

SUMMARY
We conducted comprehensive integrative molecular analyses of the complete set of tumors in The Cancer Genome Atlas (TCGA), consisting of approximately 10,000 specimens and representing 33 types of cancer. We performed molecular clustering using data on chromosome-arm-level aneuploidy, DNA hypermethylation, mRNA, and miRNA expression levels and reverse-phase protein arrays, of which all, except for aneuploidy, revealed clustering primarily organized by histology, tissue type, or anatomic origin. The influence of cell type was evident in DNA-methylation-based clustering, even after excluding sites with known preexisting tissue-type-specific methylation. Integrative clustering further emphasized the dominant role of cell-of-origin patterns. Molecular similarities among histologically or anatomically related cancer types provide a basis for focused pan-cancer analyses, such as pan-gastrointestinal, pan-gynecological, pan-kidney, and pan-squamous cancers, and those related by stemness features, which in turn may inform strategies for future therapeutic development.
In Brief
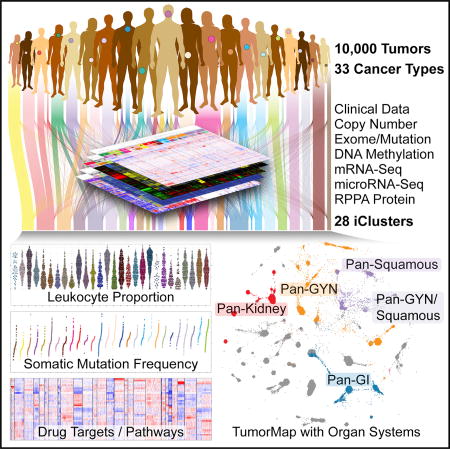
Comprehensive, integrated molecular analysis identifies molecular relationships across a large diverse set of human cancers, suggesting future directions for exploring clinical actionability in cancer treatment.
INTRODUCTION
Genomic and other molecular analyses across many types of cancer have revealed a striking diversity of genomic aberrations, altered signaling pathways, and oncogenic processes. We hypothesized that this diversity arises from endogenous factors, such as developmental and differentiation programs and epigenetic states of the originating cells, in conjunction with exogenous factors, such as mutagenic exposures, pathogens, and inflammation. Here, we performed an integrative analysis of approximately 10,000 human samples representing 33 different cancers, to provide the first comprehensive view of the molecular factors that distinguish different neoplasms in The Cancer Genome Atlas (TCGA).
In 2014, TCGA Research Network reported an interim analysis of 3,527 tumors from 12 different cancer types (Pan-Cancer-12), integrating six genome-wide platforms that assayed tumor DNA (exome sequencing, DNA methylation, and copy number), RNA (mRNA and microRNA sequencing), and a cancer-relevant set of proteins and phosphoproteins (Hoadley et al., 2014). The analysis tested the hypothesis that molecular signatures might provide a taxonomy that differed from the current organ- and tissue-histology-based pathology classification (Hoadley et al., 2014). This effort extended beyond cancer subtype classification by individual molecular platforms by employing an integrated clustering algorithm to identify higher-level structures and relationships. These integrated subtypes shared mutations, copy-number alterations, pathway commonalities, and micro-environment characteristics that appeared influential in the new molecular taxonomy, beyond any phenotypic contributions from tumor stage or tissue of origin. We estimated that at least one in ten cancer patients might be classified (and perhaps treated) differently using such a molecular taxonomy, rather than the current histopathology-based classification.
Given that the earlier analysis included only a third of the final set of TCGA tumors, it seemed appropriate to analyze all 33 tumor types (called the PanCancer Atlas) to address the intriguing questions left unanswered: whether the inclusion of many more tumors and tumor types enhances the number of cross-tissue associations, produces additional convergent and/or divergent integrated molecular subtypes, and significantly increases the fraction of cancer patients whose classification or treatment might be affected by this new taxonomic approach.
We present a new PanCancer Atlas integrative analysis using iCluster (Shen et al., 2009, 2012) identifying 28 distinct molecular subtypes arising from the 33 different tumor types analyzed across at least four different TCGA platforms. We confirmed significant taxonomic divergences from and convergences with the routinely used clinical tumor classification system. We employed a new 2D visualization approach, TumorMap (Newton et al., 2017), to intepret the relationships between the samples and iClusters. The PanCancer Atlas molecular classification also provides a rationale for several TCGA analyses based on organ systems or differentiation states, including pan-gastrointestinal (GI) (Liu et al., 2018), pan-gynecological (gyn) (Berger et al., 2018), pan-kidney (Ricketts et al., 2018), pan-squamous (Campbell et al., 2018), and cancer stemness features (Malta et al., 2018).
RESULTS
Specimens and Tumor Types
This PanCancer study encompassed 11,286 tumor samples from 33 cancer types, for which molecular data were available from at least one of the five assay platforms. Of these, 9,759 had complete data for 4 platforms: aneuploidy, DNA methylation, mRNA and miRNA. RPPA protein data were available for a subset of samples (7,858). Hematologic and lymphatic malignancies included acute myeloid leukemia (LAML), lymphoid neoplasm diffuse large B cell lymphoma(DLBC), and thymoma(THYM). Solid tumor types were from gynecologic (ovarian [OV], uterine corpus endometrial carcinoma [UCEC], cervical squamous cell carcinoma and endocervical adenocarcinoma [CESC], and breast invasive carcinoma [BRCA]), urologic (bladder urothelial carcinoma [BLCA], prostate adenocarcinoma [PRAD], testicular germ cell tumors [TGCT], kidney renal clear cell carcinoma [KIRC], kidney chromophobe [KICH], and kidney renal papillary cell carcinoma [KIRP]), endocrine (thyroid carcinoma [THCA] and adrenocortical carcinoma [ACC]), core gastrointestinal (esophageal carcinoma [ESCA], stomach adenocarcinoma [STAD], colon adenocarcinoma [COAD], and rectum adenocarcinoma [READ]), developmental gastrointestinal (liver hepatocellular carcinoma [LIHC], pancreatic adenocarcinoma [PAAD], and cholangiocarcinoma [CHOL]),head and neck (head and neck squamous cell carcinoma [HNSC]), and thoracic (lung adenocarcinoma [LUAD], lung squamous cell carcinoma [LUSC], and mesothelioma [MESO]) organ systems. Cancers of the central nervous system (glioblastoma multiforme [GBM] and brain lower-grade glioma [LGG]) and soft tissue (sarcoma [SARC] and uterine carcinosarcoma [UCS]) were represented, as were cancers from neural-crest-derived tissues, such as pheochromocytoma and paraganglioma (PCPG), and melanocytic cancers of the skin (skin cutaneous melanoma [SKCM]) and eye (uveal melanoma [UVM]). (For a complete list of the TCGA cancer-type abbreviations, please see https://gdc.cancer.gov/resources-tcga-users/tcga-code-tables/tcga-study-abbreviations.)
Clustering by Individual Platforms
We explored the sample groupings from each individual assay platform. Using aneuploidy (AN), CpG hypermethylation (METH), mRNA (MRNA), miRNA (MIR), and protein (P), the resultant number of groups ranged from 10 to 25 (Figure 1). While cell-of-origin was a dominant feature of the classification, we observed tumors from different cancer types grouping and samples within a cancer type dispersing across groups.
Figure 1. Platform-Specific Classification of 10,000 TCGA Cancer Tumor Samples across 33 Cancer Types.

(A) Aneuploidy (AN). Unsupervised consensus clustering of 10,522 tumors and chromosomal arm-level amplifications or deletions.
(B) DNA hypermethylation (METH). Clustering of cancer-associated DNA methylation profiles in 10,814 tumors at 1,035 CpG sites lacking DNA methylation in normal tissues (left) and leukocytes (right). DNA methylation β-values are represented as a color gradient from low (blue) to high (red).
(C) mRNA (MRNA). Unsupervised consensus clustering of 10,165 tumors and variably expressed genes.
(D) microRNA (MIR). Unsupervised hierarchical clustering of 743 expressed mature strands in 10,170 tumors.
(E) Protein (P). Unsupervised hierarchical clustering of 7,858 tumor samples from 32 cancer types across 216 cancer-relevant proteins and phosphoproteins. Tumor types are color-coded as shown in the lower-right corner.
Hierarchical clustering of 10,522 samples by chromosome arm-level aneuploidy yielded ten groups (Figure 1A; Table S1). Samples were split mainly by those with few alterations (AN7), those with moderate alterations (AN6,8-10), and those with many alterations (AN1-5). Over one-third of the samples displayed relatively sparse aneuploidy in AN7; these were enriched for THCA, LAML, PRAD, and THYM. We observed more distinct clustering by cell-of-origin among higher-aneuploid tumors. For example, AN2, characterized by chromosome (chr) 13 gain and chr18 loss, was strongly enriched for gastrointestinal tumors (COAD, READ, and STAD), and chromosomal instability (CIN) ESCA. Consistent with previous results (Hoadley et al., 2014), squamous (lung, head and neck, and esophageal) tumors clustered together by aneuploidy patterns, particularly 3p loss and 3q gain (AN3).
Unsupervised clustering of 10,814 tumors using DNA methylation data with 3,139 CpG sites that were hypermethylated in at least one tumor type identified 25 groups. Despite the exclusion of loci known to be involved in tissue-specific DNA methylation, tumors originating from the same organ often aggregated by cancer-type-specific hypermethylation (Figure 1B; Table S2). This result suggests that cancer-associated DNA hyper-methylation in human cancers is influenced by pre-existing cell-type-specific chromatin marks or transcriptional programs, and not just by cell-type-specific DNA methylation patterns. Tumors within an organ system tended to co-cluster. Consistent with the aneuploidy analysis, squamous cell carcinomas (HNSC, ESCA, LUSC, and CESC) associated closely in METH2 and METH3. Gastrointestinal adenocarcinomas (ESCA, STAD, COAD and READ) were represented in a branch containing METH10 through METH13.
Unsupervised consensus clustering of 10,165 tumors by mRNA expression profiles identified 25 groups that contained at least 40 samples (Figure 1C; Table S3). While tumor type was a driving feature for many groups, several groups were comprised of tumors from different organ types. Samples with squamous morphology components (BLCA, CESC, ESCA, HNSC, and LUSC) grouped together. Similarly, tumors with tissue or organ similarities or proximity also grouped together. These included neuroendocrine and glioma tumors (GBM, LGG and PCPG), melanomas of the skin and eye (SKCM and UVM), clear cell and papillary renal carcinomas (KIRC and KIRP), adrenal cortical and chromophobe renal (ACC and KICH), hepatocellular and cholangiocarcinomas (LIHC and CHOL), a gastrointestinal group (COAD, READ, non-squamous ESCA, READ, and STAD), a digestive system group (PAAD, STAD, and a few ESCA), hematologic and lymphatic cancers (LAML, DLBC, and THYM), and two mixed lung cancer groups (LUAD and LUSC).
Unsupervised hierarchical clustering of miRNA expression profiles from 10,170 tumors yielded 15 groups (Figure 1D; Table S4). While six groups contained only a single cancer type, the remaining nine groups each represented a mix of cancer types. These included a squamous-enriched group (MIR2), a pan-kidney group (MIR11), and a pan-GI-enriched group (MIR6).
Hierarchical clustering of protein expression data from 7,858 samples across 32 tumor types (LAML did not have protein data) revealed ten distinct protein (P) groups (Figure 1E; Table S5). P1 (GBM, LGG) and P2 (DLBC, SARC, PCPG, UCS, THYM, and metastatic SKCM) were distinguished from the remaining 8 groups, largely corresponding to mesenchymal-like tumor types with high EMT signatures. Similar to the other individual data platforms, samples from related organ systems grouped together: luminal breast and gynecologic cancers (BRCA-Luminal, UCEC, and OV), plus some liver samples (LIHC) with high levels of ER-alpha, AR and IGFBP2 comprised the majority of the P3 and P4 groups. In addition, a pan-kidney (P6) and a pan-GI (P8) group were identified.
Integrative Clustering across Data Types
We used clustering of cluster assignments (COCA) algorithm (Hoadley et al., 2014) to assess the overlap of platform-specific memberships from each of the five molecular platforms (aneuploidy, mRNA, miRNA, DNA methylation, and RPPA) (Figure 2A). Many samples similarly grouped together by multiple platform-specific cluster memberships, both in groups that were defined by a single tumor type and in tumor types that co-clustered, such as KIRC and KIRP (pan-kidney). Gastrointestinal tumors (COAD, READ, STAD, and ESCA adenocarcinomas) co-clustered in the mRNA, miRNA, and RPPA platforms but were represented by several distinct DNA methylation clusters. Squamous histology cancers (LUSC, HNSC, CESC, ESCA, and BLCA) were similarly classified by the miRNA, mRNA and RPPA data but were further divided by the aneuploidy and DNA methylation data. Within pan-gyn cancers (BRCA, OV, UCEC, and UCS), RPPA data suggested that ovarian serous cystadenocarcinoma (OV) and UCEC (and ER+ LIHC) shared similarities at the protein level, whereas miRNA, mRNA, and DNA methylation data were grouped by their organ sites. Also of note, 13% of BRCA formed a subtype distinct from the majority of other BRCA, influenced by the mRNA and DNA methylation platforms.
Figure 2. Cross-Platform Classification Revealed Genomic, Epigenomic, and Transcriptomic Similarities and Differences across Cancer Types.

(A) COCA clusters. Membership for individual clusters for each of the five molecular platforms—aneuploidy (AN), methylation (Meth), miRNA expression (miR), mRNA, and RPPA—is displayed as a separate binary membership variable in a distinct row. For the mRNA platform, only clusters containing >40 samples were considered. Samples are labeled for membership of each platform-specific cluster (red, member; white, non-member; gray, not evaluated on the platform). Order of samples and platform-specific clusters were determined by hierarchical clustering using a binary distance matrix and average linkage. Column annotation shows cancer type and tissue organ systems of each sample; row annotations reflect the platform for each classification (bright pink, AN; purple, Meth; light turquoise, miR; dark turquoise, mRNA; orange, RPPA).
(B) iCluster. Data used for integrated analysis of iClusters. RPPA data are also included in the heatmap to visualize proteomic patterns across the integrated clusters.
(C) iCluster robustness versus composition. Pie charts show the cancer-type composition within each iCluster and the size is proportional to the membership size. The cancer type accounting for the highest proportion of members within the iCluster was considered the dominant cancer type. They coordinate of each pie center reflects this dominant cancer-type proportion; the x coordinate was determined by the iCluster silhouette width.
(D) Relationship of TCGA tumor type, iCluster, and Pan-Organ system. The Sankey diagram demonstrates the tumor-type composition of each iCluster. The pan-cancer designations are shown on the right.
While COCA showed high consistency across most data platforms, we found less concordance for aneuploidy, where more than a third of the samples were defined by few to no aneuploidy events. This group, AN7, included almost all the THCA and LAML samples, while not well defined by aneuploidy had strong concordance among the other data platforms. COCA is less powerful when the molecular patterns are not strong enough to specify a distinct group on multiple individual platforms. To complement this analysis, we explored joint clustering across all platforms simultaneously.
We performed integrative molecular subtyping with iCluster using the four most complete data types (copy number, DNA methylation, mRNA, and miRNA) across 9,759 tumor samples, identifying 28 iClusters (Figure 2B; Table S6). The relative contribution of each platform to the overall clustering was quantified by summing the different platform feature weights on the iCluster latent variables. Copy-number alterations contributed 47% to the overall integrated clustering results, followed by the transcriptome (mRNA and miRNA) at 42%, and DNA methylation at 11%.
For 16 of the tumor types, over 80% of samples grouped together in the same iCluster. Eight iClusters were dominated by a single tumor type (C24:LAML, C11:LGG [IDH1 mut], C6:OV, C8:UCEC, C12:THCA, C16:PRAD, C26:LIHC, C14:LUAD). Others contained tumors from similar or related cells or tissues: C28:pan-kidney (KIRC, KIRP), C15:SKCM/UVM-melanoma of the skin (SKCM) and eye (UVM), C23:GBM/LGG (IDH1wt), and C5:CNS/ endocrine. Six tumor types had more diverse iCluster membership, with less than 50% of tumors represented in a given iCluster (BLCA, UCS, HNSC, ESCA, STAD, and CHOL).
The pan-GI cohort separated into three iClusters (C1, C4, and C18), primarily driven by differences in DNA methylation profiles. C1:STAD (Epstein-Barr virus [EBV]-CIMP) consisted of hypermethylated EBV-associated tumors, and C18:pan-GI (MSI) consisted mostly of microsatellite instability (MSI) tumors of STAD and COAD. C4:pan-GI (CRC) was predominantly COAD and READ with chromosomal instability (CIN) and a distinct aneuploidy profile (Figure 2B). The pan-squamous cohort formed three iClusters (C10, C25, and C27). The majority of LUSC fell into C10:pan-SCC, and nearly all CESC fell into C27:pan-SCC (human papillomavirus [HPV]). Even though all squamous iClusters were characterized by chromosome 3q amplification, unique features defined C10:pan-SCC (9p deletion) and C25:pan-SCC (Chr11 amp) (Figure 2B).
Among mixed tumor type iClusters, three were defined by copy-number alterations. C7:mixed was characterized by chr9 deletion, C2:BRCA (HER2 amp) mainly consisted of ERBB2-amplified tumors (BRCA, BLCA, and STAD), and C13:mixed (Chr8 del) contained highly aneuploid tumors, including a mixture of BRCA-Basal, UCEC (CN-high subtype), UCS, and BLCA. C3 and C20 were defined by their non-tumor-cell components including immune and stromal features.
We explored the non-tumor components of the iClusters in more detail. We estimated the stromal fraction as 1 minus tumor purity and the leukocyte fraction based on DNA methylation (Figure 3). C20 had the highest median stromal fraction followed by C14:LUAD, C10:pan-SCC, and C3 (Figure 3A). Each of these iClusters also displayed elevated leukocyte fractions (Figure 3B). To estimate how much of the stromal fraction was due to immune cell infiltration, we plotted the stromal fraction versus the leukocyte fraction (Figure 3C). In C3, more of the stromal fraction was defined by leukocytes than in C20. C3 contained predominately mesenchymal cancers, which we labeled C3:mesenchymal (immune). C20 tumors were predominately mixed epithelial cancers, which we labeled C20:mixed (stromal/immune).
Figure 3. Cellularity of the Tumor Microenvironment among iCluster Samples.

(A) Stromal fraction of tumor samples. The stromal fraction, defined by subtracting tumor purity (estimated by ABSOLUTE) from one, is shown for 9,057 TCGA tumor samples, segregated by iCluster membership.
(B) Leukocyte fraction. Leukocyte fraction, estimated from DNA methylation arrays, for 9,417 tumor samples, for each iCluster, with the exception of C24:LAML and C21:DLBC.
(C) Leukocyte fraction versus stromal fraction. Points near the diagonal correspond to tumor samples in which non-tumor stromal cells are nearly all immune cells, and points away from the diagonal correspond to a more mixed or a non-immune stromal tumor microenvironment. Points in the upper-left triangle of each plot are estimation artifacts.
To characterize composition and relative homogeneity of each iCluster, we computed the dominant-cancer-type proportion within each iCluster and plotted it against the mean iCluster silhouette width, a measure of within-group homogeneity (Figure 2C). The silhouette widths ranged from −0.05 to 0.59, with the highest silhouette widths belonging to single-cancer-type-dominant iClusters (C11:LGG [IDH1 mut], C12:THCA, C16:PRAD, and C24:LAML). Interestingly, 6 of the 7 pan-organ system iClusters (pan-GI: C1, C4, C18; pan-SCC: C25, C27, and pan-kidney: C28) had similar ranges of silhouette widths to those of single cancer-type dominant iClusters, suggesting that these were as robust as the cancer-type-dominant iClusters. iClusters driven by a shared specific chromosomal alteration (e.g., C13:mixed [chr8 del]) tended to compose multiple tumor types and appeared to have among the lowest silhouette widths, suggesting substantial molecular heterogeneity.
We used a Sankey diagram to further visualize the relationship between the iCluster classification, cancer types, and organ systems (Figure 2D). Pan-kidney mapped almost entirely to C28, except for KICH, which grouped with ACC in C9, characterized by a high frequency of hypodiploid samples (Davis et al., 2014; Zheng et al., 2016). However, pan-GI, pan-gyn, and pan-squamous were distributed among multiple iClusters. C20:mixed (stromal/immune) was fairly heterogeneous, including pan-GI, pan-gyn, and pan-squamous. Pan-gyn and pan-squamous overlapped, as cervical cancer is primarily a squamous cell carcinoma. This analysis demonstrated that the iClusters were strongly influenced by the cell type of origin for the individual cancers, though this relationship was not absolute.
Tumor Maps of Organ Systems
We visualized the samples by calculating Euclidean distances between the iCluster latent variables for all sample pairs and projecting the distances onto a 2D layout with TumorMap (Figure 4A; Table S7) (Newton et al., 2017). We over laid the tumor-type colors to reveal that tumors systematically assembled along the major organ systems (Figure 4B), lending further support for the organ-system groups explored in accompanying papers (Figure 4C) (Berger et al., 2018; Campbell et al., 2018; Liu et al., 2018; Malta et al., 2018; Ricketts et al., 2018). More subtle differences within individual iClusters were apparent, potentially signifying important distinctions from the dominant cell-of-origin-associated signals. Kidney tumors separated into KICH, KIRC, and KIRP (Ricketts et al., 2018), and CIMP kidney tumors were positioned near the Pan-GI CIMP tumors, suggesting similarities driven by DNA hypermethylation data (Figure 4D). Pan-gyn subtypes displayed partial overlap (Berger et al., 2018) (Figure 4E). Pan-gyn samples were broadly distributed, accounting for at least 5% of samples in 11 of the 28 iClusters. However, the majority of cervical cancers fell into the squamous C27:pan-SCC (HPV) with HPV-positive HNSC and BLCA, whereas other samples fell primarily within C6:OV, C19:BRCA (luminal) and C8:UCEC, reflecting their cell-of-origin and hormonal dependency (Berger et al., 2018). The pan-GI tumors separated into distinct molecular subtypes represented by MSI tumors, hypermutated-SNV tumors, genome-stable tumors, CIN tumors, and EBV-associated gastric cancers (Liu et al., 2018) (Figure 4F).
Figure 4. The iCluster TumorMap.

(A–F) The map layout was computed from sample Euclidean similarity in the iCluster latent space, and similar samples are positioned in close proximity to each other. Each spot represents a single sample and is colored to represent attributes as described for each panel including (A) iCluster, (B) disease type, and (C) organ system. Organ systems highlighted include pan-kidney, red; pan-gyn, orange; pan-GI, blue; pan-squamous, purple; and those that overlap pan-gyn and pan-squamous, light purple.
(D) Subtypes from the pan-kidney analysis (Ricketts et al., 2018). Clear cell renal cell carcinoma (ccRCC), green; papillary renal cell carcinoma type 1 (PRCC T1), blue; papillary renal cell carcinoma type 2(PRCC T2), yellow; unclassified papillary renal cell carcinoma (PRCC Unc.), dark gray; CpG island methylator phenotype renal cell carcinoma (RCC-CIMP), red; and chromophobe renal cell carcinoma (ChRCC), purple.
(E) Subtypes from the pan-gyn group (Berger et al., 2018). Not hypermutated, with low copy-number changes (non-HM CNV low), red; hypermutated, with low copy-number changes (HM), blue; high levels of leukocyte infiltration (immune), green; low AR or PR expression (AR/PR low), orange; and high androgen receptor (AR) or progesterone receptor (PR) expression (AR/PR high), dark gray.
(F) Subtypes from the pan-GI group (Liu et al., 2018). High Epstein-Barr virus (EBV) burden, red; microsatellite instability (MSI), blue; hypermutated without MSI (HM-SNV), gold; chromosomal instability tumors (CIN), purple; and genome stable (GS) with low aneuploidy, green. The gray dots represent non-highlighted diseases.
The TumorMap landscape showed that tumors with similar pathologic classification tended to assemble together, even though histopathologic information was not used in the map generation (Figure 5A). This result underscores the influence of the cell of origin on the molecular patterns observed in cancer and provides further support for the pan-squamous sub-analysis (Campbell et al., 2018). Immune-signaling subtypes identified in Thorsson et al. (2018) also co-localized on the TumorMap, indicating relationships between the iClusters, histopathology, and the types of immune infiltration (Figure 5B). Pan-squamous tumors shared predominant wound healing and interferon (IFN)-gamma-dominant immune signatures.
Figure 5. Sample Characteristics in the Context of the iCluster TumorMap.

(A–D) The TumorMap layout is as described for Figure 4.
(A) Histopathology. Colors indicate major histopathology types. Adenocarcinoma, yellow; squamous cell carcinoma, purple; other carcinomas, green; sarcomas, light blue; leukemias, dark blue; lymphomas, magenta; and other, red.
(B) Immune subtypes. Wound-healing group, red; IFN-gamma, yellow; inflammatory group, green; lymphocyte-depleted, light blue; immunologically quiescent, dark blue; and transforming growth factor (TGF)-beta activity, magenta.
(C and D) Stemness signatures for (C) mRNA and (D) DNA methylation from Malta et al. (2018) are displayed. Increasing red colors indicate increasing stemness index.
Cancer stemness has been proposed as a possible mechanism for treatment resistance and as a driver of the ability of subpopulations to repopulate new metastatic niches (Jin et al., 2017). Two stemness indices (Malta et al., 2018), based on mRNA expression and on DNA methylation data, revealed aggregation of high stemness tumors across distinct regions of the TumorMap (Figures 5C and 5D). TGCT showed strong enrichment of both signatures while others, such as LAML, showed strong enrichment only for the mRNA-based signature.
Mutational Assessment of iClusters
We did not use tumor mutation data in generating iClusters due to sparsity of mutations; however, we did use mutational burden and signatures for characterization. Overall somatic mutation burden varied among iClusters. Melanomas and lung adenocarcinomas have been shown to have relatively high mutation rates, and we observed similar results with C15:SKCM/UVM and C14:LUAD (Lawrence et al., 2013). Pan-GI and pan-squamous were also associated with overall higher somatic mutational burdens (Figure 6A). Mutation frequencies varied widely within the two iClusters with the most diverse tumor compositions: C3:mesenchymal (immune) and C20:mixed (stromal/immune). Mutational signatures (Covington et al., 2016) also varied among iClusters. Expected signatures were apparent, such as enrichment for UVB signatures in C15:SKCM/UVM, smoking in C14:LUAD, and POLE mutation in hypermutated samples of C8:UCEC and C4:pan-GI (CRC) (Figure 6B). We also found enhanced signatures in a few of our pan-organ groups such as C18:pan-GI (MSI), which showed enrichment of known (CpG, toxins) and unknown mutational signatures, some of which are likely related to the high proportion of mismatch-repair deficient tumors in this group (Figure 6B).
Figure 6. Mutation Patterns of iClusters.

(A) Somatic mutation frequency (log10) per iCluster sorted by median mutations per megabase. Somatic mutation frequencies were calculated using a filtered MC3 mutation annotation file to determine the total number of mutations per sample, normalized by whole-exome sequencing coverage as described in Knijnenburg et al. (2018). Bars represent median mutation frequency for each iCluster.
(B) Mutational signatures (Covington et al., 2016) enriched in iClusters. Mutational signature scores were scaled per sample by the overall mutation rate. The means of scaled signature scores were calculated for each iCluster and log10-transformed. Hierarchical clustered data are displayed in the heatmap (blue, low; red, high).
Pathway Characteristics of the PanCancer iCluster Subtypes
We compared the PARADIGM-inferred activation of ~19,000 pathway features (Vaske et al., 2010), as well as expression-based scores of 22 gene programs defined previously (Hoadley et al., 2014), and 18 canonical targetable pathways, to identify differential pathway characteristics across the 28 iClusters (Figure 7; Table S8). C28:pan-kidney was characterized by high hypoxia signaling, retinoid metabolism, low proliferation, PPAR-RXR pathway and immune-related signaling, including immune checkpoints PD-1 and CTLA4. However, KICH co-clustered with ACC in C9:ACC/KICH, lacking hypoxic and immune signals and showing low activity in nearly all pathways. Both these tumor types have previously been characterized as hypodiploid (Davis et al., 2014; Zheng et al., 2016).
Figure 7. Pathway Features Characterizing the PanCancer-33 iCluster Subtypes.

(A) PARADIGM pathway heatmap. Regulatory nodes with differential PARADIGM-inferred pathway levels (IPL) with at least 15 downstream regulatory targets with differential inferred activities between iClusters are shown for one versus rest comparisons. Samples are arranged by iCluster order; regulatory nodes are hierarchically clustered using 1-Pearson correlation as distance and average linkage. Red-blue intensities represent median-centered IPLs from low (blue) to high (red).
(B) Gene programs and canonical pathway values. The 22 Gene Programs (Hoadley et al., 2014) and 20 pathway signatures reflecting drug targets and canonical pathways (found in Table S4 of Hoadley et al. [2014]) were hierarchically clustered using 1-Pearson distance and complete linkage and are shown with samples arranged by iCluster subtypes in numerical order. Red-blue intensities represent signature scores from low (blue) to high (red).
Despite having very different cancer type compositions, the pan-squamous iClusters C10:pan-SCC, C25:pan-SCC (chr11 amp), and C27:pan-SCC (HPV) shared many pathway characteristics. All had high levels of squamous-cell-related signaling (dNp63 and TAp63 complexes and GP6), proliferation-related pathways, relatively high hypoxia, immune-related signaling, and high basal signaling.
Although the Pan-GI iClusters C1:STAD (EBV-CIMP), C4:pan-GI (CRC), and C18:pan-GI (MSI) shared some common characteristics such as relatively high proliferation signaling, these iClusters diverged in some respects. Immune-related signaling was high in C1:STAD (EBV-CIMP) and C18:pan-GI (MSI), but not in C4:pan-GI (CRC). In addition, C20:mixed (stromal/immune) contained 32% Pan-GI samples and also displayed strong immune-related signaling. Beta-catenin/cell-cell adhesion signaling appeared high in C4:pan-GI (CRC), C18:pan-GI (MSI), and C20:mixed (stromal/immune), but not in the smaller C1:STAD (EBV-CIMP).
Most UCS co-clustered with a subset of Basal BRCA, UCEC and BLCA in C13:mixed (chr8 del), with high basal signaling and proliferation in the absence of immune activation. Interestingly, another subset of Basal breast cancers co-clustered with squamous cancers in the C20:mixed (stromal/immune), which also had high basal signaling and proliferation, but activated immune signaling. OV and UCEC shared a number of pathway similarities with cervical cancers and a subset of Basal breast cancers despite falling into different iClusters. These similarities included high proliferation and DNA repair pathways and basal signaling. Although the estrogen-signaling gene program (GP7) was very high in the breast cancer iClusters C2:BRCA (HER2 amp) and C19:BRCA (luminal), that program did not appear to be high in the other gynecological cancers.
DISCUSSION
With nearly three times more tumors and tumor types profiled in this PanCancer Atlas analysis, we were able to detect more integrated molecular subtypes than we had reported in the original Pan-Cancer-12 analysis (Hoadley et al., 2014). We first performed unsupervised consensus clustering of tumor profiles from each of the 5 platforms, revealing from 10 to 25 platform-specific molecular subsets within ~10,000 tumors, each showing significant compositional heterogeneity based on classical tumor taxonomy (Figure 1). Aneuploidy classifications were weakly consistent with other classifications, in part due to low numbers of arm-level copy-number events in one-third of the tumors. We explored cross-platform cluster relationships using COCA and employed iCluster to integrate the multiplatform molecular data simultaneously into a final 28-cluster solution.
While a third of iClusters were mostly homogeneous for a single tumor type, the other two-thirds showed varying degrees of heterogeneity. The most diverse group, C20:mixed (stromal/ immune), contained a remarkable 25 tumor types (Figures 2C and 2D). Most of the heterogeneous iClusters, including C20:mixed (stromal/immune), contained tumor types that fell within four major cell-of-origin, or organ system, patterns (Figure 2D): pan-GI, pan-gyn, pan-squamous, and pan-kidney. Individual cluster assignments, COCA, and iCluster-determined molecular subsets were concordant, and confirmed the multi-platform co-clustering of different kidney malignancies (pan-kidney), various gastrointestinal malignancies (pan-GI), diverse squamous cell malignancies (pan-squamous) and most gynecological malignancies (pan-gyn) into molecular subgroups, each with subordinate platform-specific subsets (Figure 2A). Consequently, these four major cell-of-origin patterns are the subject of separate in-depth reports detailing their distinguishing genomic and molecular features (Berger et al., 2018; Campbell et al., 2018; Liu et al., 2018; Malta et al., 2018; Ricketts et al., 2018). These iCluster assignments have potential clinical utility, and their multi-platform basis suggests that this new subclassification system might further improve the management of the 1%–3% of all cancer patients newly diagnosed with cancer of unknown primary (CUP). Using either RNA (Hainsworth et al., 2013) or DNA methylation (Moran et al., 2016) profiling has recently led to improved patient outcomes by better defining the tissues of origin for this diverse group of life-threatening malignancies.
While separate spatial co-localization of the four major cell-of-origin patterns was generally evident in the TumorMap visualization (Figure 4), heterogeneity was also apparent between subsets within these individual iClusters, even those with generally similar tumor type, organ system, and histopathology. This indicates that while iCluster groupings were strongly influenced by organ and cell-of-origin patterns, this influence did not fully determine their molecular groupings such as seen in our largest and most heterogeneous iCluster, C20:mixed (stromal/immune), which contained 25 of our 33 tumor types. The spatial relationships of C20:mixed (stromal/immune) tumors to C10:pan-SCC and C13:mixed (chr8 del) tumors may be determined in part by their different mRNA and DNA methylation-based stemness signatures (Figures 5C and 5D).
Interrogation of individual iClusters for their differentiating PARADIGM pathway features, canonical pathways, and gene programs amenable to drug targeting identified strong immune-related signaling features for both C3:mesenchymal (immune) and C20:mixed (stromal/immune) tumors, suggesting that they may share potential susceptibility to immunotherapy. We noted that C20:mixed (stromal/immune) and C3:mesenchymal (immune) tumors were commonly enriched for gene programs representing PD1, CTLA4, and GP2-T cell/B cell activation (Figure 7B), indicating that new therapies targeting these specific immune pathways might be appropriate. Another potentially clinically relevant similarity was upregulation of different druggable growth factor signaling pathways (Figure 7B). In particular, our PARADIGM analysis showed that C3:mesenchymal (immune) and C20:mixed (stromal/immune) tumors shared upregulated JAK2/STAT1,3,6 signaling with C14:LUAD tumors and C10:pan-SCC, pointing to the possibility of treating these diverse iCluster tumors with JAK-STAT agents currently approved to treat rheumatoid arthritis, myelofibrosis, polycythemia vera, and other non-malignant diseases (Banerjee et al., 2017).
Compared to the seemingly discohesive groupings of the 17 heterogeneous iClusters, the 11 most homogeneous iClusters (C6:OV, C8:UCEC, C11:LGG [IDH1 mut], C12:THCA, C14:LUAD, C15:SKCM/UVM, C16:PRAD, C19:BRCA [luminal], C21:DLBC, C24:LAML, C26:LIHC) had higher silhouette widths, uniform tumor types, and histopathologies, but showed surprising degrees of spatial discohesion in the TumorMap. These anatomically homogeneous iClusters also showed mixed types of immune infiltration and variable degrees of stemness, attesting to their underlying molecular heterogeneity, as previously reported (Cancer Genome Atlas Network, 2015; Cancer Genome Atlas Research Network, 2011, 2012, 2014a, 2014b, 2015a, 2015b, 2017; Cancer Genome Atlas Research Network et al., 2013a, 2013b; Robertson et al., 2017).
While malignancies arising from the same anatomical site have traditionally been treated clinically as a single entity, histologic and molecular sub-classifications are now routinely used to determine treatments for subtypes of lung, breast, gastrointestinal, skin and bone marrow derived malignancies. As drugs become increasingly clinically available to target such cancer-driving pathway targets as ALK, EGFR, ERBB2, ERα, KIT, BRAF, and ABL1, the traditional system of anatomic cancer classification should be supplemented by a classification system based on molecular alterations shared by tumors across different tissue types (Hoadley et al., 2014; Saunders et al., 2012). This concept has led to the development of so-called basket or umbrella trials, such as the NCI-MATCH study, to investigate the feasibility and validity of this new clinical approach (Ramos et al., 2015). However, exceptions that challenge this concept have also become apparent from such notable examples as the unpredictable clinical responses to a potent BRAF inhibitor across diverse malignancies all expressing the same BRAF mutation (Saunders et al., 2012). Integrated molecular tumor profiling such as described here, and in our previous Pan-Cancer-12 analysis, may improve basket-trial design by considering both mutations and oncogenic signaling pathways along with consideration of each tumor’s tissue-specific or cell-of-origin context (Hoadley et al., 2014).
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACT FOR REAGENT AND RESOURCE SHARING
-
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Subjects
-
METHOD DETAILS
Sample Processing
Pathology Review
Somatic Copy-Number Alterations
DNA methylation
RNA Data Batch Correction
mRNA
miRNA
Protein
Integrative clustering with iCluster
Cancer Immune Subtypes
Leukocyte and Stromal Fraction Estimates
TumorMap
PARADIGM
Gene Programs/Canonical pathways
QUANTIFICATION AND STATISTICAL ANALYSES
DATA AND SOFTWARE AVAILABILITY
Key Resources Table
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Peter W. Laird (Peter.Laird@vai.org). Sequence data hosted at the GDC is under controlled access. Details for gaining access can be found at (https://gdc.cancer.gov/access-data/data-access-processes-and-tools).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Subjects
Tumor tissue, adjacent normal tissue, and normal whole blood samples were obtained from patients at contributing centers with informed consent according to their local Institutional Review Boards (IRBs, see below). Biospecimens were centrally processed and DNA, RNA, and protein were distributed to TCGA analysis centers.
TCGA Project Management has collected necessary human subjects documentation to ensure the project complies with 45-CFR-46 (the “Common Rule”). The program has obtained documentation from every contributing clinical site to verify that IRB approval has been obtained to participate in TCGA. Such documented approval may include one or more of the following:
An IRB-approved protocol with Informed Consent specific to TCGA or a substantially similar program. In the latter case, if the protocol was not TCGA-specific, the clinical site PI provided a further finding from the IRB that the already-approved protocol is sufficient to participate in TCGA.
A TCGA-specific IRB waiver has been granted.
A TCGA-specific letter that the IRB considers one of the exemptions in 45-CFR-46 applicable. The two most common exemptions cited were that the research falls under 46.102(f)(2) or 46.101(b)(4). Both exempt requirements for informed consent, because the received data and material do not contain directly identifiable private information.
A TCGA-specific letter that the IRB does not consider the use of these data and materials to be human subjects research. This was most common for collections in which the donors were deceased.
A total of 11,188 patients were analyzed in TCGA with at least one molecular-profiling platform. This study contained both males and females with inclusions of genders dependent on tumor types. There were 5,769 females, 5,282 males and 137 missing information about gender. TCGA’s goal was to characterize adult human tumors; therefore, the vast majority are over the age of 18. However, there are 20 samples that are under the age of 18 that had tissue submitted prior to clinical data. Age was missing for 188 patients. The range of ages was 10 – 90 (maxed 90 for protection of human subjects) with a median age of diagnosis of 60 years of age.
METHOD DETAILS
Sample Processing
RNA and DNA were extracted from tumor and adjacent normal tissue specimens using a modification of the DNA/RNA AllPrep kit (QIAGEN). The flow-through from the QIAGEN DNA column was processed using a mirVana miRNA Isolation Kit (Ambion). This latter step generated RNA preparations that included RNA < 200 nt suitable for miRNA analysis. DNA was extracted from blood using the QiaAmp Blood Midi Kit (QIAGEN). Each specimen was quantified by measuring Abs260 with a UV spectrophotometer or by PicoGreen assay. DNA specimens were resolved by 1% agarose gel electrophoresis to confirm high molecular weight fragments. A custom Sequenom SNP panel or the AmpFISTR Identifiler (Applied Biosystems) was utilized to verify that tumor DNA and germline DNA were derived from the same patient. Five hundred nanograms of each tumor and normal DNA were sent to QIAGEN for REPLI-g whole genome amplification using a 100 µg reaction scale. Only specimens yielding a minimum of 6.9 µg of tumor DNA, 5.15 µg RNA, and 4.9 µg of germline DNA were included in this study. RNA was analyzed via the RNA6000 Nano assay (Agilent) for determination of an RNA Integrity Number (RIN), and only the cases with RIN > 7.0 were included in this study.
Pathology Review
Samples were systematically evaluated by pathologists to confirm the histopathologic diagnosis and any variant histology, using the criteria of the most recent edition of the WHO / IARC Classification of Tumors relevant to each cancer type. All tumor samples were assessed for tumor content (percent tumor nuclei). Any non-concordant diagnoses among the pathologists were re-reviewed and resolution achieved after discussion.
Somatic Copy-Number Alterations
Somatic copy-number data were generated on Affymetrix SNP 6.0 arrays using standard protocols from the Genome Analysis Platform of the Broad Institute (McCarroll et al., 2008). Briefly, preliminary copy number at each probe locus was inferred by Birdseed analysis of raw .CEL files (Korn et al., 2008). Tangent normalization was then used to further refine genome-wide copy-number estimates (https://www.broadinstitute.org/cancer/cga/copynumber_pipeline). Segmented copy-number data were generated using Circular Binary Segmentation (Olshen et al., 2004). Regions corresponding to germline copy-number alterations were removed by applying filters generated from normal samples. Gene-level copy number was generated by GISTIC 2.0 analysis (Mermel et al., 2011). Purity and ploidy estimates were calculated using ABSOLUTE (Carter et al., 2012).
Chromosome arm-level copy-number calls were determined by clustering breakpoint locations and fraction of arm altered (further detailed in Taylor et al., 2018). Hierarchical clustering was performed using a metric of Manhattan distance and Ward2 methods for 10,522 samples; this analysis identified 10 groups (Figure 1A). Aneuploidy scores reflect the overall aneuploidy burden, and the range varied across tumor types. Most AN groups represented a mix of tumor types; however, tumor types with specific aneuploidy patterns defined unique groups like AN9 enriched with GBM, characterized by chr7 gain and chr10 loss, and AN10 enriched for TGCT, which all displayed chromosome ploidies greater than 2.
Cervical squamous tumors clustered in high aneuploidy clusters AN1 and AN5. These clusters were also enriched for other Pan-gyn tumors, including ovarian, high-copy number endometrial, and uterine carcinosarcoma (Cherniack et al., 2017). Gynecologic tumors with fewer copy-number alterations including Luminal breast cancers and other endometrial tumors grouped separately in low aneuploidy clusters AN7 and AN8, respectively.
DNA methylation
Illumina Infinium DNA methylation arrays were used to obtain DNA methylation profiles of 10,814 tumors from 33 tumor types and 1,064 histologically normal tumor-adjacent tissue specimens representing 24 different tissue types. Data from two generations of Infinium arrays, HumanMethylation27 (HM27) and HumanMethylation450 (HM450), were merged to generate a dataset for 22,601 probes shared between two platforms. To minimize systematic platform-specific effects, we normalized the HM27 data against the HM450 data using a probe-by-probe proportional rescaling method. During data generation, a single technical replicate of the same cell line control sample from either of two different DNA extractions (TCGA-07-0227/TCGA-AV-A03D) was included on each plate as a control, and measured 44/198 times and 12/169 times on HM27 and HM450, respectively. These repeated-measurements were therefore used for rescaling of the HM27 data to be comparable to HM450. For each probe within each platform, we computed the median β-value across all technical replicates of each of the two TCGA IDs. We then combined the two extractions by taking the mean of the two medians obtained for each of the two replicate TCGA IDs, and obtained a single summarized DNA methylation readout (β-value) for the corresponding probe i for each platform, noted as and respectively. We then applied a constrained (within the range of 0 to 1 for β-values) linear rescaling of the HM27 data for each probe and for each patient’s sample using and . When the HM27 β-value of a patient’s sample j for probe i was smaller than the mean of median replicate samples on the HM27 for that probe, we linearly rescaled the HM27 β-value Betahm27,i,j in the (0, Betahm27,i,j) space; and when Betahm27,i,j was greater, we linearly rescaled the HM27 beta value Betahm27,i,j in the (Betahm27,i,j, 1) space; This translates into the following mathematical computation: , if ; and , if . After the between-platform normalization, we further excluded 779 probes that still showed a consistent platform difference (mean β-value difference greater than or equal to 0.1) in six or more tumor types.
Unsupervised clustering was performed based on promoter CpG sites that did not exhibit tissue-specific DNA methylation, but that acquired hypermethylation in cancer. We used DNA methylation data from the histologically normal tissues and leukocytes to identify 11,275 sites that lacked tissue-specific DNA methylation (mean β-value < 0.2 in any tissue type and β-value > 0.3 in no more than five samples across the entire set). To minimize the influence of variable tumor purity levels on a clustering result, we dichotomized the data using a β-value of ≥ 0.3 to define positive DNA methylation and < 0.3 to specify lack of methylation. The dichotomization not only ameliorated the effect of tumor sample purity on the clustering, but also removed a great portion of residual batch/platform effects that are mostly reflected in small variations near the two ends of the range of β-values. For clustering analysis of tumors, we selected 3,139 CpG sites that were methylated at a β-value of ≥ 0.3 in more than 10% of tumors within any of the 33 cancer types. We performed unsupervised clustering of 10,814 tumors using hierarchical clustering with Ward’s method to cluster the distance matrix computed with the Jaccard index. The dendrogram was cut at different levels, and resulting clusters were evaluated for associations with tumor types and subtypes. The heatmap was generated using the original β-values for the top one-third (n = 1,035) of the most variability methylated CpGs across tumors (Figure 1B). We chose 25 clusters for the subsequent cross-platform analyses. We noted that a fraction of ESCA and STAD was found in METH9 with LUAD and PAAD, a result that may be related to the low tumor cellularity of the cancers in this cluster. Three types of renal cell carcinomas, including KIRC, (KIRP and KICH, aligned together in METH19, which interestingly also included THYM and THCA. Pan-GYN tumors separated into three major groups, which appeared to reflect molecular subtypes within each tumor type. Luminal and HER2 breast (BRCA-Luminal) and subtypes of UCEC lacking CIN organized into METH 4, 5 and 6. OV and UCEC with CIN-high grouped together in METH 22 and 23. Finally, Basal-like BRCA was found in METH 24 and 25.
RNA Data Batch Correction
The expression data for mRNA and miRNA were batch-corrected to adjust for platform differences between the GAII and HiSeq Illumina sequencers. For mRNA, additional adjustments were made for different sequencing centers (The University of North Carolina [UNC] and British Columbia Cancer Agency [BCCA]) and a plate effect observed in PRAD. For the mRNA data, first batch 312 and 320 PRAD were adjusted to remove batch effects. UNC GA samples (UCEC, COAD, READ) were adjusted to the UNC HiSeq data. Genes with mostly zero reads or with residual batch effects (~10% of genes) were removed from the adjusted samples and replaced with NAs. A similar adjustment was made for BCCA GAII-sequenced samples (LAML, STAD, ESCA) to HiSeq. Genes were adjusted using a novel algorithm called EB++; a variant of the Empirical Bayes / ComBat algorithm with training and testing features added.
The miRNA data were batch-corrected for GAII and HiSeq, as well as for two library construction protocols (MultiMACS and Direct). Weakly expressed miRNAs were filtered by requiring miRNA mature strands to be expressed with an RPM of at least 10 in 10% of primary tumors in each TCGA project resulting in 743 miRNAs across all 32 projects (miRNA sequencing was not performed on GBM). The EB++ method was used to correct the Direct protocol to the MultiMACs protocol and the GAII to the HiSeq protocol similar to what was done for mRNA.
mRNA
Upper quartile normalized RSEM data for batch-corrected mRNA gene expression were used for analysis. The matrix was filtered for genes expressed in 60% or more of the samples. Unsupervised consensus clustering using Consensus Cluster Plus (Wilkerson and Hayes, 2010) was performed on 10,165 tumors with 15,363 genes. At K = 43, we identified 25 major groups with at least 40 samples per group (Figure 1C). Many of the sample groups contained > 90% of a single tumor type or subtype. These included OV, PRAD, THCA, BRCA-Luminal, BRCA-Basal, LUAD, BLCA, CESC, UCEC, MESO, and TGCT. As observed in our previous publication (Hoadley et al., 2014), Basal-like breast cancer split out as a separate group from the estrogen receptor (ER)-positive and HER2-positive breast cancers.
miRNA
We analyzed batch-corrected, normalized abundance (i.e., reads per million, RPM) data for 743 expressed mature strands (of 1212 miRBase v16 strands). The data matrix contained abundance profiles for 10,170 tumor samples. We hierarchically clustered the data matrix with the pheatmap R package, using row-scaling, Pearson correlation coefficients for a distance metric, and ward.D2 clustering.
Unsupervised hierarchical clustering of batch-corrected miRNA mature-strand expression profiles from 10,170 tumors yielded a 15-group solution (Figure 1D). We observed six tumor-type-specific clusters. MIR5 contained OV, MIR8 BRCA, MIR12 LGG, MIR13 LIHC, MIR14 THCA, and MIR15 PRAD. Two clusters contained samples from two diseases. MIR7 contained two blood cancers: DLBC and LAML, while MIR10 contained two types of melanomas: SKCM and UVM. MIR11 contained only the three kidney tumors: KICH, KIRC and KIRP.
Each of the remaining 6 clusters contained at least four cancer types. MIR1 was largely UCEC, with substantial BRCA and BLCA, plus smaller numbers of 6 other cancers. MIR2 contained predominantly squamous carcinomas including HNSC, LUSC, CESC and BLCA, with smaller numbers of ESCA, LUAD, and minor BRCA and SARC. MIR3 contained largely PCPG, with SARC and ACC, and smaller numbers of 8 other cancer types. MIR6, the Pan-GI group, was largely COAD and STAD, but also had substantial PAAD, READ and ESCA, with smaller numbers of CHOL and LIHC. MIR4 was largely TGCT, with THYM and BLCA, with smaller numbers of LIHC and SKCM. MIR9 was largely LUAD and SARC, with smaller numbers of MESO and LUSC.
Protein
Protein expression data were available for 7,858 samples from 32 of the 33 tumor types (LAML data were never generated) across 216 proteins and phosphoproteins. The data were generated using the reverse phase protein array (RPPA) platform. We used batch effects-corrected RPPA data and median-centered them in both directions. We then clustered them using hierarchical clustering from the R function hclust() with 1 – Pearson’s correlation coefficient as the distance metric and Ward as the linkage function. The 10 clusters were obtained by cutting the dendrogram using the cutree() function in R.
Hierarchical clustering of protein expression data revealed 10 distinct Protein (P) groups (Figure 1E). The dendrogram first separated P1 and P2 from the remaining 8 clusters, which largely corresponded with the separation between mesenchymal-like tumor types with high EMT signatures versus tumor types with low EMT signatures, respectively. Cluster 1 consisted of the brain cancers (GBM, LGG), whereas cluster 2 contained DLBC, SARC, PCPG, UCS, THYM and metastatic SKCM. Those 2 clusters were characterized by low levels of E-cadherin, EPPK1, RAB25 and Claudin 7. The brain cancers had high levels of PKC-alpha, phosphoPKC-alpha, PKC-delta, ERK2, PEA15 and acetyl-A tubulin.
P3 and P4 consisted mainly of the Luminal breast and gynecologic cancers (BRCA-Lum8, UCEC7, OV), plus some liver samples (LIHC). The clusters had high levels of ER-alpha, AR and IGFBP2. Interestingly, the LIHC samples in P4 had high levels of ER-alpha as well, whereas those LIHC samples not in P4 had low ER-alpha levels. P6 was a Pan-kidney cluster with KIRC, KIRP and ACC and was characterized by high levels of EMT based on low expression of the negative EMT markers E-cadherin, RAB25 and Claudin 7, as well as low IGFBP2, FASN and Cyclin B1, and high GAPDH, CD26, and phosphoNDRG1. P8 was a Pan-GI cluster consisting of most of the colorectal (COAD/READ) and gastric cancer (STAD) samples. In contrast to the Pan-kidney group, the Pan-GI group had a very low EMT signature with high expression of RAB25, EPPK1 and Claudin 7. Other distinguishing features of the cluster included high levels of cleaved CASPASE 7, TFRC, MYH11, TIGAR, and beta catenin. P9 and P10 were the most diverse and included some samples from most of the tumor types. P10, in particular, had an enrichment of the squamous cancers with large proportions of HNSC, LUSC, CESC, CHOL, and BLCA. This cluster had high levels of PAI1, cleaved CASPASE 7, ANNEXIN1, TFRC, P16INK4A, ASNS, Cyclin B1, Cyclin E1, FASN and FOXM1.
Integrative clustering with iCluster
The iCluster clustering algorithm formulates the problem of subgroup discovery as a joint multivariate regression of multiple data types with reference to a set of common latent variables, which represent the underlying 28 tumor subtypes (Mo et al., 2013; Shen et al., 2009, 2012). Four molecular platforms - SCNA, DNA methylation, mRNA expression, and miRNA expression were used as input. Data were pre-processed using the following procedures: For mRNA, and mature-strand miRNA sequence data, poorly expressed genes were excluded based on median-normalized counts, and variance filtering led to a list of reduced features for clustering. mRNA and miRNA expression features were log2 transformed, normalized and scaled before using them as an input to iCluster. Pre-processing led to 3,217 mRNA and 382 miRNA features. Pre-processed DNA methylation data were obtained from the methylation merged HM27 and HM450 platform datasets and included 3,139 hypermethylation features. Circular Binary Segmented (CBS) SCNA data were further reduced to a set of 3,105 non-redundant regions as described (Mo et al., 2013).
Cancer Immune Subtypes
To characterize the commonality and diversity of intratumoral immune states, we scored 160 published immune expression signatures on all available TCGA PanCancerAtlas tumor samples, and performed cluster analysis to identify similarity modules of multiple immune signature sets. The 160 immune expression signatures were selected based on extensive literature search, utilizing diverse resources considered to be reliable and comprehensive, based on expert opinions of immuno-oncologists (Thorsson et al., 2018). Eighty-three signatures were derived in the context of immune response studies in cancer, and the remaining 77 are of general validity for immunity. TCGA RNA-seq values from the PanCancer Atlas normalized gene expression matrix were scored for each of the 160 identified gene expression signatures using single-sample gene set enrichment (ssGSEA) analysis, using the R package GSVA. Clusters of similar signature scores were identified by weighted gene correlation network analysis (WGCNA) (Langfelder and Horvath, 2008). Based on the WGCNA analysis, five immuno-oncology-related immune expression signatures: activation of macrophages/monocytes (Beck et al., 2009), overall lymphocyte infiltration (dominated by T and B cells) (Calabrò et al., 2009), TGF-β response (Teschendorff et al., 2010), IFN-γ response (Wolf et al., 2014), and wound healing (Chang et al., 2004), robustly reproduced co-clustering of the immune signature sets, and were selected to perform cluster analysis of all cancer types, with the exception of hematologic neoplasias (acute myeloid leukemia, LAML; diffuse large B cell lymphoma, DLBC; and thymoma, THYM). Clustering of tumor samples scored on these five signatures was performed using model-based clustering, using the mclust R package (Scrucca et al., 2016), with the number of clusters, K, determined by maximization of Bayesian Information Criterion (BIC). Maximal BIC was found with a six-cluster solution, and the six resulting clusters C1-C6 (with 2416, 2591, 2397, 1157, 385 and 180 cases, respectively) were characterized by a distinct distribution of scores over the five representative signatures, and effectively categorized each TCGA sample as belonging to one of six cancer “immune subtypes,” namely Wound Healing (C1), IFN-γ Dominant (C2), Inflammatory (C3), Lymphocyte Depleted (C4), Immunologically Quiet (C5), or TGF-β Dominant (C6). Additional details are found in Thorsson et al. (2018). The designations C1-C6 of immune subtypes were made independently from iCluster designations in the current work.
Leukocyte and Stromal Fraction Estimates
Overall leukocyte content in 10,814 TCGA tumor aliquots was assessed by identifying DNA methylation probes with the greatest differences between pure leukocyte cells and normal tissue, then estimating leukocyte content using a mixture model. From Illumina Infinium DNA methylation platform arrays HumanMethylation450, 2000 loci were identified (200 for HumanMethylation27) that were the most differentially methylated between leukocyte and normal tissues, 1000 in each direction. For each locus i, assuming two populations (j), for each sample we have the following equation:
Using the tumor with the least evidence of leukocyte methylation as a surrogate for the beta value (β) for each locus in the pure tumor, 2000 estimates were made, solving for π. We took the mode of 200 estimates to avoid loci that violate the assumptions. Using the estimated π and the measured β for tumor and leukocyte, with the same linear model, we solved for β (deconvoluted value) extracting the leukocyte fraction (LF).
Stromal fraction (SF) was defined as the total non-tumor cellular component, obtained by subtracting tumor purity from unity. Tumor purity was generated using ABSOLUTE (Carter et al., 2012) as detailed in Taylor et al., 2018.
TumorMap
We used the latent iCluster space (Table S7) to calculate Euclidean similarity between every pair of samples, where Euclidean similarity = (1 / (1 + Euclidean_distance)) (https://tumormap.ucsc.edu/). The distances were used as input to generate a 2D layout of the samples using the physics-based Distributed Recursive (Graph) Layout method (Alencar and Polley, 2011), previously known as VxOrd (Davidson et al., 2001). DrL layout engine was used with each sample’s 28 most similar neighbors. DrL’s default settings were used for “edge cutting” and “intermediate output interval” parameters, 0.8 and 0, respectively. Sample lists for attributes (GI, gyn, kidney, stemness, squamous) were obtained from other working groups.
PARADIGM
The PARADIGM algorithm with the interaction-learning update (Chu et al., 2014; Vaske et al., 2010) was used to infer protein activities in the context of gene regulatory pathways, based on gene expression and copy-number data. The method uses a set of interactions from several sources (NCI-PID, Reactome, and KEGG) and superimposes them into a single network (SuperPathway). The SuperPathway contained 7,369 proteins, 9,354 multi-protein complexes, 2,092 families, and 592 cellular processes connected by 45,315 interactions. The PARADIGM algorithm was applied to 9,829 tumors with platform-corrected expression data and gene-level copy-number alteration data from 33 cancer types to infer the integrated pathway levels (IPLs) of the 19,504 SuperPathway features.
Pathway features characterizing each iCluster were identified by comparing each iCluster versus all others using the t test and Wilcoxon Rank sum test with Benjamini-Hochberg (BH) false discovery rate (FDR) correction. An initial minimum variation filter (at least 1 sample with absolute activity > 0.05) was applied; and the 15,502 features passing the minimum variation feature were considered in this analysis. Features deemed significant (FDR corrected p < 0.05) by both tests and showing an absolute difference in group means > 0.05 were selected. The selected pathway features were assessed for interconnectivity; regulatory nodes with differential inferred IPLs that also had at least 15 differential downstream regulatory targets were identified.
Gene Programs/Canonical pathways
Twenty-two Gene Programs and 20 additional pathways were used to characterize the molecular, signaling, and pathway level characteristics of the iCluster-based subtypes. The Gene Programs were identified in a previous PanCancer analysis of 12 tumor types, by 1) assembling 6,898 gene signatures documented to contain gene sets that are coexpressed, coamplified, or function together; 2) applying a bimodality filter to select only those signatures with bimodal (ON/OFF) expression; and 3) performing weighted gene correlation network-based clustering (WGCNA) to identify a non-redundant set of expression modules/programs (see Hoadley et al. [2014] and associated SI, Section 5, for details). These Gene Programs were evaluated in the PanCancer-33 dataset by averaging the top most-correlated signatures from each module (Table S9). The 20 additional pathways represent known drug targets or/and canonical cancer pathways (Table S4 of Hoadley et al. [2014]) and were evaluated as the mean expression level of pathway genes.
QUANTIFICATION AND STATISTICAL ANALYSES
Quantitative and statistical methods are noted above according to their respective technologies and analytic approaches.
DATA AND SOFTWARE AVAILABILITY
The raw data, processed data and clinical data can be found at the legacy archive of the GDC (https://portal.gdc.cancer.gov/ legacy-archive/search/f) and the PancanAtlas publication page (https://gdc.cancer.gov/about-data/publications/pancanatlas). The mutation data can be found here: (https://gdc.cancer.gov/about-data/publications/mc3-2017). TCGA data can also be explored through the Broad Institute FireBrowse portal (http://gdac.broadinstitute.org) and the Memorial Sloan Kettering Cancer Center cBioPortal (http://www.cbioportal.org). Details for software availability are in the Key Resource Table.
Supplementary Material
Highlights.
An integrative data clustering method is applied to reclassify human tumors
Cell-of-origin influences, but does not fully determine, tumor classification
Immune features and copy-number aberrations define the most mixed tumor groups
Multi-cancer groups reveal new features with potential clinical utility
Acknowledgments
We are grateful to the patients and families who contributed to this study. We also thank the NCI TCGA Program Office and NHGRI coupterpart for organizational and logistical support. This work was supported by NIH grants (U54 HG003273, U54 HG003067, U54 HG003079, U24 CA143799, U24 CA143835, U24 CA143840, U24 CA143843, U24 CA143845, U24 CA143848, U24 CA143858, U24 CA143866, U24 CA143867, U24 CA143882, U24 CA143883, U24 CA144025, and P30 CA016672).
DECLARATION OF INTERESTS
Michael Seiler, Peter G. Smith, Ping Zhu, Silvia Buonamici, and Lihua Yu are employees of H3 Biomedicine, Inc. Parts of this work are the subject of a patent application: WO2017040526 titled “Splice variants associated with neomorphic sf3b1 mutants.” Shouyoung Peng, Anant A. Agrawal, James Palacino, and Teng Teng are employees of H3 Biomedicine, Inc. Andrew D. Cherniack, Ashton C. Berger, and Galen F. Gao receive research support from Bayer Pharmaceuticals. Gordon B. Mills serves on the External Scientific Review Board of Astrazeneca. Anil Sood is on the Scientific Advisory Board for Kiyatec and is a shareholder in BioPath. Jonathan S. Serody receives funding from Merck, Inc. Kyle R. Covington is an employee of Castle Biosciences, Inc. Preethi H. Gunaratne is founder, CSO, and shareholder of NextmiRNA Therapeutics. Christina Yauisa part-time employee/consultant at NantOmics. Franz X. Schaub isan employee and shareholder of SEngine Precision Medicine, Inc. Carla Grandori is an employee, founder, and shareholder of SEngine Precision Medicine, Inc. Robert N. Eisenman is a member of the Scientific Advisory Boards and shareholder of Shenogen Pharma and Kronos Bio. Daniel J. Weisenberger is a consultant for Zymo Research Corporation. Joshua M. Stuart is the founder of Five3 Genomics and shareholder of NantOmics. Marc T. Goodman receives research support from Merck, Inc. Andrew J. Gentles is a consultant for Cibermed. Charles M. Perou is an equity stock holder, consultant, and Board of Directors member of BioClassifier and GeneCentric Diagnostics and is also listed as an inventor on patent applications on the Breast PAM50 and Lung Cancer Subtyping assays. Matthew Meyerson receives research support from Bayer Pharmaceuticals; is an equity holder in, consultant for, and Scientific Advisory Board chair for OrigiMed; and is an inventor of a patent for EGFR mutation diagnosis in lung cancer, licensed to LabCorp. Eduard Porta-Pardo is an inventor of a patent for domainXplorer. Han Liang is a shareholder and scientific advisor of Precision Scientific and Eagle Nebula. Da Yang is an inventor on a pending patent application describing the use of antisense oligonucleotides against specific lncRNA sequence as diagnostic and therapeutic tools. Yonghong Xiao was an employee and shareholder of TESARO, Inc. Bin Feng is an employee and shareholder of TESARO, Inc. Carter Van Waes received research funding for the study of IAP inhibitor ASTX660 through a Cooperative Agreement between NIDCD, NIH, and Astex Pharmaceuticals. Raunaq Malhotra is an employee and shareholder of Seven Bridges, Inc. Peter W. Laird serves on the Scientific Advisory Board for AnchorDx. Joel Tepper is a consultant at EMD Serono. Kenneth Wang serves on the Advisory Board for Boston Scientific, Microtech, and Olympus. Andrea Califano is a founder, shareholder, and advisory board member of DarwinHealth, Inc. and a shareholder and advisory board member of Tempus, Inc. Toni K. Choueiri serves as needed on advisory boards for Bristol-Myers Squibb, Merck, and Roche. Lawrence Kwong receives research support from Array BioPharma. Sharon E. Plon is a member of the Scientific Advisory Board for Baylor Genetics Laboratory. Beth Y. Karlan serves on the Advisory Board of Invitae.
Footnotes
Supplemental Information includes nine tables and can be found with this article online at https://doi.org/10.1016/j.cell.2018.03.022.
AUTHOR CONTRIBUTIONS
Conceptualization: K.A.H., J.M.S., C.C.B., and P.W.L. Data Curation: K.A.H., A.D.C., V.T., R.A., R.B., and T.H. Formal Analysis: K.A.H., C.Y., T.H., D.M.W., E.D., R.S., A.M.T., A.D.C., V.T., R.A., R.B., C.K.W., F.S.-V., A.G.R., M.S.L., and T.M.M. Composition of Figures and Graphical Abstract: T.H., A.G.R., D.M.W., C.Y., and P.W.L. Writing – Original Draft: K.A.H., C.Y., T.H., D.M.W., A.J.L., A.M.T., V.T., R.A., M.W., A.G.R., B.G.S., C.C.B., and P.W.L. Writing – Review & Editing: K.A.H., C.Y., T.H., D.M.W., A.J.L., E.D., R.S., A.M.T., A.D.C., V.T., R.A., R.B., C.K.W., M.W., F.S.-V., A.G.R., B.G.S., M.S.L., H.N., T.M.M., J.M.S., C.C.B., and P.W.L. Supervision: K.A.H., and P.W.L.
References
- Alencar A, Polley T. DrL (VxOrd) 2011 http://wiki.cns.iu.edu/pages/viewpage.action?pageId=1704113.
- Banerjee S, Biehl A, Gadina M, Hasni S, Schwartz DM. JAK-STAT signaling as a target for inflammatory and autoimmune diseases: current and future prospects. Drugs. 2017;77:521–546. doi: 10.1007/s40265-017-0701-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck AH, Espinosa I, Edris B, Li R, Montgomery K, Zhu S, Varma S, Marinelli RJ, van deRijn M, West RB. The macrophage colony-stimulating factor 1 response signature in breast carcinoma. Clin. Cancer Res. 2009;15:778–787. doi: 10.1158/1078-0432.CCR-08-1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger AC, Korkut A, Kanchi RS, Hegde AM, Lenoir W, Liu W, Liu Y, Fan H, Shen H, Ravikumar V, et al. A comprehensive Pan-Cancer molecular study of gynecologic and breast cancers. Cancer Cell. 2018;33 doi: 10.1016/j.ccell.2018.03.014. https://doi.org/10.1016/j.ccell.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrò A, Beissbarth T, Kuner R, Stojanov M, Benner A, Asslaber M, Ploner F, Zatloukal K, Samonigg H, Poustka A, Sültmann H. Effects of infiltrating lymphocytes and estrogen receptor on gene expression and prognosis in breast cancer. Breast Cancer Res. Treat. 2009;116:69–77. doi: 10.1007/s10549-008-0105-3. [DOI] [PubMed] [Google Scholar]
- Campbell JD, Yau C, Bowlby R, Liu Y, Brennan K, Fan H, Taylor AM, Wang C, Walter V, Akbani E, et al. Genomic, pathway network, and immunologic features distinguishing squamous carcinomas. Cell Rep. 2018;23 doi: 10.1016/j.celrep.2018.03.063. https://doi.org/10.1016/j.celrep.2018.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell. 2015;161:1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, Benz CC, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013a;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Hoadley K, Triche TJ, Jr, Laird PW, Baty JD, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013b;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014a;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell. 2014b;159:676–690. doi: 10.1016/j.cell.2014.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell. 2015a;163:1011–1025. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA, Rheinbay E, Miller CR, Vitucci M, Morozova O, et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N. Engl. J. Med. 2015b;372:2481–2498. doi: 10.1056/NEJMoa1402121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017;169:1327–1341. e23. doi: 10.1016/j.cell.2017.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HY, Sneddon JB, Alizadeh AA, Sood R, West RB, Montgomery K, Chi JT, van de Rijn M, Botstein D, Brown PO. Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Biol. 2004;2:E7. doi: 10.1371/journal.pbio.0020007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherniack AD, Shen H, Walter V, Stewart C, Murray BA, Bowlby R, Hu X, Ling S, Soslow RA, Broaddus RR, et al. Cancer Genome Atlas Research Network. Integrated molecular characterization of uterine carcinosarcoma. Cancer Cell. 2017;31:411–423. doi: 10.1016/j.ccell.2017.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J, Sadeghi S, Raymond A, Jackman SD, Nip KM, Mar R, Mohamadi H, Butterfield YS, Robertson AG, Birol I. BioBloom tools: fast, accurate and memory-efficient host species sequence screening using bloom filters. Bioinformatics. 2014;30:3402–3404. doi: 10.1093/bioinformatics/btu558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington K, Shinbrot E, Wheeler DA. Mutation signatures reveal biological processes in human cancer. bioRxiv. 2016 https://doi.org/10.1101/036541.
- Davidson GS, Wylie BN, Boyack KW. IEEE Information Visualization 2001, INFOVIS 2001. IEEE; 2001. Cluster stability and the use of noise in interpretation of clustering. [Google Scholar]
- Davis CF, Ricketts CJ, Wang M, Yang L, Cherniack AD, Shen H, Buhay C, Kang H, Kim SC, Fahey CC, et al. The Cancer Genome Atlas Research Network. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell. 2014;26:319–330. doi: 10.1016/j.ccr.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hainsworth JD, Rubin MS, Spigel DR, Boccia RV, Raby S, Quinn R, Greco FA. Molecular gene expression profiling to predict the tissue of origin and direct site-specific therapy in patients with carcinoma of unknown primary site: a prospective trial of the Sarah Cannon research institute. J. Clin. Oncol. 2013;31:217–223. doi: 10.1200/JCO.2012.43.3755. [DOI] [PubMed] [Google Scholar]
- Hoadley KA, Yau C, Wolf DM, Cherniack AD, Tamborero D, Ng S, Leiserson MDM, Niu B, McLellan MD, Uzunangelov V, et al. Cancer Genome Atlas Research Network. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. 2014;158:929–944. doi: 10.1016/j.cell.2014.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Jin X, Kim H. Cancer stem cells and differentiation therapy. Tumour Biol. 2017;39 doi: 10.1177/1010428317729933. 1010428317729933. [DOI] [PubMed] [Google Scholar]
- Knijnenburg T, Wang L, Zimmermann M, Chambwe N, Gao G, Cherniack A, Fan H, Shen H, Way G, Greene C, et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency Across The Cancer Genome Atlas. Cell Rep. 2018;23 doi: 10.1016/j.celrep.2018.03.076. https://doi.org/10.1016/j.celrep.2018.03.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn JM, Kuruvilla FG, McCarroll SA, Wysoker A, Nemesh J, Cawley S, Hubbell E, Veitch J, Collins PJ, Darvishi K, et al. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat. Genet. 2008;40:1253–1260. doi: 10.1038/ng.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Sethi NS, Hinoue T, Schneider BG, Cherniack AD, Sanchez-Vega F, Seoane JA, Farshidfar F, Bowlby R, Islam M, et al. Comparative molecular analysis of gastrointestinal adenocarcinomas. Cancer Cell. 2018;33 doi: 10.1016/j.ccell.2018.03.010. https://doi.org/10.1016/j.ccell.2018.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malta TM, Sokolov A, Gentles AJ, Burzykowski T, Poisson L, Weinstein JN, Kaminska B, Huelsken J, Omberg L, Gevaert O, et al. Comprehensive analysis of cancer stemness. Cell. 2018;173 [Google Scholar]
- McCarroll SA, Kuruvilla FG, Korn JM, Cawley S, Nemesh J, Wysoker A, Shapero MH, de Bakker PI, Maller JB, Kirby A, et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat. Genet. 2008;40:1166–1174. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo Q, Wang S, Seshan VE, Olshen AB, Schultz N, Sander C, Powers RS, Ladanyi M, Shen R. Pattern discovery and cancer gene identification in integrated cancer genomic data. Proc. Natl. Acad. Sci. USA. 2013;110:4245–4250. doi: 10.1073/pnas.1208949110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran S, Martínez-Cardús A, Sayols S, Musulén E, Balañá C, Estival-Gonzalez A, Moutinho C, Heyn H, Diaz-Lagares A, de Moura MC, et al. Epigenetic profiling to classify cancer of unknown primary: a multicentre, retrospective analysis. Lancet Oncol. 2016;17:1386–1395. doi: 10.1016/S1470-2045(16)30297-2. [DOI] [PubMed] [Google Scholar]
- Newton Y, Novak AM, Swatloski T, McColl DC, Chopra S, Graim K, Weinstein AS, Baertsch R, Salama SR, Ellrott K, et al. TumorMap: exploring the molecular similarities of cancer samples in an interactive portal. Cancer Res. 2017;77:e111–e114. doi: 10.1158/0008-5472.CAN-17-0580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- Ramos AH, Lichtenstein L, Gupta M, Lawrence MS, Pugh TJ, Saksena G, Meyerson M, Getz G. Oncotator: cancer variant annotation tool. Hum. Mutat. 2015;36:E2423–E2429. doi: 10.1002/humu.22771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricketts CJ, De Cubas AA, Fan H, Smith CC, Lang M, Reznik E, Bowlby R, Gibb EA, Akbani R, Beroukhim R, et al. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell Rep. 2018;23 doi: 10.1016/j.celrep.2018.06.032. https://doi.org/10.1016/j.celrep.2018.03.075. [DOI] [PubMed] [Google Scholar]
- Robertson AG, Shih J, Yau C, Gibb EA, Oba J, Mungall KL, Hess JM, Uzunangelov V, Walter V, Danilova L, et al. Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell. 2017;32:204–220. e15. doi: 10.1016/j.ccell.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- Scrucca L, Fop M, Murphy TB, Raftery AE. mclust 5: clustering, classification and density estimation using gaussian finite mixture models. R J. 2016;8:289–317. [PMC free article] [PubMed] [Google Scholar]
- Shen R, Olshen AB, Ladanyi M. Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics. 2009;25:2906–2912. doi: 10.1093/bioinformatics/btp543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen R, Mo Q, Schultz N, Seshan VE, Olshen AB, Huse J, Ladanyi M, Sander C. Integrative subtype discovery in glioblastoma using iCluster. PLoS ONE. 2012;7:e35236. doi: 10.1371/journal.pone.0035236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Shih J, Ha G, Gao GF, Zhang X, Berger AC, Schumacher SE, Wang C, Hu H, Liu J, et al. Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell. 2018;33 doi: 10.1016/j.ccell.2018.03.007. https://doi.org/10.1016/j.ccell.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teschendorff AE, Gomez S, Arenas A, El-Ashry D, Schmidt M, Gehrmann M, Caldas C. Improved prognostic classification of breast cancer defined by antagonistic activation patterns of immune response pathway modules. BMC Cancer. 2010;10:604. doi: 10.1186/1471-2407-10-604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Yang T-HO, Porta-Pardo E, Gao G, Plaisier CL, Eddy JA, et al. The immune landscape of cancer. Immunity. 2018;48 https://doi.org/10.1016/j.immuni.2018.03.023. [Google Scholar]
- Vaske CJ, Benz SC, Sanborn JZ, Earl D, Szeto C, Zhu J, Haussler D, Stuart JM. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics. 2010;26:i237–i245. doi: 10.1093/bioinformatics/btq182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26:1572–1573. doi: 10.1093/bioinformatics/btq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf DM, Lenburg ME, Yau C, Boudreau A, van ’t Veer LJ. Gene co-expression modules as clinically relevant hallmarks of breast cancer diversity. PLoS ONE. 2014;9:e88309. doi: 10.1371/journal.pone.0088309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Cherniack AD, Dewal N, Moffitt RA, Danilova L, Murray BA, Lerario AM, Else T, Knijnenburg TA, Ciriello G, et al. Cancer Genome Atlas Research Network. Comprehensive pan-genomic characterization of adrenocortical carcinoma. Cancer Cell. 2016;29:723–736. doi: 10.1016/j.ccell.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.