.080713. [DOI] [PubMed] [Google Scholar]
- McNeely SC, Belshoff AC, Taylor BF, Fan TW, McCabe MJ, Jr, Pinhas AR, et al. Sensitivity to sodium arsenite in human melanoma cells depends upon susceptibility to arsenite-induced mitotic arrest. Toxicology and applied pharmacology. 2008;229:252–261. doi: 10.1016/j.taap.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghaddaskho F, Eyvani H, Ghadami M, Tavakkoly-Bazzaz J, Alimoghaddam K, Ghavamzadeh A, et al. Demethylation and alterations in the expression level of the cell cycle-related genes as possible mechanisms in arsenic trioxide-induced cell cycle arrest in human breast cancer cells. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2017;39 doi: 10.1177/1010428317692255. 1010428317692255. [DOI] [PubMed] [Google Scholar]
- Muenyi CS, Trivedi AP, Helm CW, States JC. Cisplatin plus sodium arsenite and hyperthermia induces pseudo-g1 associated apoptotic cell death in ovarian cancer cells. Toxicological sciences: an official journal of the Society of Toxicology. 2014;139:74–82. doi: 10.1093/toxsci/kfu029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitani H, Lygerou Z. Control of DNA replication licensing in a cell cycle. Genes to cells: devoted to molecular & cellular mechanisms. 2002;7:523–534. doi: 10.1046/j.1365-2443.2002.00544.x. [DOI] [PubMed] [Google Scholar]
- Nohara K, Ao K, Miyamoto Y, Suzuki T, Imaizumi S, Tateishi Y, et al. Arsenite-induced thymus atrophy is mediated by cell cycle arrest: A characteristic downregulation of e2f-related genes revealed by a microarray approach. Toxicological sciences: an official journal of the Society of Toxicology. 2008;101:226–238. doi: 10.1093/toxsci/kfm268. [DOI] [PubMed] [Google Scholar]
- Parvez F, Chen Y, Brandt-Rauf PW, Bernard A, Dumont X, Slavkovich V, et al. Nonmalignant respiratory effects of chronic arsenic exposure from drinking water among never-smokers in bangladesh. Environmental health perspectives. 2008;116:190–195. doi: 10.1289/ehp.9507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi J, Kumagai Y, Sun G, Yamauchi H, Yoshida T, Iso H, et al. Decreased serum concentrations of nitric oxide metabolites among chinese in an endemic area of chronic arsenic poisoning in inner mongolia. Free radical biology & medicine. 2000;28:1137–1142. doi: 10.1016/s0891-5849(00)00209-4. [DOI] [PubMed] [Google Scholar]
- Pi J, Diwan BA, Sun Y, Liu J, Qu W, He Y, et al. Arsenic-induced malignant transformation of human keratinocytes: Involvement of nrf2. Free Radic Biol Med. 2008;45:651–658. doi: 10.1016/j.freeradbiomed.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozo-Molina G, Ponciano-Gomez A, Rivera-Gonzalez GC, Hernandez-Zavala A, Garrido E. Arsenic-induced s phase cell cycle lengthening is associated with ros generation, p53 signaling and cdc25a expression. Chemico-biological interactions. 2015;238:170–179. doi: 10.1016/j.cbi.2015.06.040. [DOI] [PubMed] [Google Scholar]
- Ren X, McHale CM, Skibola CF, Smith AH, Smith MT, Zhang L. An emerging role for epigenetic dysregulation in arsenic toxicity and carcinogenesis. Environmental health perspectives. 2011;119:11–19. doi: 10.1289/ehp.1002114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AH, Steinmaus CM. Health effects of arsenic and chromium in drinking water: Recent human findings. Annual review of public health. 2009;30:107–122. doi: 10.1146/annurev.publhealth.031308.100143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- States JC, Reiners JJ, Jr, Pounds JG, Kaplan DJ, Beauerle BD, McNeely SC, et al. Arsenite disrupts mitosis and induces apoptosis in sv40-transformed human skin fibroblasts. Toxicology and applied pharmacology. 2002;180:83–91. doi: 10.1006/taap.2002.9376. [DOI] [PubMed] [Google Scholar]
- Sun Y, Pi J, Wang X, Tokar EJ, Liu J, Waalkes MP. Aberrant cytokeratin expression during arsenic-induced acquired malignant phenotype in human hacat keratinocytes consistent with epidermal carcinogenesis. Toxicology. 2009;262:162–170. doi: 10.1016/j.tox.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BF, McNeely SC, Miller HL, Lehmann GM, McCabe MJ, Jr, States JC. P53 suppression of arsenite-induced mitotic catastrophe is mediated by p21cip1/waf1. The Journal of pharmacology and experimental therapeutics. 2006;318:142–151. doi: 10.1124/jpet.106.103077. [DOI] [PubMed] [Google Scholar]
- Treas J, Tyagi T, Singh KP. Chronic exposure to arsenic, estrogen, and their combination causes increased growth and transformation in human prostate epithelial cells potentially by hypermethylation-mediated silencing of mlh1. The Prostate. 2013;73:1660–1672. doi: 10.1002/pros.22701. [DOI] [PubMed] [Google Scholar]
- Tseng WP. Effects and dose–response relationships of skin cancer and blackfoot disease with arsenic. Environmental health perspectives. 1977;19:109–119. doi: 10.1289/ehp.7719109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman GA, Liu X, Loiacono NJ, Kline J, Factor-Litvak P, van Geen A, et al. A cross-sectional study of well water arsenic and child iq in maine schoolchildren. Environmental health: a global access science source. 2014;13:23. doi: 10.1186/1476-069X-13-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. Guidelines for drinking-water quality third edition incorporating the first and second addenda. 2008;1 recommendations. [PubMed] [Google Scholar]
- Wright RO, Amarasiriwardena C, Woolf AD, Jim R, Bellinger DC. Neuropsychological correlates of hair arsenic, manganese, and cadmium levels in school-age children residing near a hazardous waste site. Neurotoxicology. 2006;27:210–216. doi: 10.1016/j.neuro.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Marshall G, Ferreccio C, Steinmaus C, Liaw J, Bates M, et al. Kidney cancer mortality: Fifty-year latency patterns related to arsenic exposure. Epidemiology. 2010;21:103–108. doi: 10.1097/EDE.0b013e3181c21e46. [DOI] [PubMed] [Google Scholar]
- Zhang A, Feng H, Yang G, Pan X, Jiang X, Huang X, et al. Unventilated indoor coal-fired stoves in guizhou province, china: Cellular and genetic damage in villagers exposed to arsenic in food and air. Environmental health perspectives. 2007;115:653–658. doi: 10.1289/ehp.9272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Ohnishi K, Shigeno K, Fujisawa S, Naito K, Nakamura S, et al. The induction of apoptosis and cell cycle arrest by arsenic trioxide in lymphoid neoplasms. Leukemia. 1998;12:1383–1391. doi: 10.1038/sj.leu.2401112. [DOI] [PubMed] [Google Scholar]
- Zheng N, Schulman BA, Song L, Miller JJ, Jeffrey PD, Wang P, et al. Structure of the cul1-rbx1-skp1-f boxskp2 scf ubiquitin ligase complex. Nature. 2002;416:703–709. doi: 10.1038/416703a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.