

Abstract
Until recently, the dual roles of mitochondria in ATP production (bioenergetics) and apoptosis (cell life/death decision) were thought to be separate. New evidence points to a more intimate link between these two functions, mediated by the remodeling of the mitochondrial ultra-structure during apoptosis. While most of the key molecular players that regulate this process have been identified (primarily membrane proteins), the exact mechanisms by which they function are not yet understood. Because resistance to apoptosis is a hallmark of cancer, and because ultimately all chemotherapies are believed to result directly or indirectly in induction of apoptosis, a better understanding of the biophysical processes involved may lead to new avenues for therapy.
Keywords: Mitochondria, apoptosis, cytochrome C, cristae
The dual role of mitochondria in energetics and apoptosis
The mitochondria is a double membraned organelle believed to have been integrated into modern eukaryotes via symbiosis of a proteobactirum into an anaerobic pre-eukaryotic (host) cell 1.5-2 billion years ago1. According to modern thinking (pioneered by Mitchell2), an essential role of mitochondria is to produce adenosine triphosphate (ATP) via oxidative phosphorylation (OXPHOS). In this process, the chemical energy stored in nutrients (carbohydrates, fats, etc.) is converted to an electrochemical gradient across the inner mitochondrial membrane via the electron transport chain (ETC) complexes. This electrochemical gradient acts as a “storage” of energy. ATP synthase uses this stored energy to convert adenosine diphosphate (ADP) to ATP. This bioenergetic picture of the role of mitochondria is now widely accepted. A second role of mitochondria is in the so-called “intrinsic” apoptosis pathway. This pathway converges (figuratively and literally) at the membrane of the mitochondria. Upon certain cell death signals (such as reactive oxygen species (ROS), DNA damage, etc), the outer membrane of mitochondria becomes permeable enough to release the soluble hemeprotein cytochrome C (cytc), as well as Smac/Diablo, endonuclease G, and other intermembrane space proteins, which irreversibly activate downstream caspases to carry out the apoptosis process.
This article summarizes several recent experimental results which indicate that the mitochondrial ultrastructure3 is intimately involved in the relationship between the bioenergetic4 and apoptotic 5,6,7,8 (cell life/death decision) roles of mitochondria. The discussion summarizes recent advances in 1) New understanding of the role of ultrastructure and the location of membrane complexes on mitochondrial bioenergetics; 2) New understanding of the role of ultrastructure in apoptosis; 3) An emerging, albeit incomplete, model for the relationship between 1 and 2. The article is written from a biophysical perspective. The main message of this article is that one must approach mitochondria and cancer from this perspective in order to completely understand apoptosis and, hence, cancer; a classical DNA sequencing/biochemical pathway analysis approach is insufficient for a complete and useful understanding of the system. Since resistance to apoptosis is a hallmark of cancer9, a better understanding of the biophysical processes involved in apoptosis may lead to new avenues for therapy. Existing therapeutic trials targeting this mechanism are also summarized.
This message is substantiated with the following key (“tentpole”) points 1) The mitochondria sustain a membrane potential which is not uniform along the inner membrane due to its ultra-structure and distribution of membrane proteins involved in the OXPHOS process; 2) the change in the ultrastructure (“remodeling”) occurs during both metabolic changes and apoptosis; 3) A growing body of recent evidence indicates that the bioenergetic and apoptotic functions are linked through this ultrastructure, and 4) A comprehensive model that explains this link is only in its early stages and incomplete, 5) since this is a dynamic biophysical, active electrophysiological system, with localized pH and voltage gradients mediated by and affected by a hierarchical organization of active components (ionic, protein, membrane, organelle ultrastructure, and organelle-organelle interactions), there is significant technological challenge in pinpointing the location, function, and mechanism of all the components (membrane proteins, inner and outer membrane, inner membrane structure, and membrane pore formation during apoptosis), and 6) in spite of these challenges, pharmacological manipulation of apoptosis based on current understanding is a successful route for targeted therapies and indirectly is the target of all chemotherapies, making studies of this complicated system a priority for the field of cancer.
Bioenergetics: Electron Transport Chain and Cristae
The sources and sinks of the membrane potential are not uniformly distributed along the inner membrane
In 1961, Mitchell postulated2 that energy is converted from chemical energy in carbohydrates and fats to an electrochemical gradient at the mitochondrial inner membrane, and that this electrochemical gradient is the energy source with drives ATP synthesis. Now known as the chemi-osmotic hypothesis (Box 1), this model is widely accepted. While the molecular constituents of the ETC complexes and ATP synthase are mostly identified, his model says nothing about their spatial distribution within the organelle, a key missing component (Figure 1). The mitochondrial inner membrane is invaginated into a dynamically changing cristae ultra-structure (Figure 2). (Anatomically/histologically the tubular type of mitochondria has different cristae types.) It seems reasonable to ask: where are the ETC complexes and ATP synthases within this organelle, and why?
Box 1. Mitchell's Chemiosmotic Hypothesis.
In 1961, Mitchell postulated2 that energy is converted from chemical energy in carbohydrates and fats to an electrochemical gradient at the mitochondrial inner membrane (proton motive force Δp) given by:
| Eq. 1 |
where ΔΨm is the membrane potential and ΔpH is the pH difference across the membrane. Throughout the years, this model became verified and the current model has 4 electron transport chain (ETC) complexes (CI-CIV) that pump protons across the inner membrane to create this gradient (Figure 1). The ETC components energetically couple pumping of protons across the inner membrane to oxidation of the chemical components of the Krebs cycle. This proton pumping creates a charge differential due to the positive charge of the proton, and hence a membrane potential across the inner membrane capacitance according to the well known relationship between the charge on a capacitor Q and the voltage drop across the capacitor V: Q=CV, where C is the capacitance of the membrane. (Here V is synonymous with ΔΨm.) However, it also creates a pH gradient across the membrane as more protons end up outside the membrane. ATP synthase (CV) is a complex which uses this electrochemical energy stored in the membrane to convert ADP + Pi to ATP, hence coupling oxidation of nutrients to phosphorylation of ADP (OXPHOS).
cytc is a soluble protein that plays a key role shuttling electrons from CIII to CIV
cytc is a reducible, soluble heme-protein that plays the role of electron shuttle in the ETC. It is reduced by CIII, and then subsequently reduces CIV. It also plays a key role in apoptosis, as will be discussed below. The location of cytc is believed to be 85% within the cristae22, shown in Figure 2 schematically.
Figure 1.
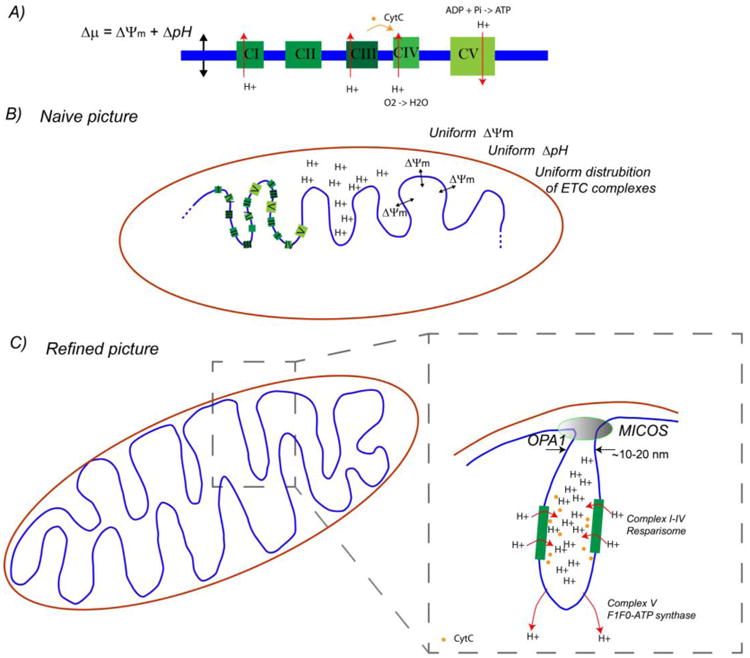
A) Electron transport chain (ETC, top). B) Naive picture of distribution of the ETC components assumes they are uniformly distributed along the mitochondrial inner membrane (blue), and that the electrochemical potential also is uniform. Because technology does not exist to directly measure these quantities accurately and with nanoscale spatial resolution, this naive picture has persisted for many years. In the refined picture, put together through indirect observations over that last few years, the spatial distribution of the electrophysiology seems to be highly ordered and hierarchically organized. The coupled electrophysiology and biochemistry, although directly related to metabolism and apoptosis (hence cancer), remain to be explored and explained in detail.
Figure 2.
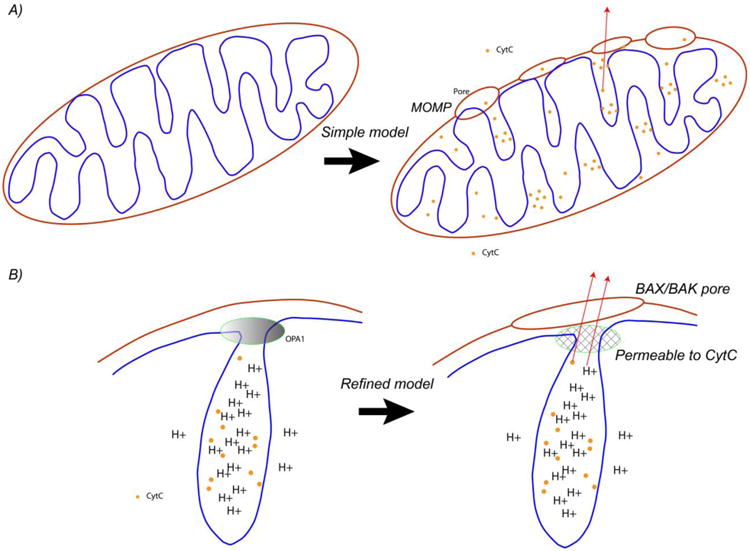
A) Process of mitochondrial outer membrane permeabilization gives rise to a BAX/BAK pore in the outer membrane, followed by cytc release, triggering caspases and irreversibly committing the cell to apoptosis. B) A more refined model takes into account that the vast majority of CtyC is stored in the cristae, and that the cristae junctions contain protein complexes which normally block cytc release, including MICOS and OPA1 oligomers. The exact mechanism by which the cristae junctions become permeable to cytc is unknown, hence the figure indicates a generic, permeable mesh.
ATP synthase forms a proton “sink” at the folds of the cristae
Recently, high resolution transmission electron microscopy (TEM) experiments have observed that the ATP synthase complex in vivo is distributed as dimers along the inner folds of the cristae10,11 (Figure 1). Super resolution optical microscopy supports this conclusion 12. In vitro studies of vesicles with monomeric vs. dimeric ATP synthase demonstrate that the dimerization induces a strong curvature to the membrane11. Furthermore, yeast and human knock-out cell lines which disable dimerization of ATP synthase result in mitochondrial morphologies without cristae structures13,14. Therefore, the dimerization of ATP synthase clearly plays a causative role in the folding of cristae.
What is the biophysical mechanism for the relationship between the membrane curvature and the dimerization of ATP synthases? One hypothesis is that the pH gradient generated by the ATP synthase causes membrane curvature. Indeed, membrane curvature of isolated liposomes is observed in the presence of pH gradient15. However, how the dimerization of ATP synthase causes folding remains to be elucidated in detail.
What is the physiological role of the dimerization and localization at the tips of the cristae? One hypothesis is that the ATP synthase acts as a “sink” of protons10 and hence generates a gradient of pH along the membrane. This would cause the cristae to have a higher pH gradient than the rest of the inter membrane space. Evidence for this hypothesis is presented in our recent work in more detail below.
The ETC complexes form supercomplexes along the walls of the inner cristae
The ETC complexes (CI-CIV, Box 1) which participate in pumping protons from the matrix to the inner membrane space have also recently observed (via immuno-gold TEM microscopy16) to be localized inside the cristae, and not in the rest of the inter membrane space. Furthermore, rather than being randomly distributed along the inner membrane inside the cristae, there is increasing evidence that complexes are localized near each other into supercomplexes called “resparisomes” 17. Therefore, it seems all the “action” is inside the cristae, in terms of both pumping protons (ETC complexes) and ATP synthesis (ATP synthase). One hypothesis is that the complexes are located close to the ATP synthase dimers to maximize the transfer of protons. However, it should be noted that the evidence for these findings is somewhat indirect. To date there are no demonstrated technologies to measure either the local membrane potential, pH gradient, nor the local distribution of the complexes in live cells with the accuracy needed for determination of their location within a single mitochondria or even along a single cristae (Box 3).
Box 3. How is mitochondrial Δp = ΔΨm – 59 Δ pH and structure measured?
Mitochondrial membrane potential ΔΨm is usually measured via fluorescence54. Most charged species are hydrophilic, but by distributing a charge some moieties become hydrophobic/lipophilic, e.g. Tetramethylrhodamine, ethyl ester (TMRE), allowing it to cross the otherwise impermeable inner membrane. When such a species is fluorescent, its concentration can be qualitatively determined through a measurement of either fluorescence intensity or spectrum. When there is a membrane potential, the charged species will be distributed across the membrane via the Nernst relation, n1/n2∼ e˄{-ΔΨm/kT}. In mitochondria with ΔΨm ∼ 100 mV this gives a 100-1000× increased concentration inside, which is usually measured as a surrogate for the membrane potential. However, the limited # of photons and the sensitivity of mitochondria to bright light generated ROS limits the temporal resolution to ∼ 100 ms55. The spatial resolution is also limited by the wavelength of light, of order the size of the mitochondria itself. Mitochondria may be only a single pixel in diameter but they can be many pixels long.
The pH, while an important component of Δp, has long been neglected until recently23. GFP proteins can be integrated into the genome and targeted to the mitochondrial compartment for in vivo real time pH measurements56. Very recently24, we have applied nanotechnology using a sensitive graphene electrode to assay extra mitochondrial pH undergoing MOMP in real time (Figure I, from ref24). This has the advantage that it can be scaled with micro fabrication for massively parallel assays. These new electronic techniques may enable high temporal resolution assays of mitochondrial electrophysiology with single mitochondria probed with single nanowire/nanotube structure55. In addition, while none of the optical methods above have high enough spatial resolution for individual ion channel activity, new nanoelectronic technologies in the future may enable this48.
Two methods are used to study mitochondrial structure: electron microscopy and super resolution optical microscopy. Electron microscopy always require fixation and therefore only capture a snapshot of mitochondrial structure. Super resolution optical microscopy also may require fixations. This, in addition to the necessary sample preparation and low throughput, has severely limited progress in the field.
For electrophysiology, patch clamp approaches have been successful only with mitoplasts57. (A mitoplast is a mitochondrion that has been stripped of its outer membrane leaving the inner membrane intact.) Patching the intact organelle has proved challenging, because the inner membrane structure is smaller than pipette diameters.
Box 3 Figure I.
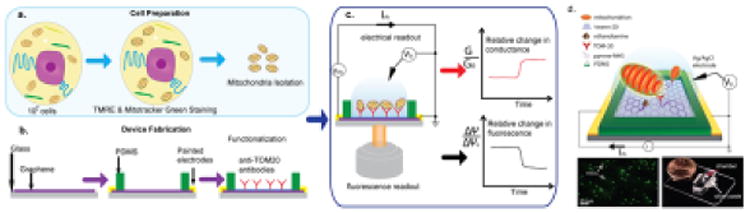
(a) Mitochondria are isolated and loaded on (b) a pre-functionalized devices with anti-TOM20 antibodies to immobilize the mitochondria. The PDMS (polydimethylsiloxane) chamber is for fluid handling.(c) After a brief incubation, mitochondrial external pH is measured with the graphene electrode, and membrane potential is measured via florescence with TMRE. e) 3d schematic of the setup, optical micrograph showing mitochondria labeled with mitotracker green dye, and optical micrograph of macroscopic device on cover slide.
A series of protein complexes maintains the mitochondrial ultra-structure (cristae structure)
MICOS
A complex of proteins called MICOS18,19 (mitochondrial contact site and cristae organizing system) staples the cristae together at the junctions. This large (> 1 Mda) heterooligomeric protein complex, conserved from yeast to humans, has at least 7 identified units (Mic10, Mic12, Mic19, Mic25, Mic26, Mic27, Mic60). Experiments (including both electron microscopy and super resolution20 optical microscopy) have shown MICOS to be localized at cristae junctions21.
OPA1
Mitochondria constantly undergo fission and fusion. Mitochondrial fusion is mediated by two proteins on the outer membrane (the mitofusins MFN1 And MFN2), and the inner membrane protein optic atrophy 1 (OPA1), named after its pathological consequences (degeneration of retinal ganglion cells sometimes follow by blindness) when mutated in the human inherited disease dominant optic atrophy. It is believed that OPA1 oligomerizes at the cristae junctions to form a diffusion barrier which traps cytc stores inside the cristae (see below). However, the exact mechanism of this is not known. (Do they form a circle at the entry to an individual cristae, or do they form long ridges along the length of extended cristae?) In addition, this may also trap protons, as they are generated by the ETC inside the cristae at high concentration. This will be discussed below in the context of apoptosis as well. For now the most consistent model has the cytc and H+ concentration higher inside cristae than the the rest of the inter membrane space, as shown in Figure 2. Experimental evidence for the cytc stores is in ref 22. Direct experimental evidence for the high H+ concentration is lacking because researchers have long focused on the membrane potential component of Δp, neglecting to study the ΔpH 23, but we recently demonstrated indirect evidence 24, discussed below.
Humans have eight different isoforms of OPA1, including long form and short forms (L-OPA1, S-OPA1). These are present in nearly equamolar forms under basal conditions25. The L form can be cleaved to the S form by OMA1 when the mitochondrial membrane potential is low26. OMA1 normally is degraded by proteolysis, but in low membrane potential situations, OMA1 is stabilized and survives long enough to cleave L-OPA1 to S-OPA1. Thus, OPA pools are determined by the bioenergetic parameter ΔΨm. This provides an important clue that relates a metabolic parameter to a protein involved in regulation of mitochondrial ultra-structure, but the detailed mechanism of this relationship is not clear yet.
The cristae structure “remodels” in response to the metabolic needs of the cell
Already over 50 years ago, Hackenbrock27 observed “condensation” of rat liver mitochondria under different metabolic conditions. New, roughly 40 years later, this topic is finding renewed interest because of new findings about the relevance to apoptosis and cancer. In 2013 Cogliati et al 28 showed that changes in cristae structure affect metabolic efficiency of the ETC components and respirasomes. In 2014, Patten et al 29 showed that OPA1-dependent cristae modulation is essential for adaption to metabolic conditions. Thus, OPA and cristae structure, separate from their role in apoptosis below, play a crucial role in response to metabolic needs of a cell.
The spatial distribution of the electrochemical gradient along the mitochondrial inner membrane: Knowns, unknowns, and prospects
It is clear that the spatial distribution of the electro physiologically active ETC components is organized in hierarchies (dimers, supercomplexes), and that these change in respond to the metabolic needs of the cell. If the “sources” of the electrochemical potential Δp are non-uniformly distributed, how is the pH and ΔΨm gradient distributed and how is this related to (via cause or effect) the sources? One hypothesis is that the pH inside the pockets is low to maximize “efficiency”. If this is true, how do the junctions hold in the pH since protons are small and highly mobile? Next, if the pH sink is at the folds (ATP synthase dimers), what is the pH gradient along the inner membrane and what is it between the cristae and the rest of the inter membrane space? A recent green fluorescence protein (GFP) based experiment demonstrated a pH difference of 0.3 between ATP synthase and CIV of the ETC30, consistent with the expectation of a gradient of the pH due to the spatial separation of the sink (ATP synthase) and source (ETC complex resparisomes) of protons. However, because of the technological difficulty for high spatial resolution studies of electrophysiology, that is the extent of our experimental knowledge at this time.
One can create biophysical models, but the size of the Debye length is comparable to the ultrastructure, making traditional electrochemical concepts challenging to apply in a straightforward manner, and requiring a more detailed mathematical approach31 Although the spatial distribution is non-uniform for the ETC complexes, pH, and (presumably) ΔΨm, a detailed model, biophysical / physiological understanding, and method to measure these quantities is still lacking. Without experimental methods to validate these models, their utility at the moment is limited. Because the spatial and temporal dynamics may be coupled to metabolism, and apoptosis, it is worth further investigations of models and tests. Just as for propagation of the action potential along neurons (the Hodgkins-Huxley model), the electrically active ETC complexes may be governed by a non-trivial equivalent circuit model governing the propagation of a voltage wave. It is entirely possible that a model similar to the Hodgkins-Huxley model, albeit undiscovered, can account for the temporal and spatial dynamics of the mitochondrial electrophysiology. Tantalizing evidence for this already exists (Box 4).
Box 4. What is the role of the mitochondrial permeability transition pore in apoptosis?
The mitochondrial permeability transition pore (mtPTP) is a name given to a complex of membrane proteins at the inner and outer membrane which are believed to form a large pore. The molecular identity of each of the components is controversial and not completely known58,59,60 despite intense effort in the area for over 10 years. Although there are many ion channels, pumps, transporters, etc. in mitochondria (many of which can be assayed individually in suspended lipid bilayers), the interrelationship among these and their contribution to the mtPTP is still largely unresolved. A lot of this uncertainty stems from a lack of tools to measure electrophysiology at the nanoscale with high temporal resolution (Figure II from 61). What is known is that when mitochondria depolarize in response to ROS (e.g. light induced free radicals) and calcium overload, the membrane potential of the entire organelle (as measured by TMRE fluorescence intensity) “flickers” on and off before dropping to zero (Figure II and 62). This flicker is believed to be the transient opening and closing of the mtPTP However, because TMRE measurements are limited in time resolution to ∼ 100 ms at best55, and spatial resolution to one voxel ∼ one mitochondria, little more is known about the electrophysiology of the mtPTP in vivo.
What regulates mtPTP? In the absence of direct patch clamp experiments in vivo, many additional clues can be gleaned by in vitro patch clamp of a variety of potential components of the mtPTP This include ATP synthase, BAX/BAK, Adenine nucleotide translocator (ANT), cyclophilin D, etc. A vast literature documents inducers of mPTP opening.
What is the physiological role of the mtPTP? It is known to be heavily involved in ischemia/reperfusion injury in the brain and heart, but calcium overload is not a known mechanism of apoptosis. One possibility it that under conditions of prolonged oxidative stress or cellular Ca+ overload, short openings of mtPTP might serve as an emergency release of accumulated Ca+ ions a and a mechanism allowing the partial dissipation of ΔΨm, reducing ROS generation. However, once the inner membrane is compromised via the mtPTP, the organelle ruptures via osmotic swelling and this does release cytc triggering apoptosis. The original discovery studies of the mtPTP over 10 years ago implicated it in vivo as a significant participant in apoptosis but since then researchers have questioned if is is necessary for the mtPTP to open in order for apoptosis to occur. The most recent detailed studies of BH3 proteins, BAX/BAK oligomerization to form MOMP, and cristae remodeling do not seem to indicate a role for the mtPTP in apoptosis
Are pH and ROS flicker63 related to membrane potential flicker? Recent work has shown that, in addition to membrane potential flicker (measured via TMRE), pH may also flicker and the mitochondria release “pufs” or ROS. These recent experiments are done using genetically engineered GFP technology. In fact the frequency of these flashes has been correlated with lifespan in some organisms64.
Box 4 Figure II.
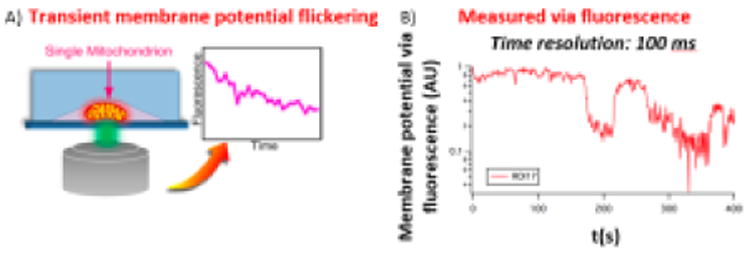
A) Single, vital, isolated mitochondria in a nano-fluidic channel, labeled with TMRE (schematic). B) TMRE fluorescence intensity vs. time indicating flickering of the membrane potential prior to complete depolarization.
Apoptosis
Mitochondrial stores of cytc are released during apoptosis
The release of the mitochondrial stores of cytc into the cytosol is one of the key events in apoptosis (Box 2). During mitochondrial outer membrane permeabilization (MOMP), the outer membrane becomes permeable with pores allowing high molecular weight proteins such as cytc to spill into the cytosol. While cytc is the key protein, other co-factors are released, such as Smac/Diablo and endonuclease G. However, because cytc is the only one also involved in the ETC, it is a candidate to explain the link between bioenergetics and apoptosis. The extent to which these secondary proteins are involved in bioenergetics is unknown. It was initially believed that the MOMP process led to the release of cytc directly.
Box 2. Apoptosis pathway, MOMP, and cancer chemotherapies.
In the mitochondrial pathway of cell death (apoptosis), various upstream effectors (DNA damage, ROS, stress, cell death signals, etc.) result in cell apoptosis. The point of no return is an abrupt event that occurs at the membranes of the mitochondria: The organelle is eventually ruptured (or changed in some other way) and releases the stores of CtyC into the cytosol. This release of cytc irreversibly activates the caspases which carry out the apoptotic process to completion. Resistance to apoptosis is a hallmark of cancer9. In fact, eventually, every chemotherapy is believed to result in apoptosis as its final mechanism of action, regardless of the initial point of attack49. Because the mitochondrial pathway is highly regulated, it is a clear target for pharmacological manipulation. Because of its potential clinical significance, extensive efforts have been made to understand the mechanisms of cytc release and its regulation. However, because of the small size of the organelle, the details of the release process are still far from understood. Since 85% of the cytc is contained in the cristae22 , the morphology of mitochondria is a key parameter to study.
Mitochondrial stores of cytc are releases during apoptosis
The release of the mitochondrial stores of cytc into the cytoplasm is one of the key events in apoptosis. One of the processes that has been well studied but is still not completely understood50 is the permeabilization of the mitochondrial outer membrane (Mitochondrial Outer Membrane Permeabilization (MOMP)). Historically, the finding of the BCL-2 gene that is implicated as an anti-apoptosis gene in b cell lymphoma, led to the study of a family of over 20 related proteins which share 4 homology domains (BH1-4). During MOMP, the outer membrane becomes permeable with pores allowing high molecular weight proteins such as cytc to pass. Thus it was initially believed that the MOMP process led to the release of cytc directly. Later, more refined thinking about CR remodeling followed.
The MOMP process is the result of oligomerization of BAX (BCL-2 antagonist killer 1) or BAK (BCL-2 associated × protein) proteins on the mitochondrial outer membrane. The details of this process are still being worked out. For example, structural studies of BAX/BAK oligomerization with AFM have demonstrated that, at least in some circumstances, BAX/BAK proteins oligomerize to form pores. With superresolution optical microscopy, Jakobs et al have imaged MOMP as large lipidic pores up to >100 nm in diameter51. These always occurred with cytc release, making it impossible to dissect the order or cause/effect relationship. Their work is consistent with lipidic pores observed in liposomes with TEM51 as well as AFM studies52.
Furthermore, the regulation of BAX/BAK mediated MOMP has been shown to be positive and negatively affected by the BCL-2 family of proteins. Although several competing models for the details of how this regulation occur, it is clear both in vitro and in vivo that MOMP (hence apoptosis) can be regulated by the BCL-2 family of proteins. Furthermore, the development of several chemotherapies that manipulate this pathway by creating so-called BH3 mimetics has resulted in some clinical significant results. For example, venetoclax (ABT-199) has been developed as a chemical inhibitor of the anti-apoptosis BCL-2 gene. It has received FDA approval for use in patients with chronic lymphocytic leukaemia (CLL) with the 17p deletion. Additional clinical trials (Phase I-III) are underway for CLL, non-Hodgkin lymphomas (including diffuse large B-cell lymphoma, mantle cell lymphoma, follicular lymphoma), acute myeloid leukaemia, multiple myeloma, systemic lupus erythematosus, and breast cancer53.
cytc must be released from the cristae: Is cristae remodeling required?
On closer inspection, the release of cytc following MOMP poses a conceptual problem, as, the stores of cytc were shown over 15 years ago to predominately reside in the cristae, and not the inner luminal space. Therefore, if MOMP allows the release of cytc, it must be free to pass through the cristae junctions. However, if this were true, the cytc would not be concentrated in the cristae. Therefore, in order for the cytc to be released (a pre-condition for apoptosis), one hypothesis is that cristae remodeling must occur to open the junctions.
Scorrano found in 2002 that cristae remodeling occurs during apoptosis (opening the neck from 18 nm to 56 nm)22 , and found this was caused by the peptide BID (a pro-apoptotic member of the ‘BH3-only’ subset of the BCL-2 family of proteins, Box 3) in a way that was independent of BAX/BAK oligomerization. BAX/BAK are proteins believed to oligomerize, forming pores with permeabilize the outer membrane (hence MOMP), Box 3. Thus, MOMP was claimed to be a different process than cristae remodeling. Both MOMP and cristae remodeling were found to be necessary for cytc release, as cristae remodeling allows cytc to escape from the cristae into the rest of the inner membrane space, and MOMP enables cytc to escape from the inner membrane space, cross the outer membrane (through the MOMP pores), and into the cytosol. This followed by work of Frezza in 200632 who showed that OPA1 prevented cytc release and hence is the glue which held the cristae together. (Additional work supporting this the importance of cristae junction opening to release cytc was also presented in Cipolat33.) Oligerimization of OPA1 was found to be the mechanism that regulates apoptosis by maintaining the tightness of the cristae junctions.
Sun 34 found that cytc release can occur prior to cristae remodeling, and that mitochondrial swelling occurs only late in apoptosis after the release of cytc and the loss of mitochondrial membrane potential. Follow on studies by Yamaguchi et al 35 found that OPA1 oligomerization indeed blocks cytc release from the cristae, and that the disassembly of the OPA1 oligomers caused cytc release. This release of cytc was found even though the diameter of the cristae junctions was halved (from 18 nm to 9 nm) during apoptosis. Although both Scorrano and Yamaguchi found BAX/BAK oligomerization was required for cytc release through the outer membrane, only Yamaguchi found BAX/BAK and BH3 containing BID were required for cytc release from the cristae junctions.
Regardless, both find the cytc permeability in the cristae junctions must change to enable release of cytc, so the physical size of the junction seems to be unrelated to the cytc permeability. One possibility is that OPA1 oligomers form a mesh impermeable to cytc across the cristae junction which is controlling cytc diffusion across the mesh, independent of the size of the cristae junction. These more refined hypothesis are shown in Figure 2.
Why is there such discrepancy between results? One of the most important factors is that TEM studies provide only a snapshot of a dynamic process. A second factor is that the processes may occur during preparation of the sample for TEM imaging36, thus masking the effect to be imaged or significantly perturbing the “frozen” state from the in vivo, live state that is desired to be studied. Static structure studies have advanced significantly, even garnering the Nobel prize in 2017 37. However, this discrepancy illustrates that, in order to better understand the dynamic and functional properties of mitochondria, it is crucial to develop dynamic, real-time imaging technologies that can probe ultrastructure and electrophysiology at the nanoscale in vivo.
What is the mechanism that allows cytc to be released from the cristae and cause apoptosis? To answer this, one must look towards the link between bioenergetics and apoptosis in more detail. Although extensive literature exists on both, the combination has not been reviewed until now.
The link between apoptosis and bioenergetics
Experimental evidence from key biophysical parameters
Whole cells
Whole cells in culture can be triggered to cause apoptosis using a variety of means, e.g. exposure to staurosporine, actinomycin, etoposide, UV, etc. During this process, the cytosolic pH, mitochondrial membrane potential ΔΨm, and respiration rate can be continuously monitored, giving insight to the role of mitochondria and bioenergetics in the process of apoptosis.
Upon induction of apoptosis in cultured cells, cytc is released into the cytosol, and the bioenergetic processes slow down or halt: The respiration rate is suppressed 38,39, membrane potential drops 38, and the cytosolic pH drops slightly 40. The drop of the membrane potential and respiration rate is consistent a model where the loss of cytc causes the ETC to stall (i.e. OXPHOS to stop) due to the lack of cytc in the mitochondria to act as an electron shuttle in the ETC. The pH change will be discussed below.
If the apoptosis process is initiated but caspase inhibitors are present, then release of cytc into the cytosol does not activate caspases and the apoptosis process is thus blocked. In this situation, the measurement of the respiration rate and membrane potential remains constant 38, even though cytc is released from the mitochondria. Upon permeabilization of the cell plasma membrane by digitonon, the membrane potential and respiration rate drop 38; these can be restored by the introduction of exogenous cytc into the buffer, which restores the respiration rate38 and ΔΨm 39. These data suggest that the loss of cytc from the cytosol is what causes OXPHOS to stop, as it can be “rescued” by exogenous cytc. (pH was not measured in these experiments). They also suggest that the caspases play an important (as yet undetermined) role in halting the OXPHOS process.
Isolated mitochondria
MOMP can be directly triggered in isolated mitochondria, or in mitochondria inside digitonon permeabilized cells, by the addition of BIM or tBid to the suspension medium. In isolated mitochondria, upon induction of MOMP, metabolic assays show decreased respiration rate 39,41 and membrane potential 39,41,24, as well as a reduction in the buffer pH 24. When exogenous cytc is introduced back into the buffer, these parameters are restored (when measured 39,41,24). These data support a model where the loss of cytc from the mitochondria stall the ETC. The pH change will be discussed below. For permeabilized cells, the data are similar: upon Bid induced MOMP, ΔΨm goes down 42, which is recovered upon addition of exogenous cytc into the buffer 38. It should be noted that peptides have 100-1000 fold less activity than full length proteins, suggesting great caution is required when interpreting the results of experiments using peptides.
Emerging Model
An early indication of the link between membrane potential and apoptosis was found in 200343, but that was before OPA and MICOS were well understood, and so it was purely phenomenological at the time. The role of OPA1 in cytc release is by now well established, but the mechanism by which OPA1 oligomers was dissembled was not known. The discovery of the dependence of OMA1 cleavage of OPA1 on ΔΨm 26 has given a critical insight into the relationship between these parameters. In particular, OMA1 suppression prevents cytc release 44. Therefore, cleavage of OPA1 must be required for cytc release. The relationship between ΔΨm, OPA, and cristae junction remodeling provided the missing link which gave rise to the following model (Figure 3):
Figure 3.
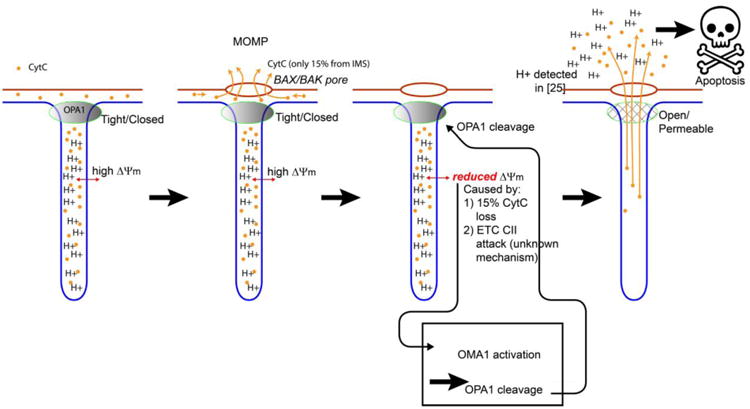
Emerging model of how cytc is released. A) Healthy mitochondria. 85% of the cytc is contained in the cristae and cannot escape. Because of the localization of the ETC resparisomes (not shown) in the cristae, the H+ concentration is high there. B) On MOMP, BAX/BAK pores form in the outer mitochondrial membrane (OMM, red). This releases a fraction of the CtyC into the cytosol; the remainder is still trapped by the tightly closed cristae junctions. C) The mitochondrial membrane potential drops in response to either 1) loss of 15% of CtyC24 or 2) Enzymatic attack on complex II (CII) of the ETC by an unknown enzyme, in response to MOMP via an unknown mechanism 45. This drop in ΔΨm causes OMA1 activation, which cleaves the long form of OPA1. D) The cleaving of OPA1 opens the cristae junctions to allow complete release of cytc, triggering apoptosis. H+ is also released causing slight acidification of the cytosol24, which may enhance the activity of the executioner caspases triggered by cytc.
The initial step in apoptosis of MOMP (BAX/BAK oligomerization) causes lipidic and/or protenactaneous pores in the mitochondrial outer membrane. This allows the small fraction of CtyC stored in the inner membrane space to be released into the cytosol, hence reducing the membrane potential ΔΨm slightly by reducing the activity of the ETC. Another, parallel pathway which reduces ΔΨm after MOMP is degradation of CII of the ETC. Although the mechanism by which CII is degraded after MOMP is not known, evidence45 confirms the model that the membrane potential degradation (actually ETC CII degradation) occurs upstream of OMA1 activation. Once OMA1 is activated due to reduction of ΔΨm, it can cleave OPA1. Cleavage of OPA1 (the glue that holds the cristae junctions together) results in disassembly of the cristae junctions. Disassembly of the cristae junctions allows complete release of cytc, as well as release of the high proton concentration inside the cristae, into the cytosol (whole cells) or buffer (isolated mitochondria). This model provides the critical link between a bioenergetic parameter (ΔΨm), mitochondrial ultra-structure (cristae remodeling), and apoptosis.
This model also explains why, in several different experiments reviewed above, addition of exogenous cytc to the medium surrounding the mitochondria would sustain the respiration required for maintaining the inter membrane space pH and membrane potential thus stabilizing OPA1 and blocking detectable changes in proton and cytc release.
DRP plays a significant role in cristae remodeling
Dynamin-related protein 1 (DRP1) is a cytosolic protein recruited to the mitochondrial outer membrane during mitochondrial fission. Its role in fission has been well studied. Although its activity is believed to be localized at the mitochondria outer membrane only, recent evidence suggests that DRP1 also has a role in cristae remodeling. Superresolution optical microscopy studies have shown BAX/BAK rings form on the outer membrane46. However, in DRP knockdown cells in the same work, these rings (which seemingly indicate large holes in the OMM) are not followed by cytc release. More recently, Otera 47 has shown that DRP-1 knockout cells completely resist CR and cytc release during apoptosis, and in these cell lines OPA1 oligomers were disassembled but the cristae were not remodeled. How DRP, which mediates mitochondrial fission, causes cytc release or assists remains to be elucidated.
Concluding Remarks
The morphology of mitochondria is clearly critically important in apoptosis. There are serious technical challenges to dissect this ultra-structure, as the structures are much smaller than the wavelength of light and just barely addressable by newly developed deep-sub wavelength microscopies (Box 3). Most of these high resolution tools (including TEM) provide static, structural information. What is needed is information about the real-time, dynamic processes, and in additional to structural information, also electrophysiological (electrical) such as: membrane potentials, proton currents and fluxes, and pH gradients, with resolution much smaller than the size of the structure, i.e. at the 1 nm length scale (see outstanding questions). Real time nano-probes such as AFM based tools, quantum dots, or even nanoelectronic probes 48,24, may enable future dissection of this electrophysiological active organelle. Just as the electrophysiological active neurons in the central nervous system give rise to information processing and human thought, the electrophysiological active mitochondria give rise to cellular energy as well as life/death decisions. A clear path forward is to develop new technology to assay the electrophysiological function of this organelle at the nanoscale in real time.
Trends box.
The elements of the electron transport chain are not uniformly distributed along the inner membrane. ATP synthase (Complex V) forms dimers. The other 4 complexes form resparisomes.
The mitochondrial inner membrane is folded into cristae, which remodel in response to metabolic demand as well as cell death pathway signals (apoptosis). The cristae both effect and are effected by the electron transport chain.
The membrane potential and pH gradient are not uniform along the inner membrane. The cristae create pockets of acidic regions, and the pH changes up to 0.3 between the ATP synthase (which consumes the energy stored in the membrane) and the resparisomes.
The spatial and temporal dependence of these quantities is not possible to assay with current technology.
Changes in the mitochondrial cristae (cristae remodeling) are an important step in apoptosis, and a potential target for future pharmacological manipulation.
Outstanding questions box.
Electrophysiology: How does the mitochondrial membrane potential and pH gradient change in space and time, and is there a physiological significance to this?
Ultrastructure: In what ways does the mitochondrial ultra-structure change during apoptosis, and what is the mechanism for protein families that regulate and control this process?
Cancer: Can an increased understanding of the link between bioenergetics and apoptosis be exploited for pharmacological manipulation of apoptosis?
Future directions: How can non-invasive technologies be developed to peer inside the nanoscale structures of mitochondria and monitor their electrophysiological and structural activities in real time
Acknowledgments
This work was funded by the National Cancer Institute grant # IMAT R33CA183384. Extensive input based on an extremely productive long term collaboration from D. Wallace regarding the models and mechanisms have been instrumental in formulating the concepts presented in this paper.
Glossary
- Apoptosis
A systematic, ordered process of programmed cell death in response to external stimuli or internal stress
- ETC
Electron transport chain. A series of protein complexes on the mitochondrial inner membrane which pump protons across the membrane
- IM Inner membrane
Inner membrane of the mitochondria, a double- membraned organelle, impermeable to small molecules
- OM Outer membrane
Outer membrane of the mitochondria, a double- membraned organelle, permeable to small molecules
- IBM
Inner boundary membrane
- CM
Cristae membrane
- TMRE
Tetramethylrhodamine, ethyl ester, a fluorescent dye used to track membrane potential
- CJ, Cristae junction
Narrowing of invaginations of the inner membrane
- GFP
Green fluorescent protein
- ROS
Reactive Oxygen Species
- cytc Cytochrome C
A heme-protein which has the dual role of shuttling electrons in the ETC and also activating caspaces in the cytosol once released from the mitochondria to carry out the process of apoptosis
- ATP synthase (CV)
a complex which uses this electrochemical energy stored in the membrane to convert ADP + Pi to ATP hence coupling oxidation of nutrients to phosphorylation of ADP (OXPHOS)
- MOMP
Mitochondrial outer membrane permeabilization, believed to be caused by oligomerization of BAX/BAK proteins at the outer membrane, triggered by BID.
- BID
a pro-apoptotic member of the ‘BH3-only’ subset of the BCL-2 family of proteins
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lane N, Martin W. The Energetics of Genome Complexity. Nature. 2010;467:929–934. doi: 10.1038/nature09486. [DOI] [PubMed] [Google Scholar]
- 2.Mitchell P. Coupling of Phosphorylation to Electron and Hydrogen Transfer by a Chemi-Osmotic Type of Mechanism. Nature. 1961;191:144–148. doi: 10.1038/191144a0. [DOI] [PubMed] [Google Scholar]
- 3.Jayashankar V, Mueller IA, Rafelski SM. Shaping the Multi-Scale Architecture of Mitochondria. Curr Opin Cell Biol. 2016;38:45–51. doi: 10.1016/j.ceb.2016.02.006. [DOI] [PubMed] [Google Scholar]
- 4.Cogliati S, Enriquez JA, Scorrano L. Mitochondrial Cristae: Where Beauty Meets Functionality. Trends Biochem Sci. 2016;41:261–273. doi: 10.1016/j.tibs.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 5.Kasahara A, Scorrano L. Mitochondria: From Cell Death Executioners to Regulators of Cell Differentiation. Trends Cell Biol. 2014;24:761–770. doi: 10.1016/j.tcb.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 6.Perkins GA, Ellisman MH. Remodeling of Mitochondria in Apoptosis. In: Hockenbery DM, editor. Mitochondria and Cell Death. Springer; New York, NY: 2016. pp. 85–110. [Google Scholar]
- 7.Otera H, Mihara K. Mitochondrial Dynamics: Functional Link with Apoptosis. Int J Cell Biol. 2012;2012:821676. doi: 10.1155/2012/821676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cleland MM, Youle RJ. Mitochondrial Dynamics and Apoptosis. In: Lu B, editor. Mitochondrial Dynamics and Neurodegeneration. Springer; Netherlands: Dordrecht: 2011. pp. 109–138. [Google Scholar]
- 9.Hanahan D, Weinberg R. Hallmarks of Cancer: The next Generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 10.Davies KM, Strauss M, Daum B, Kief JH, Osiewacz HD, Rycovska A, Zickermann V, Kuhlbrandt W. Macromolecular Organization of ATP Synthase and Complex I in Whole Mitochondria. Proc Natl Acad Sci. 2011;108:14121–14126. doi: 10.1073/pnas.1103621108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Strauss M, Hofhaus G, Schröder RR, Kühlbrandt W. Dimer Ribbons of ATP Synthase Shape the Inner Mitochondrial Membrane. EMBO J. 2008;27:1154–1160. doi: 10.1038/emboj.2008.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klotzsch E, Smorodchenko A, Löfler L, Moldzio R, Parkinson E, Schütz GJ, Pohl EE. Superresolution Microscopy Reveals Spatial Separation of UCP4 and F0F1 -ATP Synthase in Neuronal Mitochondria. Proc Natl Acad Sci. 2015;112:130–135. doi: 10.1073/pnas.1415261112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paumard P, Vaillier J, Coulary B, Schaeffer J, Soubannier V, Mueller DM, Brèthes D, di Rago JP, Velours J. The ATP Synthase Is Involved in Generating Mitochondrial Cristae Morphology. EMBO J. 2002;21:221–230. doi: 10.1093/emboj/21.3.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Habersetzer J, Larrieu I, Priault M, Salin B, Rossignol R, Brethes D, Paumard P. Human F1F0 ATP Synthase, Mitochondrial Ultrastructure and OXPHOS Impairment: A (Super-)Complex Matter? PLoS One. 2013;8:e75429. doi: 10.1371/journal.pone.0075429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bitbol AF, Puff N, Sakuma Y, Imai M, Fournier JB, Angelova MI. Lipid Membrane Deformation in Response to a Local pH Modification: Theory and Experiments. Soft Matter. 2012;8:6073–6082. [Google Scholar]
- 16.Wilkens V, Kohl W, Busch K. Restricted Diffusion of OXPHOS Complexes in Dynamic Mitochondria Delays Their Exchange between Cristae and Engenders a Transitory Mosaic Distribution. J Cell Sci. 2013;126:103–116. doi: 10.1242/jcs.108852. [DOI] [PubMed] [Google Scholar]
- 17.Acín-Pérez R, Fernández-Silva P, Peleato ML, Pérez-Martos A, Enriquez JA. Respiratory Active Mitochondrial Supercomplexes. Mol Cell. 2008;32:529–539. doi: 10.1016/j.molcel.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 18.Pfanner N, van der Laan M, Amati P, Capaldi RA, Caudy AA, Chacinska A, Darshi M, Deckers M, Hoppins S, Icho T, Jakobs S, Ji J, Kozjak-Pavlovic V, Meisinger C, Odgren PR, Park SK, Rehling P, Reichert AS, Sheikh MS, Taylor SS, Tsuchida N, van der Bliek AM, van der Klei IJ, Weissman JS, Westermann B, Zha J, Neupert W, Nunnari J. Uniform Nomenclature for the Mitochondrial Contact Site and Cristae Organizing System. J Cell Biol. 2014;204:1083–1086. doi: 10.1083/jcb.201401006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Der Laan M, Bohnert M, Wiedemann N, Pfanner N. Role of MINOS in Mitochondrial Membrane Architecture and Biogenesis. Trends Cell Biol. 2012;22:185–192. doi: 10.1016/j.tcb.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 20.Jakobs S, Wurm CA. Super-Resolution Microscopy of Mitochondria. Curr Opin Chem Biol. 2014;20:9–15. doi: 10.1016/j.cbpa.2014.03.019. [DOI] [PubMed] [Google Scholar]
- 21.Jans DC, Wurm Ca, Riedel D, Wenzel D, Stagge F, Deckers M, Rehling P, Jakobs S. STED Super-Resolution Microscopy Reveals an Array of MINOS Clusters along Human Mitochondria. Proc Natl Acad Sci U S A. 2013;110:8936–8941. doi: 10.1073/pnas.1301820110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA, Korsmeyer SJ. A Distinct Pathway Remodels Mitochondrial Cristae and Mobilizes Cytochrome c during Apoptosis. Dev Cell. 2002;2:55–67. doi: 10.1016/s1534-5807(01)00116-2. [DOI] [PubMed] [Google Scholar]
- 23.Santo-Domingo J, Demaurex N. The Renaissance of Mitochondrial pH. J Gen Physiol. 2012;139:391–393. doi: 10.1085/jgp.201110767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pham TD, Pham PQ, Li J, Letai AG, Wallace DC, Burke PJ. Cristae Remodeling Causes Acidification Detected by Integrated Graphene Sensor during Mitochondrial Outer Membrane Permeabilization. Sci Rep. 2016;6:35907. doi: 10.1038/srep35907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.MacVicar T, Langer T. OPA1 Processing in Cell Death and Disease – the Long and Short of It. J Cell Sci. 2016;129:2297–2306. doi: 10.1242/jcs.159186. [DOI] [PubMed] [Google Scholar]
- 26.Head B, Griparic L, Amiri M, Gandre-Babbe S, van der Bliek AM. Inducible Proteolytic Inactivation of OPA1 Mediated by the OMA1 Protease in Mammalian Cells. J Cell Biol. 2009;187:959–966. doi: 10.1083/jcb.200906083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hackenbrock CR. Ultrastructural Bases for Metabolically Linked Mechanical Activity in Mitochondria. J Cell Biol. 1966;30:269–297. doi: 10.1083/jcb.30.2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC, Perales-Clemente E, Salviati L, Fernandez-Silva P, Enriquez JA, Scorrano L. Mitochondrial Cristae Shape Determines Respiratory Chain Supercomplexes Assembly and Respiratory Efficiency. Cell. 2013;155:160–171. doi: 10.1016/j.cell.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patten DA, Wong J, Khacho M, Soubannier V, Mailloux RJ, Pilon-Larose K, MacLaurin JG, Park DS, McBride HM, Trinkle-Mulcahy L, Harper ME, Germain M, Slack RS. OPA1-Dependent Cristae Modulation Is Essential for Cellular Adaptation to Metabolic Demand. EMBO J. 2014;33:2676–2691. doi: 10.15252/embj.201488349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rieger B, Junge W, Busch KB. Lateral pH Gradient between OXPHOS Complex IV and F0F1 ATP-Synthase in Folded Mitochondrial Membranes. Nat Commun. 2014;5:3103. doi: 10.1038/ncomms4103. [DOI] [PubMed] [Google Scholar]
- 31.Song DH, Park J, Maurer LL, Lu W, Philbert MA, Sastry AM. Biophysical Significance of the Inner Mitochondrial Membrane Structure on the Electrochemical Potential of Mitochondria. Phys Rev E. 2013;88:62723. doi: 10.1103/PhysRevE.88.062723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, Scorrano L. OPA1 Controls Apoptotic Cristae Remodeling Independently from Mitochondrial Fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 33.Cipolat S, Rudka T, Hartmann D, Costa V, Serneels L, Craessaerts K, Metzger K, Frezza C, Annaert W, D'Adamio L, Derks C, Dejaegere T, Pellegrini L, D'Hooge R, Scorrano L, De Strooper B. Mitochondrial Rhomboid PARL Regulates Cytochrome c Release during Apoptosis via OPA1-Dependent Cristae Remodeling. Cell. 2006;126:163–175. doi: 10.1016/j.cell.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 34.Sun MG, Williams J, Munoz-Pinedo C, Perkins GA, Brown JM, Ellisman MH, Green DR, Frey TG. Correlated Three-Dimensional Light and Electron Microscopy Reveals Transformation of Mitochondria during Apoptosis. Nat Cell Biol. 2007;9:1057–1072. doi: 10.1038/ncb1630. [DOI] [PubMed] [Google Scholar]
- 35.Yamaguchi R, Lartigue L, Perkins G, Scott RT, Dixit A, Kushnareva Y, Kuwana T, Ellisman MH, Newmeyer DD. Opa1-Mediated Cristae Opening Is Bax/Bak and BH3 Dependent, Required for Apoptosis, and Independent of Bak Oligomerization. Mol Cell. 2008;31:557–569. doi: 10.1016/j.molcel.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamaguchi R, Perkins G. Dynamics of Mitochondrial Structure during Apoptosis and the Enigma of Opa1. Biochim Biophys Acta - Bioenerg. 2009;1787:963–972. doi: 10.1016/j.bbabio.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sciences, T.R.S.A of. Scientific Background on the Nobel Prize in Chemistry 2017 The Development of Cryo-Electron Microscopy. 2017 [Google Scholar]
- 38.Waterhouse NJ, Goldstein JC, Von Ahsen O, Schuler M, Newmeyer DD, Green DR. Cytochrome c Maintains Mitochondrial Transmembrane Potential and ATP Generation after Outer Mitochondrial Membrane Permeabilization during the Apoptotic Process. J Cell Biol. 2001;153:319–328. doi: 10.1083/jcb.153.2.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mootha VK, Wei MC, Buttle KF, Scorrano L, Panoutsakopoulou V, Mannella CA, Korsmeyer SJ. A Reversible Component of Mitochondrial Respiratory Dysfunction in Apoptosis Can Be Rescued by Exogenous Cytochrome c. EMBO J. 2001;20:661–671. doi: 10.1093/emboj/20.4.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matsuyama S, Llopis J, Deveraux QL, Tsien RY, Reed JC. Changes in Intramitochondrial and Cytosolic pH: Early Events That Modulate Caspase Activation during Apoptosis. Nat Cell Biol. 2000;2:318–325. doi: 10.1038/35014006. [DOI] [PubMed] [Google Scholar]
- 41.Giordano A, Calvani M, Petillo O, Grippo P, Tuccillo F, Melone MAB, Bonelli P, Calarco A, Peluso G. tBid Induces Alterations of Mitochondrial Fatty Acid Oxidation Flux by Malonyl-CoA-Independent Inhibition of Carnitine Palmitoyltransferase-1. Cell Death Differ. 2005;12:603–613. doi: 10.1038/sj.cdd.4401636. [DOI] [PubMed] [Google Scholar]
- 42.Ryan J, Letai A. BH3 Profiling in Whole Cells by Fluorimeter or FACS. Methods. 2013;61:156–164. doi: 10.1016/j.ymeth.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gottlieb E, Armour SM, Harris MH, Thompson CB. Mitochondrial Membrane Potential Regulates Matrix Configuration and Cytochrome c Release during Apoptosis. Cell Death Differ. 2003;10:709–717. doi: 10.1038/sj.cdd.4401231. [DOI] [PubMed] [Google Scholar]
- 44.Jiang X, Jiang H, Shen Z, Wang X. Activation of Mitochondrial Protease OMA1 by Bax and Bak Promotes Cytochrome c Release during Apoptosis. Proc Natl Acad Sci. 2014;111:14782–14787. doi: 10.1073/pnas.1417253111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang X, Li L, Ying Z, Pan C, Huang S, Li L, Dai M, Yan B, Li M, Jiang H, Chen S, Zhang Z, Wang X. A Small Molecule That Protects the Integrity of the Electron Transfer Chain Blocks the Mitochondrial Apoptotic Pathway. Molecular Cell. 2016:229–239. doi: 10.1016/j.molcel.2016.06.016. [DOI] [PubMed] [Google Scholar]
- 46.Große L, Wurm CA, Brüser C, Neumann D, Jans DC, Jakobs S. Bax Assembles into Large Ring-like Structures Remodeling the Mitochondrial Outer Membrane in Apoptosis. EMBO J. 2016;35:402–413. doi: 10.15252/embj.201592789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Otera H, Miyata N, Kuge O, Mihara K. Drp1-Dependent Mitochondrial Fission via MiD49/51 Is Essential for Apoptotic Cristae Remodeling. J Cell Biol. 2016;212:531–544. doi: 10.1083/jcb.201508099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou W, Wang YY, Lim TS, Pham T, Jain D, Burke PJ. Detection of Single Ion Channel Activity with Carbon Nanotubes. Sci Rep. 2015;5:9208. doi: 10.1038/srep09208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sarosiek KA, Chonghaile TN, Letai A. Mitochondria: Gatekeepers of Response to Chemotherapy. Trends Cell Biol. 2013;23:612–619. doi: 10.1016/j.tcb.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shamas-Din A, Kale J, Leber B, Andrews DW. Mechanisms of Action of Bcl-2 Family Proteins. Cold Spring Harb. Perspect Biol. 2013;5:a008714. doi: 10.1101/cshperspect.a008714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schafer B, Quispe J, Choudhary V, Chipuk JE, Ajero TG, Du H, Schneiter R, Kuwana T. Mitochondrial Outer Membrane Proteins Assist Bid in Bax-Mediated Lipidic Pore Formation. Mol Biol Cell. 2009;20:2276–2285. doi: 10.1091/mbc.E08-10-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Salvador-Gallego R, Mund M, Cosentino K, Schneider J, Unsay J, Schraermeyer U, Engelhardt J, Ries J, García-Sáez AJ. Bax Assembly into Rings and Arcs in Apoptotic Mitochondria Is Linked to Membrane Pores. EMBO J. 2016;35:389–401. doi: 10.15252/embj.201593384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Deeks ED. Venetoclax: First Global Approval. Drugs. 2016;76:979–987. doi: 10.1007/s40265-016-0596-x. [DOI] [PubMed] [Google Scholar]
- 54.Brand MD, Nicholls DG. Assessing Mitochondrial Dysfunction in Cells. Biochem J. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zand K, Pham TDA, Li J, Zhou W, Wallace DC, Burke PJ. Resistive Flow Sensing of Vital Mitochondria with Nanoelectrodes. Mitochondrion. 2017 doi: 10.1016/j.mito.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Santo-Domingo J, Giacomello M, Poburko D, Scorrano L, Demaurex N. OPA1 Promotes pH Flashes That Spread between Contiguous Mitochondria without Matrix Protein Exchange. EMBO J. 2013;32:1927–1940. doi: 10.1038/emboj.2013.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bertholet AM, Kazak L, Chouchani ET, Bogaczyńska MG, Paranjpe I, Wainwright GL, Bétourné A, Kajimura S, Spiegelman BM, Kirichok Y. Mitochondrial Patch Clamp of Beige Adipocytes Reveals UCP1-Positive and UCP1-Negative Cells Both Exhibiting Futile Creatine Cycling. Cell Metab. 2017;25:811–822.e4. doi: 10.1016/j.cmet.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bernardi P. The Mitochondrial Permeability Transition Pore: A Mystery Solved? Front Physiol. 2013;4:95. doi: 10.3389/fphys.2013.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.He J, Ford HC, Carroll J, Ding S, Fearnley IM, Walker JE. Persistence of the Mitochondrial Permeability Transition in the Absence of Subunit c of Human ATP Synthase. Proc Natl Acad Sci. 2017;114:201702357. doi: 10.1073/pnas.1702357114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shanmughapriya S, Rajan S, Hoffman NE, Higgins AM, Tomar D, Nemani N, Hines KJ, Smith DJ, Eguchi A, Vallem S, Shaikh F, Cheung M, Leonard NJ, Stolakis RS, Wolfers MP, Ibetti J, Chuprun JK, Jog NR, Houser SR, Koch WJ, Elrod JW, Madesh M. SPG7 Is an Essential and Conserved Component of the Mitochondrial Permeability Transition Pore. Mol Cell. 2015;60:47–62. doi: 10.1016/j.molcel.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zand K, Pham T, Davila A, Wallace DC, Burke PJ. Nanofluidic Platform for Single Mitochondria Analysis Using Fluorescence Microscopy. Anal Chem. 2013;85:6018–6025. doi: 10.1021/ac4010088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huser J, Blatter LA. Fluctuations in Mitochondrial Membrane Potential Caused by Repetitive Gating of the Permeability Transition Pore. Biochem J. 1999;343:311–317. [PMC free article] [PubMed] [Google Scholar]
- 63.Wang W, Gong G, Wang X, Wei-lapierre L, Al WET. Mitochondrial Flash : Integrative Reactive Oxygen Species and pH Signals in Cell and Organelle Biology. 2016 doi: 10.1089/ars.2016.6739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shen EZ, Song CQ, Lin Y, Zhang WH, Su PF, Liu WY, Zhang P, Xu J, Lin N, Zhan C, Wang X, Shyr Y, Cheng H, Dong MQ. Mitoflash Frequency in Early Adulthood Predicts Lifespan in Caenorhabditis Elegans. Nature. 2014;508:128–132. doi: 10.1038/nature13012. [DOI] [PubMed] [Google Scholar]