

Abstract
Immunotherapy with T cells genetically modified to express chimeric antigen receptors (CARs) that target tumor-associated molecules have impressive efficacy in hematological malignancies. The field has now embraced the challenge of applying this approach to treat common epithelial malignancies, which make up the majority of cancer cases but evade immunologic attack by a variety of subversive mechanisms. Here, we review the principles that have guided CAR T cell design and the extraordinary clinical results being achieved in B cell malignancies targeting CD19 with a single infusion of engineered T cells. This success has raised expectations that CAR T cells can be applied to solid tumors, but numerous obstacles must be overcome to achieve the success observed in hematologic cancers. Potential solutions driven by advances in genetic engineering, synthetic biology, T cell biology, and improved tumor models that recapitulate the obstacles in human tumors are discussed.
Introduction
Innovations in gene transfer and adoptive T cell transfer (ACT) have converged in a novel approach to cancer therapy in which a patient's T cells are genetically modified to express synthetic chimeric antigen receptors (CARs) that redirect T cell specificity toward tumor-associated antigens. CAR T cells have shown remarkable success in some hematologic malignancies and serve as an example of how advances in immunology can inform a new class of cancer therapeutics (1). Here, we review the principles underlying CAR T cell therapy and discuss obstacles to further improve results in hematologic cancers and extend this approach to common cancers that are the major cause of cancer mortality.
Principles of CAR Design and T Cell Engineering
A CAR is a synthetic construct that, when expressed in T cells, mimics T cell receptor activation and redirects specificity and effector function toward a specified antigen. For cancer therapy, this is accomplished by linking an extracellular ligand-binding domain specific for a tumor cell surface antigen to an intracellular signaling module that activates T cells upon antigen binding. The earliest “first-generation” CARs contained only a CD3ζ or Fc receptor gamma signaling domain (2), and the addition of one (second generation) or more (third generation) costimulatory domains such as CD28, 4-1BB, or OX40 induced more cytokine production and T cell proliferation (3-5). The constellation of signaling modules in a CAR is usually selected based on analysis of tumor recognition in vitro and in preclinical in vivo models(6-8), and advances in synthetic biology are likely to improve upon constructs currently in clinical trials. For example, strategies for small molecule-mediated regulatory control of CAR expression (9), combinatorial antigen sensing (10), targeted integration of the CAR transgene into defined loci (11), logic gating of CAR recognition to improve tumor selectivity (12, 13), and suicide mechanisms for targeted elimination of transferred T cells (14, 15) have been described and could provide more potent and safe CARs.
The immune cell chassis used to express a CAR is most commonly a T cell derived from the peripheral blood. Peripheral T cells can be broadly divided by surface phenotype into naïve (TN), memory (TM), and effector (TE) subsets. TM are further subdivided into memory stem (TSCM), central memory (TCM), effector memory (TEM), and tissue resident memory (TRM) cells, each of which has a distinct role in protective immunity (16-18). Current data supports a progressive differentiation model such that activation of TN by antigen gives rise to long-lived TSCM and TCM that can self-renew and provide proliferating populations of shorter-lived TEM and TE cells (19-21). This understanding has led several groups to focus on defining the starting population of T cells that are genetically modified with CARs and used for ACT, initially in preclinical models and subsequently in clinical trials (22-27). Accumulating data suggest that engineering less differentiated TN and/or TCM cells, or culturing T cells in conditions that preserve these phenotypes, provides CAR T cell products with superior persistence in vivo (22-28). Thus, as with CAR design, cell product composition can be manipulated to improve potency and potentially reduce toxicity by providing consistent proliferation and persistence after ACT.
Clinical Efficacy: B Cell Malignancies and Beyond
Clinical trials of CAR T cells have proceeded rapidly in B cell malignancies. B cell malignancies are an attractive target for CAR T cells because they express B cell lineage-specific molecules such as CD19, CD20, and CD22 that are not expressed on other tissues, and preclinical data demonstrated that human B cell tumors could be eradicated in immune-compromised mice treated with CAR T cells (29-32). To prepare CAR T cell products for treatment of patients, T cells are obtained from the blood, activated in vitro to facilitate gene insertion, and modified to express the CAR by viral or non-viral gene delivery. CAR T cells are then re-infused into the patient, often after the administration of lymphodepleting chemotherapy to promote engraftment and proliferation of transferred cells (Figure 1). Initial reports in patients with relapsed and/or refractory chronic lymphocytic leukemia (CLL), acute lymphocytic leukemia (ALL) and non-Hodgkin lymphoma (NHL) showed remarkable antitumor effects of CD19-specific CAR T cells (33-36). Subsequent larger phase 1/2 trials at a number of centers confirmed the high level of efficacy of CAR T cells, particularly in ALL where complete remission (CR) rates of 70 to 93% are achieved (26-28, 37-40). CAR T cells administered in these studies varied in T cell subset composition, method of gene delivery, cell manufacturing platform, and used either CD28/CD3ζ or 4-1BB/CD3ζ costimulatory domains. Further studies are necessary to determine optimal product characteristics and CAR design.
Figure 1. Adoptive cell therapy with chimeric antigen receptor (CAR)-modified T cells.

A majority of patients with CLL and NHL demonstrate tumor regression after treatment with CD19 CAR T cells; however, the CR rates for these lymph node based malignancies are lower than for ALL (27, 28, 34, 41-43). Defining the reasons for incomplete response in CLL and NHL is important to improve outcomes and is the subject of ongoing research. Combination therapy with checkpoint inhibitors, cytokines, modulators of the tumor microenvironment, improved CAR design, and/or further genetic modifications of the T cells are being studied to improve efficacy. The initial response rates in patients with refractory leukemia and lymphoma are impressive, but the durability of responses will only be established with longer follow-up. CRs have been reported for up to 56 months after CD19 CAR T cell therapy; and CR continues even after the disappearance of functional CD19 CAR T cells and recovery of normal B cells(44). Understanding the factors that correlate with long-term CR or with relapse will be critical to enhancing the efficacy of CAR T cell therapy.
The eradication of large tumor burdens by CAR T cells is not accomplished without toxicity. Cytokine release syndrome (CRS) is a common complication initiated by release of IFN-γ, TNF-α, and IL-2 by activated CD19 CAR T cells is associated with fever, hemodynamic compromise, and macrophage activation with production of IL-6 and additional cytokines (45). The severity of CRS correlates with tumor burden, and interventions to block IL-6 signaling, or to suppress cytokine production by immune cells with dexamethasone, are the mainstays of therapy. Algorithms for timing interventions based on clinical and laboratory parameters are rapidly evolving. Neurologic adverse events are observed concurrent with or following CRS in a subset of patients treated with CD19 CAR T cells, and rare fatal cases have occurred. The pathogenesis of neurotoxicity remains to be elucidated: current data suggest that cytokines released by activated T cells play a role by affecting endothelial integrity(46). Finally, an anticipated side effect of targeting CD19 is that normal CD19+ B cells are eliminated. Transient and even prolonged loss of normal B cells can be managed clinically. However, vector designs that permit elimination of CAR T cells and restoration of B cell numbers are effective in animal models and could be applied in patients that achieve durable remission of their malignancy and have persisting CAR T cells (14).
The success of CAR T cells in ALL, CLL, and NHL has encouraged translation of this approach to other malignancies. CARs have been designed to target molecules such as CD123 and Lewis Y on acute myeloid leukemia (AML) (47, 48). However, none of the targets are as attractive as CD19 due to their expression on other critical hematopoietic cells and/or lack of uniform expression on the tumor. Multiple myeloma expresses several candidate molecules to target with CAR T cells including BCMA and CS1(49, 50), and early clinical data targeting BCMA is promising (51). The application of CAR T cells to common solid tumors has proceeded cautiously following a fatal toxicity in a patient treated with a high dose of ERBB2-specific CAR T cells due to recognition of normal epithelial cells (52). Subsequent studies in patients with glioblastoma and sarcoma suggested it may be possible to target ERBB2 safely, although antitumor efficacy was limited in these studies (53, 54). In glioblastoma, a dramatic response was observed after local intracranial administration of CAR T cells specific for IL13Ra2, and activity of systemically administered CAR T cells specific for EGFRvIII has been reported (55, 56). CAR T cells targeting gD2 have shown activity in patients with Ewing's sarcoma and neuroblastoma (57, 58). Trials using CAR T cells to target mesothelin, Muc16, Muc1, and ROR1 are in progress (59-63) but, as discussed below, may need to overcome unique obstacles compared to hematologic malignancies.
Barriers to CAR-T Cell Efficacy and Potential Solutions
Tumor Antigen Loss
A challenge for CAR T cell therapy in solid tumors is identifying target antigens expressed homogeneously throughout the tumor and not on normal vital tissues. The success of CAR-T cells in B cell malignancies targeting CD19 is tempered by outgrowth of CD19- tumor cells in some ALL patients (26, 64, 65). Few targets with homogeneous expression on epithelial cancers have been identified, and outgrowth of antigen-null tumor cells after CAR T cell therapy is an anticipated resistance mechanism. A strategy to circumvent tumor escape is to target multiple antigens simultaneously, such that only tumor cells that lack expression of all target molecules would escape an anti-tumor immune response (66). One way to target multiple antigens is to use promiscuous receptors as the antigen-binding portion of the CAR. NKG2D CARs, for example, target multiple ligands expressed on both tumor cells and immunosuppressive cells, while CARs using the promiscuous ErbB ligand T1E as the extracellular domain can bind multiple ErbB1-based homo- and hetero-dimers that are often overexpressed in tumors(67, 68). Another strategy is to link multiple scFvs in tandem. Several groups have demonstrated that co-targeting CD20 or CD123 in addition to CD19 with bi-specific CAR T cells eliminates CD19 loss variants and is superior to targeting CD19 alone in xenograft models (69, 70). Bi-specific CAR T cells were also superior to mono-specific CAR T cells when targeting antigens with non-uniform expression on solid tumors, such as Muc1 and PSCA for pancreatic tumors, or Her2 and IL-13Ra2 for glioblastoma (71, 72). Of interest, bi-specific T cells showed superior activity in vivo compared to 1:1 mixtures of mono-specific CAR T cells targeting the same antigens, although the mechanism behind functional superiority remains unclear. Bi-specific CARs showed enhanced ZAP70 phosphorylation and downstream signaling when both target antigens were engaged, suggesting that dual-positive tumor cells activate bi-specific CAR T cells more efficiently (72). CD19, CD20 and CD22 are attractive for multivalent targeting because they are often co-expressed on B cell malignancies, but identifying other pairs of tumor-associated antigens that are co-expressed on common epithelial tumors but not normal tissues remains a challenge.
Minimizing the escape of antigen-null tumors may also depend on the ability of CAR T cells to induce epitope spreading and engage an endogenous immune response against other tumor-associated antigens. It is possible that CAR T cell-mediated lysis of tumor cells will result in release and cross-presentation of other tumor antigens to endogenous T cells, resulting in a more effective polyclonal anti-tumor response. Some preclinical studies using CAR T cells have demonstrated epitope spreading and even resistance to rechallenge with antigen-null tumors, suggesting the development of immunological memory to other tumor-associated antigens, although this has yet to be demonstrated in patients(73, 74). Mesothelin-targeting CAR T cells were reported to induce humoral epitope spreading in some patients, though not to antigens overexpressed by the tumor or involved in tumorigenesis(60). Co-treatment of CAR T cells with modulators that enhance cross-presentation or activation of the endogenous immune system may enhance the probability of epitope spreading. For example, CAR T cells secreting IL-12 were able to activate macrophages that mediated elimination of antigen-negative tumor cells in preclinical models(75). Likewise, T cells engineered to express CD40L may better activate cross-presenting CD8a+ dendritic cells, while those expressing 4-1BBL can provide direct co-stimulation to bystander tumor-specific T cells(76, 77). Future studies will be needed to determine whether CAR T cells can be engineered to better engage an endogenous anti-tumor response and whether this can help combat tumor heterogeneity more effectively.
Toxicity to Normal Tissues
Because few truly tumor-specific targets have been identified, applying principles in synthetic biology that might enable CAR T cells to discriminate between tumor and normal cells expressing the same antigen could improve both the efficacy and safety of therapy. Tuning the affinity of the CAR scFv can allow T cells to distinguish between antigens that are overexpressed on tumor cells but expressed at lower levels on normal cells (78). Tumor antigens thought to be unsafe to target due to wide normal tissue expression, thus, may be targetable if expression levels are sufficiently higher on tumor versus normal cells. CARs designed from scFvs targeting either CD38 or EGFR with ∼1000-fold reduced affinity conferred effective lysis of tumor cells but spared antigen-positive normal cells (79, 80). Whether this could truly achieve discrimination of tumor and normal cells based on level of antigen expression in clinical settings without the outgrowth of antigen low tumor cells remains to be determined.
Another strategy to increase tumor-specificity is to use “AND” logic gates that require recognition of two different antigens on the same target cell to elicit full CAR T cell activation (13). The success of this strategy requires identifying antigen pairs that are selectively co-expressed on tumor cells but not normal tissues. One implementation of this strategy is to split the CD3ζ signaling and CD28 co-stimulatory domains across separate receptors, with each signaling domain linked to an scFv specific for a different antigen (81). However, several studies employing such split-receptor systems have found that CD3ζ signaling alone is sufficient to induce some T cell effector functions, including lysis of single-positive cells(66, 81), suggesting toxicity to single-positive normal tissues may not be avoided. This problem could be solved by using a low-affinity scFv that is incapable of inducing T cell activation when linked only to the CD3ζ signaling domain (13). Development of such dual-signaling CAR T cells is likely to require further optimization of each individual scFv. Other aspects of antigen pairs, such as size and ability to co-localize in the synapse, might also affect their ability to properly activate T cells.
Several groups have built constructs in which CAR expression is regulated by a drug-inducible promoter or in which the recognition domain and signaling domain are only associated together in the presence of a small molecule dimerizer (9, 82, 83). CAR T cells can be transiently activated in vivo by drug administration, and their activity can theoretically be halted if toxicity occurs by withdrawal of the drug. Alternately, timing and location of drug delivery can be adjusted to minimize toxicity. For example, Her2-specific CAR T cells induced rapid pulmonary toxicity as a consequence of recognition of Her2+ cells in the lung (52). If a CAR-inducing drug was delivered several days after infusion, when the majority of intravenously infused T cells have migrated out of the lung, toxicity to normal cells in the lung might be diminished or averted. Likewise, by delivering the drug locally rather than systemically, CAR T cell activity could be restricted to particular tissue compartments.
Engineering T cells in which CAR expression is regulated by input signals found primarily in the tumor microenvironment (TME) is another potential strategy. Tumors are often hypoxic, and oxygen-sensitive CAR T cells have been designed by fusing the CAR to a subdomain of the hypoxia transcription factor HIF-1α that is sensitive to protein degradation under normoxic conditions (84). Likewise, Roybal et al. engineered a synthetic Notch (“synNotch”) receptor that upon recognition of one tumor-associated antigen releases a transcription factor that induces expression of a CAR specific for a second tumor-associated antigen (10). The advantage of these approaches is that CAR expression is restricted to the local tumor environment, minimizing the potential for off-tumor toxicity. However, as with “AND” gate CAR T cells, clinical application of the synNotch strategy requires the identification of two tumor-associated antigens that are co-expressed in the tumor but do not overlap in their normal tissue expression. It is also uncertain whether the kinetics of CAR degradation after signal 1 is disengaged will be sufficiently rapid to minimize off-tumor toxicity. Despite these limitations, both approaches can theoretically improve tumor-selectivity, and may prove advantageous in settings where antigen-positive tumor cells and normal tissues are spatially segregated, such that CAR expression is fully degraded before T cells that leave the tumor site encounter antigen-positive normal cells.
Trafficking to Solid Tumors
Analysis of the TME has identified a variety of obstacles such as trafficking, immunosuppressive molecules and cells, and immune checkpoints that CAR T cells will need to overcome to be effective in solid tumors (Figure 2). The efficacy of CAR T cells in hematological malignancies in part may reflect efficient access to tumor cells in the bone marrow and lymph nodes where T cells normally traffic. Recognition of solid tumors requires egress from the blood into the tumor site, and many malignancies evolve such that T cell infiltration is actively impeded (85-87). In situations where the tumor is localized, regional rather than systemic administration of CAR T cells might be effective. Intracranial delivery has been shown to be safe and to have antitumor activity in glioblastoma (56), and intra-pleural delivery of CAR T cells was superior to systemic administration in preclinical studies of human pleural malignancy (88).
Figure 2. Barriers to CAR T cell therapy for solid tumors.
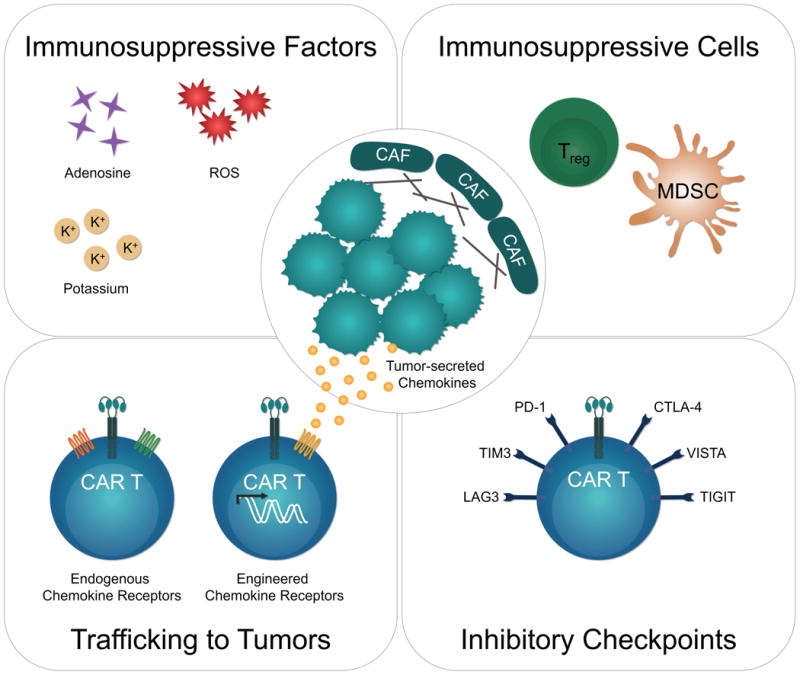
Bottom left: CAR T cell trafficking depends on expression of receptors for chemokines secreted by the tumor. CAR T cells endogenously express chemokine receptors like CXCR3 and CCR5, but their cognate ligands are often not highly expressed by solid tumors. CAR T cells can be engineered to express receptors (e.g. CCR2, CCR4) for chemokines naturally secreted by the tumor to improve trafficking to tumors. Bottom right: Antigen-activated CAR T cells in the tumor microenvironment up-regulate expression of inhibitory receptors which can lead to T cell dysfunction. Upper left: Tumor microenvironments are rich in factors like adenosine, extracellular potassium, and reactive oxygen species (ROS), which can inhibit T cells directly or indirectly. Upper right: Immunosuppressive cells like regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) can promote tumor growth and inhibit T cell activity both directly and indirectly. Cancer-associated fibroblasts (CAF) deposit extracellular matrix to limit T cell penetration and can recruit other immunosuppressive cells.
Improved understanding of mechanisms that promote or exclude T cell infiltration into tumors are likely to create opportunities to improve CAR T cell trafficking, either by additional genetic modification of T cells (89), or by combining CAR T cells with oncolytic viruses or other strategies that promote inflammation at the tumor site (90),(91). CAR T cells can be engineered to express receptors like CCR2 and CCR4 that are specific for chemokines naturally overexpressed by tumors, enabling them to traffic more efficiently to tumors (92, 93) (94). Rather than custom engineering T cells to the chemokine profile of individual tumors, a more generalizable strategy is to induce tumors to secrete chemokines that CAR T cells are already responsive to. An oncolytic virus has been used to deliver the chemokine CCL5 (RANTES) to the tumor. CAR-T cells already express receptors (CCR1, CCR3, and CCR5) for CCL5, and combination therapy with CCL5-expressing oncolytic virus and CAR T cells synergistically improved survival and tumor clearance in preclinical models (90).
CAR T cell access to tumors might also be improved by combining adoptive therapy with drugs that induce immunogenic cell death (ICD) of tumor cells. Unlike physiological cell death, in which dying cells are cleared without an inflammatory response, ICD induces release of damage-associated molecular patterns (DAMPs), which directly activate dendritic cells to secrete T cell-attracting chemokines and cross-present tumor antigens (95). Local radiotherapy and certain chemotherapeutic agents induce ICD and activate endogenous T cell responses to tumor antigens (96, 97). These modalities also inhibit or eliminate immunosuppressive cell subsets in the TME, resulting in an overall shift to a pro-inflammatory state and improved immune responses (96-98) (99). A similar regimen may improve CAR T cell infiltration by inducing production of chemokines and creating a favorable environment for CAR T cell function. Unlike genetic engineering-based approaches, such combination therapy has the advantage of modulating multiple immune pathways at once.
The differentiation state of the T cells selected for CAR modification can also influence CAR T cell function and migratory properties in vivo. Central memory T cells (TCM) have superior anti-tumor function relative to effector memory T cells (TEM) in xenograft models of hematological malignancies due to superior persistence and proliferation (22, 24, 25). However, TEM express higher levels of chemokine receptors and adhesion molecules required for homing to inflamed peripheral tissues and may be better poised to enter solid tumor sites. Despite these attributes, a recent study demonstrated that in vitro-generated TEM expressing a gp100-specific TCR were less effective on a per-cell basis than TCM of the same antigen-specificity against B16 tumors (23). Superior activity was dependent on the ability of TCM to traffic first to secondary lymphoid organs rather than peripheral tissues, which may be necessary to engage antigen-presenting cells (APCs) in tumor-draining lymph nodes. CAR T cells, however, do not depend on interactions with APCs for activation, and one study demonstrated that CAR T cells engineered from CCR7- T cells accumulated better within solid tumors than those derived from CCR7+ T cells (100). These CAR T cells were more prone to activation-induced cell death (AICD), but when CD28 and OX40 co-stimulation were incorporated into the CAR construct, AICD was reduced such that CCR7- CAR T cells were more effective at clearing tumors than CCR7+ CAR T cells. Thus, the best T cell subset for CAR T cell therapy for solid tumors may differ from the subset suited for hematological malignancies, or from the subset used for TCR-based T cell therapy. Further research is likely to define genetic manipulations of specific T cell subsets that endow the cells with homing and functional properties needed to infiltrate and effectively target solid tumors.
Overcoming the Immunosuppressive Tumor Microenvironment
Migration of CAR T cells into tumor sites is not sufficient to ensure tumor destruction because of the immunosuppressive TME (Figure 2). Low pH, hypoxia, an absence of vital nutrients, and stromal and immune cells that release suppressive factors are characteristic of the TME and inhibit T cells. Additionally, tumor and infiltrating cells may express inhibitory receptor ligands like PD-L1 that can directly suppress tumor-specific T cells.
Several groups have attempted to enhance CAR T cell activity by combining ACT with modulators of the TME. A promising avenue is the use of checkpoint inhibitors that target the PD-1/PD-L1 or CTLA-4 pathways, which alone have shown efficacy in some cancers (101). Responsiveness to checkpoint blockade was improved by enhancing priming of tumor-specific T cells and might logically be combined with adoptive transfer of CAR T cells, although the risk of toxicity to normal tissues may be increased (99) (102, 103). Other groups have engineered CAR T cells to secrete anti-PD-L1 antibodies (104), knocked out PD-1 and Lag3 using CRISPR (105-107), or co-expressed “switch receptors” linking the PD-1 ectodomain to the CD28 endodomain such that engagement of PD-L1 delivers an activating rather than inhibitory signal to the T cell (108, 109). Anti-CTLA-4 antibodies can also boost endogenous T cell responses to tumors, but the context in which they might improve CAR T cell responses is unclear. CTLA-4 inhibits T cell responses in part by competing with CD28 for binding to CD80/CD86 on dendritic cells and by physically excluding CD28 from the synapse (110). Thus, CAR T cells with a CD28 signaling endodomain may not be intrinsically affected by CTLA-4 regulation. This is supported by a study demonstrating that shRNA-mediated knockdown of CTLA-4 improved the function of first generation CAR T cells in vivo but not second generation CAR T cells with CD28 signaling domains (111). However, anti-CTLA-4 antibodies also promote immune responses in a cell extrinsic fashion by depleting CTLA-4+ Treg cells(112, 113), which may benefit CAR T cells. In addition to inhibitory receptor expression, T cell dysfunction may be acquired in the TME by dysregulation of signaling pathways through upregulation of SHP-1 or diacylglycerol kinase (DGK), and pharmacologic inhibition of these enzymes can improve the anti-tumor function of CAR T cells(114, 115).
Overcoming immunosuppressive cells in the TME is likely to be necessary for CAR T cell efficacy. Depletion of regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) with blocking antibodies or genetic manipulation has improved the efficacy of T cell therapy in animal models(116-118). Cancer-associated fibroblasts (CAFs), which comprise a majority of tumor stromal cells and express high levels of fibroblast activation protein (FAP), play a central role in establishing the immunosuppressive microenvironment and depositing extracellular matrix (ECM) proteins to limit T cell penetration. Targeting CAFs with FAP-specific CAR T cells or engineering CAR T cells to secrete ECM-degrading enzymes improves their ability to infiltrate and lyse tumors (73, 119). Alternatively, engineering CAR T cells to express the pro-inflammatory cytokine IL-12 can modulate the TME and promote recruitment and activation of macrophages (73, 75, 120, 121).
A number of studies have focused on improving CAR T cell activity by altering their metabolic profiles to enhance cell function in hostile environments. Tumors are often characterized by high levels of adenosine and reactive oxygen species (ROS), both of which directly impair T cell responses (122, 123). Knocking down the adenosine 2A receptor with shRNA or co-transducing T cells with catalase to enable breakdown of ROS significantly improved CAR T cell persistence and function in vivo (124, 125). Likewise, tumors display elevated levels of extracellular potassium that directly impair TCR-driven Akt-mTOR phosphorylation and effector function. Engineering T cells to overexpress a potassium channel to enable greater potassium efflux effectively undoes this mode of suppression and improves T cell function within the tumor (126).
Overall, CAR T cells face a number of hurdles in combating solid tumors, but some obstacles may be easier to overcome than others. Advances in genetic engineering are proceeding rapidly, and our ability to engineer CARs that, for example, target multiple antigens to overcome tumor heterogeneity and antigen-loss or that co-express modulators of the TME are now undergoing clinical evaluation. On the other hand, one of the highest barriers to success may be the ability of CAR T cells to infiltrate solid tumors. A number of studies have shown that the success of checkpoint inhibitors depends on the presence of tumor-infiltrating T cells, and strategies aimed at enhancing T cell infiltration can overcome tumor resistance to checkpoint inhibition(99, 127). Increasing CAR T cell migration to tumors could potentially increase the response of tumors to other combination therapies as well, such as those aimed at targeting immunosuppressive cells, enhancing CAR survival, and activating the endogenous immune response. Thus, efficient infiltration of tumors is likely to be a rate-limiting step for CAR T cell therapy.
Moving Beyond Empirical Testing
Given the myriad ways in which tumors can suppress T cells, the number of genetic manipulations and combination therapies that could be tested in the clinic are seemingly limitless. A challenge for the field is the need for faithful preclinical models to screen therapeutic combinations before clinical translation. Better tools to analyze post-treatment biopsies will also help maximize our understanding of what resistance mechanisms may evolve and inform the design of future combination therapies.
Models for CAR T Cell Therapy
A challenge for preclinical studies evaluating the efficacy of CAR T cells is having clinically relevant models that recapitulate the obstacles in human solid tumors (Figure 3). Most studies have relied on transplanted human tumor xenografts in immune-compromised NOD/SCID/γc-/- (NSG) mice that lack T cells, NK cells and B cells. The NSG model allows rapid analysis of human T cell recognition of tumor cells in vivo and is useful to evaluate T cell persistence and effector function. However, NSG models fail to develop a clinically relevant TME and do not inform the safety of targets that lack epitope homology and/or normal tissue expression. ACT is also studied in immune-competent syngeneic mouse models but a majority of these models implant tumor cells into foreign anatomical sites where the tumors grow rapidly and do not co-evolve with the host immune system in the same way as human tumors (128). Moreover, endogenous anti-tumor immune responses are higher in transplanted than autochthonous tumors for the same disease, suggesting the implantation process artificially increases immunogenicity (129, 130).
Figure 3. Comparison of mouse models for human solid tumors.
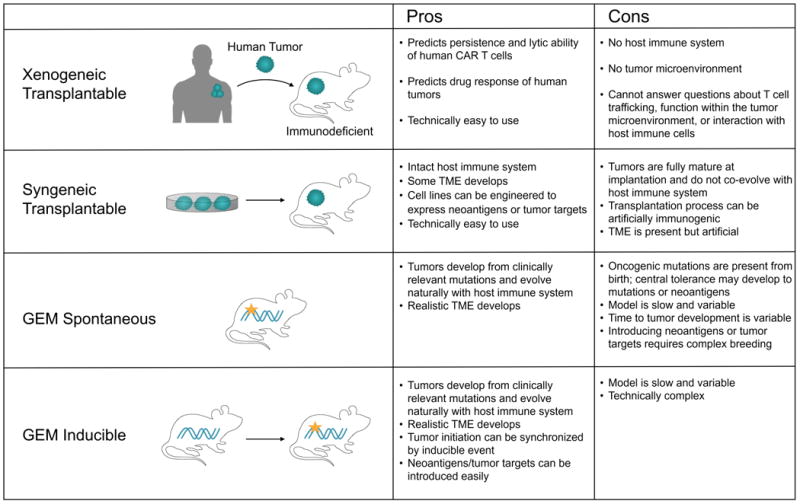
An alternative to transplantable models is to induce malignant transformation in normal cells in situ with defined oncogenic events. Such genetically-engineered mouse (GEM) models recapitulate tumor initiation, progression, and the genetic and histopathological characteristics of human cancers (128). Oncogenic mutations in Kras and p53 can be introduced at birth through a Cre/lox system, such as in the KPC (KrasLSL-G12D/+; p53f/f; Pdx1-Cre) model of pancreatic adenocarcinoma, where a Pdx1-driven Cre restricts the mutations to the pancreas (131). A disadvantage of tissue-restricted Cre expression is that cancer is induced throughout the entire tissue, and the presence of mutations from birth may affect central tolerance and influence the evolution of immune responses differently than if mutations were acquired postnatally. To address this, Tyler Jacks' group developed an inducible “KP” GEM model of lung adenocarcinoma in which intratracheal infection with Cre-expressing lentivirus initiates p53 deletion and KrasG12D activation in individual lung epithelial cells (132). Importantly, this model mimics both the development and therapeutic response of human lung adenocarcinomas (99). A drawback of GEM models, however, is their relative lack of CAR targets and neoantigens relative to carcinogen-induced models and human cancer(133). However, model antigens can easily be introduced in the KP model by engineering the lentivirus, and exposing Kras-mutant GEM mice to tobacco smoke can induce a more realistic mutational landscape.
GEM models that reflect the TME of human tumors may give more accurate estimates of treatment efficacy and offer insight into resistance mechanisms that evolve and pathways to target with combination therapy (134). For example, recent studies have used GEM models to study Tregs in tumor development and test strategies for Treg inhibition. One approach to Treg inhibition may be to block IL-35, an immunosuppressive cytokine secreted by tumor-resident Tregs that promotes effector T cell exhaustion in part by promoting expression of inhibitory receptors like PD-1, Tim3, and Lag3 (135). Interestingly, this model accurately predicted that IL-35 and PD-1 blockade would not synergize since IL-35 overexpression and PD-1 upregulation are part of the same suppressive pathway. Thus, it is anticipated that GEM models will be useful for studying impediments to CAR T cell therapy of solid tumors and for identifying rational combination therapies for clinical translation.
Advances in Immune Monitoring of Clinical Trials
Identifying methods to enhance CAR T cell therapy will be assisted by discovery-driven approaches to clinical trials. Collecting tumor biopsies and blood pre- and post-treatment, enables thorough analysis of tumors by flow cytometry, immunohistochemistry, and unbiased genome-wide RNA sequencing and can identify correlates of clinical success or failure. Response to anti-CTLA-4 therapy of localized bladder cancer, for example, was associated with up-regulation of ICOS on T cells(136, 137); subsequent studies in mice demonstrated that ICOS expression was required for the efficacy of anti-CTLA-4 in vivo and that activation of the ICOS/ICOSL pathway synergistically enhanced response to anti-CTLA-4 (138, 139). Similar analysis of pre- and post-treatment biopsies and transcriptomic and epigenetic analysis of CD19 CAR T cells are being performed to identify mechanisms of resistance in the approximately 50% of NHL patients that do not achieve a complete remission, and where CD19 loss is not the mechanism of escape (27, 28, 42, 140).
New technologies with improved sensitivity and systems-analysis will facilitate the identification of pathways associated with therapy response or resistance. Single-cell RNA sequencing can provide unbiased insight into tumor responses, revealing differences in gene transcription that may be obscured by heterogeneity at the cell population level. At the protein level, methods such as CyTOF allow analysis of 40 (and up to 100 theoretically) proteins simultaneously, providing high resolution of cell phenotype, and can be coupled to immunohistochemical methods to obtain spatial information of proteins and protein modifications at subcellular resolution (141). Additionally, the development of multiplex immunohistochemistry allows detection of multiple biomarkers simultaneously on tumor biopsies and visualization of cell subsets with tissue architecture preserved. Integrating longitudinal data from gene expression, epigenetics, flow and mass spectrometry, and IHC will provide a comprehensive understanding of patient responses to therapy and should guide the development of rational, rather than empirical, combinations.
Conclusions
Progress in immune-based therapies is improving outcomes for many patients with advanced malignancies. The development of CAR T cells represents a convergence of insights from multiple scientific fields, but success has thus far been limited to B cell malignancies. Extending this approach to other cancers will require the development of strategies based on understanding the obstacles posed by tumor heterogeneity and the tumor microenvironment that is emerging from sophisticated analytic tools and superior models. These strategies will take advantage of our unprecedented ability to genetically manipulate T cells to confer novel functions, enabling them to target tumor cells and persist and function in hostile circumstances.
Acknowledgments
This work was supported by grants from the National Institutes of Health CA136551 and CA114536 (S.R.R.), and by the Cancer Research Institute (SS).
References
- 1.Sadelain M, Riviere I, Riddell S. Therapeutic T cell engineering. Nature. 2017;545:423–431. doi: 10.1038/nature22395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. 1993;90:720–724. doi: 10.1073/pnas.90.2.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat Biotechnol. 2002;20:70–75. doi: 10.1038/nbt0102-70. [DOI] [PubMed] [Google Scholar]
- 4.Kershaw MH, Teng MW, Smyth MJ, Darcy PK. Supernatural T cells: genetic modification of T cells for cancer therapy. Nat Rev Immunol. 2005;5:928–940. doi: 10.1038/nri1729. [DOI] [PubMed] [Google Scholar]
- 5.Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, Campana D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18:676–684. doi: 10.1038/sj.leu.2403302. [DOI] [PubMed] [Google Scholar]
- 6.Srivastava S, Riddell SR. Engineering CAR-T cells: Design concepts. Trends Immunol. 2015;36:494–502. doi: 10.1016/j.it.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kawalekar OU, O'Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD, Jr, Patel PR, Guedan S, Scholler J, Keith B, Snyder NW, Blair IA, Milone MC, June CH. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity. 2016;44:380–390. doi: 10.1016/j.immuni.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 8.Zhao Z, Condomines M, van der Stegen SJ, Perna F, Kloss CC, Gunset G, Plotkin J, Sadelain M. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell. 2015;28:415–428. doi: 10.1016/j.ccell.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu CY, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science. 2015;350:aab4077. doi: 10.1126/science.aab4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, Lim WA. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell. 2016;164:770–779. doi: 10.1016/j.cell.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, Odak A, Gonen M, Sadelain M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543:113–117. doi: 10.1038/nature21405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. 2013;5:215ra172. doi: 10.1126/scitranslmed.3006597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31:71–75. doi: 10.1038/nbt.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paszkiewicz PJ, Frassle SP, Srivastava S, Sommermeyer D, Hudecek M, Drexler I, Sadelain M, Liu L, Jensen MC, Riddell SR, Busch DH. Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J Clin Invest. 2016;126:4262–4272. doi: 10.1172/JCI84813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diaconu I, Ballard B, Zhang M, Chen Y, West J, Dotti G, Savoldo B. Inducible Caspase-9 Selectively Modulates the Toxicities of CD19-Specific Chimeric Antigen Receptor-Modified T Cells. Mol Ther. 2017;25:580–592. doi: 10.1016/j.ymthe.2017.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer. 2012;12:671–684. doi: 10.1038/nrc3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, Almeida JR, Gostick E, Yu Z, Carpenito C, Wang E, Douek DC, Price DA, June CH, Marincola FM, Roederer M, Restifo NP. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17:1290–1297. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sathaliyawala T, Kubota M, Yudanin N, Turner D, Camp P, Thome JJ, Bickham KL, Lerner H, Goldstein M, Sykes M, Kato T, Farber DL. Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity. 2013;38:187–197. doi: 10.1016/j.immuni.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerlach C, Rohr JC, Perie L, van Rooij N, van Heijst JW, Velds A, Urbanus J, Naik SH, Jacobs H, Beltman JB, de Boer RJ, Schumacher TN. Heterogeneous differentiation patterns of individual CD8+ T cells. Science. 2013;340:635–639. doi: 10.1126/science.1235487. [DOI] [PubMed] [Google Scholar]
- 20.Buchholz VR, Flossdorf M, Hensel I, Kretschmer L, Weissbrich B, Graf P, Verschoor A, Schiemann M, Hofer T, Busch DH. Disparate individual fates compose robust CD8+ T cell immunity. Science. 2013;340:630–635. doi: 10.1126/science.1235454. [DOI] [PubMed] [Google Scholar]
- 21.Graef P, Buchholz VR, Stemberger C, Flossdorf M, Henkel L, Schiemann M, Drexler I, Hofer T, Riddell SR, Busch DH. Serial transfer of single-cell-derived immunocompetence reveals stemness of CD8(+) central memory T cells. Immunity. 2014;41:116–126. doi: 10.1016/j.immuni.2014.05.018. [DOI] [PubMed] [Google Scholar]
- 22.Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008;118:294–305. doi: 10.1172/JCI32103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, Palmer DC, Antony PA, Hwang ST, Rosenberg SA, Waldmann TA, Restifo NP. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci U S A. 2005;102:9571–9576. doi: 10.1073/pnas.0503726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, Riddell SR. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. 2016;30:492–500. doi: 10.1038/leu.2015.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang X, Wong CW, Urak R, Taus E, Aguilar B, Chang WC, Mardiros A, Budde LE, Brown CE, Berger C, Forman SJ, Jensen MC. Comparison of naive and central memory derived CD8+ effector cell engraftment fitness and function following adoptive transfer. Oncoimmunology. 2016;5:e1072671. doi: 10.1080/2162402X.2015.1072671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, Sommermeyer D, Melville K, Pender B, Budiarto TM, Robinson E, Steevens NN, Chaney C, Soma L, Chen X, Yeung C, Wood B, Li D, Cao J, Heimfeld S, Jensen MC, Riddell SR, Maloney DG. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126:2123–2138. doi: 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turtle CJ, Hay KA, Hanafi LA, Li D, Cherian S, Chen X, Wood B, Lozanski A, Byrd JC, Heimfeld S, Riddell SR, Maloney DG. Durable Molecular Remissions in Chronic Lymphocytic Leukemia Treated With CD19-Specific Chimeric Antigen Receptor-Modified T Cells After Failure of Ibrutinib. J Clin Oncol. 2017;35:3010–3020. doi: 10.1200/JCO.2017.72.8519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turtle CJ, Hanafi LA, Berger C, Hudecek M, Pender B, Robinson E, Hawkins R, Chaney C, Cherian S, Chen X, Soma L, Wood B, Li D, Heimfeld S, Riddell SR, Maloney DG. Immunotherapy of non-Hodgkin's lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med. 2016;8:355ra116. doi: 10.1126/scitranslmed.aaf8621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kowolik CM, Topp MS, Gonzalez S, Pfeiffer T, Olivares S, Gonzalez N, Smith DD, Forman SJ, Jensen MC, Cooper LJ. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. 2006;66:10995–11004. doi: 10.1158/0008-5472.CAN-06-0160. [DOI] [PubMed] [Google Scholar]
- 30.Brentjens RJ, Santos E, Nikhamin Y, Yeh R, Matsushita M, La Perle K, Quintas-Cardama A, Larson SM, Sadelain M. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin Cancer Res. 2007;13:5426–5435. doi: 10.1158/1078-0432.CCR-07-0674. [DOI] [PubMed] [Google Scholar]
- 31.Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, Samanta M, Lakhal M, Gloss B, Danet-Desnoyers G, Campana D, Riley JL, Grupp SA, June CH. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17:1453–1464. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, Jensen MC, Riddell SR. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res. 2015;3:125–135. doi: 10.1158/2326-6066.CIR-14-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska M, Borquez-Ojeda O, Qu J, Wasielewska T, He Q, Bernal Y, Rijo IV, Hedvat C, Kobos R, Curran K, Steinherz P, Jurcic J, Rosenblat T, Maslak P, Frattini M, Sadelain M. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra138. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes MS, Sherry RM, Yang JC, Kammula US, Devillier L, Carpenter R, Nathan DA, Morgan RA, Laurencot C, Rosenberg SA. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, Maric I, Raffeld M, Nathan DA, Lanier BJ, Morgan RA, Rosenberg SA. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, Qu J, Wasielewska T, He Q, Fink M, Shinglot H, Youssif M, Satter M, Wang Y, Hosey J, Quintanilla H, Halton E, Bernal Y, Bouhassira DC, Arcila ME, Gonen M, Roboz GJ, Maslak P, Douer D, Frattini MG, Giralt S, Sadelain M, Brentjens R. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra225. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, Bleakley M, Brown C, Mgebroff S, Kelly-Spratt KS, Hoglund V, Lindgren C, Oron AP, Li D, Riddell SR, Park JR, Jensen MC. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129:3322–3331. doi: 10.1182/blood-2017-02-769208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN, Steinberg SM, Stroncek D, Tschernia N, Yuan C, Zhang H, Zhang L, Rosenberg SA, Wayne AS, Mackall CL. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, Shen A, Teachey DT, Levine BL, June CH, Porter DL, Grupp SA. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, Yang JC, Phan GQ, Hughes MS, Sherry RM, Raffeld M, Feldman S, Lu L, Li YF, Ngo LT, Goy A, Feldman T, Spaner DE, Wang ML, Chen CC, Kranick SM, Nath A, Nathan DA, Morton KE, Toomey MA, Rosenberg SA. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33:540–549. doi: 10.1200/JCO.2014.56.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Locke FL, Neelapu SS, Bartlett NL, Siddiqi T, Chavez JC, Hosing CM, Ghobadi A, Budde LE, Bot A, Rossi JM, Jiang Y, Xue AX, Elias M, Aycock J, Wiezorek J, Go WY. Phase 1 Results of ZUMA-1: A Multicenter Study of KTE-C19 Anti-CD19 CAR T Cell Therapy in Refractory Aggressive Lymphoma. Mol Ther. 2017;25:285–295. doi: 10.1016/j.ymthe.2016.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kochenderfer JN, Somerville RPT, Lu T, Shi V, Bot A, Rossi J, Xue A, Goff SL, Yang JC, Sherry RM, Klebanoff CA, Kammula US, Sherman M, Perez A, Yuan CM, Feldman T, Friedberg JW, Roschewski MJ, Feldman SA, McIntyre L, Toomey MA, Rosenberg SA. Lymphoma Remissions Caused by Anti-CD19 Chimeric Antigen Receptor T Cells Are Associated With High Serum Interleukin-15 Levels. J Clin Oncol. 2017;35:1803–1813. doi: 10.1200/JCO.2016.71.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kochenderfer JN, Somerville RPT, Lu T, Yang JC, Sherry RM, Feldman SA, McIntyre L, Bot A, Rossi J, Lam N, Rosenberg SA. Long-Duration Complete Remissions of Diffuse Large B Cell Lymphoma after Anti-CD19 Chimeric Antigen Receptor T Cell Therapy. Mol Ther. 2017;25:2245–2253. doi: 10.1016/j.ymthe.2017.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, Grupp SA, Mackall CL. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124:188–195. doi: 10.1182/blood-2014-05-552729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gust J, H KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, Yeung C, Liles WC, Wurfel M, Lopez JA, Chen J, Chung D, Harju-Baker S, Ozpolat T, Tozer-Fink K, Riddell SR, Maloney DG, Turtle CJ. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discovery. Oct 12; doi: 10.1158/2159-8290.CD-17-0698. In press. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ritchie DS, Neeson PJ, Khot A, Peinert S, Tai T, Tainton K, Chen K, Shin M, Wall DM, Honemann D, Gambell P, Westerman DA, Haurat J, Westwood JA, Scott AM, Kravets L, Dickinson M, Trapani JA, Smyth MJ, Darcy PK, Kershaw MH, Prince HM. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol Ther. 2013;21:2122–2129. doi: 10.1038/mt.2013.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tettamanti S, Marin V, Pizzitola I, Magnani CF, Giordano Attianese GM, Cribioli E, Maltese F, Galimberti S, Lopez AF, Biondi A, Bonnet D, Biagi E. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor. Br J Haematol. 2013;161:389–401. doi: 10.1111/bjh.12282. [DOI] [PubMed] [Google Scholar]
- 49.Carpenter RO, Evbuomwan MO, Pittaluga S, Rose JJ, Raffeld M, Yang S, Gress RE, Hakim FT, Kochenderfer JN. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin Cancer Res. 2013;19:2048–2060. doi: 10.1158/1078-0432.CCR-12-2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chu J, He S, Deng Y, Zhang J, Peng Y, Hughes T, Yi L, Kwon CH, Wang QE, Devine SM, He X, Bai XF, Hofmeister CC, Yu J. Genetic modification of T cells redirected toward CS1 enhances eradication of myeloma cells. Clin Cancer Res. 2014;20:3989–4000. doi: 10.1158/1078-0432.CCR-13-2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ali SA, Shi V, Maric I, Wang M, Stroncek DF, Rose JJ, Brudno JN, Stetler-Stevenson M, Feldman SA, Hansen BG, Fellowes VS, Hakim FT, Gress RE, Kochenderfer JN. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood. 2016;128:1688–1700. doi: 10.1182/blood-2016-04-711903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, Robertson C, Gray TL, Diouf O, Wakefield A, Ghazi A, Gerken C, Yi Z, Ashoori A, Wu MF, Liu H, Rooney C, Dotti G, Gee A, Su J, Kew Y, Baskin D, Zhang YJ, New P, Grilley B, Stojakovic M, Hicks J, Powell SZ, Brenner MK, Heslop HE, Grossman R, Wels WS, Gottschalk S. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017;3:1094–1101. doi: 10.1001/jamaoncol.2017.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, Liu E, Dakhova O, Ashoori A, Corder A, Gray T, Wu MF, Liu H, Hicks J, Rainusso N, Dotti G, Mei Z, Grilley B, Gee A, Rooney CM, Brenner MK, Heslop HE, Wels WS, Wang LL, Anderson P, Gottschalk S. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J Clin Oncol. 2015;33:1688–1696. doi: 10.1200/JCO.2014.58.0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, Martinez-Lage M, Brem S, Maloney E, Shen A, Isaacs R, Mohan S, Plesa G, Lacey SF, Navenot JM, Zheng Z, Levine BL, Okada H, June CH, Brogdon JL, Maus MV. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9 doi: 10.1126/scitranslmed.aaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J, Simpson J, Kurien A, Priceman SJ, Wang X, Harshbarger TL, D'Apuzzo M, Ressler JA, Jensen MC, Barish ME, Chen M, Portnow J, Forman SJ, Badie B. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med. 2016;375:2561–2569. doi: 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, Liu H, Wu MF, Mei Z, Gee A, Mehta B, Zhang H, Mahmood N, Tashiro H, Heslop HE, Dotti G, Rooney CM, Brenner MK. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol Ther. 2017;25:2214–2224. doi: 10.1016/j.ymthe.2017.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, Huls MH, Liu E, Gee AP, Mei Z, Yvon E, Weiss HL, Liu H, Rooney CM, Heslop HE, Brenner MK. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Balakrishnan A, Goodpaster T, Randolph-Habecker J, Hoffstrom BG, Jalikis FG, Koch LK, Berger C, Kosasih PL, Rajan A, Sommermeyer D, Porter PL, Riddell SR. Analysis of ROR1 Protein Expression in Human Cancer and Normal Tissues. Clin Cancer Res. 2017;23:3061–3071. doi: 10.1158/1078-0432.CCR-16-2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, Chew A, Zhao Y, Levine BL, Albelda SM, Kalos M, June CH. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. 2014;2:112–120. doi: 10.1158/2326-6066.CIR-13-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Berger C, Sommermeyer D, Hudecek M, Berger M, Balakrishnan A, Paszkiewicz PJ, Kosasih PL, Rader C, Riddell SR. Safety of targeting ROR1 in primates with chimeric antigen receptor-modified T cells. Cancer Immunol Res. 2015;3:206–216. doi: 10.1158/2326-6066.CIR-14-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Koneru M, O'Cearbhaill R, Pendharkar S, Spriggs DR, Brentjens RJ. A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16(ecto) directed chimeric antigen receptors for recurrent ovarian cancer. J Transl Med. 2015;13:102. doi: 10.1186/s12967-015-0460-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wei X, Lai Y, Li J, Qin L, Xu Y, Zhao R, Li B, Lin S, Wang S, Wu Q, Liang Q, Peng M, Yu F, Li Y, Zhang X, Wu Y, Liu P, Pei D, Yao Y, Li P. PSCA and MUC1 in non-small-cell lung cancer as targets of chimeric antigen receptor T cells. Oncoimmunology. 2017;6:e1284722. doi: 10.1080/2162402X.2017.1284722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gardner R, Wu D, Cherian S, Fang M, Hanafi LA, Finney O, Smithers H, Jensen MC, Riddell SR, Maloney DG, Turtle CJ. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood. 2016;127:2406–2410. doi: 10.1182/blood-2015-08-665547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, Sussman R, Lanauze C, Ruella M, Gazzara MR, Martinez NM, Harrington CT, Chung EY, Perazzelli J, Hofmann TJ, Maude SL, Raman P, Barrera A, Gill S, Lacey SF, Melenhorst JJ, Allman D, Jacoby E, Fry T, Mackall C, Barash Y, Lynch KW, Maris JM, Grupp SA, Thomas-Tikhonenko A. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015;5:1282–1295. doi: 10.1158/2159-8290.CD-15-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Grada Z, Hegde M, Byrd T, Shaffer DR, Ghazi A, Brawley VS, Corder A, Schonfeld K, Koch J, Dotti G, Heslop HE, Gottschalk S, Wels WS, Baker ML, Ahmed N. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol Ther Nucleic Acids. 2013;2:e105. doi: 10.1038/mtna.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Davies DM, Foster J, Van Der Stegen SJ, Parente-Pereira AC, Chiapero-Stanke L, Delinassios GJ, Burbridge SE, Kao V, Liu Z, Bosshard-Carter L, Van Schalkwyk MC, Box C, Eccles SA, Mather SJ, Wilkie S, Maher J. Flexible targeting of ErbB dimers that drive tumorigenesis by using genetically engineered T cells. Mol Med. 2012;18:565–576. doi: 10.2119/molmed.2011.00493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sentman CL, Meehan KR. NKG2D CARs as cell therapy for cancer. Cancer J. 2014;20:156–159. doi: 10.1097/PPO.0000000000000029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ruella M, Barrett DM, Kenderian SS, Shestova O, Hofmann TJ, Perazzelli J, Klichinsky M, Aikawa V, Nazimuddin F, Kozlowski M, Scholler J, Lacey SF, Melenhorst JJ, Morrissette JJ, Christian DA, Hunter CA, Kalos M, Porter DL, June CH, Grupp SA, Gill S. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J Clin Invest. 2016;126:3814–3826. doi: 10.1172/JCI87366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schneider D, Xiong Y, Wu D, Nlle V, Schmitz S, Haso W, Kaiser A, Dropulic B, Orentas RJ. A tandem CD19/CD20 CAR lentiviral vector drives on-target and off-target antigen modulation in leukemia cell lines. J Immunother Cancer. 2017;5:42. doi: 10.1186/s40425-017-0246-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Anurathapan U, Chan RC, Hindi HF, Mucharla R, Bajgain P, Hayes BC, Fisher WE, Heslop HE, Rooney CM, Brenner MK, Leen AM, Vera JF. Kinetics of tumor destruction by chimeric antigen receptor-modified T cells. Mol Ther. 2014;22:623–633. doi: 10.1038/mt.2013.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hegde M, Corder A, Chow KK, Mukherjee M, Ashoori A, Kew Y, Zhang YJ, Baskin DS, Merchant FA, Brawley VS, Byrd TT, Krebs S, Wu MF, Liu H, Heslop HE, Gottschalk S, Yvon E, Ahmed N. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol Ther. 2013;21:2087–2101. doi: 10.1038/mt.2013.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, Antzis M, Cotner CE, Johnson LA, Durham AC, Solomides CC, June CH, Pure E, Albelda SM. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. 2014;2:154–166. doi: 10.1158/2326-6066.CIR-13-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang T, Wu MR, Sentman CL. An NKp30-based chimeric antigen receptor promotes T cell effector functions and antitumor efficacy in vivo. J Immunol. 2012;189:2290–2299. doi: 10.4049/jimmunol.1103495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chmielewski M, Kopecky C, Hombach AA, Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011;71:5697–5706. doi: 10.1158/0008-5472.CAN-11-0103. [DOI] [PubMed] [Google Scholar]
- 76.Curran KJ, Seinstra BA, Nikhamin Y, Yeh R, Usachenko Y, van Leeuwen DG, Purdon T, Pegram HJ, Brentjens RJ. Enhancing antitumor efficacy of chimeric antigen receptor T cells through constitutive CD40L expression. Mol Ther. 2015;23:769–778. doi: 10.1038/mt.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stephan MT, Ponomarev V, Brentjens RJ, Chang AH, Dobrenkov KV, Heller G, Sadelain M. T cell-encoded CD80 and 4-1BBL induce auto- and transcostimulation, resulting in potent tumor rejection. Nat Med. 2007;13:1440–1449. doi: 10.1038/nm1676. [DOI] [PubMed] [Google Scholar]
- 78.Cao Y, Marks JD, Huang Q, Rudnick SI, Xiong C, Hittelman WN, Wen X, Marks JW, Cheung LH, Boland K, Li C, Adams GP, Rosenblum MG. Single-chain antibody-based immunotoxins targeting Her2/neu: design optimization and impact of affinity on antitumor efficacy and off-target toxicity. Mol Cancer Ther. 2012;11:143–153. doi: 10.1158/1535-7163.MCT-11-0519. [DOI] [PubMed] [Google Scholar]
- 79.Drent E, Themeli M, Poels R, de Jong-Korlaar R, Yuan H, de Bruijn J, Martens ACM, Zweegman S, van de Donk N, Groen RWJ, Lokhorst HM, Mutis T. A Rational Strategy for Reducing On-Target Off-Tumor Effects of CD38-Chimeric Antigen Receptors by Affinity Optimization. Mol Ther. 2017;25:1946–1958. doi: 10.1016/j.ymthe.2017.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, Cogdill AP, Li N, Ramones M, Granda B, Zhou L, Loew A, Young RM, June CH, Zhao Y. Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res. 2015;75:3596–3607. doi: 10.1158/0008-5472.CAN-15-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wilkie S, van Schalkwyk MC, Hobbs S, Davies DM, van der Stegen SJ, Pereira AC, Burbridge SE, Box C, Eccles SA, Maher J. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol. 2012;32:1059–1070. doi: 10.1007/s10875-012-9689-9. [DOI] [PubMed] [Google Scholar]
- 82.Juillerat A, Marechal A, Filhol JM, Valton J, Duclert A, Poirot L, Duchateau P. Design of chimeric antigen receptors with integrated controllable transient functions. Sci Rep. 2016;6:18950. doi: 10.1038/srep18950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sakemura R, Terakura S, Watanabe K, Julamanee J, Takagi E, Miyao K, Koyama D, Goto T, Hanajiri R, Nishida T, Murata M, Kiyoi H. A Tet-On Inducible System for Controlling CD19-Chimeric Antigen Receptor Expression upon Drug Administration. Cancer Immunol Res. 2016;4:658–668. doi: 10.1158/2326-6066.CIR-16-0043. [DOI] [PubMed] [Google Scholar]
- 84.Juillerat A, Marechal A, Filhol JM, Valogne Y, Valton J, Duclert A, Duchateau P, Poirot L. An oxygen sensitive self-decision making engineered CAR T-cell. Sci Rep. 2017;7:39833. doi: 10.1038/srep39833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–235. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 86.Spranger S, Dai D, Horton B, Gajewski TF. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell. 2017;31:711–723 e714. doi: 10.1016/j.ccell.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kohanbash G, Carrera DA, Shrivastav S, Ahn BJ, Jahan N, Mazor T, Chheda ZS, Downey KM, Watchmaker PB, Beppler C, Warta R, Amankulor NA, Herold-Mende C, Costello JF, Okada H. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J Clin Invest. 2017;127:1425–1437. doi: 10.1172/JCI90644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, Jones DR, Sadelain M. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med. 2014;6:261ra151. doi: 10.1126/scitranslmed.3010162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Newick K, O'Brien S, Sun J, Kapoor V, Maceyko S, Lo A, Pure E, Moon E, Albelda SM. Augmentation of CAR T-cell Trafficking and Antitumor Efficacy by Blocking Protein Kinase A Localization. Cancer Immunol Res. 2016;4:541–551. doi: 10.1158/2326-6066.CIR-15-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nishio N, Diaconu I, Liu H, Cerullo V, Caruana I, Hoyos V, Bouchier-Hayes L, Savoldo B, Dotti G. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. 2014;74:5195–5205. doi: 10.1158/0008-5472.CAN-14-0697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wu X, Giobbie-Hurder A, Liao X, Lawrence D, McDermott D, Zhou J, Rodig S, Hodi FS. VEGF Neutralization Plus CTLA-4 Blockade Alters Soluble and Cellular Factors Associated with Enhancing Lymphocyte Infiltration and Humoral Recognition in Melanoma. Cancer Immunol Res. 2016;4:858–868. doi: 10.1158/2326-6066.CIR-16-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Asai H, Fujiwara H, An J, Ochi T, Miyazaki Y, Nagai K, Okamoto S, Mineno J, Kuzushima K, Shiku H, Inoue H, Yasukawa M. Co-introduced functional CCR2 potentiates in vivo anti-lung cancer functionality mediated by T cells double gene-modified to express WT1-specific T-cell receptor. PLoS One. 2013;8:e56820. doi: 10.1371/journal.pone.0056820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, Foster AE. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. 2010;33:780–788. doi: 10.1097/CJI.0b013e3181ee6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, Heslop HE, Brenner MK, Dotti G, Savoldo B. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 2009;113:6392–6402. doi: 10.1182/blood-2009-03-209650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97–111. doi: 10.1038/nri.2016.107. [DOI] [PubMed] [Google Scholar]
- 96.Derer A, Deloch L, Rubner Y, Fietkau R, Frey B, Gaipl US. Radio-Immunotherapy-Induced Immunogenic Cancer Cells as Basis for Induction of Systemic Anti-Tumor Immune Responses - Pre-Clinical Evidence and Ongoing Clinical Applications. Front Immunol. 2015;6:505. doi: 10.3389/fimmu.2015.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol. 2008;8:59–73. doi: 10.1038/nri2216. [DOI] [PubMed] [Google Scholar]
- 98.North RJ. Cyclophosphamide-facilitated adoptive immunotherapy of an established tumor depends on elimination of tumor-induced suppressor T cells. J Exp Med. 1982;155:1063–1074. doi: 10.1084/jem.155.4.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier-Colame V, Newton A, Redouane Y, Lin YJ, Wojtkiewicz G, Iwamoto Y, Mino-Kenudson M, Huynh TG, Hynes RO, Freeman GJ, Kroemer G, Zitvogel L, Weissleder R, Pittet MJ. Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity. 2016;44:343–354. doi: 10.1016/j.immuni.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hombach AA, Chmielewski M, Rappl G, Abken H. Adoptive immunotherapy with redirected T cells produces CCR7- cells that are trapped in the periphery and benefit from combined CD28-OX40 costimulation. Hum Gene Ther. 2013;24:259–269. doi: 10.1089/hum.2012.247. [DOI] [PubMed] [Google Scholar]
- 101.Korman AJ, Peggs KS, Allison JP. Checkpoint blockade in cancer immunotherapy. Adv Immunol. 2006;90:297–339. doi: 10.1016/S0065-2776(06)90008-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, Chow MT, Smyth MJ, Kershaw MH, Darcy PK. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. 2013;19:5636–5646. doi: 10.1158/1078-0432.CCR-13-0458. [DOI] [PubMed] [Google Scholar]
- 103.John LB, Kershaw MH, Darcy PK. Blockade of PD-1 immunosuppression boosts CAR T-cell therapy. Oncoimmunology. 2013;2:e26286. doi: 10.4161/onci.26286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Suarez ER, Chang de K, Sun J, Sui J, Freeman GJ, Signoretti S, Zhu Q, Marasco WA. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget. 2016;7:34341–34355. doi: 10.18632/oncotarget.9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang Y, Zhang X, Cheng C, Mu W, Liu X, Li N, Wei X, Liu X, Xia C, Wang H. CRISPR-Cas9 mediated LAG-3 disruption in CAR-T cells. Front Med. 2017 Jun 17; doi: 10.1007/s11684-017-0543-6. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 106.Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin Cancer Res. 2017;23:2255–2266. doi: 10.1158/1078-0432.CCR-16-1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, Marson A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. 2017;7:737. doi: 10.1038/s41598-017-00462-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liu X, Ranganathan R, Jiang S, Fang C, Sun J, Kim S, Newick K, Lo A, June CH, Zhao Y, Moon EK. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res. 2016;76:1578–1590. doi: 10.1158/0008-5472.CAN-15-2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Prosser ME, Brown CE, Shami AF, Forman SJ, Jensen MC. Tumor PD-L1 co-stimulates primary human CD8(+) cytotoxic T cells modified to express a PD1:CD28 chimeric receptor. Mol Immunol. 2012;51:263–272. doi: 10.1016/j.molimm.2012.03.023. [DOI] [PubMed] [Google Scholar]
- 110.Yokosuka T, Kobayashi W, Takamatsu M, Sakata-Sogawa K, Zeng H, Hashimoto-Tane A, Yagita H, Tokunaga M, Saito T. Spatiotemporal basis of CTLA-4 costimulatory molecule-mediated negative regulation of T cell activation. Immunity. 2010;33:326–339. doi: 10.1016/j.immuni.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 111.Condomines M, Arnason J, Benjamin R, Gunset G, Plotkin J, Sadelain M. Tumor-Targeted Human T Cells Expressing CD28-Based Chimeric Antigen Receptors Circumvent CTLA-4 Inhibition. PLoS One. 2015;10:e0130518. doi: 10.1371/journal.pone.0130518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, Michielin O, Weide B, Romero P, Speiser DE. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci U S A. 2015;112:6140–6145. doi: 10.1073/pnas.1417320112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, Peggs KS, Ravetch JV, Allison JP, Quezada SA. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. 2013;210:1695–1710. doi: 10.1084/jem.20130579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Moon EK, Wang LC, Dolfi DV, Wilson CB, Ranganathan R, Sun J, Kapoor V, Scholler J, Pure E, Milone MC, June CH, Riley JL, Wherry EJ, Albelda SM. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res. 2014;20:4262–4273. doi: 10.1158/1078-0432.CCR-13-2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Riese MJ, Wang LC, Moon EK, Joshi RP, Ranganathan A, June CH, Koretzky GA, Albelda SM. Enhanced effector responses in activated CD8+ T cells deficient in diacylglycerol kinases. Cancer Res. 2013;73:3566–3577. doi: 10.1158/0008-5472.CAN-12-3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Burga RA, Thorn M, Point GR, Guha P, Nguyen CT, Licata LA, DeMatteo RP, Ayala A, Joseph Espat N, Junghans RP, Katz SC. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol Immunother. 2015;64:817–829. doi: 10.1007/s00262-015-1692-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Long AH, Highfill SL, Cui Y, Smith JP, Walker AJ, Ramakrishna S, El-Etriby R, Galli S, Tsokos MG, Orentas RJ, Mackall CL. Reduction of MDSCs with All-trans Retinoic Acid Improves CAR Therapy Efficacy for Sarcomas. Cancer Immunol Res. 2016;4:869–880. doi: 10.1158/2326-6066.CIR-15-0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhou Q, Munger ME, Highfill SL, Tolar J, Weigel BJ, Riddle M, Sharpe AH, Vallera DA, Azuma M, Levine BL, June CH, Murphy WJ, Munn DH, Blazar BR. Program death-1 signaling and regulatory T cells collaborate to resist the function of adoptively transferred cytotoxic T lymphocytes in advanced acute myeloid leukemia. Blood. 2010;116:2484–2493. doi: 10.1182/blood-2010-03-275446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, Ittmann MM, Marchetti D, Dotti G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. 2015;21:524–529. doi: 10.1038/nm.3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kerkar SP, Muranski P, Kaiser A, Boni A, Sanchez-Perez L, Yu Z, Palmer DC, Reger RN, Borman ZA, Zhang L, Morgan RA, Gattinoni L, Rosenberg SA, Trinchieri G, Restifo NP. Tumor-specific CD8+ T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Cancer Res. 2010;70:6725–6734. doi: 10.1158/0008-5472.CAN-10-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, Brentjens RJ. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119:4133–4141. doi: 10.1182/blood-2011-12-400044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hoskin DW, Mader JS, Furlong SJ, Conrad DM, Blay J. Inhibition of T cell and natural killer cell function by adenosine and its contribution to immune evasion by tumor cells (Review) Int J Oncol. 2008;32:527–535. [PubMed] [Google Scholar]
- 123.Hildeman DA, Mitchell T, Kappler J, Marrack P. T cell apoptosis and reactive oxygen species. J Clin Invest. 2003;111:575–581. doi: 10.1172/JCI18007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Beavis PA, Henderson MA, Giuffrida L, Mills JK, Sek K, Cross RS, Davenport AJ, John LB, Mardiana S, Slaney CY, Johnstone RW, Trapani JA, Stagg J, Loi S, Kats L, Gyorki D, Kershaw MH, Darcy PK. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J Clin Invest. 2017;127:929–941. doi: 10.1172/JCI89455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ligtenberg MA, Mougiakakos D, Mukhopadhyay M, Witt K, Lladser A, Chmielewski M, Riet T, Abken H, Kiessling R. Coexpressed Catalase Protects Chimeric Antigen Receptor-Redirected T Cells as well as Bystander Cells from Oxidative Stress-Induced Loss of Antitumor Activity. J Immunol. 2016;196:759–766. doi: 10.4049/jimmunol.1401710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Eil R, Vodnala SK, Clever D, Klebanoff CA, Sukumar M, Pan JH, Palmer DC, Gros A, Yamamoto TN, Patel SJ, Guittard GC, Yu Z, Carbonaro V, Okkenhaug K, Schrump DS, Linehan WM, Roychoudhuri R, Restifo NP. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature. 2016;537:539–543. doi: 10.1038/nature19364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tang H, Wang Y, Chlewicki LK, Zhang Y, Guo J, Liang W, Wang J, Wang X, Fu YX. Facilitating T Cell Infiltration in Tumor Microenvironment Overcomes Resistance to PD-L1 Blockade. Cancer Cell. 2016;29:285–296. doi: 10.1016/j.ccell.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.DuPage M, Jacks T. Genetically engineered mouse models of cancer reveal new insights about the antitumor immune response. Curr Opin Immunol. 2013;25:192–199. doi: 10.1016/j.coi.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.DuPage M, Cheung AF, Mazumdar C, Winslow MM, Bronson R, Schmidt LM, Crowley D, Chen J, Jacks T. Endogenous T cell responses to antigens expressed in lung adenocarcinomas delay malignant tumor progression. Cancer Cell. 2011;19:72–85. doi: 10.1016/j.ccr.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Garbe AI, Vermeer B, Gamrekelashvili J, von Wasielewski R, Greten FR, Westendorf AM, Buer J, Schmid RM, Manns MP, Korangy F, Greten TF. Genetically induced pancreatic adenocarcinoma is highly immunogenic and causes spontaneous tumor-specific immune responses. Cancer Res. 2006;66:508–516. doi: 10.1158/0008-5472.CAN-05-2383. [DOI] [PubMed] [Google Scholar]
- 131.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 132.DuPage M, Dooley AL, Jacks T. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc. 2009;4:1064–1072. doi: 10.1038/nprot.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.McFadden DG, Politi K, Bhutkar A, Chen FK, Song X, Pirun M, Santiago PM, Kim-Kiselak C, Platt JT, Lee E, Hodges E, Rosebrock AP, Bronson RT, Socci ND, Hannon GJ, Jacks T, Varmus H. Mutational landscape of EGFR-, MYC-, and Kras-driven genetically engineered mouse models of lung adenocarcinoma. Proc Natl Acad Sci U S A. 2016;113:E6409–E6417. doi: 10.1073/pnas.1613601113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nakasone ES, Askautrud HA, Kees T, Park JH, Plaks V, Ewald AJ, Fein M, Rasch MG, Tan YX, Qiu J, Park J, Sinha P, Bissell MJ, Frengen E, Werb Z, Egeblad M. Imaging tumor-stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell. 2012;21:488–503. doi: 10.1016/j.ccr.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Turnis ME, Sawant DV, Szymczak-Workman AL, Andrews LP, Delgoffe GM, Yano H, Beres AJ, Vogel P, Workman CJ, Vignali DA. Interleukin-35 Limits Anti-Tumor Immunity. Immunity. 2016;44:316–329. doi: 10.1016/j.immuni.2016.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Liakou CI, Kamat A, Tang DN, Chen H, Sun J, Troncoso P, Logothetis C, Sharma P. CTLA-4 blockade increases IFNgamma-producing CD4+ICOShi cells to shift the ratio of effector to regulatory T cells in cancer patients. Proc Natl Acad Sci U S A. 2008;105:14987–14992. doi: 10.1073/pnas.0806075105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chen H, Liakou CI, Kamat A, Pettaway C, Ward JF, Tang DN, Sun J, Jungbluth AA, Troncoso P, Logothetis C, Sharma P. Anti-CTLA-4 therapy results in higher CD4+ICOShi T cell frequency and IFN-gamma levels in both nonmalignant and malignant prostate tissues. Proc Natl Acad Sci U S A. 2009;106:2729–2734. doi: 10.1073/pnas.0813175106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Fu T, He Q, Sharma P. The ICOS/ICOSL pathway is required for optimal antitumor responses mediated by anti-CTLA-4 therapy. Cancer Res. 2011;71:5445–5454. doi: 10.1158/0008-5472.CAN-11-1138. [DOI] [PubMed] [Google Scholar]
- 139.Zamarin D, Holmgaard RB, Ricca J, Plitt T, Palese P, Sharma P, Merghoub T, Wolchok JD, Allison JP. Intratumoral modulation of the inducible co-stimulator ICOS by recombinant oncolytic virus promotes systemic anti-tumour immunity. Nat Commun. 2017;8:14340. doi: 10.1038/ncomms14340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, Bagg A, Marcucci KT, Shen A, Gonzalez V, Ambrose D, Grupp SA, Chew A, Zheng Z, Milone MC, Levine BL, Melenhorst JJ, June CH. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7:303ra139. doi: 10.1126/scitranslmed.aac5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Giesen C, Wang HA, Schapiro D, Zivanovic N, Jacobs A, Hattendorf B, Schuffler PJ, Grolimund D, Buhmann JM, Brandt S, Varga Z, Wild PJ, Gunther D, Bodenmiller B. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat Methods. 2014;11:417–422. doi: 10.1038/nmeth.2869. [DOI] [PubMed] [Google Scholar]