

Abstract
Healthy individuals have few goblet cells in their airways, but in patients with hypersecretory diseases goblet-cell upregulation results in mucus hypersecretion, airway plugging, and death. Multiple stimuli produce hypersecretion via epidermal growth factor receptor (EGFR) expression and activation, causing goblet-cell metaplasia from Clara cells by a process of cell differentiation. These cells are also believed to be the cells of origin of non-small-cell lung cancer, but this occurs via cell multiplication. The mechanisms that determine which pathway is chosen are critical but largely unknown. Although no effective therapy exists for hypersecretion at present, the EGFR cascade suggests methods for effective therapeutic intervention.
Keywords: activated neutrophils, airway goblet cell, asthma, chronic intubation, cigarette smoke, nasal polyp, oxygen free radical, tumor necrosis factor-α
Introduction
Epidermal growth factor (EGF) was discovered by Cohen, and he and his colleagues subsequently extended our knowledge of the mechanisms of action of EGF and its receptor EGFR [1]. EGFR is a 170 kDa membrane glycoprotein, which is activated by ligands such as EGF, transforming growth factor (TGF)-α, heparin-binding EGF, amphiregulin, betacellulin, and epiregulin. These proteins are synthesized as transmembrane precursors and are cleaved proteolytically by metalloproteases to release the mature growth factor, which can interact with EGFR and cause its activation.
Epidermal growth factor expression and activation causes goblet-cell metaplasia in airways
The discovery that a human epidermoid (A-431) cancer cell line contains high concentrations of EGFR led to extensive investigation into the role of the EGFR cascade in epithelial cell multiplication (cancer). Although growth factors can act as transforming proteins, it was recently hypothesized that EGFR activation may also be involved in epithelial differentiation into mucin-containing goblet cells by specific inflammatory mediators. The hypothesis was supported by the following observations. First, mucosubstances can be detected in dysplastic lesions and in foci of carcinoma in situ in human airways, and in tracheal lesions induced by carcinogens in animals [2]. The coexistence of mucin-containing cells and cancer cells suggests the possibility of a common progenitor. Second, Clara cells (also called 'nongranulated secretory cells') are believed to be the progenitor cells for bronchiolar carcinoma [3], and various studies also implicate these cells as precursors of goblet cells [4].
Takeyama et al [4] hypothesized that EGFR expression and activation would result in mucin expression and goblet-cell metaplasia. First, NCI-H292 cells (an epidermoid carcinoma cell line that expresses EGFR constitutively) were studied. EGFR ligands (EGF, TGF-α) caused expression of the MUC5AC mucin (a predominant airway mucin) gene. Interaction of EGFR with its ligands led to EGFR tyrosine kinase phosphorylation, and a selective EGFR tyrosine kinase inhibitor (BIBX1522) blocked MUC5AC expression induced by EGFR ligands, thereby implicating EGFR activation in mucin production. Second, the airways of pathogen-free rats do not express EGFR constitutively, and contain few goblet cells [4]. Instillation of tumor necrosis factor (TNF)-α induced EGFR expression in the airway epithelium, and subsequent instillation of EGFR ligand (TGF-α) induced mucin expression. BIBX1522 prevented this response in a dose-dependent manner, implicating EGFR activation in the goblet-cell response. A diagram of this EGFR cascade that is responsible for mucin production is provided (Fig. 1).
Figure 1.
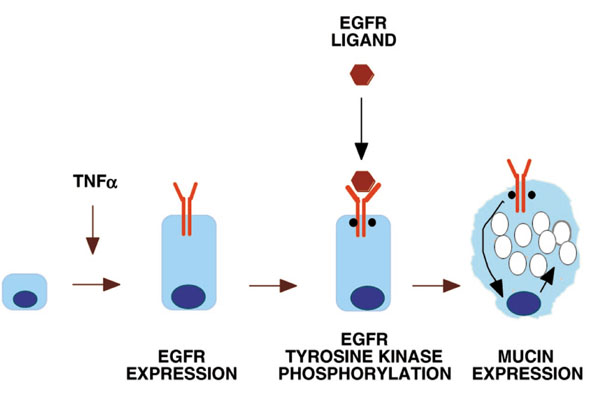
The mechanism of EGFR expression and activation. Stimulation of airway epithelial cells with TNF-α causes EGFR expression (the extracellular and intracellular parts of EGFR are shown). EGFR ligands (produced by epithelial or nearby cells) bind to EGFR, resulting in EGFR tyrosine phosphorylation and a subsequent downstream cascade, which causes mucin gene and protein expression.
Epithelial cell multiplication versus cell differentiation
In cancer cells, EGFR activation leads to cell multiplication. In normal airway epithelium, however, goblet cells appear to form from precursor (Clara) cells in the epithelium. Thus, in Sendai virus-induced goblet-cell metaplasia in pathogen-free rats, developing mucin-containing cells did not incorporate [3H]thymidine, suggesting that cell mitosis was not involved in synthesis of mucin mRNA [5]. In endotoxin-induced goblet-cell metaplasia in rat nasal septum, pretreatment with colchicine (which causes metaphase blockade) did not inhibit the production of goblet cells [6]. The mitotic rate and the total number of epithelial cells was unchanged, which led Shimizu et al [6] to conclude that the new goblet cells were produced by direct conversion of Clara cells. Finally, EGFR expression and activation causes goblet-cell metaplasia without changing the total number of epithelial cells, indicating that the number of goblet cells increases and the number of Clara cells decreases equivalently, thus implicating a differentiation process in goblet-cell development [4].
Stimuli for mucin synthesis: role of the epidermal growth factor receptor cascade
Many chronic inflammatory diseases of the airway are associated with mucus hypersecretion; this may contribute to asthma mortality [7,8]. Hypersecretion is also associated with nasal polyps [9], and in cystic fibrosis hypersecretion is associated with bacterial infections, especially with Pseudomonas aeruginosa [10]. In addition, cigarette smoking is a major cause of death in chronic obstructive pulmonary disease (COPD) [11], and the airways of smokers contain more goblet cells than do those of nonsmokers [12]. Exposure to cigarette smoke results in hypersecretion in the airways [13]. Orotracheal intubation-induced injury and other types of mechanical damage to the airway epithelium also result in mucus hypersecretion [14,15,16]. Neutrophils and their products are implicated in cystic fibrosis [10], COPD [11], and acute severe asthma [17].
Asthma and nasal polyps
The secretory state of the airways may vary considerably among asthmatic persons. Amishima et al [18] reported increased EGFR expression in submucosal glands and the surface epithelium of lungs removed surgically from asthmatic persons, although the severity of disease was not assessed. Takeyama et al [19] found increased numbers of goblet cells in biopsy specimens removed from the proximal airways of mildly asthmatic persons as compared with specimens from healthy nonasthmatic persons. EGFR and MUC5AC gene expression was increased, but varied, among the asthmatic persons. There was, however, a positive correlation between EGFR immunoreactivity and the amount of mucin staining, and Takeyama et al [19] suggested that the variability was related to the degree of 'activity' of the disease.
Nasal polyp epithelium contains increased numbers of goblet cells [9]. Specimens from healthy persons did not express EGFR strongly while four out of eight polyp specimens showed strong EGFR expression. TNF-α, which induces EGFR expression [4], was found (mostly in eosinophils) in polyps that express EGFR, suggesting that TNF-α in eosinophils may be responsible for EGFR expression in polyps. Burgel et al [9] also noted that neutrophils were more concentrated in the epithelium of EGFR-positive than in the epithelium of EGFR-negative polyps. It has been speculated that EGFR activation in polyps could occur via the release of TGF-α from eosinophils [20] or by transactivation of EGFR by oxygen free radicals released from neutrophils [21].
These clinical studies have limitations, but their findings suggest that goblet-cell metaplasia could result from an interaction among multiple cells (eg epithelial cells, eosinophils, and neutrophils).
Clinical studies may be informative, but carefully controlled studies in animals and in cells in vitro may also provide important insights into basic mechanisms. For instance, allergic sensitization with ovalbumin causes goblet-cell metaplasia in rodents [22], and the T-helper-2 cytokines interleukin-4 and interleukin-13 have been implicated in this process [23]. In interleukin-5-knockout mice, ovalbumin sensitization no longer leads to eosinophil recruitment after allergen exposure, but goblet-cell metaplasia still occurs [24]. Cohn et al [24] concluded that eosinophils were not required for allergic goblet-cell metaplasia, although neutrophils were not evaluated. Takeyama et al [4] showed that ovalbumin sensitization induced goblet-cell metaplasia and EGFR-positive staining in the epithelium, and that selective EGFR inhibitors prevented this process, implicating EGFR.
Furthermore, an antibody to the interleukin-4 receptor was reported [25] to prevent ovalbumin-induced goblet-cell metaplasia. EGFR inhibitors were also effective, leading Shim et al [26] to suggest that T-helper-2 cytokines activate an EGFR cascade. Selective EGFR inhibitors also prevented interleukin-13 goblet-cell metaplasia. IL-13 instillation also resulted in neutrophil recruitment into the airways [26], and the infiltrating neutrophils expressed TNF-α. An interleukin-8-blocking antibody inhibits interleukin-13-induced neutrophil recruitment and mucin production. Thus, interleukin-13 causes goblet-cell metaplasia indirectly by neutrophil recruitment and activation, perhaps releasing oxygen free radicals that then cause EGFR transactivation and goblet-cell metaplasia [21].
These studies show the importance of the EGFR cascade in allergic mucus hypersecretion. They also implicate neutrophils in allergic goblet-cell metaplasia in rodents. Perhaps neutrophils have been ignored previously in such allergic responses because allergens cause early recruitment of neutrophils, which initiate an EGFR gene and protein cascade, which in turn initiates mucin gene and protein expression. Neutrophils disappear from the airways when goblet-cell metaplasia is obvious (24-48 h later), although they appear to be important in experimental allergen-induced mucus hypersecretion.
Cigarette smoke and activated neutrophils
Cigarette smoke exposure in animals also results in mucus hypersecretion [13]. Takeyama et al [27] reported that exposure to cigarette smoke upregulated EGFR mRNA expression and induced EGFR-specific tyrosine phosphorylation, resulting in upregulation of MUC5AC mucin mRNA and protein production, effects that were inhibited completely by selective EGFR tyrosine kinase inhibitors. Approximately half of the response was also inhibited by antioxidants, implicating a role for oxygen free radicals, although the remainder could have been due to other substances such as acrolein [28]. Neutrophil infiltration in airways is characteristic of patients with COPD, and activated neutrophils increase EGFR tyrosine phosphorylation and subsequent MUC5AC expression at both the mRNA and protein levels in NCI-H292 cells. These effects can be blocked by selective EGFR inhibitors. Neutrophil supernatant-induced EGFR tyrosine kinase phosphorylation and MUC5AC synthesis is also inhibited by antioxidants [21]. These results implicate oxidative stress produced by neutrophils in mucin synthesis in airways.
Mechanical 'wounding'
EGFR enhances repair of sheep tracheal epithelial injury [29], and morphologically damaged bronchial epithelial repair is accelerated by EGFR activation [30]. The authors of those studies did not comment on goblet-cell metaplasia. However, in horses, orotracheal intubation results in mucus hypersecretion [14], and mechanical denudation of hamster airways results in secretory-cell metaplasia [15]. Lee et al [16] instilled irregular agarose plugs into rat airways to produce irritation, which also resulted in a profound increase in goblet cells. Plugged bronchi showed EGFR upregulation, and selective EGFR inhibitors prevented agarose-induced goblet-cell metaplasia. Peribronchial infiltration with neutrophils was also observed in plugged airways. Cyclophosphamide prevented agarose-induced neutrophil recruitment and goblet-cell metaplasia. Activated neutrophils produce TNF-α, and an anti-TNF-α antibody also prevented agarose-induced goblet-cell metaplasia. These findings implicate neutrophils and TNF-α in wound-induced EGFR activation and goblet-cell metaplasia.
These studies of mechanical damage to airways may be of clinical relevance for two reasons. First, epithelial damage is believed by some investigators to occur in asthma, and this damage could induce mucus hypersecretion. Second, because chronic intubation leads to mucus hypersecretion in horses [14], a similar effect is likely to occur in humans. It would be difficult to differentiate secretions generated in the lower airways and transported to the trachea from those secretions generated at the site of intubation. Perhaps during the day the secretions would be coughed up and aspirated, but during sleep cough is suppressed and mucus produced in the upper airways would be aspirated into the lungs, leading to mucus plugging and impairing gas exchange. The importance of such a mechanism should be evaluated in patients in the intensive care environment.
Bacterial infection
Mucus hypersecretion is characteristic of cystic fibrosis [10], and P. aeruginosa infections are associated with deterioration and death from cystic fibrosis. This has led to studies of the effect of Gram-negative bacterial products (including endotoxin) on mucin production; for example, Escherichia coli endotoxin increases epithelial mucosubstances [31], causes goblet-cell metaplasia in rat nose [32], and increases mucin synthesis in the lower airways [33]. Li et al [34] reported that P. aeruginosa activates the c-Src-Ras-MAPK kinase signaling pathway, leading to activation of nuclear factor-κB, which in turn activates mucin synthesis [35]. Kohri et al [36] reported that selective EGFR inhibitors prevent P. aeruginosa-induced mucin synthesis in human airway epithelial (NCI-H292) cells, again implicating EGFR activation.
Conclusion
Many chronic inflammatory airway diseases (eg asthma, cystic fibrosis, COPD, and nasal polyps) are associated with mucus hypersecretion. A wide variety of stimuli (eg allergens, bacteria, mechanical injury, cigarette smoke, and cytokines and activated neutrophils) cause the airway epithelium to differentiate into mucin-producing (goblet) cells via activation of an EGFR cascade. Airways of healthy individuals contain few goblet cells, but development of mature goblet cells de novo occurs within 3 days [16] and degranulation occurs within minutes [37]. In peripheral airways, this may lead to mucus plugging [38], which may not cause early symptoms and may be difficult to diagnose, but may progress rapidly to impairment of gas exchange and death. Mucus hypersecretion probably fluctuates with inflammation in disease.
Effective therapy for hypersecretion does not currently exist, but the novel pathway involved in EGFR expression and activation suggests new approaches to therapy for mucus hypersecretion.
Abbreviations
COPD = chronic obstructive pulmonary disease; EGF(R) = epidermal growth factor (receptor); MAPK = mitogen-activated protein kinase; TGF = transforming growth factor; TNF = tumor necrosis factor.
References
- Cohen S. Epidermal growth factor. Nobel Lecture. Biosci Rep. 1986;6:1017–1028. doi: 10.1007/BF01141022. [DOI] [PubMed] [Google Scholar]
- McDowell EM, Trump BF. Histogenesis of preneoplastic lesions in tracheobronchial epithelium. Surv Synth Pathol Res. 1983;2:235–279. [Google Scholar]
- Greenberg SD, Smith MN, Spjut HJ. Bronchiolo-alveolar carcinoma-cell of origin. Am J Clin Pathol. 1975;63:153–167. doi: 10.1093/ajcp/63.2.153. [DOI] [PubMed] [Google Scholar]
- Takeyama K, Dabbagh K, Lee H-M, Agusti C, Lausier JA, Ueki IF, Grattan KM, Nadel JA. Epidermal growth factor system regulates mucin production in airways. Proc Natl Acad Sci USA. 1999;96:3081–3086. doi: 10.1073/pnas.96.6.3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royce FH, Basbaum CB. Analysis of thymidine incorporation by airway epithelial cells in a rat model of chronic bronchitis [abstract]. Am Rev Respir Dis. 1990;141:A107. [Google Scholar]
- Shimizu T, Takahashi Y, Kawaguchi S, Sakakura Y. Hypertrophic and metaplastic changes of goblet cells in rat nasal epithelium induced by endotoxin. Am J Respir Crit Care Med. 1996;153:1412–1418. doi: 10.1164/ajrccm.153.4.8616574. [DOI] [PubMed] [Google Scholar]
- Cardell BS, Pearson RSB. Death in asthmatics. Thorax. 1959;14:341–352. [Google Scholar]
- Aikawa T, Shimura S, Sasaki H, Ebina M, Takishima T. Marked goblet cell hyperplasia with mucus accumulation in the airways of patients who died of severe asthma attack. Chest. 1992;1101:916–921. doi: 10.1378/chest.101.4.916. [DOI] [PubMed] [Google Scholar]
- Burgel P-R, Escudier E, Coste A, Dao-Pick T, Ueki IF, Takeyama K, Shim JJ, Murr AH, Nadel JA. Relation of EGF receptor expression to goblet cell hyperplasia in nasal polyps. J Allergy Clin Immunol. 2000;106:705–712. doi: 10.1067/mai.2000.109823. [DOI] [PubMed] [Google Scholar]
- Boucher RC, Knowles MR, Yankaskas JR. Cystic fibrosis.7. Textbook of Respiratory Medicine, 3rd ed. Edited by Murray JF, Nadel JA. Philadelphia, PA: WB Saunders Co; 2000. pp. 1291–1323.
- Piquette CA, Rennard SI, Snider GL. Chronic bronchitis and emphysema. Textbook of Respiratory Medicine, 3rd ed. Edited by Murray JF, Nadel JA. Philadelphia, PA: WB Saunders Co; 2000. pp. 1187–1245.
- Cosio MG, Hale KA, Niewoehner DE. Morphologic and morphometric effects of prolonged cigarette smoking on the small airways. Am Rev Respir Dis. 1980;122:265–271. doi: 10.1164/arrd.1980.122.2.265. [DOI] [PubMed] [Google Scholar]
- Coles SJ, Levine LR, Reid L. Hypersecretion of mucus glycoproteins in rat airways induced by tobacco smoke. Am J Pathol. 1979;94:459–471. [PMC free article] [PubMed] [Google Scholar]
- Heath RB, Steffey EP, Thurmon JC, Wertz EM, Meagher DM, Hyppa T, Van Slyke GL. Laryngotracheal lesions following routine orotracheal intubation in the horse. Equine Vet J. 1989;21:434–437. doi: 10.1111/j.2042-3306.1989.tb02190.x. [DOI] [PubMed] [Google Scholar]
- Keenan KP, Wilson TS, McDowell EM. Regeneration of hamster tracheal epithelium after mechanical injury. IV. Histochemical, immunocytochemical and ultrastructural studies. Virchows Arch [Cell Pathol] 1983;43:213–240. doi: 10.1007/BF02932958. [DOI] [PubMed] [Google Scholar]
- Lee H-M, Takeyama K, Dabbagh K, Lausier JA, Ueki IF, Nadel JA. Agarose plug instillation causes goblet cell metaplasia by activating EGF receptors in rat airways. Am J Physiol Lung Cell Mol Physiol. 2000;278:L185–L192. doi: 10.1152/ajplung.2000.278.1.L185. [DOI] [PubMed] [Google Scholar]
- Fahy JV, Kim KW, Liu J, Boushey HA. Prominent neutrophilic inflammation in sputum from subjects with asthma exacerbation. J Allergy Clin Immunol. 1995;95:843–852. doi: 10.1016/s0091-6749(95)70128-1. [DOI] [PubMed] [Google Scholar]
- Amishima M, Munakata M, Nasuhara Y, Sato A, Takahashi T, Homma Y, Kawakami Y. Expression of epidermal growth factor and epidermal growth factor receptor immunoreactivity in the asthmatic human airway. Am J Respir Crit Care Med. 1998;57:1907–1912. doi: 10.1164/ajrccm.157.6.9609040. [DOI] [PubMed] [Google Scholar]
- Takeyama K, Fahy JV, Nadel JA. Relationship of epidermal growth factor receptors to airway goblet cell production. Am J Respir Crit Care Med. 2001;163:1–6. doi: 10.1164/ajrccm.163.2.2001038. [DOI] [PubMed] [Google Scholar]
- Elovic A, Wong DTW, Weller PF, Matossian K, Galli SJ. Expression of transforming growth factors-alpha and beta-1 messenger RNA and product by eosinophils in nasal polyps. J Allergy Clin Immunol. 1994;93:864–869. doi: 10.1016/0091-6749(94)90379-4. [DOI] [PubMed] [Google Scholar]
- Takeyama K, Dabbagh K, Shim JJ, Dao-Pick T, Ueki IF, Nadel JA. Oxidative stress causes mucin synthesis via transactivation of epidermal growth factor receptor: role of neutrophils. J Immunol. 2000;164:1546–1552. doi: 10.4049/jimmunol.164.3.1546. [DOI] [PubMed] [Google Scholar]
- Blyth DI, Pedrick MS, Savage TJ, Hessel EM, Fattah D. Lung inflammation and epithelial changes in a murine model of atopic asthma. Am J Respir Cell Mol Biol. 1996;14:425–438. doi: 10.1165/ajrcmb.14.5.8624247. [DOI] [PubMed] [Google Scholar]
- Grunig G, Warnock M, Wakil A, Venkayya R, Brombacher F, Rennick DM, Sheppard D, Mohrs M, Donaldson DD, Locksley RM, Corry DB. Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 1999;282:2261–2263. doi: 10.1126/science.282.5397.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn L, Homer RJ, MacLeod H, Mohrs M, Brombacher F, Bottomly K. Th2-induced airway mucus production is dependent on IL-4Rα, but not on eosinophils. J Immunol. 1999;162:6178–6183. [PubMed] [Google Scholar]
- Gavett SH, O'Hearn DJ, Karp CL, Patel EA, Schofield BH, Finkelman FD, Wills-Karp M. Interleukin-4 receptor blockade prevents airway responses induced by antigen challenge in mice. Am J Physiol Lung Cell Mol Physiol. 1997;272:L253–L261. doi: 10.1152/ajplung.1997.272.2.L253. [DOI] [PubMed] [Google Scholar]
- Shim JJ, Dabbagh K, Ueki IF, Dao-Pick T, Burgel P-R, Takeyama K, Tam DC-W, Nadel JA. Interleukin-13 induces mucus production by stimulating epidermal growth factor receptors and by activating neutrophils. Am J Physiol Lung Cell Mol Physiol. 2001;280:L134–L140. doi: 10.1152/ajplung.2001.280.1.L134. [DOI] [PubMed] [Google Scholar]
- Takeyama K, Jung B, Shim JJ, Burgel P-R, Dao-Pick T, Ueki IF, Protin U, Kroschel P, Nadel JA. Activation of epidermal growth factor receptors is responsible for mucin synthesis induced by cigarette smoke. Am J Physiol Lung Cell Mol Physiol. 2001;280:L165–L172. doi: 10.1152/ajplung.2001.280.1.L165. [DOI] [PubMed] [Google Scholar]
- Borchers MT, Wert SE, Leikauf GD. Acrolein-induced MUC5AC expression in rat airways. Am J Physiol Lung Cell Mol Physiol. 1998;274:L573–L581. doi: 10.1152/ajplung.1998.274.4.L573. [DOI] [PubMed] [Google Scholar]
- Barrow RE, Wang C, Evans MJ, Herndon DN. Growth factors accelerate epithelial repair in sheep trachea. Lung. 1993;171:335–344. doi: 10.1007/BF00165699. [DOI] [PubMed] [Google Scholar]
- Puddicombe SM, Polosa R, Richter A, Krishna MT, Howarth PH, Holgate ST, Davis DE. Involvement of the epidermal growth factor receptor in epithelial repair in asthma. FASEB J. 2000;14:1362–1374. doi: 10.1096/fj.14.10.1362. [DOI] [PubMed] [Google Scholar]
- Harkema JR, Hotchkiss JA. In vivo effects of endotoxin on intraepithelial mucosubstances in rat pulmonary airways. Quantitative histochemistry. Am J Pathol. 1992;141:307–317. [PMC free article] [PubMed] [Google Scholar]
- Shimizu T, Takahashi Y, Kawaguchi S, Sakakura Y. Hypertrophic and metaplastic changes of goblet cells in rat nasal epithelium induced by endotoxin. Am J Respir Crit Care Med. 1996;153:1412–1418. doi: 10.1164/ajrccm.153.4.8616574. [DOI] [PubMed] [Google Scholar]
- Steiger D, Hotchkiss J, Bajaj L, Harkema J, Basbaum C. Concurrent increases in the storage and release of mucin-like molecules by rat airway epithelial cells in response to bacterial endotoxin. Am J Respir Cell Mol Biol. 1995;12:307–314. doi: 10.1165/ajrcmb.12.3.7873197. [DOI] [PubMed] [Google Scholar]
- Li J-D, Dohrman AF, Gallup M, Miyata S, Gum JR, Kim YS, Nadel JA, Prince A, Basbaum CB. Transcriptional activation of mucin by Pseudomonas aeruginosa lipopolysaccharide in the pathogenesis of cystic fibrosis lung disease. Proc Natl Acad Sci USA. 1997;94:967–972. doi: 10.1073/pnas.94.3.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J-D, Fang W, Gallup M, Kim J-H, Gum J, Kim Y, Basbaum C. Activation of NF-κB via a Src-dependent Ras-MAPK-pp90rsk pathway is required for Pseudomonas aeruginosa -induced mucin overproduction in epithelial cells. Proc Natl Acad Sci USA. 1998;95:5718–5723. doi: 10.1073/pnas.95.10.5718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohri K, Ueki IF, Shim JJ, Burgel P-R, Oh YM, Tam DC, Dao-Pick T, Nadel JA. Pseudomonas aeruginosa (PA) exoproducts induce mucin MUC5AC production via expression and activation of epidermal growth factor receptor (EGFR). Am J Respir Crit Care Med. 2001.
- Verdugo P. Goblet cells secretion and mucogenesis. Annu Rev Physiol. 1990;52:157–176. doi: 10.1146/annurev.ph.52.030190.001105. [DOI] [PubMed] [Google Scholar]
- Shimura S, Andoh Y, Haraguchi M, Shirato K. Continuity of airway goblet cells and intraluminal mucus in the airways of patients with bronchial asthma. Eur Respir J. 1996;9:1395–1401. doi: 10.1183/09031936.96.09071395. [DOI] [PubMed] [Google Scholar]