

Abstract
Diffuse intrinsic pontine glioma (DIPG) is a deadly paediatric brain cancer. Transient response to radiation, ineffective chemotherapeutic agents and aggressive biology result in rapid progression of symptoms and a dismal prognosis. Increased availability of tumour tissue has enabled the identification of histone gene aberrations, genetic driver mutations and methylation changes, which have resulted in molecular and phenotypic subgrouping. However, many of the underlying mechanisms of DIPG oncogenesis remain unexplained. It is hoped that more representative in vitro and preclinical models–using both xenografted material and genetically engineered mice–will enable the development of novel chemotherapeutic agents and strategies for targeted drug delivery. This review provides a clinical overview of DIPG, the barriers to progress in developing effective treatment, updates on drug development and preclinical models, and an introduction to new technologies aimed at enhancing drug delivery.
Keywords: Glioma, Paediatric, DIPG, Brainstem tumor, Focused ultrasound
INTRODUCTION
Diffuse intrinsic pontine gliomas (DIPG) are paediatric highgrade gliomas (pHGG) characterised by infiltrative tumours of the brainstem. DIPGs are histologically astrocytomas, have a peak onset of 6–9 years (adolescents and adults can also be affected) and account for 10–20% of all paediatric brain tumours [45]. DIPGs are the leading cause of brain tumour deaths in children [33]. Median overall survival (OS) is between 8 to 12 months, and OS is approximately 30% at 1 year, 10% at 2 years and less than 1% at 5 years from diagnosis [23,45,49,51]. The combination of a short clinical history (<6 months) and typical appearances on magnetic resonance imaging (MRI) are usually diagnostic [21]. Neurological symptoms can vary based on the extent and specific location of the lesion within the pons but over 50% present with the classical triad of cranial nerve deficits (facial asymmetry and diplopia), cerebellar signs (ataxia, dysmetria, and dysarthria) and long tract signs (hyperreflexia, upward Babinski and decreased strength) [54]. A highly sensitive predictor of DIPG is a primary presentation of abducens palsy in a young child [20]. In the absence of an exophytic component that extends posteriorly to cause obstructive hydrocephalus (<10%), raised intracranial pressure is usually not observed. DIPG tumours can spread along fiber tracts to local sites such as the cerebellum and thalamus but rarely metastasise to distant sites.
Radiation is the only treatment with proven efficacy (albeit limited) in prolonging progression-free survival (PFS) [64]. It can also provide transient symptomatic improvement and improved neurologic function [33]. The current radiotherapy dose in children with DIPG is typically 54 Gy (54/1.8 Gy) over 6 weeks [67]. A shorter hypofractionated radiation therapy regimen delivering 39 Gy (39/3 Gy) over 13 days has been proposed for newly diagnosed DIPG as it was shown to lessen the treatment burden whilst being non-inferior to conventional radiotherapy (54/1.8 Gy) with respect to OS and PFS [29].
There are currently no proven chemotherapeutic agents that have been shown to increase PFS or OS [51]. Numerous clinical trials aimed at investigating existing alkylating agents (e.g., cisplatin, temozolomide, carboplatin), alone or neo-adjuvant to radiation, have failed [2,13,28]. Agents targeting other biologic and molecular pathways, such as erlotinib (EGFR inhibitor), tamoxifen (oestrogen-receptor modifier) and bevacizumab (VEGF inhibitor) have also been shown to be ineffective, alone or in combination with radiotherapy [33,44]. Radiosensitisers such as carbogen and motexafin gadolinium also have no proven survival benefit [1,7].
In part, these failures have been due to a poor understanding of the underlying molecular and cellular biology, an underappreciation of the tumour heterogeneity (paucity of primary tissue), and a lack of representative in vitro and in vivo models [23]. Many trials have also failed due to a long-held assumption that the biological properties of paediatric DIPGs are similar to adult high grade gliomas (HGG) [35,50]. It is becoming increasingly clear however, that DIPG differs markedly from adult HGG, and even from supratentorial pHGG, at a phenotypic and molecular level [11,35].
This review aims to outline ongoing and historical barriers to advancing treatment, provide an overview of molecular profiling and preclinical models, and appraise the latest research into novel chemotherapeutic agents and advances in enhanced drug delivery technologies–including, but not limited to, magnetic resonance guided focused ultrasound (MRgFUS).
SIGNIFICANT BARRIERS TO PROGRESS
Significant challenges exist in DIPG management. Principal to this is the eloquent brainstem location of the tumour which results in unacceptable risks of morbidity and mortality from surgical debulking. Aligned with this, the infiltrative nature of the tumour makes complete resection impossible. Additionally, the absence of effective drug treatment that changes the natural history of the disease, and hallmark imaging suggest that even biopsy offered little diagnostic or therapeutic advantage to the patient. The lack of primary tissue from either biopsy or debulking has contributed to the longstanding poor understanding of the underlying biology and a lack of representative experimental model systems [23]. However, there has been an increased tendency more recently to perform stereotactic biopsy as it has been shown to be a minimally morbid procedure with high diagnostic yield [9,52]. Most centres would support a biopsy in suspected DIPG patients with atypical imaging and/or in the context of an approved clinical trial where tissue would help stratify participants into trial arms [51,63]. In addition to more postmortem tissue donations, the increased quantity and quality of fresh biopsy tissue has enabled a number of molecular studies to be performed robustly and reproducibly [45], and preclinical models to be developed [32]. Biological information gleaned from biopsy tissue has also informed clinical trial treatment regimens [3].
MRI in DIPG typically reveals a T1 hypointense, T2 hyperintense tumour centred in and involving >50% of the pons, with associated signal change (Fig. 1). Perhaps most interestingly, and not in keeping with other HGGs, there is absent or irregular contrast enhancement [25,47]. This implies a preservation of the blood-brain barrier (BBB). It may also explain, in part, why chemotherapeutic agents previously shown to be effective in other gliomas have failed in DIPG–although there are surely also biological differences [60]. Therefore, improving drug delivery, either through structural adaptation of drugs or disruption of the BBB itself, are required in DIPG if sufficient intratumoural doses are to be achieved.
Fig. 1.
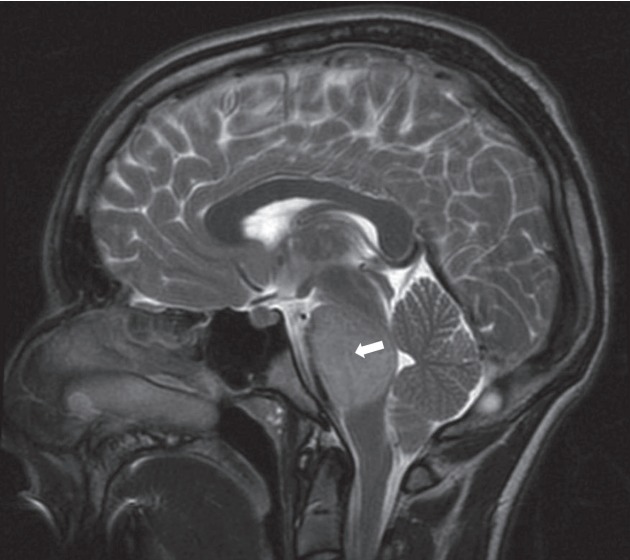
Magnetic resonance imaging (T2) showing lesion expanding the pons(white arrow), with associated signal change.
MOLECULAR CLASSIFICATION and SUBGROPING
Recent identification of a specific point mutation in H3F3A (encoding histone 3.3) and HIST1H3B (encoding histone 3.1) genes–leading to the substitution of a lysine by a methionine at position 27 (K27M)–has enabled the molecular classification of DIPGs [11,34,65]. A profound epigenetic change was found to occur in 95% of DIPG samples with the H3-K27M mutation, shown to be due to impaired function of polycomb repressive complex 2 (PRC2) methyltransferase. The result is global hypomethylation of H3K27 [5,12]. The H3-K27M mutation was used to define a new entity in the most recent 2016 World Health Organization classification of central nervous system tumours [35]. Recent epigenetic data have shown that despite a major reduction in trimethylation of H3K27 secondary to impaired PRC2 function, some residual PRC2 activity is maintained in DIPG cells and may in fact be required to repress neuronal differentiation and function, ultimately maintaining cellular proliferation [48]. This complicates the biology somewhat but potentially exposes another therapeutic target.
Patients with tumours carrying a H3F3A mutation had a significantly poorer response to radiotherapy, relapsed earlier and had more metastatic recurrences than those with HIST1H3B mutations [11]. This pro-metastatic H3F3A cohort was characterised by increased PDGFRA gene expression and a proneural/oligodendroglial phenotype whereas the HIST1H3B genotype was characterised by a pro-angiogenic/hypoxic gene expression signature and a mesenchymal/astrocytic phenotype [11]. The HIST1H3B and H3F3A mutations are mutually exclusive. Such an improved understanding of the differences in the natural history of DIPG molecular subtypes has proven very useful to clinicians in prognosticating, and empowering patients.
In addition to the histone 3 variant mutations, other recurrent driver mutations have also been identified. Mutations in the ACVR1 gene exist concurrently with H3 mutations in 20–30% of DIPGs [8]. This mutation has been shown to ultimately upregulate the bone morphogenetic protein developmental signalling pathway [59]. It is not directly clear what the role of the ACVR1 mutation is in DIPG oncogenesis. Mutations in TP53, a known tumour suppressor gene, exists in up to 77% of DIPGs [54]. A further important genetic aberration in DIPG involves amplification of components of the receptor tyrosine kinase/Ras/phosphatidylinositol-3 pathway, especially platelet-derived growth factor receptor A (PDGFRA) [54].
Recently, three molecular subgroups–MYCN, Silent and H3K27M–have been defined by integrating DNA methylation analyses, whole genome sequencing, histopathological and clinical data [8]. The MYCN subgroup is characterised by highgrade histology, chromothripsis (thousands of chromosomal rearrangements occurring as a single event in localised and confined regions) on chromosome 2p, and DNA hypermethylation. The Silent subgroup is characterised by a lower genomic mutational burden than the other 2 subgroups. The H3K27M subgroup is the most prevalent in DIPG and is characterised by the histone mutations above, as well as by global DNA hypomethylation and multiple concurrent mutations (TP53, PAX3, PGFRA, EGFR, ATRX, NF1, PPM1D, PIK3CA, TERT, NTRK, IL-13RA2, PARP1, PTEN, CCND1/2/3, CDK4/6 and MET) [45].
The current understanding between the interrelated DIPG subgroups is shown in Fig. 2. Perhaps most importantly, it is hoped that such molecular classification and subgrouping will identify distinct cellular origins and biological drivers of tumorigenicity that will lead to effective DIPG-specific therapies [31].
Fig. 2.
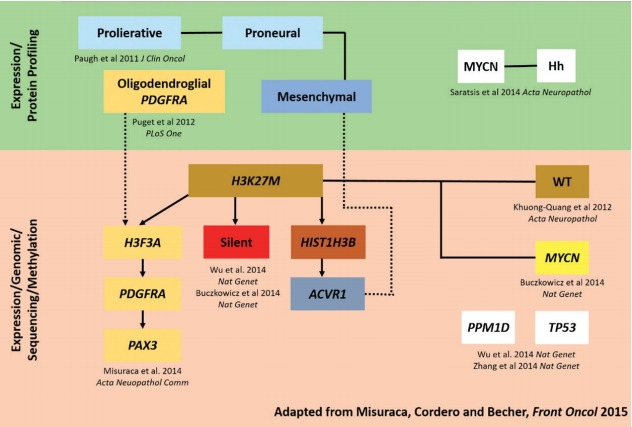
DIPG subgrouping map showing the current understanding between the interrelated DIPG subgroups (adapted from Misuraca et al. [41]). DIPG : diffuse intrinsic pontine glioma.
PRECLINICAL MOUSE MODELS OF DIPG
The identification of recurrent driver mutations has enabled the development of preclinical mouse models. As with all models, strengths and caveats exist, but work in other cancers has demonstrated that validated preclinical models are essential in drug screening and the development of novel therapeutics. There are now well established orthotopic xenograft and genetically engineered mouse models (GEMMs) of DIPG.
The earliest preclinical models used cell-derived orthotopic xenografts (CDOXs) and patient-derived orthotopic xenografts (PDOXs) from autopsy tissue. CDOX models were generated by stereotactic intracranial injection of tumour-initiating cells cultured from DIPG autopsy tissue harbouring the H3K27M driver mutation [43]. The advantage of a CDOX model is that the cells can be manipulated to express luciferase and tagged with fluorescent reporter proteins, allowing for longitudinal tumour growth monitoring and cell tracing, respectively.
PDOX models have been developed more recently from direct injection of autopsy DIPG cells [32]. The advantage of developing PDOX models is that the cells are not subject to the clonal selective pressures exerted by in vitro cell culture. Both these CDOX and PDOX models showed the characteristic DIPG infiltrative phenotype. The disadvantage of both models is that autopsy tissue is not treatment-naïve. This means that the models may not be representative of the patient at the time of presentation. To exemplify this, the average mutation rate detected at the time of autopsy is 3.12 somatic mutations per megabase [8] while it is 0.76 at diagnosis [59]. Recently, a group aimed to overcome this by developing preclinical models (both CDOX and PDOX) using treatment-naïve fresh tissue attained at the time of diagnostic biopsy [49]. These models more faithfully recapitulate the histological and MRI features seen in DIPG patients at the time of diagnosis. They also demonstrate a typically invasive phenotype, harbour the H3K27M trimethylation loss genotype in vivo and cover more of the spectrum of histone modifications and lineage markers observed in patients. Moreover, the mice presented with neurological symptoms and a natural history similar to that observed in DIPG patients. One previous problem with using xenografted DIPG tissue had been the development of malignant murine tumours rather than human tumours [10]. These latest models do not develop such tumours, representing an advancement on previous DIPG preclinical models. The results of future studies using these models, and their translational to clinical practice, will ultimately determine their true value.
The development of models using treatment-naïve and autopsy tissue is complementary–the former allowing for testing of strategies more applicable to the early stages of the disease, whilst the latter is more ideal for the end stages of DIPG, recurrence and resistance [49]. Moving from one to the other also has the potential to better delineate the evolution of the disease.
Another class of preclinical models, GEMMs, work on the principle of introducing a limited number of genetic alterations to induce in vivo oncogenesis. The advantage of these models is the ability to study tumours arising in the natural environment of immune-proficient animals (CDOX and PDOX animal models must be immune-deficient to prevent rejection of the xenografted material). One such model that shows promise in DIPG is the replication-competent avian sarcoma–leucosis virus long terminal repeat with splice acceptor/tumour virus A (RCAS-Tva) system [61]. This system can produce brainstem tumours that harbour the three most common genetic aberrations observed in DIPG–H3K27M, TP53 and PDGFRA [4]. This model has been used to study a multi-kinase inhibitor (BMS-754807) but no survival benefit was shown [24]. It is difficult to evaluate the drug’s effectiveness against the tumour biology, however, since the authors identified that drug delivery was below the known IC-50 (the concentration of an inhibitor at which response is reduced by half)–most likely due to BBB impermeability.
The RCAS-Tva GEMM has also been used to study the in vivo biology of DIPG. In contrast to supratentorial glioma, high levels of PAX3 expression have been implicated in the oncogenesis of brainstem gliomas such as DIPG [40]. This has further defined a new subset of DIPG and subsequently a novel mouse model that aims to represent this genotype [41,42].
Another new development in DIPG modelling uses neural progenitors (derived from human pluripotent stem cells) cotransduced in vitro with common DIPG alterations (PDGFRA, TP53 and H3F3A) [22]. These cells can then be studied in vitro or transplanted into rodents, and the resultant oncogenesis studied.
Recently, a group has used electroporation to transduce mouse brainstem neural progenitor cells with K27M DNA in utero [46]. The resultant progeny developed highly proliferative, diffusely spreading lesions that had the hallmark molecular and histopathological features of pHGG. These tumours could then be serially engrafted in recipient mice and used for targeted drug screening. This novel mouse model might provide insights into in utero drivers of neurodevelopmental change that result in oncogenic transformation to pHGG postnatally.
In terms of xenograft material delivery, there have also been developments on the technical side of preclinical models in DIPG. A recent publication replaced the stereotactic frame traditionally used to deliver cells into CDOX and PDOX models with a guide-screw bolt [36]. The main advantage of this technique is the ability to leave the bolt in place and use it as a conduit for delivery of intratumoural chemotherapy, antibodies, and gene or viral therapies.
Clearly much progress has been made in DIPG preclinical modelling, but developments are still required to represent other oncogenic drivers such as ACVR1 and MYCN to fully cover the subtypes.
NOVEL CHEMOTHERAPEUTIC AGENTS
Building on the increased knowledge of the H3K27M DIPG epigenetic landscape, members of the bromo- and extra-terminal (BET) domain family have been shown to be important in the role of translating the hypomethylation changes into a disease phenotype [48]. By targeting this relationship, a recent study using a BET inhibitor (JQ1) to treat mice xenografted with human DIPG H3K27M cell lines showed tumour regression [48]. Furthermore, BET inhibitors have been shown to cross the BBB.
There have also been promising results investigating the multi-histone deacetylase inhibitor panobinostat, alone and in combination with the histone demethylase inhibitor GSKJ4, in patient-derived DIPG in vitro and orthotopic xenograft models [23,26].
Another approach investigated the reactivation of p16 (CDKN2A), a tumour suppressor gene that has been shown to be inactivated in the H3F3A subgroup of DIPG patients. The study showed that the administration of a DNA methylation inhibitor restored p16 levels with resultant cytotoxicity to murine tumour cells [14]. Clinical trials are now required to investigate all these approaches further.
Since PDGFRA is one of the most frequent mutations in DIPG, its downstream effector pathway PI3K/AKT/mTOR has been targeted with single agents [66]. However, this has proved ineffective, primarily due to the development of resistance. The latter was linked to activation of the related Ras/Raf/MEK/ERK pathway. Therefore, combinatorial therapy (perifosine–a PI3K/AKT inhibitor and trametinib–a MEK inhibitor) targeting both pathways has recently been tested [66]. Results show a reduction in in vitro cell viability. Validation in vivo and progression to clinical trials is now required. In a separate phase I trial recently reported, perifosine was combined with temsirolimus (an mTOR inhibitor) to treat a range of recurrent and refractory paediatric high-grade solid tumours, DIPG amongst them. Results show that this combination was generally well tolerated, with no dose limiting toxicity i.e., safe and feasible. Further trials will investigate efficacy.
BIOMARKER DISCOVERY
There are currently no clinically validated biomarkers in paediatric or adult gliomas that enable non-invasive screening, treatment response monitoring, or detection of recurrence. In DIPG patients, the tumour-associated proteins cyclophilin A and dimethylarginase 1 have been identified in the cerebrospinal fluid. They are both upregulated when compared to controls [53]. Early results from the same authors show that these proteins may also be expressed in serum and urine, but the numbers studied were low. Further work is required to validate these results in larger numbers, and prove the sensitivity and specificity required to develop these findings into a clinically useful biomarker.
ADVANCES IN DRUG DELIVERY
Convection-enhanced delivery (CED) is a technique first established in the 1990s that aims to bypass the BBB by delivering drugs directly to their intended target [6]. It relies on accurate surgical placement of a microcatheter (or multiple microcatheters) directly into the tumour. The catheter(s) are connected to a pump which exerts a small, repetitive hydraulic pressure that delivers the drug in solution [68]. CED with various therapeutic agents have been trialled numerous times in patients with supratentorial high-grade gliomas, with some success [68]. As DIPG is a relatively compact tumour and has no definitive treatment, it is an attractive option for evaluating CED. In vivo safety of CED has been established in both small and large animals. This led to a phase I trial of CED of124I-8H9 radio-labelled monoclonal antibody in children with DIPG that had been previously irradiated [58]. This study demonstrated that CED is well tolerated and safe. Subsequently, there are now a few clinical trials in progress of brainstem CED in children with DIPG using targeted macromolecules such as antibodies and immunotoxins. There is also an ongoing neurosurgical trial (Memorial Sloan Kettering Cancer Center and Weill Cornell Medical College, trial number NCT01502917) evaluating a neuronavigational tool (iPlan Flow, BrainLAB AG, Feldkirchen, Germany) specifically designed to work with CED microcatheter placement in the brainstem.
MRgFUS (Fig. 3A) has its origins in high intensity focused ultrasound (HIFU), a modality that is routinely used in ablative procedures to treat diseases such as Parkinson’s disease and essential tremor [17,38]. In the presence of intravenously administered microbubbles, and with a lower frequency of ultrasound than used in HIFU, MRgFUS can be used to permeabilise the BBB without lasting tissue injury [19,27,37]. The preformed microbubbles reduce the extent of BBB tissue damage by 1) limiting the interaction between the endothelial cells and the sonic waves, and 2) allowing for a lower amount of acoustic energy [30,62]. MRg-FUS exerts several mechano-biological effects on the microbubbles, including inertial cavitation, oscillation, and growth (Fig. 3B). This, in turn, results in shock waves and acoustic streaming that increase surrounding shear stress [39]. It is not entirely clear which of these mechanisms is more important in BBB disruption, or the underlying molecular changes that occur at a cellular level, but the resultant increase in paracellular transport and transcellular active transport provide a window of opportunity (around 4–6 hours) for drug delivery [55-57] (Fig. 3C). MRg-FUS enhanced delivery has been shown to significantly increase brain intratumoural drug concentrations and concomitantly increased cell death in preclinical models [15,18,19]. We have recently demonstrated that MRgFUS and microbubble disruption of the BBB is both safe and feasible (manuscript under review); in addition, this technique is efficacious in concentrating cisplatinum concentrations within the pons. Aside from acting as an adjuvant delivery mechanism, MRgFUS may also enable repurposing of BBB impermeable drugs known to work in other cancers, and the administration of higher local doses whilst minimising adverse systemic toxicity [16].
Fig. 3.

A : Magnetic resonance guided focused ultrasound preclinical system. B : Schematic of focused ultrasound (FUS) delivery causing temporary breach of blood-brain barrier within tumour. C : Schematic of FUS effect on microbubbles within blood vessel and resultant breach of BBB allowing delivery of gold nanoparticles (GNPs). MRI : magnetic resonance imaging , BBB : blood-brain barrier.
CONCLUSION
DIPG is a fatal childhood brain cancer that has devastating consequences for patients, their caregivers and families. Despite decades of clinical trials and research, little or no progress has been made in improving the lives of affected patients, or changing the natural history of the disease. Recent advances in molecular profiling techniques allied to increased availability of tissue through post-mortem donation and biopsy has significantly improved our understanding of the origin and biological features of DIPG. This has enabled the field to move from a purely histology-based characterisation to a more sophisticated molecular-based classification and subgrouping. The discovery that the majority of DIPG tumours have epigenetic dysregulation as a result of histone mutations has meant that epigenetic modifying agents are emerging as a promising class of therapeutic agents [5,23,26]. More work is required to identify and validate these mechanisms as effective therapeutic targets. There also remain a significant portion of DIPGs that do not carry histone or ACVR1 mutations. Further molecular profiling is required to identify the driver mutational signature underlying this Silent subgroup. Ongoing efforts to develop new in vitro and preclinical models to represent this subgroup will hopefully bring forward new information. Aside from the novel chemotherapeutic agents currently under evaluation, perhaps the most exciting developments are in the field of enhanced drug delivery. Advances in ultrasound and convection technology show significant promise in overcoming a long-held obstacle to effective drug targeting of DIPG–the BBB. Establishing a clinically proven enhanced drug delivery system, such as MRgFUS, will enable drug repurposing screens and the re-evaluation of drugs that have previously failed in clinical trials–perhaps due to poor target IC-50. Given the significant recent advances in other tumours that for many years were also therapeutically stagnant, such as medulloblastoma, there is renewed hope that DIPG is on the verge of a new, more promising era.
Acknowledgments
R.K.M. acknowledges funding from the Leeds Cancer Research UK-Centre. This review was supported by grants from the Canadian Institutes of Health Research, Curing Kids Cancer Foundation, Brainchild, Meagan’s Walk, The Cure Starts Now, and Aidan’s Avengers of the Diffuse Intrinsic Pontine Glioma Collaborative.
Footnotes
No potential conflict of interest relevant to this article was reported.
INFORMED CONSENT
This type of study does not require informed consent.
References
- 1.Aquino-Parsons C, Hukin J, Green A. Concurrent carbogen and radiation therapy in children with high-risk brainstem gliomas. Pediatr Blood Cancer. 2008;50:397–399. doi: 10.1002/pbc.21057. [DOI] [PubMed] [Google Scholar]
- 2.Bailey S, Howman A, Wheatley K, Wherton D, Boota N, Pizer B, et al. Diffuse intrinsic pontine glioma treated with prolonged temozolomide and radiotherapy--results of a United Kingdom phase II trial (CNS 200704) Eur J Cancer. 2013;49:3856–3862. doi: 10.1016/j.ejca.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartels U, Hawkins C, Vézina G, Kun L, Souweidane M, Bouffet E. Proceedings of the diffuse intrinsic pontine glioma (DIPG) Toronto Think Tank: advancing basic and translational research and cooperation in DIPG. J Neurooncol. 2011;105:119–125. doi: 10.1007/s11060-011-0704-4. [DOI] [PubMed] [Google Scholar]
- 4.Becher OJ, Hambardzumyan D, Walker TR, Helmy K, Nazarian J, Albrecht S, et al. Preclinical evaluation of radiation and perifosine in a genetically and histologically accurate model of brainstem glioma. Cancer Res. 2010;70:2548–2557. doi: 10.1158/0008-5472.CAN-09-2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DT, Kool M, et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell. 2013;24:660–672. doi: 10.1016/j.ccr.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 6.Bobo RH, Laske DW, Akbasak A, Morrison PF, Dedrick RL, Oldfield EH. Convection-enhanced delivery of macromolecules in the brain. Proc Natl Acad Sci U S A. 1994;91:2076–2080. doi: 10.1073/pnas.91.6.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bradley KA, Zhou T, McNall-Knapp RY, Jakacki RI, Levy AS, Vezina G, et al. Motexafin-gadolinium and involved field radiation therapy for intrinsic pontine glioma of childhood: a children’s oncology group phase 2 study. Int J Radiat Oncol Biol Phys. 2013;85:e55–e60. doi: 10.1016/j.ijrobp.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buczkowicz P, Hoeman C, Rakopoulos P, Pajovic S, Letourneau L, Dzamba M, et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet. 2014;46:451–456. doi: 10.1038/ng.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cage TA, Samagh SP, Mueller S, Nicolaides T, Haas-Kogan D, Prados M, et al. Feasibility, safety, and indications for surgical biopsy of intrinsic brainstem tumors in children. Childs Nerv Syst. 2013;29:1313–1319. doi: 10.1007/s00381-013-2101-0. [DOI] [PubMed] [Google Scholar]
- 10.Caretti V, Sewing AC, Lagerweij T, Schellen P, Bugiani M, Jansen MH, et al. Human pontine glioma cells can induce murine tumors. Acta Neuropathol. 2014;127:897–909. doi: 10.1007/s00401-014-1272-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Castel D, Philippe C, Calmon R, Le Dret L, Truffaux N, Boddaert N, et al. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol. 2015;130:815–827. doi: 10.1007/s00401-015-1478-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan KM, Fang D, Gan H, Hashizume R, Yu C, Schroeder M, et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013;27:985–990. doi: 10.1101/gad.217778.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chassot A, Canale S, Varlet P, Puget S, Roujeau T, Negretti L, et al. Radiotherapy with concurrent and adjuvant temozolomide in children with newly diagnosed diffuse intrinsic pontine glioma. J Neurooncol. 2012;106:399–407. doi: 10.1007/s11060-011-0681-7. [DOI] [PubMed] [Google Scholar]
- 14.Cordero FJ, Huang Z, Grenier C, He X, Hu G, McLendon RE, et al. Histone H3.3K27M represses p16 to accelerate gliomagenesis in a murine model of DIPG. Mol Cancer Res. 2017;15:1243–1254. doi: 10.1158/1541-7786.MCR-16-0389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diaz RJ, McVeigh PZ, O’Reilly MA, Burrell K, Bebenek M, Smith C, et al. Focused ultrasound delivery of Raman nanoparticles across the bloodbrain barrier: potential for targeting experimental brain tumors. Nanomedicine. 2014;10:1075–1087. doi: 10.1016/j.nano.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doolittle ND, Anderson CP, Bleyer WA, Cairncross JG, Cloughesy T, Eck SL, et al. Importance of dose intensity in neuro-oncology clinical trials: summary report of the sixth annual meeting of the Blood-Brain Barrier Disruption Consortium. Neuro Oncol. 2001;3:46–54. doi: 10.1093/neuonc/3.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elias WJ, Huss D, Voss T, Loomba J, Khaled M, Zadicario E, et al. A pilot study of focused ultrasound thalamotomy for essential tremor. N Engl J Med. 2013;369:640–648. doi: 10.1056/NEJMoa1300962. [DOI] [PubMed] [Google Scholar]
- 18.Etame AB, Diaz RJ, O’Reilly MA, Smith CA, Mainprize TG, Hynynen K, et al. Enhanced delivery of gold nanoparticles with therapeutic potential into the brain using MRI-guided focused ultrasound. Nanomedicine. 2012;8:1133–1142. doi: 10.1016/j.nano.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Etame AB, Diaz RJ, Smith CA, Mainprize TG, Hynynen K, Rutka JT. Focused ultrasound disruption of the blood-brain barrier: a new frontier for therapeutic delivery in molecular neurooncology. Neurosurg Focus. 2012;32:E3. doi: 10.3171/2011.10.FOCUS11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fisher PG, Breiter SN, Carson BS, Wharam MD, Williams JA, Weingart JD, et al. A clinicopathologic reappraisal of brain stem tumor classification. Identification of pilocystic astrocytoma and fibrillary astrocytoma as distinct entities. Cancer. 2000;89:1569–1576. doi: 10.1002/1097-0142(20001001)89:7<1569::aid-cncr22>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 21.Freeman CR, Perilongo G. Chemotherapy for brain stem gliomas. Childs Nerv Syst. 1999;15:545–553. doi: 10.1007/s003810050542. [DOI] [PubMed] [Google Scholar]
- 22.Funato K, Major T, Lewis PW, Allis CD, Tabar V. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science. 2014;346:1529–1533. doi: 10.1126/science.1253799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grasso CS, Tang Y, Truffaux N, Berlow NE, Liu L, Debily MA, et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat Med. 2015;21:555–559. doi: 10.1038/nm.3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Halvorson KG, Barton KL, Schroeder K, Misuraca KL, Hoeman C, Chung A, et al. A high-throughput in vitro drug screen in a genetically engineered mouse model of diffuse intrinsic pontine glioma identifies BMS-754807 as a promising therapeutic agent. PLoS One. 2015;10:e0118926. doi: 10.1371/journal.pone.0118926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hankinson TC, Campagna EJ, Foreman NK, Handler MH. Interpretation of magnetic resonance images in diffuse intrinsic pontine glioma: a survey of pediatric neurosurgeons. J Neurosurg Pediatr. 2011;8:97–102. doi: 10.3171/2011.4.PEDS1180. [DOI] [PubMed] [Google Scholar]
- 26.Hashizume R, Andor N, Ihara Y, Lerner R, Gan H, Chen X, et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat Med. 2014;20:1394–1396. doi: 10.1038/nm.3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hynynen K, McDannold N, Vykhodtseva N, Jolesz FA. Noninvasive MR imaging-guided focal opening of the blood-brain barrier in rabbits. Radiology. 2001;220:640–646. doi: 10.1148/radiol.2202001804. [DOI] [PubMed] [Google Scholar]
- 28.Jansen MH, van Vuurden DG, Vandertop WP, Kaspers GJ. Diffuse intrinsic pontine gliomas: a systematic update on clinical trials and biology. Cancer Treat Rev. 2012;38:27–35. doi: 10.1016/j.ctrv.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 29.Janssens GO, Jansen MH, Lauwers SJ, Nowak PJ, Oldenburger FR, Bouffet E, et al. Hypofractionation vs conventional radiation therapy for newly diagnosed diffuse intrinsic pontine glioma: a matched-cohort analysis. Int J Radiat Oncol. 2013;85:315–320. doi: 10.1016/j.ijrobp.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 30.Jolesz FA, Hynynen KH. MRI-Guided Focused Ultrasound Surgery. ed 1. Boca Raton: CRC Press; 2007. [Google Scholar]
- 31.Jones C, Karajannis MA, Jones DTW, Kieran MW, Monje M, Baker SJ, et al. Pediatric high-grade glioma: biologically and clinically in need of new thinking. Neuro Oncol. 2017;19:153–161. doi: 10.1093/neuonc/now101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kambhampati M, Perez JP, Yadavilli S, Saratsis AM, Hill AD, Ho CY, et al. A standardized autopsy procurement allows for the comprehensive study of DIPG biology. Oncotarget. 2015;6:12740–12747. doi: 10.18632/oncotarget.3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kebudi R, Cakir FB. Management of diffuse pontine gliomas in children: recent developments. Paediatr Drugs. 2013;15:351–362. doi: 10.1007/s40272-013-0033-5. [DOI] [PubMed] [Google Scholar]
- 34.Khuong-Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012;124:439–447. doi: 10.1007/s00401-012-0998-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131:803–820. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- 36.Marigil M, Martinez-Velez N, Domínguez PD, Idoate MA, Xipell E, Patiño-García A, et al. Development of a DIPG orthotopic model in mice using an implantable guide-screw system. PLoS One. 2017;12:e0170501. doi: 10.1371/journal.pone.0170501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marquet F, Tung YS, Teichert T, Ferrera VP, Konofagou EE. Noninvasive, transient and selective blood-brain barrier opening in non-human primates in vivo. PLoS One. 2011;6:e22598. doi: 10.1371/journal.pone.0022598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin E, Jeanmonod D, Morel A, Zadicario E, Werner B. High-intensity focused ultrasound for noninvasive functional neurosurgery. Ann Neurol. 2009;66:858–861. doi: 10.1002/ana.21801. [DOI] [PubMed] [Google Scholar]
- 39.Miller DL. Particle gathering and microstreaming near ultrasonically activated gas-filled micropores. J Acoust Soc Am. 1988;84:1378–1387. doi: 10.1121/1.396636. [DOI] [PubMed] [Google Scholar]
- 40.Misuraca KL, Barton KL, Chung A, Diaz AK, Conway SJ, Corcoran DL, et al. Pax3 expression enhances PDGF-B-induced brainstem gliomagenesis and characterizes a subset of brainstem glioma. Acta Neuropathol Commun. 2014;2:134. doi: 10.1186/s40478-014-0134-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Misuraca KL, Cordero FJ, Becher OJ. Pre-clinical models of diffuse intrinsic pontine glioma. Front Oncol. 2015;5:172. doi: 10.3389/fonc.2015.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Misuraca KL, Hu G, Barton KL, Chung A, Becher OJ. A novel mouse model of diffuse intrinsic pontine glioma initiated in Pax3-expressing cells. Neoplasia. 2016;18:60–70. doi: 10.1016/j.neo.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Monje M, Mitra SS, Freret ME, Raveh TB, Kim J, Masek M, et al. Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proc Natl Acad Sci U S A. 2011;108:4453–4458. doi: 10.1073/pnas.1101657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Narayana A, Kunnakkat S, Chacko-Mathew J, Gardner S, Karajannis M, Raza S, et al. Bevacizumab in recurrent high-grade pediatric gliomas. Neuro Oncol. 2010;12:985–990. doi: 10.1093/neuonc/noq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Panditharatna E, Yaeger K, Kilburn LB, Packer RJ, Nazarian J. Clinicopathology of diffuse intrinsic pontine glioma and its redefined genomic and epigenomic landscape. Cancer Genet. 2015;208:367–373. doi: 10.1016/j.cancergen.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 46.Pathania M, De Jay N, Maestro N, Harutyunyan AS, Nitarska J, Pahlavan P, et al. H3.3K27M cooperates with Trp53 loss and PDGFRA gain in mouse embryonic neural progenitor cells to induce invasive high-grade gliomas. Cancer Cell. 2017;32:684–700.e9. doi: 10.1016/j.ccell.2017.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paugh BS, Broniscer A, Qu C, Miller CP, Zhang J, Tatevossian RG, et al. Genome-wide analyses identify recurrent amplifications of receptor tyrosine kinases and cell-cycle regulatory genes in diffuse intrinsic pontine glioma. J Clin Oncol. 2011;29:3999–4006. doi: 10.1200/JCO.2011.35.5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Piunti A, Hashizume R, Morgan MA, Bartom ET, Horbinski CM, Marshall SA, et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat Med. 2017;23:493–500. doi: 10.1038/nm.4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Plessier A, Le Dret L, Varlet P, Beccaria K, Lacombe J, Mériaux S, et al. New in vivo avatars of diffuse intrinsic pontine gliomas (DIPG) from stereotactic biopsies performed at diagnosis. Oncotarget. 2017;8:52543–52559. doi: 10.18632/oncotarget.15002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Puget S, Philippe C, Bax DA, Job B, Varlet P, Junier MP, et al. Mesenchymal transition and PDGFRA amplification/mutation are key distinct oncogenic events in pediatric diffuse intrinsic pontine gliomas. PLoS One. 2012;7:e30313. doi: 10.1371/journal.pone.0030313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robison NJ, Kieran MW. Diffuse intrinsic pontine glioma: a reassessment. J Neurooncol. 2014;119:7–15. doi: 10.1007/s11060-014-1448-8. [DOI] [PubMed] [Google Scholar]
- 52.Roujeau T, Machado G, Garnett MR, Miquel C, Puget S, Geoerger B, et al. Stereotactic biopsy of diffuse pontine lesions in children. J Neurosurg. 2007;107(1 Suppl):1–4. doi: 10.3171/PED-07/07/001. [DOI] [PubMed] [Google Scholar]
- 53.Saratsis AM, Yadavilli S, Magge S, Rood BR, Perez J, Hill DA, et al. Insights into pediatric diffuse intrinsic pontine glioma through proteomic analysis of cerebrospinal fluid. Neuro Oncol. 2012;14:547–560. doi: 10.1093/neuonc/nos067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schroeder KM, Hoeman CM, Becher OJ. Children are not just little adults: recent advances in understanding of diffuse intrinsic pontine glioma biology. Pediatr Res. 2014;75:205–209. doi: 10.1038/pr.2013.194. [DOI] [PubMed] [Google Scholar]
- 55.Shang X, Wang P, Liu Y, Zhang Z, Xue Y. Mechanism of low-frequency ultrasound in opening blood-tumor barrier by tight junction. J Mol Neurosci. 2011;43:364–369. doi: 10.1007/s12031-010-9451-9. [DOI] [PubMed] [Google Scholar]
- 56.Sheikov N, McDannold N, Jolesz F, Zhang YZ, Tam K, Hynynen K. Brain arterioles show more active vesicular transport of blood-borne tracer molecules than capillaries and venules after focused ultrasound-evoked opening of the blood-brain barrier. Ultrasound Med Biol. 2006;32:1399–1409. doi: 10.1016/j.ultrasmedbio.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 57.Sheikov N, McDannold N, Sharma S, Hynynen K. Effect of focused ultrasound applied with an ultrasound contrast agent on the tight junctional integrity of the brain microvascular endothelium. Ultrasound Med Biol. 2008;34:1093–1104. doi: 10.1016/j.ultrasmedbio.2007.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Souweidane MM, Kramer K, Pandit-Taskar N, Zanzonico P, Zhou Z, Donzelli M, et al. A phase I study of convection enhanced delivery (CED) of 124I-8H9 radio-labeled monoclonal antibody in children with diffuse intrinsic pontine glioma (DIPG) J Clin Oncol. 2017;35(15 suppl):2010–2010. [Google Scholar]
- 59.Taylor KR, Mackay A, Truffaux N, Butterfield YS, Morozova O, Philippe C, et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet. 2014;46:457–461. doi: 10.1038/ng.2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Veringa SJ, Biesmans D, van Vuurden DG, Jansen MH, Wedekind LE, Horsman I, et al. In vitro drug response and efflux transporters associated with drug resistance in pediatric high grade glioma and diffuse intrinsic pontine glioma. PLoS One. 2013;8:e61512. doi: 10.1371/journal.pone.0061512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.von Werder A, Seidler B, Schmid RM, Schneider G, Saur D. Production of avian retroviruses and tissue-specific somatic retroviral gene transfer in vivo using the RCAS/TVA system. Nat Protoc. 2012;7:1167–1183. doi: 10.1038/nprot.2012.060. [DOI] [PubMed] [Google Scholar]
- 62.Vykhodtseva NI, Hynynen K, Damianou C. Histologic effects of high intensity pulsed ultrasound exposure with subharmonic emission in rabbit brain in vivo. Ultrasound Med Biol. 1995;21:969–979. doi: 10.1016/0301-5629(95)00038-s. [DOI] [PubMed] [Google Scholar]
- 63.Walker DA, Liu J, Kieran M, Jabado N, Picton S, Packer R, et al. A multidisciplinary consensus statement concerning surgical approaches to lowgrade, high-grade astrocytomas and diffuse intrinsic pontine gliomas in childhood (CPN Paris 2011) using the Delphi method. Neuro Oncol. 2013;15:462–468. doi: 10.1093/neuonc/nos330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Warren KE. Diffuse intrinsic pontine glioma: poised for progress. Front Oncol. 2012;2:205. doi: 10.3389/fonc.2012.00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44:251–253. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu YL, Maachani UB, Schweitzer M, Singh R, Wang M, Chang R, et al. Dual inhibition of PI3K/AKT and MEK/ERK pathways induces synergistic antitumor effects in diffuse intrinsic pontine glioma cells. Transl Oncol. 2017;10:221–228. doi: 10.1016/j.tranon.2016.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zaghloul MS, Eldebawy E, Ahmed S, Mousa AG, Amin A, Refaat A, et al. Hypofractionated conformal radiotherapy for pediatric diffuse intrinsic pontine glioma (DIPG): a randomized controlled trial. Radiother Oncol. 2014;111:35–40. doi: 10.1016/j.radonc.2014.01.013. [DOI] [PubMed] [Google Scholar]
- 68.Zhou Z, Singh R, Souweidane MM. Convection-enhanced delivery for diffuse intrinsic pontine glioma treatment. Curr Neuropharmacol. 2017;15:116–128. doi: 10.2174/1570159X14666160614093615. [DOI] [PMC free article] [PubMed] [Google Scholar]