

Abstract
The outermost layer of Gram negative bacteria is composed of a lipopolysaccharide (LPS) network that forms a dense protective hydrophilic barrier against entry of hydrophobic drugs. At low μM concentrations, a large family of cationic polypeptides known as antimicrobial peptides (AMPs) is able to penetrate the LPS layer and permeabilize the outer membrane (OM) and the cytoplasmic membrane (CM), causing cell death. Cecropin A is a well-studied cationic AMP from moth. Here a battery of time-resolved, single-cell microscopy experiments explores how deletion of sugar layers and/or phosphoryl negative charges from the core oligosaccharide layer (core OS) of K12 E. coli alters the timing of OM and CM permeabilization induced by Cecropin A. Deletion of sugar layers, or phosphoryl charges, or both from the core OS shortens the time to the onset of OM permeabilization to periplasmic GFP and also the lag time between OM permeabilization and CM permeabilization. Meanwhile, the 12-hr minimum inhibitory concentration (MIC) changes only twofold with core OS alterations. The results suggest a two-step model in which the core oligosaccharide layers act as a kinetic barrier to penetration of Cecropin A to the lipid A outer leaflet of the OM. Once a threshold concentration has built up at the lipid A leaflet, nucleation occurs and the OM is locally permeabilized to GFP and, by inference, to Cecropin A. Whenever Cecropin A permeabilizes the OM, CM permeabilization always follows, and cell growth subsequently halts and never recovers on the 45 min observation timescale.
Keywords: Antimicrobial peptides, Cecropin A, lipopolysaccharide layer, core oligosaccharide mutations, single-cell timing measurements, deep rough mutants
Graphical abstract
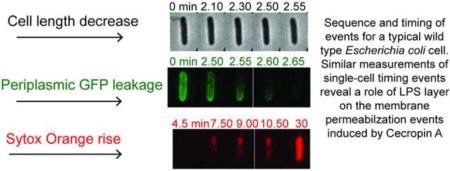
1. Introduction
Gram negative bacteria such as E. coli are notoriously difficult to kill with hydrophobic drugs due to the lipopolysaccharide (LPS) layer that forms the outer leaflet of the outer membrane (OM) [1–4]. This dense polysaccharide layer acts as a barrier to penetration by drugs and to dissolution by neutral and anionic detergents. In general, the LPS layer of E. coli comprises three sub-layers (Fig. 1): the outermost, structurally diverse O-antigen; the core oligosaccharide (core OS), a short chain of sugar residues with two anionic phosphoryl substituents; and lipid A, a hydrophobic membrane anchor which includes two additional phosphoryl charges [5]. E. coli strains lacking an O-antigen layer, such as the K12 strain studied here, are termed rough mutants [6]. The density and impermeability of the LPS layer evidently relies on the presence of divalent cations Mg2+ and Ca2+, which serve to stabilize the phosphate charges in both the core OS and lipid A and form electrostatic links across molecules [1, 4]. It is well known that polycationic agents such as polymyxins and polylysines at sub-MIC concentrations, as well as chelating agents that bind Mg2+ and Ca2+, can enhance drug permeability [4]. The interpretation is that displacement of the stabilizing divalent cations from the LPS layer facilitates access of the drugs to the hydrophobic tail region of the lipid A leaflet, lowering the threshold drug concentration for penetration of the OM and access to the periplasm. At sufficiently high concentration, many cationic agents by themselves can penetrate the OM, enter the periplasm, and permeabilize the cytoplasmic membrane (CM), eventually causing a variety of downstream events leading to cell death [7].
Figure 1.
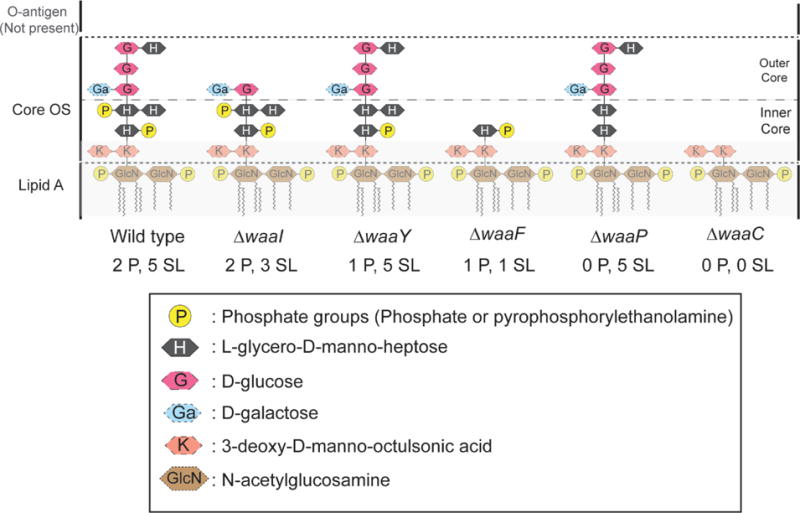
Cartoon representation of the LPS layer of E. coli K12. Each mutation of WT BW25113 (Table 1) alters the core OS sugar layers and phosphoryl charges as shown. The notation (n P, m SL) denotes the presence of n phosphoryl charges on heptose layers and m total sugar layers above the Kdo layer. Adapted from Ref. [8].
Antimicrobial peptides (AMPs, also known as host-defense peptides) comprise a large family of short polypeptides (usually <40 amino acids, with net positive charge of +1 to +12) that display wide-ranging antibacterial activity against both Gram-negative and Gram-positive bacteria without causing significant damage to eukaryotic cells [9]. The polyanionic nature of the outer layer of the cell envelope is believed to explain the selective activity of cationic AMPs against bacterial cells. In contrast, eukaryotic cells carry minimal negative charge in the outer leaflet of the plasma membrane. For example, the red blood cells used in standard toxicity assays have no anionic lipids in the outer leaflet [10].
We have used single-cell fluorescence microscopy to resolve in time individual steps in the attack of AMPs on live bacterial cells [7]. Our studies of LL-37 [11, 12], Cecropin A [13], CM15 [14], and melittin [15] have focused on K12 E. coli. In the typical experiment, plated cells that export GFP to the periplasm are growing normally in a continuous flow of fresh, aerated medium. At t = 0, we introduce a constant concentration of AMP into the flow. For each cell, the microscopy then reveals the timing of events including the onset of permeabilization of the OM to periplasmic GFP, the onset of cell shrinkage, and the onset of permeabilization of the CM to a DNA stain in the Sytox family. The same experiments directly determine the duration of the leakage of GFP out of the periplasm and the duration of the entry of Sytox into the cytoplasm, providing some measure of the degree of OM and CM permeability induced by the AMP.
Here we undertake a systematic study of the effect of alterations of the core OS layer of K12 E. coli on the timing and degree of membrane permeabilization events induced by the cationic AMP Cecropin A. Cecropin A (KWKLFKKIEKVGQNIRDGIIKAGPAVAVVGQATQIAKNH2) is a 37-aa, net +6 charge peptide from moth, with positive residues concentrated near the N-terminus and hydrophobic residues concentrated near the C-terminus of the chain [16]. Early studies of the effects of Cecropin A on phospholipid vesicles [17, 18] and on E. coli cells [19] implicated membrane permeabilization as a key step in the mechanism of antibacterial action. Here we study the effects of OM perturbations on the timing of OM and CM permeabilization events.
The core OS alterations under study (Fig. 1) include truncation of some of the five sugar layers outside the Kdo layer, deletion of one or both of the phosphoryl charges on the heptose layers, or combinations of these. Each alteration is caused by a particular deletion mutation within the waa operon of E. coli, as summarized in Table 1. The specific effects of each mutation have been well characterized by detailed biochemical studies that have elucidated the sequence of steps in LPS biosynthesis (summarized in Fig. S1) [20–23].
Table 1.
E. coli strains.
| E. coli strain | Genotypes and phenotypes |
|---|---|
| BW25113 GFP1 WT GFP (2 P, 5 SL) |
K12 strain, no O-antigen, core OS with 2 heptose layers and 3 glucose layers above Kdo layer, both heptose layers phosphorylated |
| JW3605-1 GFP1 ΔwaaP GFP (0 P, 5 SL) |
BW25113 derivative, core OS with 2 heptose layers (HepIII missing) and 3 glucose layers above Kdo layer, core OS dephosphorylated |
| JW3596-1 GFP1 ΔwaaC GFP (0 P, 0 SL) |
BW25113 derivative, core OS lacking all five sugar layers above Kdo layer, core OS dephosphorylated |
| BW25113 WT (2 P, 5 SL) |
K12 strain, no O-antigen, core OS with 2 heptose layers and 3 glucose layers above Kdo layer, both heptose layers phosphorylated |
| JW3602-1 ΔwaaI (2 P, 3 SL) |
BW25113 derivative, core OS with 2 heptose layers and 1 glucose layer above Kdo layer, both heptose layers phosphorylated |
| JW3600-1 ΔwaaY (1 P, 5 SL) |
BW25113 derivative, core OS with 2 heptose layers and 3 glucose layers above Kdo layer, phosphorylation at HepII missing |
| JW3592-1 ΔwaaF (1 P, 1 SL) |
BW25113 derivative, core OS with 1 phosphorylated heptose layer above Kdo layer |
These strains were transformed with a plasmid expressing GFP that is exported to the periplasm for two-color imaging experiments. See Methods for details of strain construction.
We find that deletion of sugar layers or of phosphate charges from the core OS shortens both the time to the onset of OM permeabilization and also, surprisingly, the lag time between OM and CM permeabilization. Effects of deletion of both sugar layers and phosphate charges on these same timescales are roughly multiplicative. Somewhat paradoxically, the same deletions have only a minor effect on the 12-hr minimal inhibitory concentration (MIC). We interpret the data in terms of a two-step model. First, Cecropin A must penetrate the core OS layer to reach the lipid A leaflet of the OM. Sugar layers and phosphate charges within the core OS present a kinetic barrier to passage of Cecropin A, presumably due to hydrogen bonding and Coulombic interactions, respectively. Second, when a threshold concentration of Cecropin A has built up at the lipid A leaflet, a nucleation event can occur, leading to abrupt and localized OM disruption. Removal of sugar layers and of phosphate charges not only hastens the transit time of Cecropin A through the core OS, but also enhances the degree of permeability of the OM to GFP (and presumably to Cecropin A) once the disruption event has occurred. The two-step model helps to explain why the MIC is rather insensitive to core OS alterations.
2. Materials and methods
2.1 Bacterial strains, materials and growth conditions
The strains used in this study were first obtained from the Keio collection [24] of the Coli Genetic Stock Center (Yale University) maintained at the Weibel lab at UW-Madison (Table 1). Figure 1 depicts schematically the LPS layer structure of these strains [20–23]. E. coli BW25113, which is the parental WT strain in this study, has the typical K12 LPS structure. The core OS has both the Hep I and the Hep II sugars phosphorylated. For simplicity, in this paper we label each strain using the notation (n P, m SL) to identify a strain having n phosphate groups and m sugar layers above the Kdo layer in its core OS. The Kdo layer with its two negative phosphoryl charges remains constant for all strains. For example, since the WT strain has two phosphate groups in the core OS and 5 layers of sugars above Kdo (Hep I, Hep II, Glc I, Glc II, and Glc III), it is denoted WT (2 P, 5 SL). The other strains under study have fewer phosphate groups, fewer sugar layers, or both, as summarized in Table 1 and Fig. 1.
For experiments that monitor loss of periplasmic GFP, in addition to the waa mutation the strains were modified to include plasmid pJW1 as previously described [25]. This plasmid expresses the protein TorA-GFP. TorA-GFP has a 43 residue sequence from trimethylamine N-oxide (TMAO) reductase that signals the twin-arginine translocation (TAT) pathway [26]. TAT recognizes the signal and exports the properly folded GFP to the periplasm, cleaving the signal peptide in the process. The strains including periplasmic GFP are denoted WT GFP (2 P, 5 SL), GFP (0 P, 5 SL), and GFP (0 P, 0 SL).
Bulk cultures were grown in a chemically defined rich medium EZRDM [27], which contains a MOPS-buffered solution supplemented with metal ions (M2130; Teknova), glucose (2 mg/mL), amino acids and vitamins (M2104; Teknova), nitrogenous bases (M2103; Teknova), 1.32 mM K2HPO4, and 76 mM NaCl. Cell cultures were grown overnight at 30°C to stationary phase from glycerol frozen stocks. Subcultures were grown to exponential phase (OD = 0.2–0.6 at 600 nm) before sampling for imaging. The doubling times for all the strains studied are listed in Table 2.
Table 2.
Bulk doubling times and 12-hour MIC values for Cecropin A.1
| Strain | Doubling (min) time | MIC (μM) |
|---|---|---|
| WT (2 P, 5 SL) | 56 ± 2 | 0.5 |
| WT GFP (2 P, 5 SL) | 52 ± 2 | 0.5 |
| ΔwaaP GFP (0 P, 5 SL) | 69 ± 2 | 0.5 |
| ΔwaaC GFP (0 P, 0 SL) | 56 ± 3 | 0.5 |
| ΔwaaI (2 P, 3 SL) | 58 ± 1 | 0.25 |
| ΔwaaY (1 P, 5 SL) | 56 ± 2 | 0.25 |
| ΔwaaF (1 P, 1 SL) | 52 ± 1 | 0.25 |
In EZRDM growth medium at 30°C.
Cecropin A (Anaspec, catalog no. 24010, > 95% purity) was purchased as lyophilized powder and used without further purification. Sterile, ultrapure (18 MΩ) water was used to make peptide stock solutions. The DNA stain Sytox Orange (S11368) was purchased from Thermo-Fisher Scientific. The minimum inhibitory concentration (MIC) for Cecropin A against each strain was determined using a broth microdilution method as previously described [12]. The 12-hr MIC for strains, which were reproducible in three trials, are listed in Table 2.
2.2 Microfluidics Chamber for Time-Lapse Imaging
Single-cell imaging experiments were performed in a microfluidics chamber described earlier [28]. The chamber consists of a single rectilinear channel of uniform height of 50 μm and width of 6 mm, with a channel length of 11 mm. The total chamber volume is ∼10 μL. The glass coverslip was cleaned first in acetone and then through plasma oxidation. A polydimethylsiloxane (PDMS) slab containing the chamber pattern was then bonded to the coverslip. After chamber assembly, 0.01% poly-L-lysine (molecular weight >150,000 Da) was injected through the chamber for 30 min, followed by thorough rinsing with Millipore water. During imaging, the chamber was maintained at 30°C by a TC-344B dual channel temperature controller and CC-28 heating cables attached to RH-2 heater blocks (Warner Instruments). Injection of the solutions into the flow chamber was performed with a syringe pump using 1 mL NORMJECT injection syringes. The flow of fresh, aerated medium is continuous throughout the observation. This maintains a stable external bulk Cecropin A concentration and enables normal cell growth until the AMP disrupts cell membranes.
2.3 Microscopy
The single-cell imaging experiments were performed on a Nikon Eclipse Ti inverted microscope with a 100X, 1.45 N.A. phase contrast objective (Nikon). Images were further magnified 1.5X using the in-built magnification lens of the microscope. The pixel size corresponds to 106 ± 10 nm at the sample. GFP was imaged using 488 nm excitation (Coherent Sapphire laser). Sytox Orange was imaged using a 561 nm excitation (Coherent Sapphire laser). Both lasers were expanded to illuminate the field of view uniformly. Fluorescence images were detected by an EMCCD camera Andor iXon 897.
For the two-color imaging experiments, the E. coli strains transformed with plasmid pJW1 export GFP to the periplasm. Cecropin A-induced outer membrane (OM) permeabilization to GFP and cytoplasmic membrane (CM) permeabilization to the DNA stain Sytox Orange were imaged as green and red fluorescence, respectively. Emission filter HQ510/20 (Chroma) and HQ600/50M (Chroma) were used for the green and the red channel respectively. Phase contrast images enable monitoring of cell length vs time to ±50 nm precision. Interleaved images of green channel fluorescence, red channel fluorescence, and phase contrast were obtained (3 s per complete cycle) with an exposure time of 50 ms/frame. For two-color imaging, μManager software was used for data acquisition and switching of filters between frames using a LB10-NW filter wheel (Sutter). At t = 0, EZRDM with Cecropin A and 5 nM Sytox Orange is injected into the flow chamber. Cells were typically imaged for 30–45 min after injection. The sequence of permeabilization events caused by Cecropin A is monitored using the change in intensity and spatial distribution of periplasmic GFP and Sytox Orange over time. When both OM and CM are permeabilized to Sytox Orange, the stain gains access to the cytoplasm, binds to the chromosomal DNA, and becomes fluorescent. We show below that the onset of OM permeabilization to GFP is nearly coincident in time with the onset of shrinkage of cell length.
The one-color imaging experiments used red fluorescence from Sytox Orange to observe CM permeabilization. Phase contrast images again monitor cell length vs time. Time-lapse movies of 45-min duration were obtained, with red channel fluorescence and phase contrast images interleaved (3 s per complete cycle) and an exposure time of 50 ms/frame. Emission filter HQ605/75 (Chroma) was used for the red channel.
Faster, one-color imaging imaging experiments (fluorescence only) were carried out to identify the spatial pattern of OM permeabilization to GFP or of CM permeabilization to Sytox Orange [13]. Time-lapse movies of either green channel fluorescence from periplasmic GFP or red channel fluorescence from Sytox Orange were obtained at 0.5 s per cycle with an exposure time of 50 ms/frame.
2.4 Data Analysis
Andor Solis (version 4.8.3002.0) was used to acquire the images in one-color experiments. In two-color imaging experiments, images were acquired using the software μManager (version 1.4) [29]. Image J (version 1.49m) and Microcal Origin (version 9.0) were used for the data analysis. The intensity analysis was performed using a custom macro code written in ImageJ. For measuring the total fluorescence intensity in a single cell, a region of interest (ROI) enclosing the cell was drawn and the background signal was subtracted by duplicating the same ROI on an area that did not have a cell. For the intensity analysis of the periplasmic GFP signal, the time point at which the total GFP intensity of the cell minus the background intensity begins to decrease is defined as tOM for that cell; this marks the onset of GFP leakage out of the periplasm. The duration of GFP leakage through the OM to the cell surround is characterized by ΔtGFP, defined as the time taken for the GFP signal to decrease by 90% from its peak value. Similarly, for the red channel, the time point at which the background corrected Sytox Orange signal begins to rise is defined as tCM for that cell. The duration of Sytox leakage through the CM is characterized by ΔtSytox, defined as the interval over which the Sytox Orange intensity increased from 10% to 90% of its final value. Single-cell imaging enables direct determination of the lag time between OM and CM permeabilization, (tCM – tOM). The measurement process was repeated for each well-plated, well isolated cell in the field of view of the image. Transverse intensity linescans of variable width were drawn along the short y-axis of the cell to obtain information on the spatial distribution of GFP intensity.
The phase contrast images enable measurement of the tip-to-tip cell length with a precision of ±50 nm. As observed before for Cecropin A and for LL-37 [11–13], cell shrinkage consistently begins at essentially the same time (within 15 s) as the onset of loss of periplasmic GFP. In addition, for rhodamine-labeled LL-37, the degree of shrinkage closely tracked the build-up of the AMP within the periplasm. For Cecropin A, the ultimate degree of shrinkage is about 25% in length. In recent work on melittin [15], we suggested that the gradual shrinkage is caused by binding of the cationic AMP to anionic peptide crosslinks within the peptidoglycan (PG) layer. This stiffens the PG layer, enabling it to counteract the turgor pressure while axially stretched to a lesser degree. For the one-color plus phase contrast imaging of three peptide strains, we lack the GFP signal. Instead, we take the time of onset of cell shrinkage tshrink as a proxy for tOM and the duration of the shrinkage event Δtshrinkage as a proxy for ΔtGFP. In addition, we take (tCM – tshrink) as a proxy for (tCM – tOM). This is justified quantitatively using shrinkage and GFP data from the two-color measurements, as detailed in Figs. S2 and S3.
3. Results
3.1 Effects of mutations ΔwaaP (0 P, 5 SL) and ΔwaaC (0 P, 0 SL) on timing of OM and CM permeabilization
We began with two-color studies comparing WT GFP with the mutant strains GFP (0 P, 5 SL) and GFP (0 P, 0 SL). As seen in Fig. 1, these have the most profoundly altered core OS layer structure compared with the parent WT (2 P, 5 SL) strain. The MICs for the variants of the three strains expressing periplasmic GFP are all the same, 0.5 μM (Table 2). We carried out two-color, single-cell, time resolved experiments on the three strains at two different Cecropin A concentrations, 0.5 μM (1X MIC) and 0.25 μM (0.5X MIC). These experiments involve collection of sequential, time-resolved snapshots from three different channels: green fluorescence from GFP (488 nm excitation), red fluorescence from Sytox Orange (561 nm excitation), and phase contrast (cell length). Interleaved movies of snapshots with a total cycle time of 3 s are obtained for a time duration of ~30 min after the injection of Cecropin A. The timing of cell shrinkage, OM permeabilization to GFP, and CM permeabilization to Sytox Orange is determined for each individual cell. The cells are classified as “non-septating” or “septating” based on the morphology observed by phase contrast and periplasmic GFP images. The early stages of septation can be visually identified on the basis of a slight depression in an otherwise straight cylindrical cell body.
3.1.1 Cecropin A at 0.5 μM (1X MIC)
A montage of snapshots in the phase contrast, GFP, and Sytox Orange channels during the attack of Cecropin A at 0.5 μM on a typical WT GFP (2 P, 5 SL), non-septating cell is shown in Fig. 2A, B, and C. The onset of flow of Cecropin A is defined as t = 0. Plots of normalized total fluorescence intensity vs time reveal the onset of leakage of periplasmic GFP through the OM to the cell surround (tOM) and the onset of leakage of Sytox Orange through the CM to the cytoplasm (tCM, Fig. 2D). For the particular cell in Fig. 2, the plot of normalized cell length vs time reveals that cell shrinkage begins at tshrink = 2.1 min = 126 s. For the same cell, tOM = 2.3 min = 138 s and tCM = 4.7 min = 282 s. The single-cell lag time between OM and CM permeabilization is (tCM – tOM) = 2.4 min = 144 s. The duration of leakage of periplasmic GFP through the OM is ΔtGFP = 48 s; the duration of cell shrinkage is Δtshrinkage = 66 s; and the duration of leakage of Sytox Orange through the CM is ΔtSytox = 23.0 min = 1380 s.
Figure 2.
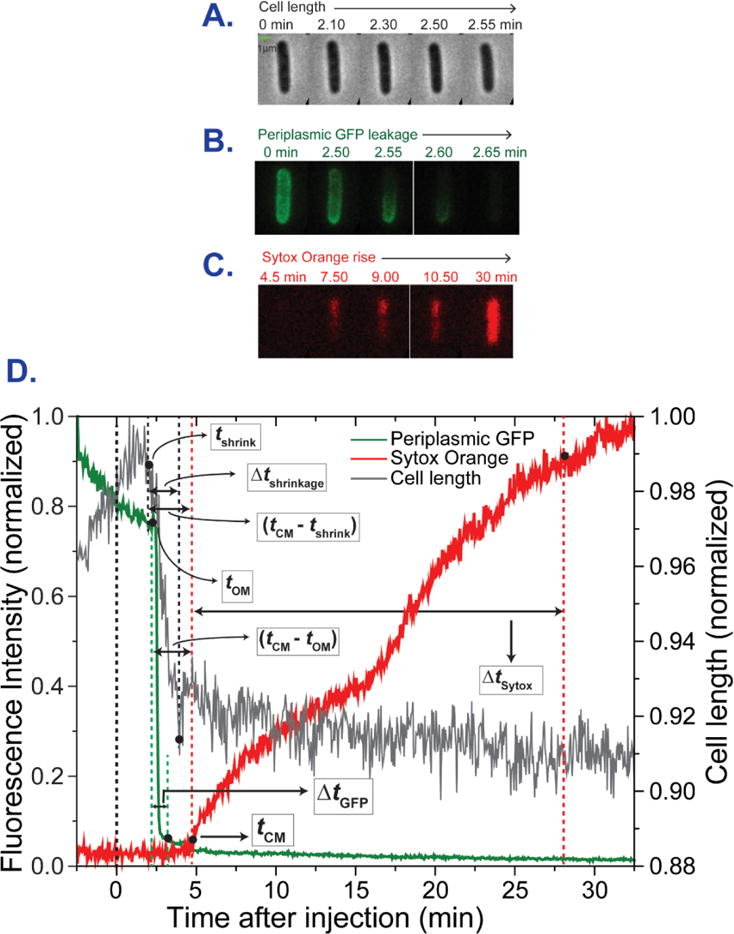
Sequence and timing of events for a typical WT GFP, non-septating cell subjected to a constant flow of 0.5 μM Cecropin A and 5 nM Sytox Orange beginning at t = 0. Three-channel, two-color imaging with 3 s cycle time. (A) Phase contrast images. (B) Green (GFP) fluorescence images. (C) Red (Sytox Orange) fluorescence images. (D) Plots of cell length (from phase contrast images), total green fluorescence (GFP), and total red fluorescence (Sytox Orange) vs time. The single-cell timing events described in the text are extracted from such plots.
Analogous timing measurements at 0.5 μM Cecropin A were carried out for dozens of cells each for all three strains. At 0.5 μM, all the cells in the field of view for the three strains exhibited both OM and CM permeabilization within the 30 min movie duration. All the cells exhibited abrupt shrinkage in cell length by ~25% (measured from the phase contrast images) beginning at essentially the same time as the onset of leakage of periplasmic GFP. Once the cells are permeabilized, cell growth never recovers.
As in the earlier study of Cecropin A, there is substantial cell-to-cell variability in the times tOM, tCM, and (tCM – tOM). The resulting timing mean values and standard errors of the mean are displayed as bar graphs in Fig. 3. Detailed numerical values are collected in Table S1. The data are presented as the mean across all cells and also as separate means for septating and non-septating cells. The same Cecropin A solution was used for all these experiments, so that the concentration was well controlled and timing comparisons across strains are valid.
Figure 3.
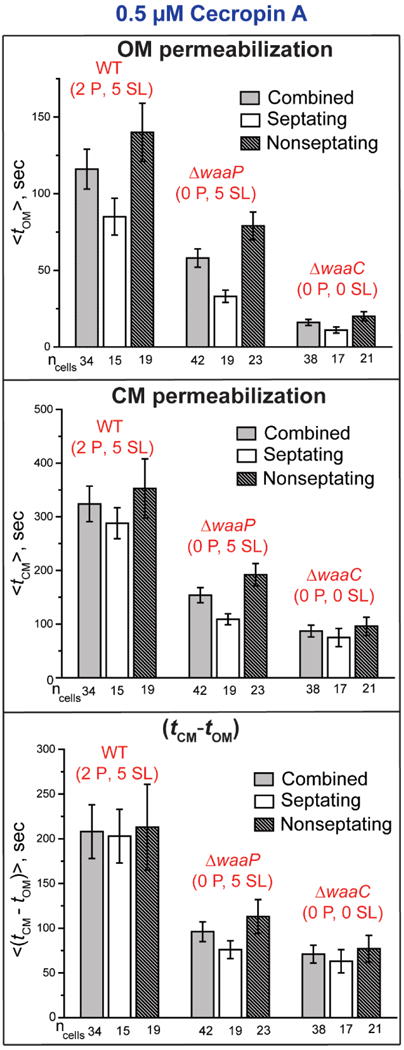
Comparison of the mean timing of onset of OM and CM permeabilization events (from two-color GFP and Sytox Orange imaging, respectively) at 0.5 μM Cecropin A across the strains WT GFP (2 P, 5 SL), GFP (0 P, 5 SL), and GFP (0 P, 0 SL). For each strain, data are shown for all cells and for septating and non-septating cells. Error bars are ± 1 SEM for the number of cells indicated. See Table S1 for detailed numerical values.
The mean across all cells <tOM> for the WT GFP (2 P, 5 SL), GFP (0 P, 5 SL), and GFP (0 P, 0 SL) strains is 116 ± 13 s (± standard error of the mean), 58 ± 6 s, and 16 ± 2 s, respectively (Table S1). Removal of two anionic phosphate groups from the core OS decreases <tOM> by a factor of two. Removal of two anionic phosphate groups and five sugar layers decreases <tOM> by a factor of seven. In all three strains, the OM of septating cells is permeabilized significantly earlier than the OM of non-septating cells. In addition, the mean duration of GFP leakage <ΔtGFP > is 93 ± 7 s, 68 ± 7 s, and 48 ± 4 s for the WT GFP (2 P, 5 SL), GFP (0 P, 5 SL), and GFP (0 P, 0 SL), respectively (Table S1). GFP leaks across the OM significantly more rapidly as phosphates are removed; additional removal of sugar layers has a modest additional effect. For each strain, the leakage duration for GFP is quite similar for septating and non-septating cells.
From the Sytox Orange results, the mean <tCM> for the WT GFP (2 P, 5 SL), GFP (0 P, 5 SL), and GFP (0 P, 0 SL) strains is 324 ± 33 s, 154 ± 14 s, and 87 ± 11 s, respectively (Table S1). Removal of two anionic phosphate groups from the core OS decreases <tCM> by a factor of two. Removal of two anionic phosphate groups and five sugar layers decreases <tCM> by a factor of 3.7. Differences in <tCM> for septating vs non-septating cells are modest. In addition, the mean duration of leakage of Sytox Orange <ΔtSytox> is 953 ± 59 s, 832 ± 30 s, and 992 ± 83 s for the WT GFP (2 P, 5 SL), GFP (0 P, 5 SL), and GFP (0 P, 0 SL) cells, respectively (Table S1). These differences are minor.
Because OM permeabilization always precedes CM permeabilization, <tCM> is dependent on <tOM> for the different strains. More informative is the mean lag time <(tCM – tOM)> between OM permeabilization and CM permeabilization. The two-color measurements provide this quantity for each cell, enabling evaluation of the single-cell average lag time. Separate measurements of two distributions using different batches of cells would yield the difference <tCM> – <tOM>. The mean across all cells <(tCM – tOM)> is 208 ± 30 s, 96 ± 11 s, and 71 ± 10 s for the WT GFP (2 P, 5 SL), GFP (0 P, 5 SL), and GFP (0 P, 0 SL) strains, respectively (Fig. 3, Table S1). Removal of two anionic phosphate groups from the core OS decreases the lag time by a factor of two. Removal of two anionic phosphate groups and five sugar layers decreases the lag time by a factor of three. It is somewhat surprising that alterations of the OM affect the lag time between OM permeabilization and subsequent CM permeabilization. Intriguingly, there is a strong positive correlation between the lag time <(tCM – tOM)> and the timescale <ΔtGFP> of leakage of periplasmic GFP across the OM. This is discussed in detail below.
In an earlier study [13], it was observed that Cecropin A induces localized OM permeabilization to GFP and localized CM permeabilization to Sytox in a spatial pattern that depended on whether or not the cell was septating. We briefly explored the possibility that deletion of phosphate charges from the core OS might alter the spatial pattern of permeabilization events. Using faster one-color imaging (0.5 s/frame) of either the loss of GFP from the periplasm or the entry of Sytox Orange into the cytoplasm (Methods), we compared the behavior of the WT (2P, 5 SL) strain and the GFP (0 P, 5 SL) strain. No obvious differences were observed. Representative examples are presented in Figs. S4, S5, S6, and S7.
3.1.2 Cecropin A at 0.25 μM (0.5X MIC)
Previous work showed that the time to the onset of both OM and CM permeabilization lengthens as the Cecropin A concentration decreases [13]. To test for differential effects of concentration due to the core OS alterations, we carried out an analogous battery of two-color experiments on the same three strains at a Cecropin A concentration of 0.25 μM, half the MIC. The mean OM and CM permeabilization timing results are displayed as bar graphs in Fig. 4, with numerical data collected in Table S2. Table 3, a subset of Table S2, compares the key quantities <tOM>, the lag time <(tCM – tOM)>, and the leakage durations <ΔtGFP> and <ΔtSytox> across the three strains at 0.25 μM Cecropin A.
Figure 4.
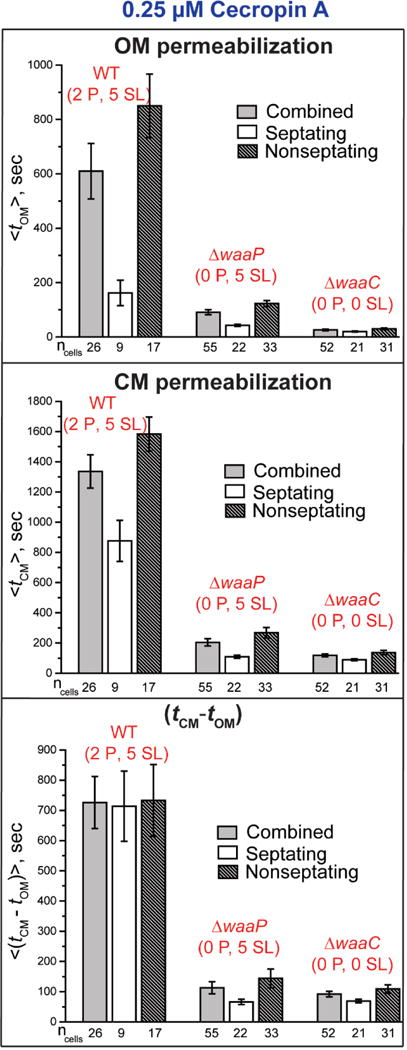
Comparison of the mean timing of onset of OM and CM permeabilization events (from two-color GFP and Sytox Orange imaging, respectively) at 0.25 μM Cecropin A across the strains WT GFP (2P, 5 SL), GFP (0 P, 5 SL), and GFP (0 P, 0 SL). For each strain, data are shown for all cells and for septating and non-septating cells. Error bars are ± 1 SEM for the number of cells indicated. See Table S2 for detailed numerical values.
Table 3.
Mean timing data for OM and CM permeabilization from two-color experiments at 0.25 μM Cecropin A.1
| Strain | <tOM> | < tCM − tOM> | <ΔtGFP> | <ΔtSytox> |
|---|---|---|---|---|
| WT GFP (2 P, 5 SL) | 610 ± 102 | 726 ± 86 | 281 ± 21 | 691 ± 77 |
| ΔwaaP GFP (0 P, 5 SL) | 91 ± 9 | 113 ± 20 | 68 ± 8 | 911 ± 21 |
| ΔwaaC GFP (0 P, 0 SL) | 26 ± 3 | 92 ± 9 | 65 ± 4 | 781 ± 28 |
Values in s, abstracted from the more detailed Table S2. Uncertainties are ±1 SEM.
In spite of all three 12-hr MICs being equal, decreasing the Cecropin A concentration to 0.25 μM had by far the greatest effect on the timing of permeabilization events for the WT GFP strain. At 0.25 μM, only 40% of the WT GFP (2 P, 5 SL) cells in the field of view of the experiments were permeabilized within 30 min of injection of Cecropin A. In sharp contrast, all the GFP (0 P, 5 SL) and all the GFP (0 P, 0 SL) cells were permeabilized within 30 min. When we observed the WT GFP cells for an additional 20 min, even then only 70% of the cells were permeabilized. Out of all the cells which showed OM permeabilization within 50 min, 90% of them showed CM permeabilization as well. Only those cells which showed both OM and CM permeabilization were used to calculate the mean values in Fig. 4 and Tables 3 and S2. The mean values for WT GFP cells at 0.25 μM should be taken as a lower bound on the true mean values.
At 0.25 μM Cecropin A, the mean <tOM> for the WT GFP (2 P, 5 SL), GFP (0 P, 5 SL), and GFP (0 P, 0 SL) strains is 610 ± 102 s, 91 ± 9 s, and 26 ± 3 s, respectively (Table S2). At this sub-MIC concentration, removal of two anionic phosphate groups from the core OS decreases <tOM> by more than a factor of seven. Removal of two anionic phosphate groups and five sugar layers decreases <tOM> by more than a factor of 20. In all three strains, the OM of septating cells is again permeabilized significantly earlier than the OM of non-septating cells. The contrast is especially strong for the WT GFP (2 P, 5 SL) and GFP (0 P, 5 SL) strains. The mean leakage duration <ΔtGFP > of periplasmic GFP through the OM is 281 ± 21 s, 68 ± 8 s, and 65 ± 4 s for the WT GFP (2 P, 5 SL), GFP (0 P, 5 SL), and GFP (0 P, 0 SL) cells, respectively (Table S2). Once again, GFP leaks across the OM significantly more rapidly as phosphoryl charges are removed. Removal of the sugar layers plus the phosphates has only a minor additional effect. Again, within each strain the leakage duration for GFP is quite similar for septating and non-septating cells.
At 0.25 μM Cecropin A, the Sytox Orange data yield the mean times <tCM> for the WT GFP (2 P, 5 SL), GFP (0 P, 5 SL), and GFP (0 P, 0 SL) strains of 1336 ± 110 s, 204 ± 24 s, and 118 ± 9 s, respectively (Table S2). Differences in <tCM> for septating vs non-septating cells are much more pronounced at 0.25 μM than they were at 0.5 μM (Fig. 4). The mean <(tCM – tOM)> is 726 ± 86 s, 113 ± 20 s, and 92 ± 9 s for the WT GFP (2 P, 5 SL), GFP (0 P, 5 SL), and GFP (0 P, 0 SL) strains, respectively (Table S2). Removal of two anionic phosphate groups from the core OS decreases the lag time by a factor of six. Additional removal of five sugar layers has little additional effect.
In Table 3, we compare the quantities <tOM>, <(tCM – tOM)>, and <ΔtGFP> for all three strains at 0.25 μM Cecropin A. At both 0.5 μM (Table S1) and 0.25 μM (Table S2), there is a positive correlation between the lag time <(tCM – tOM)> and the timescale <ΔtGFP> of leakage of periplasmic GFP across the OM. For example, at 0.5 μM the ratio <(tCM – tOM)>/<ΔtGFP> varies from 1.4 to 2.2 across the three strains. We suggest an underlying cause in the Discussion.
3.2 Effects of mutations ΔwaaI (2 P, 3 SL), ΔwaaY (1 P, 5 SL), and ΔwaaF (1 P, 1 SL) on timing of cell shrinkage and CM permeabilization at 0.25 μM Cecropin A
The three additional mutant strains (2 P, 3 SL), (1 P, 5 SL), and (1 P, 1 SL) (all without periplasmic GFP) present a somewhat more subtle set of core OS alterations for comparison with WT (2 P, 5 SL). These three strains had a slightly lower MIC than WT, 0.25 μM vs 0.5 μM (Table 2). In practice, we had difficulty finding protocols for which these strains efficiently exported GFP to the periplasm. Instead of two-color imaging, we carried out one-color imaging experiments using 0.25 μM Cecropin A and 5 nM Sytox Orange in the flow cell. When combined with the phase contrast images, this enabled single-cell measurement of the onset and duration of the cell shrinkage event (tshrink and Δtshrinkage), as well as the Sytox Orange fluorescence onset and leakage duration (tCM and ΔtSytox). The earlier two-color experiments on the GFP-containing strains (Sec. 3.1) also included phase contrast imaging. This enabled us to measure the degree of correlation between tshrink and tOM and between Δtshrinkage and ΔtGFP (Fig. S2 and Fig. S3). The correlation coefficients R2 are 0.99 and 0.73, respectively. Thus in the one-color experiments, we consider tshrink to be a good proxy for tOM and Δtshrinkage to be a semi-quantitative approximation for ΔtGFP.
We chose to use 0.25 μM Cecropin A for WT and the three mutant strains in order to hold the bulk peptide concentration constant across strains. Interleaved snapshots of Sytox Orange fluorescence and phase contrast images with a total cycle time of 3 s were obtained for 45 min after injection of Cecropin A. Similar to the two-color results at 0.25 μM, ~80% of wild-type cells shrank in cell length and were later permeabilized to Sytox Orange within 45 min. The minority of WT (2 P, 5 SL) cells that were not permeabilized kept growing in length. All the cells that shrank never grew again in length. For the (2 P, 3 SL), (1 P, 5 SL), and (1 P, 1 SL) strains, all the cells shrank and were later permeabilized to Sytox Orange within 45 min. After shrinkage, growth never resumed.
For the WT and three mutant strains at 0.25 μM Cecropin A, we measured the single-cell mean quantities <tshrink>, <tCM>, the lag time <(tCM – tshrink)>, the duration of the shrinkage event <Δtshrink>, and the duration of Sytox leakage <ΔtSytox>. These mean timing results are displayed as bar graphs in Fig. 5, with detailed numerical data collected in Table S3 and a summary of key quantities in Table 4. The timing results for the mutant strains GFP (0 P, 5 SL) and GFP (0 P, 0 SL) studied in the two-color experiments at 0.25 μM (Tables 3 and S2, Fig. 4) should not be directly compared with the results for the mutant strains lacking GFP, (2 P, 3 SL), (1 P, 5 SL), and (1 P, 1 SL), studied in the one-color experiments at the same nominal concentration (Tables 4 and S3, Fig. 5). The reason is that the timings for the WT GFP strain are 1.5–2 times longer than the corresponding timings for the WT strain (no GFP). This could be due to use of a different stock solution of Cecropin A; recall that the WT GFP timings were highly sensitive to Cecropin A concentration (Table S1 vs Table S2). Alternatively, there could be real differences in behavior between the strains expressing periplasmic GFP and those that do not. To be cautious, we restrict our comparisons to entries within Table S2 or within Table S3.
Figure 5.
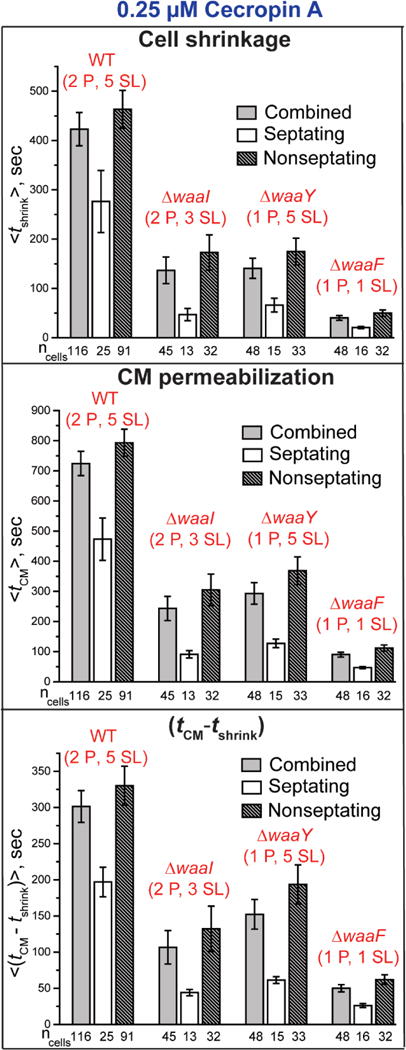
Comparison of the mean timing of the onset of cell shrinkage and CM permeabilization events (from phase contrast and Sytox Orange imaging, respectively) at 0.25 μM Cecropin A across the strains WT (2 P, 5 SL), (2 P, 3 SL), (1 P, 5 SL), and (1 P, 1 SL). For each strain, data are shown for all cells and for septating and non-septating cells. Error bars are ± 1 SEM for the number of cells indicated. See Table S3 for detailed numerical values.
Table 4.
Mean timing data for OM and CM permeabilization from one-color experiments with 0.25 μM Cecropin A.1
| Strain | <tshrink> | < tCM – tshrink> | <Δtshrinkage> | <ΔtSytox> |
|---|---|---|---|---|
| WT (2 P, 5 SL) | 423 ± 34 | 302 ± 22 | 132 ± 5 | 936 ± 43 |
| ΔwaaI (2 P, 3 SL) | 137 ± 27 | 106 ± 23 | 50 ± 4 | 1174 ± 23 |
| ΔwaaY (1 P, 5 SL) | 141 ± 20 | 152 ± 20 | 62 ± 5 | 968 ± 37 |
| ΔwaaF (1 P, 1 SL) | 40 ± 5 | 50 ± 5 | 35 ± 3 | 1186 ± 9 |
Values in s, abstracted from the more detailed Table S3. Uncertainties are ±1 SEM.
As shown in Fig. 5 and Table 4, at 0.25 μM Cecropin A removal of two sugar layers (retaining both phosphates) or removal of one phosphate group (retaining all five sugar layers) from the core OS decreases <tshrink> threefold as compared with the WT results. The same alterations decrease both <tCM – tshrink> and <Δtshrinkage> by a factor of 2-3. Removal of one phosphate and of four sugar layers has a composite effect. This more drastic alteration decreases <tshrink> by 11X, decreases <tCM – tshrink> by 6X, and decreases <Δtshrinkage> by 4X as compared with WT. Meanwhile, the Sytox Orange entry duration <ΔtSytox> remains sensibly constant across all four strains. As observed in the other strains, the septating cells are permeabilized earlier than the non-septating cells.
4. Discussion
Our previous single-cell studies of the attack of a variety of cationic AMPs on E. coli focused on the rough mutant strain MG1655 (K12), the “wild-type” strain in the present study. The AMPs have included LL-37 [11, 12], Cecropin A [13], CM15 [14], and melittin [15]. Each AMP exhibits its own set of behaviors, characterized by localized or global OM and CM permeabilization, evidence of induction of oxidative stress or lack thereof, and permanent or transient membrane permeabilization to GFP. The dependence of the attack of Cecropin A and of LL-37 on bulk AMP concentration supported a model in which a threshold surface concentration of AMP within the LPS layer is required for OM permeabilization [13, 30, 31]. A slow nucleation event then leads to abrupt and localized OM permeabilization. Studies of AMP permeabilization of synthetic lipid vesicles support an analogous nucleation concept [32–36]. The maximum concentration that can be achieved within the LPS layer should increase with bulk AMP concentration. Accordingly, at low bulk AMP concentration, OM permeabilization is not observed and all cells continue to grow. At a sufficiently high bulk AMP concentration, the surface concentration lies above the threshold for some cells, and OM permeabilization occurs for those cells on the observation timescale. For still higher bulk concentrations, OM permeabilization occurs earlier and in a greater percentage of cells, eventually reaching 100% on our observation timescale of 30-45 min. Once the OM of a particular cell is permeabilized to an AMP, we have always observed subsequent CM permeabilization and the permanent halting of cell growth.
In our experiments, the flow of AMP through the observation chamber maintains a constant bulk concentration in the cell surround, providing an effectively limitless supply of AMP. The AMP must attack bacterial cells from the outside in. The present work has explored how the timing of OM and CM permeabilization by Cecropin A varies with mutations that systematically remove 1–2 phosphate groups, or 1–5 sugar layers outside the Kdo layer, or both, from the core OS layer of MG1655 E. coli. At constant Cecropin A concentration of 0.25 μM, removal of one phosphate while leaving all five sugar layers intact, the (1 P, 5 SL) mutant in Table 4, decreases <tshrink> by at least a factor of 3 vs WT (2 P, 5 SL). Removal of two sugar layers while retaining both phosphates, the (2 P, 3 SL) mutant in Table 4, decreases <tshrink> by at least a factor of 2.5 vs WT (2 P, 5 L). The effects of removal of phosphates and removal of sugar layers are roughly multiplicative. Removal of one phosphate and of four sugar layers, the (1 P, 1 SL) mutant in Table 4, decreases <tshrink> by at least a factor of 10. Removal of both phosphates and all five sugar layers, the GFP (0 P, 0 SL) mutant in Table 3, decreases <tOM> by at least a factor of 25 vs WT GFP (2 P, 5 SL).
These same alterations of the OM also decrease the mean time to permeabilization of the CM, <tCM>. Part of this decrease is trivial. Because tOM marks the moment when Cecropin A gains access to the periplasm and begins its attack on the CM, tCM necessarily decreases as tOM decreases. However, the effects on tCM are larger than this. The additional effects are directly measured by the variation of the mean single-cell lag time <tCM – tOM> in the two-color experiments, or its proxy <tCM – tshrink> in the one-color experiments. Surprisingly, at 0.25 μM the mean lag time between OM and CM permeabilization decreases by a factor of 2–3 for removal of one phosphate or two sugar layers. It decreases by a factor of 6–7 for removal of both phosphates, of one phosphate and four sugar layers, or of both phosphates and all five sugar layers (Tables 3 and 4).
The new results suggest that we augment the threshold surface concentration model to include two steps: (1) the transit of the AMP through the core OS layer to reach the lipid A leaflet of the OM bilayer, and (2) the subsequent permeabilization of the OM once sufficient local concentration has built up at the lipid A leaflet to induce a nucleation event. In this picture, the time tOM between introduction of Cecropin A and OM permeabilization to periplasmic GFP (from two-color experiments) can be viewed as a composite of two timescales: tOM = tcore OS + tperm. Here tcore OS denotes the timescale of passage of sufficient Cecropin A through the core OS to cause build a threshold Cecropin A concentration at the lipid A leaflet; tperm is the additional nucleation time required for the permeabilization step to occur once the threshold concentration is achieved. The analogous equation tshrink = tcore OS + tperm would apply to the one-color experiments.
In the simplest view, we would expect tcore OS to depend strongly on both the bulk Cecropin A concentration and on the core OS alterations under study here. Greater bulk concentration feeds Cecropin A molecules into the core OS more rapidly. During transit through the core OS, the cationic Cecropin A peptide presumably binds transiently to both the nucleophilic –OH groups of the sugar layers and to the anionic phosphoryl groups of the heptose layers. Both types of binding interaction slow the passage of Cecropin A through the core OS. Accordingly, deletion of phosphoryl charges and/or sugar layers from the core OS should decrease tcore OS. In contrast, tperm might be expected to be fairly insensitive to the core OS mutations. In the first approximation, the Kdo and lipid A layers are not altered by the mutations studied [20, 21, 37–39]. As Cecropin A molecules penetrate the core OS layer, they bind to the phosphate charges on the lipid A headgroups. When the local concentration reaches a threshold, a nucleation event occurs and the OM is permeabilized. In this view, tperm is determined primarily by the nucleation timescale, not by the nature of the core OS layer. The two-step model attributes the measured changes in <tOM> (or its proxy <tshrink>) with core OS alterations primarily to changes in the transit time tcore OS.
This two-step model may help to explain why deletion of sugar layers and/or phosphate groups from the core OS had only a minor effect on the 12-hr MIC values. The MIC was either 0.5 μM or 0.25 μM for all the strains studied (Table 2). Such insensitivity of the MIC to core OS alterations is not uncommon among AMPs [40]. Cell-to-cell heterogeneity in OM composition and in the phase of the cell cycle suggests that each cell may have its own binding constant for Cecropin A interactions with the lipid A headgroups. Each cell may also have its own threshold concentration of Cecropin A at the lipid A leaflet to enable nucleation of the OM permeabilization step. The MIC is achieved when the bulk Cecropin A concentration becomes sufficient to cause all cells to exceed their threshold concentration at the lipid A leaflet. The primary effect of the core OS alterations is to shorten the timescale on which that threshold is achieved, given sufficient bulk AMP concentration.
Why do alterations of the core OS (part of the OM) strongly affect the lag time between OM and CM permeabilization? We suspect the answer lies in the varying degree of permeability of the OM to Cecropin A for the different alterations. Here we presume that the duration of GFP leakage from the periplasm to the cell surround (<ΔtGFP>) and the duration of the cell shrinkage event (<Δtshrinkage>) are both indicative of the degree of permeability of the OM to Cecropin A itself. These two timescales are positively correlated, as shown in Fig. S3. The cell shrinkage event occurs across all the cationic AMPs we have studied. In recent work on melittin [15], we attributed the shrinkage event to binding of the cationic AMP to the anionic peptide crosslinks of the peptidoglycan (PG) layer. This stiffens the PG layer longitudinally, enabling it to counteract the same turgor pressure at smaller axial extension. If this interpretation is essentially correct, then Δtshrinkage provides a fairly direct measure of the timescale of buildup of AMP concentration within the periplasm. The two-color measurements show that the duration of the GFP leakage event ΔtGFP correlates with Δtshrinkage (Fig. S3); the underlying reasons may be complex.
The data indeed show positive correlation between <tCM – tOM> and <ΔtGFP> in Table 3 and between <tCM – tshrink> and <Δtshrinkage> in Table 4. Our interpretation is that removal of phosphates and/or sugar layers not only hastens the onset of OM permeabilization, but also enhances the degree of permeability of the OM layer to Cecropin A once permeabilization has begun. The faster Cecropin A enters the periplasm, the faster it binds to the CM and the sooner it reaches the threshold surface concentration for CM permeabilization. More rapid entry of Cecropin A into the periplasm for the more severe truncations may arise in part from the shortening of tcore OS, but there may also be a contribution from the degree of leakiness of the localized OM disruption site. Our methods provide little insight into the latter. We observe both the OM and CM permeabilization events to be localized (SI and [13]), but fluorescence microscopy lacks sufficient spatial resolution to distinguish whether the mechanism involves toroidal pore formation or some more disordered, detergent-like process.
In contrast, <ΔtSytox>, the duration of Sytox Orange transit across the CM, remains fairly constant across strains in both the two-color experiments (Table 3) and the one-color experiments (Table 4). Evidently the core OS alterations do not affect the degree of permeability induced in the CM, only the lag time between the onset of OM permeabilization and the onset of CM permeabilization. This suggests that the core OS alterations have little or no effect on the nature of the cytoplasmic membrane disruption, as might be expected.
5. Conclusions
We have shown that deletion of either sugar layers or phosphoryl charges from the K12 E. coli core OS not only hastens the onset of both OM and CM permeabilization by Cecropin A, but also enhances the degree of permeability of the OM once the OM permeabilization event has occurred. The 12-hr MIC is affected rather little by the same deletions. This suggests that the core OS acts primarily as a kinetic barrier to passage of Cecropin A from bulk to the lipid A leaflet.
Supplementary Material
Highlights.
Single-cell, real-time observation of E. coli membrane permeabilization by Cecropin A
Deletion of phosphates or sugar layers from core oligosaccharide shortens timescale
Minimum inhibitory concentration largely unaffected by same mutations
Suggests two-step model of outer membrane permeabilization by cationic peptides
Acknowledgments
We thank Dr. Nambirajan Rangarajan for helpful discussions.
Funding
This research was supported by the National Institute of General Medical Sciences of the National Institutes of Health under awards R01GM094510 (to JCW as PI) and R01GM093265 (to JCW and Samuel Gellman as co-PIs). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- AMP
antimicrobial peptide
- OM
outer membrane
- CM
cytoplasmic membrane
- LPS
lipopolysaccharide
- core OS
core oligosaccharide
- (n P, m SL)
mutant strain with n phosphoryl charges and m sugar layers above the Kdo layer SEM: standard error of the mean
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nikaido H, Vaara M. Molecular basis of bacterial outer membrane permeability. Microbiol Rev. 1985;49:1–32. doi: 10.1128/mr.49.1.1-32.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hancock RE. Role of porins in outer membrane permeability. J Bacteriol. 1987;169:929–933. doi: 10.1128/jb.169.3.929-933.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leive L. The barrier function of the gram-negative envelope. Ann NY Acad Sci. 1974;235:109–129. doi: 10.1111/j.1749-6632.1974.tb43261.x. [DOI] [PubMed] [Google Scholar]
- 4.Vaara M. Agents that increase the permeability of the outer membrane. Microbiol Rev. 1992;56:395–411. doi: 10.1128/mr.56.3.395-411.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raetz CR, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hitchcock PJ, Leive L, Makela PH, Rietschel ET, Strittmatter W, Morrison DC. Lipopolysaccharide nomenclature–past, present, and future. J Bacteriol. 1986;166:699–705. doi: 10.1128/jb.166.3.699-705.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi H, Rangarajan N, Weisshaar JC. Lights, camera, action! Antimicrobial peptide mechanisms imaged in space and time. Trends Microbiol. 2016;24:111–122. doi: 10.1016/j.tim.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakao R, Ramstedt M, Wai SN, Uhlin BE. Enhanced biofilm formation by Escherichia coli LPS mutants defective in Hep biosynthesis. PLoS One. 2012;7:e51241. doi: 10.1371/journal.pone.0051241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brogden KA. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nature Reviews Microbiology. 2005;3:238. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 10.Virtanen JA, Cheng KH, Somerharju P. Phospholipid composition of the mammalian red cell membrane can be rationalized by a superlattice model. Proc Natl Acad Sci U S A. 1998;95:4964–4969. doi: 10.1073/pnas.95.9.4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi H, Yang Z, Weisshaar JC. Oxidative stress induced in Escherichia coli by the human antimicrobial peptide LL-37. PLoS Pathog. 2017;13:e1006481. doi: 10.1371/journal.ppat.1006481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sochacki KA, Barns KJ, Bucki R, Weisshaar JC. Real-time attack on single Escherichia coli cells by the human antimicrobial peptide LL-37. Proc Natl Acad Sci U S A. 2011;108:E77–81. doi: 10.1073/pnas.1101130108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rangarajan N, Bakshi S, Weisshaar JC. Localized permeabilization of Escherichia coli membranes by the antimicrobial peptide Cecropin A. Biochemistry. 2013;52:6584–6594. doi: 10.1021/bi400785j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi H, Yang Z, Weisshaar JC. Single-cell, real-time detection of oxidative stress induced in Escherichia coli by the antimicrobial peptide CM15. Proc Natl Acad Sci U S A. 2015;112:E303–310. doi: 10.1073/pnas.1417703112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang Z, Choi H, Weisshaar JC. Melittin-Induced Permeabilization, Re-sealing, and Re-permeabilization of Escherichia coli Membranes. Biophys J. 2018;114:1–12. doi: 10.1016/j.bpj.2017.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steiner H, Hultmark D, Engström Å, Bennich H, Boman HG. Sequence and specificity of two antibacterial proteins involved in insect immunity. Nature. 1981;292:246. doi: 10.1038/292246a0. [DOI] [PubMed] [Google Scholar]
- 17.Silvestro L, Gupta K, Weiser JN, Axelsen PH. The concentration-dependent membrane activity of Cecropin A. Biochemistry. 1997;36:11452–11460. doi: 10.1021/bi9630826. [DOI] [PubMed] [Google Scholar]
- 18.Gregory SM, Cavenaugh A, Journigan V, Pokorny A, Almeida PF. A quantitative model for the all-or-none permeabilization of phospholipid vesicles by the antimicrobial peptide Cecropin A. Biophys J. 2008;94:1667–1680. doi: 10.1529/biophysj.107.118760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silvestro L, Weiser JN, Axelsen PH. Antibacterial and antimembrane activities of Cecropin A in Escherichia coli Antimicrob. Agents Chemother. 2000;44:602–607. doi: 10.1128/aac.44.3.602-607.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pradel E, Parker CT, Schnaitman CA. Structures of the rfaB, rfaI, rfaJ, and rfaS genes of Escherichia coli K-12 and their roles in assembly of the lipopolysaccharide core. J Bacteriol. 1992;174:4736–4745. doi: 10.1128/jb.174.14.4736-4745.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yethon JA, Heinrichs DE, Monteiro MA, Perry MB, Whitfield C. Involvement of waaY, waaQ, and waaP in the modification of Escherichia coli lipopolysaccharide and their role in the formation of a stable outer membrane. J Biol Chem. 1998;273:26310–26316. doi: 10.1074/jbc.273.41.26310. [DOI] [PubMed] [Google Scholar]
- 22.Genevaux P, Bauda P, DuBow MS, Oudega B. Identification of Tn10 insertions in the rfaG, rfaP, and galU genes involved in lipopolysaccharide core biosynthesis that affect Escherichia coli adhesion. Arch Microbiol. 1999;172:1–8. doi: 10.1007/s002030050732. [DOI] [PubMed] [Google Scholar]
- 23.Yethon JA, Vinogradov E, Perry MB, Whitfield C. Mutation of the lipopolysaccharide core glycosyltransferase encoded by waaG destabilizes the outer membrane of Escherichia coli by interfering with core phosphorylation. J Bacteriol. 2000;182:5620–5623. doi: 10.1128/jb.182.19.5620-5623.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2 doi: 10.1038/msb4100050. 2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sochacki KA, Shkel IA, Record MT, Weisshaar JC. Protein diffusion in the periplasm of Escherichia coli under osmotic stress. Biophys J. 2011;100:22–31. doi: 10.1016/j.bpj.2010.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sargent F. The twin-arginine transport system: moving folded proteins across membranes. Biochem Soc Trans. 2007;35:835–847. doi: 10.1042/BST0350835. [DOI] [PubMed] [Google Scholar]
- 27.Neidhardt FC, Bloch PL, Smith DF. Culture medium for enterobacteria. J Bacteriol. 1974;119:736–747. doi: 10.1128/jb.119.3.736-747.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Christensen B, Fink J, Merrifield RB, Mauzerall D. Channel-forming properties of cecropins and related model compounds incorporated into planar lipid membranes. Proc Natl Acad Sci U S A. 1988;85:5072–5076. doi: 10.1073/pnas.85.14.5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Edelstein A, Amodaj N, Hoover K, Vale R, Stuurman N. Current Protocols In Molecular Biology. John Wiley & Sons, Inc.; 2001. Computer control of microscopes using μManager. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roversi D, Luca V, Aureli S, Park Y, Mangoni ML, Stella L. How many antimicrobial peptide molecules kill a bacterium? The case of PMAP-23. ACS Chem Biol. 2014;9:2003–2007. doi: 10.1021/cb500426r. [DOI] [PubMed] [Google Scholar]
- 31.Starr CG, He J, Wimley WC. Host cell interactions are a significant barrier to the clinical utility of peptide antibiotics. ACS Chem Biol. 2016;11:3391–3399. doi: 10.1021/acschembio.6b00843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang HW, Chen FY, Lee MT. Molecular mechanism of peptide-induced pores in membranes. Phys Rev Lett. 2004;92:198304. doi: 10.1103/PhysRevLett.92.198304. [DOI] [PubMed] [Google Scholar]
- 33.Tamba Y, Yamazaki M. Single giant unilamellar vesicle method reveals effect of antimicrobial peptide Magainin 2 on membrane permeability. Biochemistry. 2005;44:15823–15833. doi: 10.1021/bi051684w. [DOI] [PubMed] [Google Scholar]
- 34.Huang HW. Molecular mechanism of antimicrobial peptides: The origin of cooperativity, Biochimica et Biophysica Acta (BBA) Biomembranes. 2006;1758:1292–1302. doi: 10.1016/j.bbamem.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 35.Hung WC, Chen FY, Lee CC, Sun Y, Lee MT, Huang HW. Membrane-thinning effect of Curcumin. Biophys J. 2008;94:4331–4338. doi: 10.1529/biophysj.107.126888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wimley WC. Describing the mechanism of antimicrobial peptide action with the interfacial activity model. ACS Chem Biol. 2010;5:905–917. doi: 10.1021/cb1001558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen L, Coleman WG., Jr Cloning and characterization of the Escherichia coli K-12 rfa-2 (rfaC) gene, a gene required for lipopolysaccharide inner core synthesis. J Bacteriol. 1993;175:2534–2540. doi: 10.1128/jb.175.9.2534-2540.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schnaitman CA, Klena JD. Genetics of lipopolysaccharide biosynthesis in enteric bacteria. Microbiol Rev. 1993;57:655–682. doi: 10.1128/mr.57.3.655-682.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heinrichs DE, Yethon JA, Whitfield C. Molecular basis for structural diversity in the core regions of the lipopolysaccharides of Escherichia coli and Salmonella enterica. Mol Microbiol. 1998;30:221–232. doi: 10.1046/j.1365-2958.1998.01063.x. [DOI] [PubMed] [Google Scholar]
- 40.Ebbensgaard A, Mordhorst H, Overgaard MT, Nielsen CG, Aarestrup FM, Hansen EB. Comparative evaluation of the antimicrobial activity of different antimicrobial peptides against a range of pathogenic bacteria. PLoS One. 2015;10:e0144611. doi: 10.1371/journal.pone.0144611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Z, Wang J, Ren G, Li Y, Wang X. Influence of core oligosaccharide of lipopolysaccharide to outer membrane behavior of Escherichia coli. Marine Drugs. 2015;13:3325–3339. doi: 10.3390/md13063325. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.