

Abstract
The medial prefrontal cortex (mPFC) has been implicated in the extinction of emotional memories, including conditioned fear. Here we show that ventral hippocampal (vHPC) projections to the infralimbic (IL) cortex recruit parvalbumin (PV)-expressing interneurons to counter the expression of extinguished fear and promote fear relapse. Whole-cell recordings ex vivo revealed that optogenetic activation of vHPC input to amygdala projecting pyramidal neurons in the IL is dominated by feed-forward inhibition. Selectively silencing PV- but not somatostatin (SOM)-expressing interneurons in the IL eliminated vHPC-mediated inhibition. In behaving rats, pharmacogenetic activation of vHPC→IL projections impairs extinction recall, whereas silencing IL projectors diminishes fear renewal. Intra-IL infusion of GABA receptor agonists or antagonists, respectively, reproduced these effects. Together, these experiments reveal a novel circuit mechanism for the contextual control of fear, and indicate that vHPC-mediated inhibition of IL is an essential neural substrate for fear relapse.
Extinction learning is essential to cognitive-behavioral therapies in patients with trauma and stressor-related disorders, including posttraumatic stress disorder1,2. Despite the success of these approaches, extinction training does not erase traumatic memories but only dampens the expression of those memories. Consequently, work in both rats and humans has revealed that extinguished fear ‘relapses’ under a number of circumstances3,4. For example, extinction training only transiently suppresses learned fear responses, such as freezing behavior, in rats conditioned to fear an auditory conditioned stimulus (CS)5. Importantly, fear to an extinguished CS ‘renews’ if it is encountered outside the extinction context6, a phenomenon that reveals that extinction memories are context-dependent. In humans undergoing exposure therapy, the context-dependence of extinction leads to fear relapse outside of the clinic3,4. For this reason, understanding the neural circuits underlying the relapse of extinguished fear is critical for developing effective neurobehavioral interventions for trauma and stressor-related disorders.
Considerable evidence indicates that the encoding and consolidation of extinction memories requires the medial prefrontal cortex (mPFC)7–9. After extinction is learned, projections from the infralimbic (IL) cortex to the amygdala inhibit the production of conditioned fear responses, including freezing behavior10,11. The recruitment of this prefrontal-amygdala circuit is context-dependent: Fos expression in the IL is maximal in the extinction context, but reduced in other contexts (where fear relapses)12,13. This contextual control of prefrontal-amygdala inhibition appears to depend, at least in part, on the hippocampus14. Specifically, lesions or pharmacological inactivation of the hippocampus lead to a context-independent expression of extinction and prevent fear renewal15–17. This work indicates that the hippocampus drives fear renewal when an extinguished CS is encountered outside the extinction context. Indeed, the renewal of fear recruits ventral hippocampal neurons (vHPC) neurons projecting to the amygdala13,18 and engages ‘fear’ neurons in the amygdala that receive hippocampal input19,20. Moreover, optogenetic silencing of vHPC neurons projecting to the central nucleus of the amygdala, in particular, but not the basolateral amygdala prevents renewal21.
Although renewal engages neural circuits involved in fear expression, prevailing models of relapse phenomena, including renewal, posit that a loss of inhibitory control is necessary for the return of conditional responding to an extinguished CS5,14. Interestingly, we have recently observed that renewal increases Fos expression in vHPC neurons projecting to the mPFC22, suggesting that vHPC→mPFC projections might mediate the loss of inhibitory control hypothesized to yield fear relapse. Here we characterize the physiology of these projections and explore their contribution to the contextual control of extinguished fear memories.
Results
Characterization of vHPC→mPFC projections ex vivo
Hippocampal projections to the medial prefrontal cortex (mPFC) have previously been identified23, however the precise projection patterns, local circuits that are driven by these inputs, and functional role of these projections are little understood. To determine the nature of hippocampal projections to the mPFC, we expressed channelrhodopsin (ChR2) using viral mediated transduction of neurons in the area CA1 of the ventral hippocampus (vHPC; Fig. 1), the primary projection site from the hippocampus to the mPFC24. Ex-vivo whole-cell recordings from neurons in the mPFC combined with optical terminal stimulation of vHPC input revealed targeted, location-specific innervation. We found that vHPC input innervates both the infralimbic (IL) and prelimbic (PL) prefrontal cortex but with rostral and caudal PL neurons receiving significantly smaller input as compared to the IL (Supplementary Fig. 1). Within the IL, whose activity is crucial for extinction7,8, both pyramidal neurons and interneurons (Fig. 1b) received excitatory input from the vHPC (Fig. 1c,d) with no failures, low response jitter and latency, consistent with direct monosynaptic connections (Supplementary Fig. 2). Input to interneurons was large (mean excitatory postsynaptic current, EPSC, amplitude: 237 ± 66 pA; n = 9/10), and able to drive these cells to threshold (Fig. 1d, e; top). In contrast, input to pyramidal neurons was subthreshold (Fig. 1d, e; bottom), but significantly larger to layer 2/3 (L2/3) neurons with a mean EPSC amplitude of 173 ± 31 pA (n = 27/29) as compared to L5/6 neurons that had a mean EPSC amplitude of 81 ± 20 pA (n = 14/14; one-tailed Mann-Whitney test: P = 0.046, Fig. 1g).
Figure 1. Ventral hippocampal projection to the mPFC is dominated by strong local feed-forward inhibition mediated by fast-spiking interneurons in the IL.

a and e, Coronal images of terminal labeling in the IL following injection of ChR2-expressing virus in the CA1 region of the vHPC (scale bars for a and e: 1 mm); infusion site and terminal expression were confirmed by GFP expression (DAPI, blue). b, Biocytin-recovered IL neurons after recording (scale bar: 20 μm). c, Intrinsic firing properties of the corresponding interneuron (top) and pyramidal neuron (bottom). d, Voltage clamp- (left; holding potential of −70 mV in red, and −40 mV in gray) and current clamp recordings (right) of the recovered interneuron and principal neuron following optical stimulation of vHPC terminals. Note that terminal release in the interneuron caused the neuron to spike (top trace), while the pyramidal neuron displayed a small EPSP followed by inhibition. f, Synaptic response in a L2/3 principal neuron in the IL that shows a monosynaptic EPSC, followed by a feed-forward inhibitory current (blue trace). Both the EPSC and IPSC are eliminated in the presence of AMPA- and NMDA-receptor antagonists NBQX and APV (green trace). g, EPSC amplitudes (top; L2/3: n = 27; L5/6 = 14; one-tailed Mann-Whitney test: P = 0.046) and peak conductance ratios of IPSC/EPSC components (bottom; measured at −40 mV; L2/3: n = 20; L5/6 = 7; one-tailed Mann-Whitney test: P = 0.11) in L2/3 and L5/6 pyramidal neurons. h, Comparison of EPSC amplitudes in fast-spiking (top, left) and non-fast-spiking interneurons (top, right) revealed significantly larger vHPC input onto fast spiking neurons (bottom; EPSCs for FS neurons: 405 ± 178 pA, n = 5; non-FS neurons: 38 ± 13 pA; n = 8, from 5 animals; two-sided Mann-Whitney test; P = 0.0016). Current injections are shown below the firing traces. Amygdala-projecting neurons in the IL receive input from the vHPC. i, Schematic illustration of experimental setup. Retrograde markers (red RetroBeads, Lumafluor) were injected into the amygdala and ChR2-expressing virus injected into the vHPC (as described in main text). j, Typical discharge properties of amygdala-projecting pyramidal neurons (amygdala injection with retrograde marker (white bar inset: 1 mm) and recorded amygdala-projecting IL neuron (white bar inset: 20 μm) shown on the left) to positive and negative current injections (shown below traces). k, Typical voltage-clamp (left) and current clamp (right) responses to optical stimulation of vHPC input to amygdala-projecting IL pyramidal neuron. l, Comparisons of the IPSC/EPSC peak conductance ratio (left; control neurons: n = 20; projection neurons: n = 9; two-tailed t-test: t27 = 0.42; P = 0.68) and EPSC amplitudes (right) between control neurons (white) and amygdala-projecting neurons (red; control neurons: n = 16; projection neurons: n = 9; two-tailed t-test: t23 = 0.52; P = 0.61). Blue bars illustrate 470 nm light stimulation. Dots represent data from individual neurons. Error bars show mean ± s.e.m. *P<0.05; **P<0.01.
When pyramidal neurons in the IL were depolarized (Fig. 1d, bottom; grey trace), stimulation of vHPC input revealed a delayed disynaptic inhibitory postsynaptic current (IPSC; Fig. 1d, bottom; red trace) that shunted the excitatory postsynaptic potential (EPSP), and evoked a long-lasting inhibitory synaptic potential (Fig. 1d, bottom). This large disynaptic inhibition in pyramidal neurons is consistent with the strong vHPC-driven recruitment of local interneurons and feed-forward inhibition (Fig 2). All vHPC-evoked synaptic inputs were blocked by the application of AMPA/kainite- and NMDA-receptor antagonists NBQX and APV (n = 3; Fig. 1f), confirming the glutamatergic nature of this hippocampal projection. The vHPC-driven disynaptic inhibition of principal neurons was large with a peak IPSC to EPSC conductance ratio of 1.86 ± 0.67 (n = 20) in L2/3 principal neurons, and 2.44 ± 0.68 (n = 7) in L5/6 principal neurons (Fig. 1g, bottom), showing that vHPC-driven inhibition is nearly twice as large as incoming excitatory input. Interneurons in the mPFC form a diverse population of cells that can be separated by their discharge properties and expression of cellular markers25. Quantification of vHPC input to randomly selected interneurons revealed a distinct innervation pattern with fast-spiking (FS) interneurons receiving significantly larger input compared to non-FS interneurons (Fig. 1h; EPSCs for FS neurons: 405 ± 178 pA, n = 5; non-FS neurons: 38 ± 13 pA; n = 8, from 5 animals; two-sided Mann-Whitney test; P = 0.0016).
Figure 2. Selective optical activation of interneuronal subtypes in the IL evokes slow and fast inhibitory conductances onto pyramidal neurons.

a, Schematic showing experimental setup of optical stimulation of ChR2-infected PV+ interneurons in PV::Cre animals while recording synaptic responses of pyramidal neurons in the IL. b, Voltage clamp trace of an infected PV+ interneuron to a prolonged 470 nm light pulse (blue bar; 100 ms) shows a maintained inward current with initial spiking. c, Current injection (shown below the trace) reveals a typical regular fast-spiking intrinsic discharge pattern. d, Coronal section containing the IL shows PV cell labeling (DIO-ChR2-mCherry, red; DAPI, blue) and the recording site (circle). Scale bar: 1 mm. e, PV+-interneuron-driven responses in local pyramidal neurons typically evoked an IPSC containing a fast (yellow) and slow component (light green). f, Schematic of specific infection of IL SOM+ interneurons with ChR2 using SOM::Cre animals to probe SOM+-mediated inhibition in the IL. g, Example of a biocytin-recovered (green) SOM+ interneuron (DIO-mCherry, red). Scale bar: 20 μm. h, Infected SOM+ cells respond to blue light (blue bar: 100 ms) and cause a stuttering discharge pattern. i, Optical stimulation of SOM+-interneurons evoked IPSCs in local pyramidal neurons with a fast (yellow) and slow IPSCs (light green). j, The average response composition (top) was similar for both PV+- and SOM+-interneurons with either only fast IPSCs (yellow) or fast and slow IPSCs (black). Neurons with an IPSC fast and slow response showed an amplitude ratio (bottom) that is equal for both PV+(filled circle; n = 6)- and SOM+(empty circles; n = 8)-driven inhibition.
Feed-forward inhibition in vHPC→IL projections is mediated by PV+ interneurons
The vHPC-driven IPSC consisted of two components: a fast γ-aminobutyric acid (GABA)A-receptor mediated component (50–100 ms), blocked by the selective GABAA/C-receptor antagonist picrotoxin (n = 3), and a slower (150–1000 ms) component that was blocked by the selective GABAB-receptor antagonist CGP55845 (n = 4) (Supplementary Fig. 3). The fast disynaptic IPSC was present in ~88% of L2/3 principal neurons and ~62% of L5/6 neurons, whereas the slow IPSC occurred in ~60% of L2/3 and ~37% of L5/6 principal neurons (Supplementary Fig. 3c), and this inhibitory conductance is capable of suppressing spiking activity of local IL pyramidal neurons (Supplementary Fig. 3d). These results show that the impact of vHPC input to pyramidal neurons in the IL is largely inhibitory, and largest to neurons in L2/3. Pyramidal neurons in the IL that project to the BLA are primarily located in L2/326,27, so we tested whether vHPC input targets amygdala-projecting neurons in the IL, by combining optical HPC stimulation with retrograde bead injections into the basolateral and central amygdala (Fig. 1i,j). Amygdala-projecting L2/3 neurons in the IL received excitatory vHPC input (9/10) of similar amplitude as that to randomly selected IL pyramidal neurons (Fig. 1l). Moreover, amygdala-projecting neurons received dual component disynaptic IPSCs (8/9 with fast IPSC components and 3/9 with slow IPSC components) and a similar IPSC/EPSC conductance ratio (Fig. 1k,l), showing that the vHPC input to the IL modulates amygdala-projecting pyramidal neurons.
We have shown that vHPC input to the IL generates strong feed-forward inhibition onto principal neurons that project to the amygdala. To confirm that local IL interneurons are responsible for this dual component feed-forward inhibition, we first used local electrical stimulation in the presence of AMPA/kainate- and NMDA-receptor blockade (NBQX and APV) to isolate inhibitory synaptic transmission. Stimulation within the IL evoked IPSCs in principal neurons that contained fast and slow inhibitory components (Supplementary Fig. 4), confirming that local IL interneurons evoke dual component inhibition in pyramidal neurons. As with most cortical regions25, the mPFC contains a diverse population of interneurons28 and in most cortical regions, these interneurons provide fast GABAA-receptor mediated inhibition. While GABAB-receptor mediated inhibition in principal neurons has been known for many years, the predominant subtype of interneuron that mediates this synaptic current are the neurogliaform cells29,30. As principal neurons in the IL receive both fast and slow feed-forward inhibition, but the intrinsic firing properties of vHPC-innervated interneurons in the IL were different to that of neurogliaform cells29, we asked which interneuron population provides this inhibition. We therefore selectively expressed ChR2 in PV+ or SOM+ interneurons using PV::Cre and SOM::Cre mice, respectively (Fig. 2). Both cell types had a maintained inward current to prolonged optical stimulation confirming expression of ChR2 (Fig. 2b). In ex vivo acute brain slices, ChR2-transduced PV+ interneurons showed typical fast-spiking discharge patterns (Fig. 2c) while SOM+ interneurons showed a stuttering firing phenotype31 (Fig. 2g, h). Whole-cell recordings from IL pyramidal neurons revealed that optical stimulation of PV+ interneurons evoked fast GABAA-receptor mediated IPSCs in 8/8 neurons with a clear slow GABAB-receptor mediated IPSCs in 5/8 neurons. Stimulation of SOM+ interneurons evoked GABAA-receptor mediated IPSCs in 10/10 neurons, and slow GABAB-receptor mediated IPSCs in 5/10 neurons (data from 4 PV::Cre and 2 SOM::Cre animals), with the ratio of the two currents (IPSCslow/IPSCfast) evoked by PV+ interneurons being 1.57 ± 0.47 and 2.39 ± 0.74 when evoked by SOM+ interneurons (two-sided t-test: t(12) = 0.87, P = 0.40). Thus, both PV+ and SOM+ interneurons in the IL can mediate dual component IPSCs (Fig. 2j). To determine if vHPC-evoked disynaptic inhibition is mediated by one subtype of interneuron, we tested the effect of selectively silencing these interneurons on vHPC-evoked IPSCs. Using PV::Cre animals, vHPC input was labeled with ChR2, and we first used an ivermectin-gated chloride channel system (IL injection of AAV-DIO-ivermectin-GFP) to silence PV+ interneurons (Fig. 3a). In acute brain slices, application of ivermectin blocked PV+ interneuron firing to threshold current injection (Fig. 3b), confirming expression of the ivermectin-gated chloride channel. Activation of vHPC input to IL principal neurons evoked disynaptic IPSCs in L2/3 pyramidal cells with fast and slow components (Fig. 3c, black trace), and application of ivermectin to silence PV+ neurons abolished the slow GABAB-mediated component (3/3) while the fast GABAA-mediated IPSC was blocked in 1/3 recordings (Fig. 3c), showing that PV+ neurons in the IL can mediate vHPC-driven feed-forward inhibition onto principal neurons. As the pharmacological approach is intrinsically slow, to silence interneurons in the more time-locked manner, we applied a dual-optogenetic approach in which vHPC input was recruited using the light-sensitive opsin Chronos, while interneurons were silenced using ArchaerhodopsinT (ArchT; Fig. 3d, f). Chronos-driven vHPC input was excited using 420 nm light, while ArchT was specifically activated using 640 nm light (Fig. 3e). Prolonged 640 nm light pulse completely blocked Chronos-evoked spiking of infected PV+ interneurons (Fig. 3g, h; n = 3). In IL pyramidal neurons in PV::Cre animals, stimulation with 420 nm light evoked an EPSC-IPSC response (Fig. 3i, black trace), and silencing PV+ interneurons with 640 nm light during vHPC stimulation eliminated the inhibitory component while preserving the excitatory input (Fig. 3i, red trace, n = 5 from 3 animals). In contrast, silencing SOM+ interneurons using ArchT (Fig. 3j) had no effect on vHPC-evoked feed-forward inhibition (Fig. 3k; n = 6 from 2 animals). Together, these results show that vHPC input to the IL innervates both principal neurons and interneurons but is dominated by the input to PV+ interneurons, which drive large disynaptic inhibition of principal neurons in the IL to shape s.piking activity.
Figure 3. vHPC-driven feed-forward inhibition onto IL pyramidal neurons are specifically mediated by PV+ interneurons.

a, Schematic illustrating expression of ivermectin-sensitive chloride-channels (red) in IL PV+ cells to suppress local feed-forward inhibition mediated by the vHPC (using optical stimulation). b, Suppression of neural activity of PV+ neurons was confirmed by investigating intrinsic firing (left) that was inhibited in the presence of ivermectin (right; orange bar). c, Optical activation of vHPC inputs evoked a disynaptic inhibitory synaptic potential in a pyramidal cell (black trace), and application of ivermectin (30 nM) to silence PV-interneurons blocked this inhibition (orange trace). The bar graph shows the suppression of the fast and slow inhibitory conductances (number of times the fast (yellow) and slow (green) IPSC were blocked/total number of investigated neurons). d, In order to achieve better temporal control, a dual-optical approach was used to recruit vHPC inputs using Chronos (a more light sensitive opsin) at 420 nm (blue bars) and to silence local IL PV+ neurons using ArchaerhodopsinT (ArchT) at 640 nm (red bars). e, left: Chronos-infected cells showed minimal activation using 640nm light (200 ms pulses), but were strongly activated by 420 nm light. Right: In contrast, ArchT-infected PV+ neurons (bottom trace) showed a strong response to 640 nm light, whereas 420nm light (bottom trace) did not activate the proton-pump (ArchT) much. f, Examples of viral injection sites into the IL (ArchT) and vHPC (Chronos); white bar inset: 200 μm. g, Illustration showing the dual-optical approach to recruit hippocampal inputs while silencing PV+ interneurons using PV::Cre animals. h, Chronos-driven spiking activity in ArchT-infected PV+ neurons (top trace) was suppressed in the presence of a 640 nm light pulse (200 ms long, n = 3). i, The vHPC-mediated feed-forward inhibition in pyramidal neurons (black traces; vHPC stimulation: blue bar) was blocked by silencing PV+ neurons using 640 nm light during the hippocampal stimulation (red bar; n = 5). j, Illustration showing the dual-optical approach using SOM::Cre animals (instead of PV::Cre). k, vHPC-mediated inhibition in IL pyramidal neurons (black trace) was not blocked in the presence of 640 nm light (n = 6). l, The bar graph summarizes the amount of times (as the percentage) the disynaptic inhibition in principal cells was blocked during the 640 nm light presentation in PV::Cre (left; n = 5) and SOM::Cre animals (right; n = 6; two-tailed t-test: t9 = 8.47; P < 0.0001).
vHPC→IL projections mediate fear relapse
Considerable work has revealed that the IL is essential for suppressing fear after extinction learning which is mediated by its excitatory projections to the amygdala8,10,32, whereas the vHPC appears to limit fear suppression and promote fear relapse (i.e., “renewal”)16. In extinction, IL-mediated reduction of fear results from an excitatory projection from IL to the BLA, and activation of the intercalated cell clusters, that ultimately reduces output from the central amygdala10,33. Our recording results suggest that the vHPC may relieve fear suppression and promote fear relapse via feed-forward inhibition of IL principal neurons. To examine this hypothesis, we used designer receptors exclusively activated by designer drugs (DREADDs) to selectively manipulate the activity of vHPC neurons projecting to the IL during presentation of an extinguished conditioned stimulus (CS) outside of the extinction context, a situation that leads to fear renewal.
We first confirmed that systemic administration of the DREADD agonist, clozapine-N-oxide (CNO), decreases spontaneous firing in neurons expressing an inhibitory DREADD in vivo (Supplementary Fig. 5) and that silencing vHPC neurons using the same DREADD receptors attenuates the renewal of extinguished fear (Fig. 4). Specifically, CNO-treated rats expressing hM4D(Gi) receptors in the vHPC exhibited significantly less fear renewal as compared to rats that received control virus without modulating baseline contextual freezing (Fig. 4a–c). These data confirm the important role of the vHPC in fear renewal16.
Figure 4. vHPC-IL projections bidirectionally modulate fear relapse.
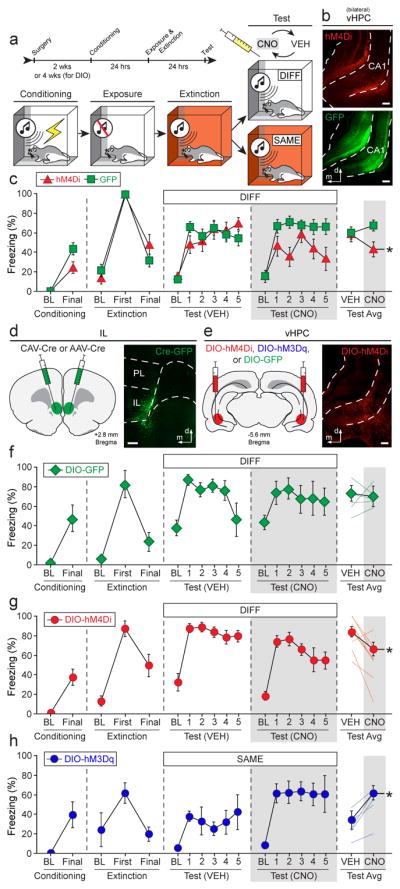
a, Behavioral design: within-subject ABA renewal (DIFF) or ABB retrieval (SAME) paradigm. b, Non-Cre-dependent hM4D(Gi)-expressing virus (top) or control virus (bottom) expression in the vHPC. c, Conditioning data show the mean percentage of freezing during the pre-conditioning baseline (BL) period and after the final conditioning trial (1-min post-shock). Extinction data show mean freezing during the pre-CS baseline period (3-min), as well as during the first and last 5-trial blocks (ITIs). Test data show mean percentage of freezing during the 10-min baseline period and during the 30-sec ITIs for each of the 5 test trials; each animal was tested after VEH or CNO administration in separate counterbalanced tests—mean freezing across all ITIs is shown in the rightmost panel. Conditioning and extinction were normal (statistical data not shown). CNO- and virus-dependent silencing of the vHPC decreased fear renewal at test (red triangles), n = 8; control animals exhibited robust renewal (green squares), n = 17 (two-way repeated measures ANOVA, drug × virus interaction: F1,23 = 5.028, P = 0.0349; two-tailed paired t-tests for average freezing [split by virus]: hM4Di: t7 = 2.783, P = 0.0272; GFP: t16 = −1.178, P = 0.2562). d, CAV2-Cre or AAV5-Cre infused into the IL (left); infusion sites confirmed by co-infusion of AAV8-hSyn-GFP (right). e, Cre-dependent (DIO) viruses expressing hM4D(Gi)-mCherry, hM3D(Gq)-mCherry, or GFP infused into the vHPC (left); mCherry expression in vHPC→IL neurons in CA1 region (right). f–h, Effects of CNO on renewal or retrieval of extinction in rats expressing GFP (f), hM4Ddi (g), or hM3Dq (h); conditioning and extinction were typical in all cases (statistical data not shown), f, CNO had no effect on fear renewal in GFP control rats, n = 5 (two-tailed paired t-test for average freezing, t4 = 0.982, P = 0.3818; light lines depict individual animals). g, CNO silencing of vHPC→IL decreased fear renewal, n = 8 (test average: t7 = 2.634, P = 0.0337); baseline freezing was not affected by CNO (t7 = 1.692, P = 0.1345). h, CNO activation of vHPC→IL led to fear relapse, n = 5, (test average: t4 = −3.535, P = 0.0241) without affecting baseline freezing (t4 = −1.377, P = 0.2405). Error bars indicate means ± s.e.m. *P < 0.05. White bar scale = 250 μm.
We next specifically manipulated vHPC neurons projecting to IL using an intersectional viral approach. We injected canine adenovirus expressing Cre-recombinase into the IL and a Cre-dependent DREADD virus or reporter control virus into the vHPC (Fig. 4d, e). Four weeks after surgery, rats underwent fear conditioning, context exposure and extinction before receiving a two-day within-subject renewal test in the conditioning context (i.e., an “ABA” design in which each rat was tested after CNO or VEH administration in a counterbalanced order; Fig. 4a). Silencing vHPC→IL projections with CNO significantly reduced freezing to the extinguished CS relative to VEH treatment, without affecting baseline freezing (Fig. 4g). This indicates that inhibiting vHPC neurons projecting to the IL attenuates fear renewal. Importantly, CNO administration did not affect fear renewal in rats that expressed a control virus (Fig. 4f), indicating that the effects of CNO (whether directly or through its conversion to clozapine34) were the result of a specific interaction with virally expressed DREADDs.
We next examined whether pharmacogenetically activating IL projectors in the vHPC would induce fear relapse within the extinction context. To this end, we expressed a Cre-dependent excitatory DREADD in vHPC neurons projecting to the IL (Fig. 4d, e). After fear conditioning and extinction, rats received a within-subject retrieval test in the extinction context (i.e., an “ABB” design in which each rat received counterbalanced CNO or VEH administration; Fig. 4a). Activation of vHPC→IL projections increased freezing elicited by the CS in the extinction context (Fig. 4h), without affecting baseline freezing. These results indicate that vHPC→IL projections inhibit the expression of extinction leading to a relapse of extinguished fear in the extinction context.
Fear relapse requires GABA receptors in the IL
Given that neuronal activity in the IL has been linked to fear suppression8, the present results suggest that vHPC-mediated GABAergic feed-forward inhibition of IL projection neurons mediates fear relapse. To test this hypothesis, we examined the role of GABAergic transmission in the IL in the expression of fear renewal. Intracranial microinfusion of muscimol (a GABAA receptor agonist) into the IL induced relapse of fear in the extinction context (Fig. 5a–c). Interestingly, IL inactivation did not affect freezing in the earliest trials of the retrieval test (Supplementary Fig. 6), which suggests spontaneous recovery of fear might mask the effects of IL inactivation when brief retrieval tests are used35. Furthermore, intracranial microinfusions of CGP55845 (a GABAB receptor antagonist), picrotoxin (a GABAA/C receptor antagonist), or both into the IL significantly reduced fear renewal as compared to vehicle treatment (Fig. 5d–f). These results reveal that GABAergic transmission in the IL regulates fear relapse.
Figure 5. Local GABA-mediated signaling in the IL gates fear renewal.

a, Behavioral design: ABB or BBB retrieval paradigm. b, Single cannula placements aimed at IL (11°). c, Percentage of freezing during the 3-min baseline (BL) and after the final trial (Final, 1-min ITI) during conditioning, during the first and final 5-trial blocks (average of 5 post-CS ITIs) during extinction, and average freezing across twenty CS-alone trials (post-CS ITIs) on the retention test. Conditioning and extinction were typical with no differences between groups (statistical data not shown). c, Intra-IL infusions of muscimol (MUSC, n = 6) induced relapse of fear in the extinction context compared to vehicle-treated rats (VEH, n = 10; t14 = 5.9, P < 0.0001). d, ABA renewal paradigm. e, Bilateral IL cannula placements (30°). f, Percentage of freezing during the 3-min baseline (BL) and after the final trial (Final, 1-min ITI) during conditioning, during the first and final 5-trial blocks (average of 5 post-CS ITIs) during extinction, and average freezing across five CS-alone trials (post-CS ITIs) on the retention test. Conditioning and extinction were typical with no differences between groups (statistical data not shown). Intra-IL infusions of CGP55845 (CGP, n = 12), picrotoxin (PTX, n = 7), or both (CGP+PTX, n = 8; VEH, n = 14) decreased fear renewal (one-way factorial ANOVA, main effect of drug, F3,37 = 12.470, P < 0.0001; post hoc Fisher’s PLSD, P = 0.0439, P < 0.0001, P < 0.0001 for CGP, PTX, and CGP+PTX vs. VEH comparisons, respectively; P = 0.0105 and P = 0.0018 for PTX and CGP+PTX vs. CGP comparisons, respectively). g, Infusions of AAV5-Cre in the IL (left) and AAV8-hSyn-DIO-hM3D(Gq)-mCherry (or non-DREADD-expressing control virus) in the vHPC (right). h, Left: hM3D(Gq)+ neurons (mCherry: red) and Fos+ nuclei (GFP: green) in vHPC (arrows = double-labeled cells; white bar inset = 50 μm); Right: percentages of Fos+ and mCherry+ cells among total mCherry+ neurons, comparing controls (n = 11: DREADD-expressing rats treated with vehicle [n = 6], non-DREADD-expressing rats treated with CNO [n = 5]) vs. DIO-hM3Dq-CNO animals (n = 6); two-tailed unpaired t-test, t15 = 9.043, P < 0.0001. i, Left: PV+ neurons (brown in soma) and Fos+ neurons (purple/black in nuclei) in PFC (arrows = double-labeled cells; black bar inset = 50 μm); Right: percentages of PV+ and Fos+ neurons of total PV+ neurons, comparing CNO-treated hM3D(Gq)-expressing animals (n = 7) vs. controls (n = 12: hM3D(Gq)-expressing animals rats treated with vehicle [n = 6], mCherry-only-expressing rats treated with CNO [n = 6]); main effect of brain region: F1,1 = 30.5, P < 0.0001; brain region × percentage interaction: F1,17 = 8.076, P = 0.0113; post hoc Fisher’s PLSD, IL: P = 0.0288 for hM3D(Gq)-CNO vs. controls. Error bars indicate means ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.0001.
We have shown that renewal of extinguished fear results from activation of IL projecting vHPC neurons. Activation of these inputs ex vivo leads to activation of PV+ interneurons and inhibition of amygdala projecting IL pyramidal neurons. These data suggest that activation of PV+ interneurons, and the resultant feed-forward GABAergic inhibition of IL pyramidal cells, mediates inhibition in the vHPC→IL circuit in behaving rats. To test this hypothesis in vivo, we again expressed excitatory hM3D(Gq) virus in vHPC neurons projecting to the IL (Fig. 5g), and examined Fos expression in vHPC projectors and infralimbic cortical PV+ interneurons after CNO injection. Significantly higher percentages of hM3D(Gq)+ neurons in the vHPC expressed Fos after CNO injection compared to the control (Fig. 5h). Importantly, this was accompanied by a significant increase in the number of Fos-positive PV+ interneurons in the IL but not PL (Fig. 5i). This reveals that pharmacogenetic activation of VH projectors to the IL recruits PV+ interneurons in intact animals, a population of neurons positioned to inhibit IL output and promote fear relapse.
Discussion
Collectively, the present data reveal that vHPC projections generate a significant feed-forward inhibition of IL pyramidal neurons by recruiting PV+ interneurons. vHPC-mediated inhibition is most pronounced in amygdala-projecting pyramidal neurons, located in layers 2 and 3 of IL. This vHPC-mediated inhibition of IL principal cells has an important role in the relapse of extinguished fear. Pharmacogenetic activation of IL projectors in the vHPC recruits PV+ interneurons in the IL and causes a relapse of extinguished fear; this effect was also observed with direct pharmacological activation of GABA receptors in IL. In contrast, either pharmacogenetic silencing of vHPC→IL projectors or antagonizing GABAergic receptors in the IL diminishes fear relapse. Together, these experiments reveal that vHPC projections to the mPFC drive feed-forward inhibition of IL principal neurons by PV+ interneurons to mediate fear relapse after extinction.
The present work is consistent with early electrophysiological analyses of vHPC-mPFC projections that have shown a strong inhibitory effect of electrical stimulation of the vHPC on mPFC neuronal activity36,37. Indeed, an inhibitory influence of the vHPC on mPFC function has been reported to influence the expression of learned fear38. However, in contrast to this work, a recent report by Spellman and colleagues suggests that the dominant effect of the vHPC in the mPFC is excitatory39. Specifically, they report that optogenetic inhibition of vHPC terminals in the mPFC reduced short-latency (10–20 ms) vHPC-evoked increases in mPFC multiunit activity in anesthetized mice. In the present work, we also observed short-latency EPSPs in IL pyramidal neurons, but this excitation was overshadowed by a dominant feed-forward inhibition that lasted hundreds of milliseconds beyond the excitatory volley (see Figure 1k). Our ex vivo experiments show that this vHPC-mediated inhibition is mediated by both GABAA and GABAB receptor-mediated currents, and inhibition of either receptor type has an impact on fear renewal (Fig. 5). Hence, while short-latency excitatory responses in IL principal cells can be driven by vHPC input, the net effect of feed-forward inhibition on amygdala-projecting neurons is likely to be inhibition over a much longer time scale36,37.
Of course, it is noteworthy that the magnitude of the renewal impairments obtained with pharmacogenetically silencing either the vHPC (Fig. 4c) or vHPC projectors to IL (Fig. 4f) was relatively modest compared to that associated with GABA antagonists infused directly into the IL (Fig. 5f); DREADD manipulations reduced, rather than eliminated renewal. This disparity might be explained by differences in the inhibitory circuits influenced by the two manipulations. For example, intracranial drug infusions affect both local inhibitory circuits and extrinsic feed-forward inhibition in the IL that includes, but is not limited to, that mediated by vHPC projections. Moreover, intersectional DREADD expression only targets a subset of vHPC neurons projecting to the IL; inhibition of this discrete population of neurons would not necessarily be expected to recapitulate the behavioural impairments associated with a broad manipulation of IL activity. Conversely, pharmacogenetic activation of vHPC projectors to IL produced significant increases in freezing (Fig 4h) that were absent during the corresponding block of test trials (i.e., trials 1–5) in rats receiving intra-IL muscimol infusions (Supplemental Fig. 6). In this case, the circuit-specific DREADD manipulation appeared to be more effective than the non-selective pharmacological manipulation in revealing a behavioral effect. In both cases, however, pharmacogenetic manipulations of vHPC projectors to IL and GABAergic manipulations of IL produce qualitatively similar outcomes, suggesting they operate on similar neural processes. Nonetheless, further experiments that directly manipulate vHPC projections onto discrete IL interneuron populations are required to determine the specific contribution of those projections to the regulation of IL function and fear expression.
The present data complement a large body of evidence in both rats and humans implicating the mPFC in the regulation of emotional responses2,40,41. The IL, in particular, is thought to dampen learned fear responses by inhibiting the amygdala42–44. Indeed, prefrontal-amygdala circuits have been posited to play an important role in both the encoding and retrieval of extinction memories10. However, it has recently been reported that optogenetically silencing the IL35 or its terminals in the amygdala45 impairs the acquisition of extinction, but does not affect the retrieval of extinction memories35. This contrasts with the present results, which reveal robust effects of IL manipulations on extinction retrieval and renewal. One possibility is that the recent optogenetic studies only targeted IL principal cells (or their projections)35,45. Consistent with this, another group has found that optogenetic inhibition of both principal cells and inhibitory interneurons in the IL produced extinction retrieval deficits; inhibition of principal cells alone did not produce a deficit46. This suggests that both inhibitory interneurons and principal cells are recruited in the regulation of extinction retrieval, which is consistent with our observations in the present study; future studies should nevertheless attempt to directly modulate the activity of interneurons of the IL during extinction retrieval and relapse.
Together, the present experiments reveal a novel hippocampal-prefrontal circuit for the context-dependent regulation of memory retrieval. Specifically, we show that the renewal of extinguished fear results from vHPC-mediated inhibition of mPFC circuits involved in suppressing fear. The engagement of this vHPC-IL circuit occurs when animals experience an extinguished CS in a context where the meaning of the CS is ambiguous (such as the original conditioning context or a novel context). We and others have previously shown that activity in the hippocampus is increased by unexpected events, including experiencing an extinguished CS outside the extinction context13,18,22,47,48. In these instances, it is likely that the hippocampus retrieves information about the context in which stimuli have previously occurred to influence prefrontal cortical processes involved in emotional regulation14. Of course, it has also been appreciated that prefrontal cortical circuits are involved in directing memory retrieval by the hippocampus based on contextual information49. Ultimately, oscillatory networks that regulate bidirectional information flow through hippocampal-prefrontal circuits may generate context-appropriate behavioral responses50.
In conclusion, we have discovered a novel circuit for fear relapse that is centered on ventral hippocampal projections to the infralimbic cortex. We show that afferent activity in IL-projecting cells of the vHPC is associated with activation of GABAergic PV+ interneurons and strong inhibition of IL projection cells. We suggest that the function of this circuit is to promote the return of fear under conditions when it is warranted, including in contexts in which an extinguished CS is potentially dangerous (i.e., the conditioning context or a novel context). The renewal of fear in these situations is adaptive in many cases, but can interfere with the efficacy of cognitive-behavioral therapies designed to limit fear after trauma. We conclude that vHPC synapses on IL interneurons are a unique target for therapeutic interventions aimed at broadening the generalization of extinction memories, thereby reducing the possibility that pathological fear relapses after therapy.
Online Methods
Subjects
Adult male Wistar rats (170–240 g), PV::Cre knock-in (C57BL/6 strain) mice (for the local optical PV stimulation, the ivermectin-receptor silencing experiments, and the dual-optical approach; 20–26 g), and SOM::Cre knock-in (C57BL/6 strain) mice (19–25 g; all animals were bred in house) were used in the ex vivo electrophysiological experiments. The animals were housed in a 12/12 h light/dark cycle with access to food and water ad libitum. Adult male Long-Evans rats (200–224 g; Blue-Spruce, Harlan) were used for the behavioral experiments. The Long-Evans rats were individually housed in a 14/10 h light/dark cycle and had access to food and water ad libitum. Rats for behavioral testing were handled for 5 days after arrival. All experimental procedures were performed in accordance with the protocols approved by the University of Queensland Animal Ethics Committee and the Animal Care and Use Committee at Texas A&M University.
Viruses and drugs
AAV5-CamKIIα-hM4D(Gi)-mCherry (3.4×10e12 vg/mL), AAV5-CamKIIα-mCherry (6.4×10e12 vg/mL), AAV5-hSyn-DIO-hM4D(Gi)-mCherry (5.7×10e12 vg/mL), AAV8-hSyn-DIO-GFP (~6×10e12 vg/mL), AAV5-hSyn-DIO-hM3D(Gq)-mCherry (6.7×10e12 vg/mL), and AAV8-CamKIIα-GFP (5.6×10e12 vg/mL) vectors were obtained from the University of North Carolina Vector Core and Addgene. CAV2-Cre (8.7×10e12 vg/mL) vectors were obtained from the Institute of Molecular Genetics of Montpellier. AAV5-CMV-HI-eGFP-Cre-WPRE-SV40 (1.62×10e13 vg/mL), AAV2/5-CAG-ChR2-Venus-WSV40 (1.1×10e13 vg/mL), AAV2/5-hSyn-hChR2(H134R)-eYFP-WPRE-hGH (1.42×10e13 vg/mL), AAV2/1-pSyn-Chronos-EGFP (2.41×10e12 vg/mL), and AAV5-EF1a-dflox-hChR2(H134R)-mCherry-WPRE (1.31×10e13 vg/mL) vectors were obtained from University of Pennsylvania Vector Core. EF1a-D20-ArchT-EGFP (1.3×10e11 vg/mL) was produced in-house at the University of Queensland. R.H. and J.E.P. packaged and provided AAVDJ/8-CamKIIα-hM4D(Gi)-mCherry (1.23×10e13 vg/mL) vectors. For use in the experiments involving designer receptors exclusively activated by designer drugs (DREADDs)51, clozapine-N-oxide (CNO) was obtained from the National Institute of Mental Health (NIMH; Chemical synthesis and drug supply program). CNO was first dissolved in DMSO and then mixed with sterile saline (VEH, 2.5% DMSO). The action of CNO has been suggested to be mediated by its metabolite, clozapine34. To control for this, CNO was also tested in a non-DREADD-expressing GFP control (Fig. 4). Drugs used in the electrophysiological experiments were obtained from Tocris Bioscience. For the behavioral experiments, muscimol (MUS) and CGP55845 (CGP) were obtained from Sigma-Aldrich, and picrotoxin (PTX) was obtained from Tocris Bioscience.
Surgical procedures
For the ex vivo electrophysiological experiments, mice (p25–45) were anesthetized with intraperitoneal injections of ketamine (10%) and xylazine (5%, 10 ml/kg; rats received an additional injection of 0.4 ml/kg of Zoletil). Animals were then placed in a stereotactic frame. An incision at the midline was made using a single edged blade. For the investigation of hippocampal projections to the mPFC, viruses (0.3–0.5 μl of AAV2/5-CAG-ChR2-Venus-WSV40 or AAV2/5-hSyn-hChR2(H134R)-eYFP-WPRE-hGH were injected in the vHPC of rats. The coordinates (all in mm) used for vHPC stereotactic injections in the rats are (anterior/posterior, medial/lateral, dorsal/ventral): −6.0, ±5.3, −4.0. For the investigation of amygdala-projecting IL neurons in rats, retrograde beads (0.3–0.5 μl; Lumafluor) were injected in the BLA (A/P, M/L [angle], D/V): −2.3, ±1.2 (22°), −9.3. For the study of local PV-projections, 1–2 μl of AAV5-EF1a-dflox-hChR2(H134R)-mCherry-WPRE were injected into the IL of PV::Cre mice (for the silencing approach using ivermectin-receptors52,53). Coordinates for the IL injections are (A/P, M/L [angle], D/V): 2.5, ±2.9 (34°), −4.5. For the dual-optogenetic approach using Chronos54 and ArchT55, AAV2/1-pSyn-Chronos-EGFP (vHPC) and EF1a-D20-ArchT-EGFP (IL) were injected using the following coordinates in either PV::Cre or SOM::Cre mice: (A/P, M/L, D/V) vHPC: −3.3, ±3.1, −3.5; IL: 1.8, ±0.4, 2.0 (the injection was targeted slightly more dorsal than usual due to issues with strong expression at the core infection site). The incision was then disinfected and closed using vetbond tissue adhesive and stitched. Baytril (0.1 μl/g) and Metacam (0.4 μl/g) were each diluted into 0.5 ml of saline and then injected subcutaneously.
For all behavioral experiments, all surgery and group assignments were randomized for cage position in the vivarium. In the experiment examining the role of DREADD-mediated inactivation of the vHPC during fear renewal (Fig. 4a–c), rats were deeply anesthetized with isoflurane (5% for induction; ~2% during surgery), and were placed into a stereotaxic apparatus for viral injection. The scalp was shaved, treated with povidone-iodine, and a small incision was made to expose the skull. Small holes were drilled in the skull and rats were bilaterally injected with either AAVDJ/8-CamKIIα-hM4D(Gi)-mCherry (n = 14) or AAV8-CamKIIα-GFP (n = 18) into the vHPC. Four bilateral injections (0.4 μl/injection) were made into the vHPC at two different A/P levels (coordinates [in mm from bregma] are shown in A/P, M/L, D/V): −5.2, ±6.0, −6.5; −5.2, ±6.0, −5.3; −6.1, ±6.0, −6.1; −6.1, ±6.0, −5.0 (all D/V coordinates are measured from dura). Viruses were injected (0.15 μl/min) using an injector connected to polyethylene tube and a Hamilton syringe (10 μl) mounted on an infusion pump. Following the infusions, virus was allowed 5–10 min for diffusion before removing the injectors. Once completed, the incision was closed and rats were returned to their homecages to recover for two weeks following surgery. Rats with unilateral, off-target, or no viral expression were excluded from the final analyses. In addition, two rats were excluded because of a technical issue during tissue collection resulting in a loss of vHPC tissue. Based on these criteria, twenty-five rats are included in the final analyses (Fig. 4a–c; hM4Di, n = 8; GFP, n = 17).
For the intersectional DREADD experiments (Fig. 4d–h), rats underwent surgery as described above. To pharmacogenetically inhibit vHPC neurons projecting to IL, rats (n = 14) were bilaterally injected with CAV2-Cre in the IL and AAV5-hSyn-DIO-hM4D(Gi)-mCherry in the vHPC. Pharmacogenetic excitation of this projection was achieved by bilaterally injecting rats (n = 8) with AAV5-CMV-HI-eGFP-Cre-WPRE-SV40 in the IL and AAV5-hSyn-DIO-hM3D(Gq)-mCherry in the vHPC. Control rats (n = 5) received CAV2-Cre in the IL and AAV8-hSyn-DIO-GFP in the vHPC. All the viruses for this experiment were injected (0.1 μl/min) as described above. Four injections (0.5 μl/injection) were made into the vHPC at the coordinates described above. One injection (~1.5–1.8 μl) was made in the IL in each hemisphere at the following coordinates (mm from bregma): +2.8 (A/P), ±3.0 (M/L), −4.9 (D/V), with a 30° angle. The viruses were allowed to diffuse for 5–10 min per injection before removing the injectors. Once completed, rats were placed back in their home cages to allow for viral expression for at least 4 weeks. Rats with unilateral, off-target, or no viral expression were excluded from the analyses. Specifically, six rats infected with AAV5-hSyn-DIO-hM4D(Gi)-mCherry and three rats infected with AAV5-hSyn-DIO-hM3D(Gq)-mCherry were excluded based on the aforementioned criteria, resulting in the final group sizes (Fig. 4d–h): DIO-GFP (n = 5), DIO-hM4Di (n = 8), DIO-hM3Dq (n = 5).
For the behavioral experiment involving pharmacological inactivation of the IL with a GABA receptor agonist (Fig. 5a–c, Supplemental Fig. 7), rats (n = 32; sixteen rats per group: vehicle [VEH] and muscimol [MUS]) were implanted with a single guide cannula (26 gauge, 8 mm; Small Parts) in the IL (A/P: +2.65, M/L: ±1.0, D/V: −4.1) at an 11° angle. Cannulas were secured to the skull with jeweler’s screws and dental cement. Stainless steel obturators (30 gauge, 9 mm; Small Parts) were placed in each guide cannula and were changed twice prior to behavioral tests. Rats recovered from surgery for one week prior to the onset of behavioral training and testing. Rats with cannulas terminating outside the borders of the IL were excluded from the final analyses. Based on these criteria, sixteen rats are included in the final analyses (Fig. 5a–c, Supplemental Fig. 7; VEH, n = 10, MUS; n = 6).
For the behavioral experiment consisting of pharmacological activation of the IL with GABA receptor antagonists (Fig. 5d–f), rats (n = 56) were implanted with bilateral guide cannulas (26 gauge, 8 mm) in the IL ([mm from bregma] A/P: +2.7, M/L: 3.0, D/V: −4.9 at 30° angle). The guide cannulas were secured as above with jeweler’s screws and dental cement. Stainless steel obturators (30 gauge, 9 mm) were inserted into the cannulas and removed and replaced with clean obturators twice prior to the behavioral tests. Rats recovered for one week following surgery in their homecages prior to the behavioral procedures. Rats with off-target cannula placements (including unilateral misses) were excluded from all of the analyses (ten vehicle [VEH] rats, four CGP55845 [CGP] rats, and one picrotoxin [PTX] rat were excluded). Accordingly, forty-one rats are included in the final analyses (Fig. 4d–f; VEH, n = 14; CGP, n = 12; PTX, n = 7; CGP+PTX, n = 8).
For those animals utilized in the Fos analyses (Fig. 5g–i), rats were bilaterally injected with AAV5-CMV-HI-eGFP-Cre-WPRE-SV40 in IL and AAV5-hSyn-DIO-hM3D(Gq)-mCherry in vHPC (n = 16; eight rats per group: VEH, CNO). As an additional control, another group of rats was bilaterally injected with AAV5-CMV-HI-eGFP-Cre-WPRE-SV40 in the IL and AAV5-hSyn-DIO-mCherry in the vHPC mCherry-CNO, DIO-hM3Dq-VEH (n = 8, CNO). Injection procedures and locations were identical to those in the DIO-DREADD experiment. As with the abovementioned experiments, rats with off-target viral infection were excluded from the final analyses. In addition, five rats were excluded because of a technical issue in collecting tissue at the level of the PFC resulting in poorly stained tissue. A total of nineteen rats were included in the final analyses for Fos analyses of the PFC (Fig. 5i; DIO-hM3Dq-CNO, n = 7; DIO-hM3Dq-VEH, n = 6; DIO-mCherry-CNO, n = 6; the latter two groups were collapsed for a total of twelve control animals). For the Fos analyses of the vHPC (Fig. 5h), another two rats were excluded due to a technical issue in collecting vHPC tissue. Accordingly, a total of seventeen rats were included in the vHPC Fos analyses (Fig. 5h; DIO-hM3Dq-CNO, n = 6; controls, n = 11: DIO-hM3Dq-VEH, n = 5; DIO-mCherry-CNO, n = 6).
Finally, for the in vivo electrophysiological recordings anesthetized rats (three total) received bilateral stereotaxic infusions (2.0 μl/per infusion; 0.3 μl/min with 5 min of diffusion time) of AAV5-CamKIIα-hM4D(Gi)-mCherry (n = 1) or AAV5-CamKIIα-mCherry (n = 2) in IL (A/P: +2.7, M/L: ±3.0, D/V: −5.1 at a 30° angle). Following viral infusions, rats were implanted with a 16-channel microelectrode array (Innovative Neurophysiology, Durham, NC) within the right hemisphere targeting the mPFC (8 channels in IL; 8 channels in PL). The 2×8 array was comprised of two rows of 50 μm diameter tungsten wires terminating at different lengths (6.9 mm for targeting PL, and 8.0 for targeting IL). From center-to-center, the wires were spaced 200 μm apart. The array was implanted such that its long axis was parallel to the A/P plane of the brain, and that its centermost wires targeted IL as follows: A/P: +2.7, ML: +0.35, −5.1 D/V (from skull surface). Dental cement was used to secure the array to the skull. Animals recovered from surgeries for two weeks prior to the onset of recordings.
Slice electrophysiology
Similar to prior reports56, electrophysiological experiments commenced at least 28 d after the virus injections to allow for terminal expression. Single-cell recordings of neurons and application of brief light pulses (470 nm; 5 ms) using an LED source (CoolLED) were performed to study the location and pharmacological properties of synaptic connections onto mPFC neurons. Only injected animals with at least one successful synaptic response at the projection site were included in the analyses. Light stimulation was kept maximal, except for cases where a decrease in light stimulation eliminated the polysynaptic component, but didn’t diminish the initial component.
Animals were anesthetized with isoflurane (1 ml; applied in enclosed container) and then decapitated. Brains were rapidly removed and placed in ice cold cutting solution containing the following chemicals (mM): NaCl 118, KCl 2.5, NaHCO3 25, glucose 10, MgCl2 4, CaCl2 0.5 and NaH2PO4 1.2. Coronal brain slices (300 μm) were prepared using a vibratome (Leica VT 1000S). Slices were allowed to recover in oxygenated (95% O2 and 5% CO2) artificial cerebrospinal fluid (aCSF) containing the following chemicals (mM): NaCl 118, KCl 2.5, NaHCO3 25, glucose 10, MgCl2 1.3, CaCl2 2.5 and NaH2PO4 1.2) at 35°C for at least 30 min, then kept at room temperature for at least another 30 min before experiments. Slices were transferred to the recording chamber as required and were continuously perfused with oxygenated aCSF through a gravity fed system and maintained at 30–32°C. In cases where either a virus or a tracer was injected, slices containing the injection sites were also kept to determine localized injection sites. Slices were visualized using an upright microscope (BX50WI, Olympus Optical, Tokyo, Japan) with a 5×NA 0.1 or 40×NA 0.8 objective and infrared and differential interference contrast optics. Fluorescent neurons were visualized by using an LED system (pE-2, CoolLED) and YFP/RFP filter sets (Olympus). Electrodes (3–7 MΩ) were filled with pipette solution containing chemicals (mM): KMeSO4 135, NaCl 7, HEPES 10, Mg2ATP 2, Na3GTP 0.3, EGTA 0.3, biocytin 8 (pH 7.3 with KOH; osmolarity ~290 mOsm/kg). Signals were recorded using a patch clamp amplifier (Multiclamp 700B, Axon instruments). Sampling rate was 20 kHz and signals were digitized at 2 kHz (Instrutech, ITC-18). All data were acquired, stored and analyzed using Axograph X (Axograph, V 1.2.1). For all the voltage-clamp recordings, neurons were held at −60 to −70 mV by injecting current if needed, and neurons with resting membrane potential above −55 mV were excluded from the analysis. For current-clamp recordings, recordings were corrected for bridge-balance and pipette resistance. All investigations were performed on the ipsilateral site of injection. Access resistance was 5–25 MΩ and was monitored throughout the experiment (neurons with access resistance changes >25% were excluded). Drugs were bath applied by using a gravity-fed system that allowed continuous change of solutions. For optogenetic stimulation, 470 nm light pulses were applied with a CoolLed system (pE-2) attached to the upright microscope (Olympus BX51WI). Five ms pulses were applied to evoke postsynaptic responses and 200 ms pulses were used to study infected neurons. Maximal light output at 470 nm was measured at 15 mW/mm2 (ThorLabs, optical power meter). Response latency was calculated by measuring the delay between the onset of the light pulse and the onset of the EPSC (5%). The response jitter was calculated by measuring the standard deviation of 8–15 individual responses to optical stimulation. Only responses > 50 pA were used for the analysis. For the dual-optical approach, a fully reflective mirror (Chroma) was selected for the stimulation, and band-pass filters (for 400 nm light, the light was selected from the 400/470 nm LED array module [LAM]; for 640nm, a bandpass filter of 641/75 nm was used in between the light source [GYR CoolLed LAM] and the microscope). 640 nm light was presented every second time during the 400 nm light pulse presentation. Electrophysiological responses were analysed using AxoGraph. For synaptic responses, an average of 5–10 traces were taken for analysis. For optogenetic stimulation, only samples with at least one synaptic response to maximal intensity light pulses were used for the statistics. Electrical stimulation (at 0.1 ms using a stimulator-box) was done by using a patch-clamp pipette filled with 3M saline and the ground loop wire was place in the bath. Intrinsic firing properties were analyzed at current injections of two-fold threshold firing. Drug application of Picrotoxin (100 μM), CGP55845 (1 mM), NBQX (20 mM) and APV (100 mM) (all from Tocris) were done through a gravity fed system. For the comparison between PL and IL and pyramidal neurons and interneurons, respectively, EPSCs were compared in animals with recordings from both regions and subtypes, respectively. Analytical tests were performed with SPSS (IBM) or Prism (GraphPad). Outliers (if applicable) were determined by Grubb’s test (http://graphpad.com/quickcalcs/Grubbs1.cfm).
In vivo electrophysiology
Two weeks after surgery, freely-moving rats underwent three 70-min single-unit recording sessions across three consecutive days (one session/day) in a standard testing chamber. Following a 10-min baseline period, rats were injected (i.p.) with CNO (1 or 3 mg/kg) or vehicle (counterbalanced) and remained in the chamber for 60 minutes. Extracellular single-unit activity was recorded automatically using a multichannel neurophysiological recording system (OmniPlex, Plexon, Dallas, TX) as previously described57. One of the recording wires (located in PL) served as a reference for the wideband signal recorded on each channel. Signals were amplified (8,000×) and digitized (40 kHz sampling rate) and saved on a computer for offline sorting and analysis. After high-pass filtering the signal at 600 Hz, waveforms were manually sorted using 2-dimensional principal component analysis (Offline Sorter, Plexon). Isolated waveforms and their respective timestamps were then imported to NeuroExplorer (Nex Technologies, Madison, AL) for further analysis. The analyses of neural activity focused on changes in spontaneous single-unit firing rate during each recording session. For each session, each neuron was z-score normalized to its 10-min baseline firing rate. Data are plotted in 20-sec bins across the duration of the session.
Behavioral apparatus
Behavioral testing was conducted in two distinct rooms in the laboratory. Eight identical conditioning chambers (30×24×21 cm; MED-Associates) in each room were used in all behavioral experiments. The chambers consisted of aluminum sidewalls and Plexiglas ceilings, rear walls, and hinged front doors. The chamber floors consisted of 19 stainless steel rods that were wired to a shock source and a solid-state grid scrambler (MED-Associates) for delivery of footshock (US). A speaker mounted on one wall of the chamber was used for delivery of the acoustic CS, and ventilation fans and house lights were installed in each chamber to allow for the manipulation of contexts. Each chamber rests on a load-cell platform that is used to record chamber displacement in response to each rat’s motor activity and is acquired online via Threshold Activity software (MED Associates). Absolute values of the load-cell voltages are computed and multiplied by 10 to yield a scale that ranges from 0 to 100. For each chamber, load-cell voltages are digitized at 5 Hz, yielding one observation every 200 ms. Freezing is quantified by computing the number of observations for each rat that has a value less than the freezing threshold (load-cell activity = 10). Freezing is only scored if the rat is immobile for at least 1 s. Because all behavioural measurements of freezing were performed using this automated system, no blinding to group assignments was necessary.
Sensory stimuli were adjusted within these chambers to generate distinct contexts (A and B). For context A, a 15-W house light mounted on the sidewall was turned on, and the white room light remained on. Ventilation fans (65 dB) were turned on, cabinet doors were left open, and the chambers were cleaned with 1% ammonium hydroxide. Rats were transported to context A in white plastic boxes. For context B, house light and white room light were all turned off, and fluorescent red room light was turned on. Ventilation fans were turned off, the cabinet doors were closed and the chambers were cleaned with 1% acetic acid. Rats were transported to context B in black plastic boxes. In each context, stainless steel pans were filled with a thin layer of the respective odors of the contexts and inserted below the grid floor. Context B in the muscimol experiment consisted of 70% ethanol odor in place of acetic acid, cupboard doors remained open, black plastic floors were placed on the grid floor, and rats were transported in 5-gallon buckets with bedding (all other features matched context B as described above).
Behavioral procedures
We used standard procedures for Pavlovian fear conditioning, extinction, and retrieval testing16,58. Rats were randomly assigned to each experimental group, and each training or testing squad was counterbalanced to equally represent each group in the squad. Additionally, rats were randomly assigned to the testing chambers so as to counterbalance the placement of group assignments in each context and squad. Freezing behavior (as a percentage of a block of time or trial[s]) served as the dependent measure of fear in all training and test sessions. For the experiment involving pharmacogenetic inactivation of the vHPC during the renewal of fear (Fig. 4a–c), animals first underwent fear conditioning consisting of five tone (CS; 10 s, 80 dB, 2 kHz)-footshock (US; 2 s, 1.0 mA) pairings with 60 s intertrial intervals (ITIs) after a 3 min baseline period in the context (context A). Rats remained in the chamber for 60 s following the final CS-US pairing, and were then returned to their homecages. On days 2–5, rats underwent extinction training in which they received context exposure to context A for 35.5 min in the morning, and 45 CS-alone trials (30 s ITIs; a total of 35.5 min) in context B in the afternoon (3 min baseline). Twenty-four hours after the final extinction session, rats received a within-subject renewal test in context A over two consecutive days; VEH or CNO (3 mg/kg) was administered 30 min prior to each test in a counterbalanced order. Each test session consisted of a 10-min baseline followed by five CS-alone (30 s ITIs) presentations; the long baseline period was included to assess any nonspecific effects of drug treatment on conditional freezing.
For the intersectional DREADD experiments, rats received fear conditioning followed by extinction as described above (only two days of extinction were administered in this experiment). Rats in the DIO-hM4Di and DIO-GFP groups received within-subject renewal tests as described above in context A; whereas DIO-hM3Dq rats received within-subject extinction retrieval tests in context B; thirty minutes before each test, rats received systemic injections of CNO (~1–3mg/kg) or VEH in a counterbalanced order. Each test session consisted of a 10-min baseline followed by five CS-alone (30 s ITIs) presentations.
For the behavioral experiment involving pharmacological blockade of the IL during the retrieval of extinction (Fig. 5a–c, Supplemental Fig. 7), animals underwent fear conditioning (identical to that described above) in either context A or context B (rats conditioned in the two contexts did not differ and were ultimately collapsed). Twenty-four hours after conditioning, rats were placed in context B and underwent a single session of extinction training (identical to that described above). Twenty-four hours after extinction, the rats underwent a retrieval test consisting of a second extinction session in context B immediately after an intra-IL infusion (0.2 μl; 0.1 μl/min for 2 min) of either SAL or MUS (0.8 mM). The procedure was identical for the experiment examining the effects of GABA receptor antagonism in the IL during fear renewal (Fig 5d–f), except that rats received intra-IL infusions (0.3 μl/hemisphere; 0.3 μl/min) of CGP (10 mM), PTX (0.33 mM), CGP+PTX cocktail (from 10 mM and 0.33 mM stocks, respectively), or SAL in context A. For the intracranial drug infusions, injection cannulas were connected via water-filled polyethylene tubing to gas-tight Hamilton syringes (10 μl) that were secured to an infusion pump (KD Scientific). Drug or vehicle was loaded into the injection cannulas and pressure-injected following the parameters described above and based on prior work (injection cannulas remained in the guides for 1 min following infusion to allow for adequate diffusion) 59.
Histology
At the conclusion of behavioral testing or in vivo recordings, rats were overdosed with sodium pentobarbital (Fatal Plus; 100 mg/ml, 0.5 ml) and were transcardially perfused with saline and 10% formalin. Brains were extracted and post-fixed in formalin solution for 24 hrs at 4° C and transferred to 20% sucrose (4° C). Brains were flash frozen with dry ice and sectioned (40 μm) on a cryostat maintained at −20° C. For non-Cre-dependent viral experiments, sections were wet-mounted to subbed slides and coverslipped with Fluoromount (Sigma-Aldrich) and imaged on a Zeiss microscope (via Axio Imager 2). Cre-dependent viral tissue was collected into wells for immunohistochemistry. For cannula-implanted rats, thionin-stained coronal sections containing IL were imaged on a Leica microscope (MZ FLIII).
Immunohistochemistry
For biocytin recovery and immunohistochemistry in the slice electrophysiology experiments, brain slices were fixed with 4% paraformaldehyde in 0.1M PBS for 40–60 min at room temperature. Slices were washed three times with 0.1M PBS, then blocked with a blocking solution containing PBS (0.1M), bovine serum albumin (3%), saponin (0.1%) and sodium azide (0.05%) for 1 h at room temperature. Slices were washed with PBS and incubated in primary antibodies including anti-TBR1 and streptavidin (Alexa fluor 488/555/647 at 1:1000, Invitrogen) for 1–3 days at room temperature or at 4°C. Sections were washed 3 times for 15 min each time in PBS and Alexa fluor-conjugated species-specific secondary antibodies (1:1000, Jackson Biosciences or Invitrogen) for at least 5 h at room temperature. After a triple wash with PBS, the brain slices were mounted in glycerol and PBS solution (50% and 50%) or PBS only for tissues containing retrograde tracers. Slices were imaged using an upright microscope (5× or 20×, Zeiss, Axio Imager) or a confocal system (20×, Zeiss LSM510). Images were produced by flattening z-stacks (1 μm intervals) to a maximum projection image using Zen 2011 software (Zeiss).
For DIO-DREADD-expressing animals, immunohistochemistry was performed on free-floating brain sections containing the vHPC. The tissue was washed three times in 1×Tris-buffered saline (TBS, pH 7.4). Brain sections were then incubated in 10% normal donkey serum (NDS) in 1×Tris-buffered saline with Tween (TBST) for 1 h at room temperature followed by two washes in TBST for 5 min. Tissue was then incubated in primary antibody solution in TBST with 2% NDS (rabbit anti-mCherry antibody at 1:2000; Abcam) for 48 h at 4°C. Brain sections were then washed three times in TBST for 10 min and were incubated in secondary antibody solution in TBST with 2% NDS (AF594 conjugated donkey anti-rabbit antibody at 1:200; Abcam) for 2 h at room temperature. Tissue was washed three times in TBS for 10 min and then was mounted on subbed slides in 0.9% saline and coverslipped with Fluoromount. Brain sections containing the IL were wet mounted to microscope slides and coverslipped with Fluoromount for imaging. For Fos immunohistochemistry, the tissue was processed exactly as for mCherry immunohistochemistry except rabbit anti-c-Fos antibody (1:5000; Millipore) and AF488-conjugated donkey anti-rabbit antibody (1:500, Life Technologies) were used for primary and secondary antibodies, respectively.
For PV/Fos dual-staining, rats were sacrificed 120 min after CNO or vehicle administration. 18 h post-fix, brains were moved into 30% sucrose solution for 3 days. 40 μm-thick brain sections were collected for staining. Brain sections were washed three times in TBST and then were incubated in 0.3% H2O2 for 15 minutes. The tissue was washed in TBS three times and was incubated in rabbit anti-c-Fos primary antibody (1:1000; Millipore) overnight. Brain tissue was washed three times in TBS followed by 1 h incubation in a biotinylated goat anti-rabbit secondary antibody (1:1000; Jackson Immunoresearch), amplification with the avidin biotin complex at 1:1000 (ABC; Vector labs), and visualization with 3, 3′ diaminobenzidine (DAB) + nickel ammonium sulfate to yield a purple/black nuclear reaction product. The tissue was subsequently incubated in a mouse anti-PV primary antibody (1:5000; Sigma) overnight and biotinylated goat anti-mouse secondary antibody (1:500; Jackson Immunoresearch) for 1 h, amplified with the avidin biotin complex at 1:1000 (ABC; Vector labs), and visualized with 3, 3′ diaminobenzidine (DAB) to yield a light brown somatic stain. Slices were mounted and coverslipped with mounting medium (Permount; Sigma), and stored at room temperature until photographed using a Zeiss microscope. Images for cell counting were generated and counted (using ImageJ) following standard procedures (including, when possible, the blinding of the experimenter to group assignments of the subjects) and based on prior work18,22. Three bilateral images of the vHPC were selected (−5.52mm, −5.76mm, and −6.00mm from Bregma) for mCherry-hM3D(Gq) and Fos double-labeling. For mPFC analyses, three bilateral images of the PL and IL were selected (PL: +4.2mm, +3.7mm and +3.2mm from bregma; IL: +3.7mm, +3.2mm and +3.00mm from bregma) for PV and Fos double-labeling.
Statistics
Paired t-tests were used to analyze inputs into mPFC subregions, interneuronal subtypes and IL sub-layers. Unpaired t-tests were used to analyse PV+-and SOM+-mediated IPSC amplitudes. Paired t-tests were used to analyze data in DIO-hM4D(Gi), DIO-GFP, and DIO-hM3D(Gq) experiments separately. Unpaired t-tests were used to analyze the PV/Fos dual-staining experiment. IL microinfusion and the non-Cre-dependent and Cre-dependent virus behavioral data were analyzed with repeated measures analysis of variance (ANOVA). Post-hoc comparisons in the form of Fisher’s protected least significant difference (PLSD) post hoc tests, which were performed after a significant overall F ratio. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those in prior reports and are typical for the field. The data distribution was assumed to be normal, but this was not formally tested. All data are represented as means ± s.e.m.
Data and code availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. No custom code was used in the current work.
Accession codes
No data with mandated deposition are included in this work.
Life Sciences Reporting Summary
Additional information regarding experimental design, reproducibility, software, and materials and reagents can be found in Life Sciences Reporting Summary.
Supplementary Material
Acknowledgments
We thank A. Woodruff for comments on the manuscript. We thank L. Xu, University of North Carolina Vector Core, University of Pennsylvania Vector Core, and the Institute of Molecular Genetics of Montpellier for producing viruses. This work was supported by the National Institutes of Health (R01MH065961 to S.M.; F31MH107113 to T.D.G.; F31MH112208 to T.F.G.), a McKnight Memory and Cognitive Disorders Award to S.M, and Australian Research Council (CE140100007) and National Health and Medical Research Grants to P.S.
Footnotes
Author contributions. S.M. and P.S. supervised all experiments. S.M., P.S., and R.M. designed the experiments. R.M., J.J., T.D.G., T.F.G., Q.W., G.M.A., and P.J.F. collected the data. R.M., J.J., T.D.G., T.F.G., Q.W., G.M.A., P.J.F., S.M., and P.S. analyzed the data. R.H. and J.E.P. generated and provided AAVDJ/8 viral vectors. T.P.L. and J.W.L. generated and provided the ivermectin construct. R.M., J.J., T.D.G., S.M., and P.S. wrote the manuscript; all authors read and edited the manuscript.
Competing financial interests. The authors declare no competing financial interests.
References
- 1.Quirk GJ, Milad MR. Neuroscience: Editing out fear. Nature. 2010;463:36–37. doi: 10.1038/463036a. [DOI] [PubMed] [Google Scholar]
- 2.Milad MR, Quirk GJ. Fear extinction as a model for translational neuroscience: ten years of progress. Annu Rev Psychol. 2012;63:129–151. doi: 10.1146/annurev.psych.121208.131631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goode TD, Maren S. Animal models of fear relapse. ILAR J. 2014;55:246–258. doi: 10.1093/ilar/ilu008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vervliet B, Craske MG, Hermans D. Fear extinction and relapse: state of the art. Annu Rev Clin Psychol. 2013;9:215–248. doi: 10.1146/annurev-clinpsy-050212-185542. [DOI] [PubMed] [Google Scholar]
- 5.Bouton ME. Context and behavioral processes in extinction. Learn Mem. 2004;11:485–494. doi: 10.1101/lm.78804. [DOI] [PubMed] [Google Scholar]
- 6.Bouton ME, Bolles RC. Contextual control of the extinction of conditioned fear. Learn Motiv. 1979;10:445–466. [Google Scholar]
- 7.Laurent V, Westbrook RF. Inactivation of the infralimbic but not the prelimbic cortex impairs consolidation and retrieval of fear extinction. Learn Mem. 2009;16:520–529. doi: 10.1101/lm.1474609. [DOI] [PubMed] [Google Scholar]
- 8.Milad MR, Quirk GJ. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature. 2002;420:70–74. doi: 10.1038/nature01138. [DOI] [PubMed] [Google Scholar]
- 9.Morgan MA, Romanski LM, LeDoux JE. Extinction of emotional learning: contribution of medial prefrontal cortex. Neurosci Lett. 1993;163:109–113. doi: 10.1016/0304-3940(93)90241-c. [DOI] [PubMed] [Google Scholar]
- 10.Likhtik E, Pelletier JG, Paz R, Paré D. Prefrontal control of the amygdala. J Neurosci. 2005;25:7429–7437. doi: 10.1523/JNEUROSCI.2314-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cho JH, Deisseroth K, Bolshakov VY. Synaptic encoding of fear extinction in mPFC-amygdala circuits. Neuron. 2013;80:1491–1507. doi: 10.1016/j.neuron.2013.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Knapska E, Maren S. Reciprocal patterns of c-Fos expression in the medial prefrontal cortex and amygdala after extinction and renewal of conditioned fear. Learn Mem. 2009;16:486–493. doi: 10.1101/lm.1463909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orsini CA, Kim JH, Knapska E, Maren S. Hippocampal and prefrontal projections to the basal amygdala mediate contextual regulation of fear after extinction. J Neurosci. 2011;31:17269–17277. doi: 10.1523/JNEUROSCI.4095-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maren S, Phan KL, Liberzon I. The contextual brain: implications for fear conditioning, extinction and psychopathology. Nat Rev Neurosci. 2013;14:417–428. doi: 10.1038/nrn3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corcoran KA, Maren S. Hippocampal inactivation disrupts contextual retrieval of fear memory after extinction. J Neurosci. 2001;21:1720–1726. doi: 10.1523/JNEUROSCI.21-05-01720.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hobin JA, Ji J, Maren S. Ventral hippocampal muscimol disrupts context-specific fear memory retrieval after extinction in rats. Hippocampus. 2006;16:174–182. doi: 10.1002/hipo.20144. [DOI] [PubMed] [Google Scholar]
- 17.Zelikowsky M, et al. Prefrontal microcircuit underlies contextual learning after hippocampal loss. Proc Natl Acad Sci U S A. 2013;110:9938–9943. doi: 10.1073/pnas.1301691110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin J, Maren S. Fear renewal preferentially activates ventral hippocampal neurons projecting to both amygdala and prefrontal cortex in rats. Sci Rep. 2015;5:8388. doi: 10.1038/srep08388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herry C, et al. Switching on and off fear by distinct neuronal circuits. Nature. 2008;454:600–606. doi: 10.1038/nature07166. [DOI] [PubMed] [Google Scholar]
- 20.Knapska E, et al. Functional anatomy of neural circuits regulating fear and extinction. Proc Natl Acad Sci U S A. 2012;109:17093–17098. doi: 10.1073/pnas.1202087109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu C, et al. Distinct hippocampal pathways mediate dissociable roles of context in memory retrieval. Cell. 2016;167:961–972.e16. doi: 10.1016/j.cell.2016.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Q, Jin J, Maren S. Renewal of extinguished fear activates ventral hippocampal neurons projecting to the prelimbic and infralimbic cortices in rats. Neurobiol Learn Mem. 2016;134(Pt A):38–43. doi: 10.1016/j.nlm.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parent MA, Wang L, Su J, Netoff T, Yuan LL. Identification of the hippocampal input to medial prefrontal cortex in vitro. Cereb Cortex. 2010;20:393–403. doi: 10.1093/cercor/bhp108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoover WB, Vertes RP. Anatomical analysis of afferent projections to the medial prefrontal cortex in the rat. Brain Struct Funct. 2007;212:149–179. doi: 10.1007/s00429-007-0150-4. [DOI] [PubMed] [Google Scholar]
- 25.Petilla Interneuron Nomenclature Group et al. Petilla terminology: nomenclature of features of GABAergic interneurons of the cerebral cortex. Nat Rev Neurosci. 2008;9:557–568. doi: 10.1038/nrn2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferreira AN, Yousuf H, Dalton S, Sheets PL. Highly differentiated cellular and circuit properties of infralimbic pyramidal neurons projecting to the periaqueductal gray and amygdala. Front Cell Neurosci. 2015;9:161. doi: 10.3389/fncel.2015.00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Little JP, Carter AG. Synaptic mechanisms underlying strong reciprocal connectivity between the medial prefrontal cortex and basolateral amygdala. J Neurosci. 2013;33:15333–15342. doi: 10.1523/JNEUROSCI.2385-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawaguchi Y, Kubota Y. Neurochemical features and synaptic connections of large physiologically-identified GABAergic cells in the rat frontal cortex. Neuroscience. 1998;85:677–701. doi: 10.1016/s0306-4522(97)00685-4. [DOI] [PubMed] [Google Scholar]
- 29.Tamás G, Lorincz A, Simon A, Szabadics J. Identified sources and targets of slow inhibition in the neocortex. Science. 2003;299:1902–1905. doi: 10.1126/science.1082053. [DOI] [PubMed] [Google Scholar]
- 30.Wozny C, Williams SR. Specificity of synaptic connectivity between layer 1 inhibitory interneurons and layer 2/3 pyramidal neurons in the rat neocortex. Cereb Cortex. 2011;21:1818–1826. doi: 10.1093/cercor/bhq257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kawaguchi Y, Kondo S. Parvalbumin, somatostatin and cholecystokinin as chemical markers for specific GABAergic interneuron types in the rat frontal cortex. J Neurocytol. 2002;31:277–287. doi: 10.1023/a:1024126110356. [DOI] [PubMed] [Google Scholar]
- 32.Bukalo O, et al. Prefrontal inputs to the amygdala instruct fear extinction memory formation. Sci Adv. 2015;1 doi: 10.1126/sciadv.1500251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duvarci S, Pare D. Amygdala microcircuits controlling learned fear. Neuron. 2014;82:966–980. doi: 10.1016/j.neuron.2014.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gomez JL, et al. Chemogenetics revealed: DREADD occupancy and activation via converted clozapine. Science. 2017;357:503–507. doi: 10.1126/science.aan2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Do-Monte FH, Manzano-Nieves G, Quiñones-Laracuente K, Ramos-Medina L, Quirk GJ. Revisiting the role of infralimbic cortex in fear extinction with optogenetics. J Neurosci. 2015;35:3607–3615. doi: 10.1523/JNEUROSCI.3137-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ishikawa A, Nakamura S. Convergence and interaction of hippocampal and amygdalar projections within the prefrontal cortex in the rat. J Neurosci. 2003;23:9987–9995. doi: 10.1523/JNEUROSCI.23-31-09987.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tierney PL, Dégenètais E, Thierry AM, Glowinski J, Gioanni Y. Influence of the hippocampus on interneurons of the rat prefrontal cortex. Eur J Neurosci. 2004;20:514–524. doi: 10.1111/j.1460-9568.2004.03501.x. [DOI] [PubMed] [Google Scholar]
- 38.Sotres-Bayon F, Sierra-Mercado D, Pardilla-Delgado E, Quirk GJ. Gating of fear in prelimbic cortex by hippocampal and amygdala inputs. Neuron. 2012;76:804–812. doi: 10.1016/j.neuron.2012.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Padilla-Coreano N, et al. Direct Ventral Hippocampal-Prefrontal Input Is Required for Anxiety-Related Neural Activity and Behavior. Neuron. 2016;89:857–866. doi: 10.1016/j.neuron.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jin J, Maren S. Prefrontal-Hippocampal Interactions in Memory and Emotion. Front Syst Neurosci. 2015;9:170. doi: 10.3389/fnsys.2015.00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shin LM, Rauch SL, Pitman RK. Amygdala, medial prefrontal cortex, and hippocampal function in PTSD. Ann N Y Acad Sci. 2006;1071:67–79. doi: 10.1196/annals.1364.007. [DOI] [PubMed] [Google Scholar]
- 42.Herry C, et al. Neuronal circuits of fear extinction. Eur J Neurosci. 2010;31:599–612. doi: 10.1111/j.1460-9568.2010.07101.x. [DOI] [PubMed] [Google Scholar]
- 43.Giustino TF, Maren S. The role of the medial prefrontal cortex in the conditioning and extinction of fear. Front Behav Neurosci. 2015;9:298. doi: 10.3389/fnbeh.2015.00298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marek R, Strobel C, Bredy TW, Sah P. The amygdala and medial prefrontal cortex: partners in the fear circuit. J Physiol (Lond) 2013;591:2381–2391. doi: 10.1113/jphysiol.2012.248575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adhikari A, et al. Basomedial amygdala mediates top-down control of anxiety and fear. Nature. 2015;527:179–185. doi: 10.1038/nature15698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim HS, Cho HY, Augustine GJ, Han JH. Selective Control of Fear Expression by Optogenetic Manipulation of Infralimbic Cortex after Extinction. Neuropsychopharmacology. 2016;41:1261–1273. doi: 10.1038/npp.2015.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maren S. Fear of the unexpected: hippocampus mediates novelty-induced return of extinguished fear in rats. Neurobiol Learn Mem. 2014;108:88–95. doi: 10.1016/j.nlm.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Knight R. Contribution of human hippocampal region to novelty detection. Nature. 1996;383:256–259. doi: 10.1038/383256a0. [DOI] [PubMed] [Google Scholar]
- 49.Eichenbaum H. Prefrontal-hippocampal interactions in episodic memory. Nat Rev Neurosci. 2017;18:547–558. doi: 10.1038/nrn.2017.74. [DOI] [PubMed] [Google Scholar]
- 50.Adhikari A, Topiwala MA, Gordon JA. Synchronized activity between the ventral hippocampus and the medial prefrontal cortex during anxiety. Neuron. 2010;65:257–269. doi: 10.1016/j.neuron.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci U S A. 2007;104:5163–5168. doi: 10.1073/pnas.0700293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lerchner W, et al. Reversible silencing of neuronal excitability in behaving mice by a genetically targeted, ivermectin-gated Cl− channel. Neuron. 2007;54:35–49. doi: 10.1016/j.neuron.2007.02.030. [DOI] [PubMed] [Google Scholar]
- 53.Lynagh T, Lynch JW. An improved ivermectin-activated chloride channel receptor for inhibiting electrical activity in defined neuronal populations. J Biol Chem. 2010;285:14890–14897. doi: 10.1074/jbc.M110.107789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Klapoetke NC, et al. Independent optical excitation of distinct neural populations. Nat Methods. 2014;11:338–346. doi: 10.1038/nmeth.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Han X, et al. A high-light sensitivity optical neural silencer: development and application to optogenetic control of non-human primate cortex. Front Syst Neurosci. 2011;5:18. doi: 10.3389/fnsys.2011.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Strobel C, Marek R, Gooch HM, Sullivan RKP, Sah P. Prefrontal and auditory input to intercalated neurons of the amygdala. Cell Rep. 2015 doi: 10.1016/j.celrep.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 57.Fitzgerald PJ, Giustino TF, Seemann JR, Maren S. Noradrenergic blockade stabilizes prefrontal activity and enables fear extinction under stress. Proc Natl Acad Sci U S A. 2015;112:E3729–37. doi: 10.1073/pnas.1500682112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Corcoran KA, Desmond TJ, Frey KA, Maren S. Hippocampal inactivation disrupts the acquisition and contextual encoding of fear extinction. J Neurosci. 2005;25:8978–8987. doi: 10.1523/JNEUROSCI.2246-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Giustino TF, et al. β-Adrenoceptor Blockade in the Basolateral Amygdala, But Not the Medial Prefrontal Cortex, Rescues the Immediate Extinction Deficit. Neuropsychopharmacology. 2017 doi: 10.1038/npp.2017.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. No custom code was used in the current work.