

Abstract
Colony-stimulating factor 1 (CSF1) is a key regulator of monocyte/macrophage differentiation that sustains the protumorigenic functions of tumor-associated macrophages (TAMs). We show that CSF1 is expressed in human melanoma, and patients with metastatic melanoma have increased CSF1 in blood compared to healthy subjects. In tumors, CSF1 expression correlated with the abundance of CD8+ T cells and CD163+ TAMs. Human melanoma cell lines consistently produced CSF1 after exposure to melanoma-specific CD8+ T cells or T cell–derived cytokines in vitro, reflecting a broadly conserved mechanism of CSF1 induction by activated CD8+ T cells. Mining of publicly available transcriptomic data sets suggested co-enrichment of CD8+ T cells with CSF1 or various TAM-specific markers in human melanoma, which was associated with nonresponsiveness to programmed cell death protein 1 (PD1) checkpoint blockade in a smaller patient cohort. Combination of anti-PD1 and anti–CSF1 receptor (CSF1R) antibodies induced the regression of BRAFV600E-driven, transplant mouse melanomas, a result that was dependent on the effective elimination of TAMs. Collectively, these data implicate CSF1 induction as a CD8+ T cell–dependent adaptive resistance mechanism and show that simultaneous CSF1R targeting may be beneficial in melanomas refractory to immune checkpoint blockade and, possibly, other T cell–based therapies.
INTRODUCTION
Tumor-associated macrophages (TAMs) frequently make up a substantial proportion of all tumor-infiltrating immune cells. TAMs play complex immunological roles in the tumor microenvironment, which vary with their activation state (1, 2). Although a spectrum of TAM phenotypes likely exists in tumors, two opposing phenotypes, denoted classically activated (or M1-like) and alternatively activated (or M2-like), have been associated with anti- and protumoral functions, respectively (3–5). Progressing tumors typically display M2-like TAM activation in response to various tumor-derived factors (1, 2). M2-polarized TAMs facilitate angiogenesis, cancer cell invasion, and metastasis and may also suppress T cells (6–8). Conversely, there is evidence for M1-like TAMs engaging tumor-antagonizing functions, and their presence and roles in tumors have been primarily characterized in the context of anticancer therapies or gene-targeting studies (9, 10). Although abundant TAM infiltrates generally associate with poor clinical outcome in most cancer types (11–13), some studies have also shown that M1-like TAMs may correlate positively with patient survival (13, 14). Macrophages make up a significant, albeit variable, proportion of the immune cell infiltrate in human melanoma (2), but their significance for clinical outcome remains unclear. Although several studies showed that macrophage abundance correlates with melanoma thickness (15–19), only one study (18) out of six examined (15–20) suggested that TAMs may correlate with poor overall survival in patients with malignant melanoma.
Colony-stimulating factor 1 [CSF1; also known as macrophage colony-stimulating factor (M-CSF)] controls proliferation, differentiation, and survival of macrophages from their precursors (21, 22). CSF1 receptor (CSF1R) signaling in TAMs may promote their acquisition of an immunosuppressive and protumorigenic, M2-like phenotype (10, 23, 24). For example, expression of CSF1 is associated with poor prognosis in tumors of the reproductive system, such as ovarian, uterine, breast, and prostate cancers (25, 26). Accordingly, preclinical studies have shown that CSF1R blockade may delay tumor growth in some mouse cancer models through the elimination or repolarization of TAMs (9, 10, 27–32). Two phase 1 clinical trials documented objective clinical responses after CSF1R blockade in patients with diffuse-type tenosynovial giant cell tumors, a degenerative condition sustained by proliferating macrophages (31–33). However, single-agent CSF1R blockade had limited activity in Hodgkin lymphoma (34, 35) and provided no benefits in glioblastoma (36), according to phase 2 and 2/3 clinical trials.
In recent years, antibodies targeting immune inhibitory receptors (“checkpoints”) have markedly improved the clinical outcome of patients with advanced melanoma. The U.S. Food and Drug Administration (FDA) approved the first anti–cytotoxic T lymphocyte–associated protein 4 (CTLA4) antibody, ipilimumab, in 2011, followed by two programmed cell death protein 1 (PD1)–specific antibodies, pembrolizumab and nivolumab, in 2014 (37). Three programmed death ligand 1 (PDL1)–specific antibodies—atezolizumab, durvalumab, and avelumab—have also been approved by the FDA (38, 39). The response to anti-PD1 therapy may correlate with prognostic baseline markers, such as expression of PDL1 in the tumors, high intratumoral T cell infiltration, or the abundance of tumor-specific T cells (40, 41). However, clinical responses are not always associated with these prognostic biomarkers, suggesting that additional mechanisms may hamper effective T cell responses in melanoma.
Generally, immune checkpoint blockade aims to support the activation of antitumor T cells (42). Two major cytokines secreted by activated CD8+ T cells are interferon-γ (IFNγ) and tumor necrosis factor–α (TNFα), which are both known to induce CSF1 expression in various cell types of the tumor microenvironment (43). This raises the possibility that a feedback mechanism conducive to T cell suppression may be instigated by T cells through CSF1 induction and the recruitment or activation of immunosuppressive and protumoral TAMs. Here, we show that CSF1 expression by melanoma cells may limit immune attack by activated, IFNγ-secreting CD8+ T cells in patients with metastatic cutaneous melanoma. Our results support the notion that concomitant blockade of the CSF1/CSF1R pathway may improve intratumoral CD8+ T cell function and melanoma treatment with immune checkpoint inhibitors, such as PD1- or PDL1-blocking antibodies. Although monotherapy with CSF1R inhibitors may have limited clinical efficacy (34, 35), its combination with immune checkpoint blockade has the potential to provide clinical benefits to patients with melanoma and, possibly, other cancer types.
RESULTS
Melanoma progression is associated with increased CSF1 concentration in peripheral blood
We measured CSF1 in the blood of 12 healthy individuals and 15 patients with distant metastases (stage IV) or locally advanced (stage III) cutaneous melanoma (table S1), staged according to the American Joint Committee on Cancer. CSF1 was significantly increased (P < 0.0001) in melanoma patients, suggesting that CSF1 production is tumor-induced (Fig. 1A). A positive correlation was observed between CSF1 and lactate dehydrogenase (LDH), a biomarker of disease burden (44), in the blood of the patients (Fig. 1B). In addition, analysis of a larger patient cohort (n = 40) showed higher concentration of blood CSF1 in patients at stage IV compared to stage IIIB (Fig. 1C). These findings indicate that CSF1 production is elevated in patients with melanoma and increases with disease progression.
Fig. 1. CSF1 is increased in blood of melanoma patients and correlates with disease progression.
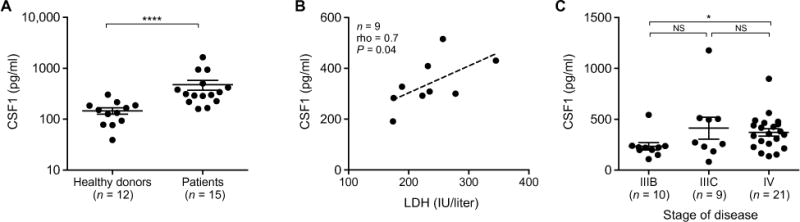
(A) CSF1 concentration in the plasma of healthy donors (n = 12) and melanoma patients (n = 15), quantified by enzyme-linked immunosorbent assay (ELISA). Data are means ± SEM. (B) Correlation between LDH and CSF1 concentration in the serum of nine melanoma patients, of which the LDH concentration was available, using Spearman’s correlation coefficient. The dashed line indicates least-squares linear fit. (C) CSF1 concentration in the serum of melanoma patients (40 samples from 27 patients analyzed at different time points of disease progression). Patients were grouped by disease stage at the time of sample withdrawal. Data are means ± SEM. n, number of samples; rho, Spearman’s correlation coefficient; NS, not significant. Statistical analysis in (A) and (C) by Mann-Whitney U test. *P < 0.05; ****P < 0.0001.
Melanoma infiltration by CD8+ T cells correlates with enrichment of CSF1+, CSF1R+, and CD163+ cells
To determine whether the expression of CSF1 is associated with the abundance of CSF1R+ macrophages in human melanoma, we examined CSF1 and CSF1R expression by chromogenic immunohistochemistry (IHC). In both primary tumors and cutaneous metastases (table S1), the expression of CSF1 correlated with that of CSF1R (Fig. 2A), suggesting a relationship between CSF1 production and TAM abundance.
Fig. 2. Tumor infiltration by CD8+ T cells correlates with enrichment of CSF1+, CSF1R+, and CD163+ cells in primary tumors and skin metastases of melanoma patients.
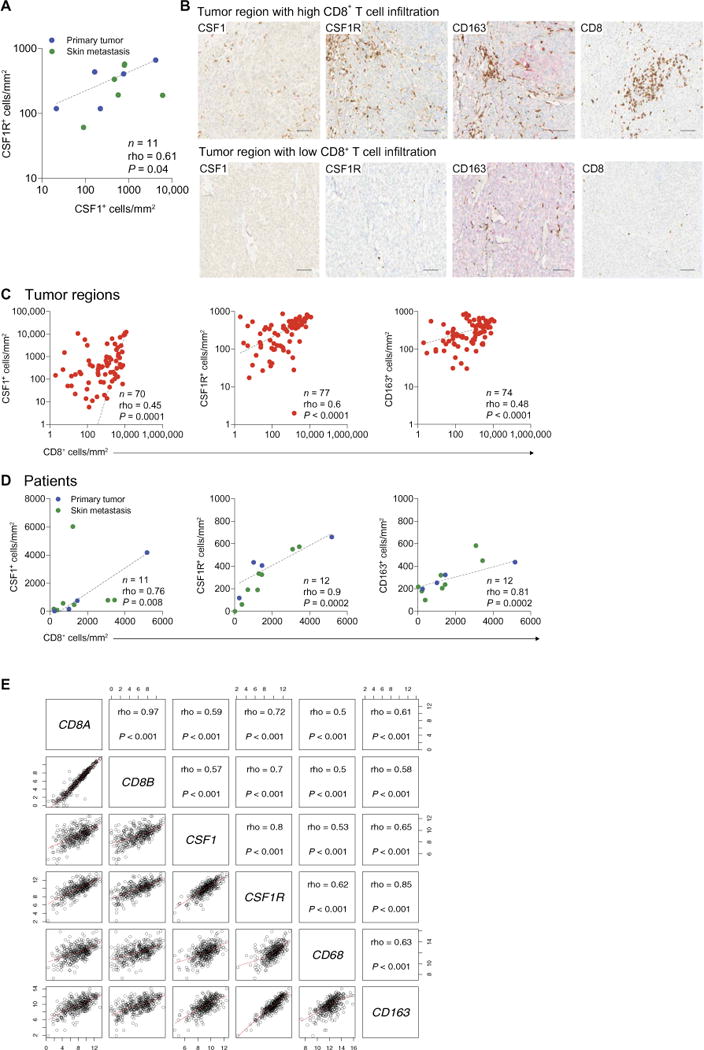
(A) Correlation between density of CSF1+ and CSF1R+ cells according to chromogenic IHC, assessed by Spearman’s correlation coefficient. The dashed line indicates log-log correlation. (B) Representative images of CSF1, CSF1R, CD163, and CD8 immunostaining selected from a tumor region with either high or low CD8+ T cell infiltration in a skin metastasis of patient LAU1283. Scale bars, 100 μm. (C and D) Spearman’s correlations of CD8+ with CSF1+ (left), CSF1R+ (middle), or CD163+ (right) cells displayed per tumor region (C) or per patient (D). Dashed lines indicate log-log correlation (C) or linear regression (D). Data are from primary melanomas and melanoma skin metastases of the patients listed in table S1. (E) Matrix of scatterplots showing correlations between CD8A, CD8B, CSF1, CSF1R, CD68, and CD163 gene expression in the SKCM metastatic cohort (n = 369) of TCGA (46). Correlation was assessed using Spearman’s correlation coefficient. Red lines indicate the local regression (LOESS) fit. P, P value; n, number of samples; rho, Spearman’s correlation coefficient.
CSF1 may promote T cell suppression by expanding immunosuppressive TAMs (45). To test this hypothesis, we quantified the expression of CD8, CSF1, CSF1R, and the M2 marker CD163 on histological sections of primary tumors and cutaneous metastases. Tumor regions with high density of tumor-infiltrating CD8+ T cells were also enriched in CSF1+ cells and CSF1R+ or CD163+ TAMs, whereas tumor regions with scant CD8+ T cell infiltrates displayed poor TAM infiltration (fig. S1 and Fig. 2, B and C). We observed these correlations in patients with either high or low CD8+ T cell infiltration in their tumors (Fig. 2D). We next examined whether the correlations between CD8+ T cells and TAMs that we observed in our patient cohort could be confirmed in an independent, larger patient cohort. Transcriptomic data of skin cutaneous melanoma (SKCM) of The Cancer Genome Atlas (TCGA) (46) showed that, in metastatic tumors, the expression of CD8A and CD8B strongly correlated with the expression of CSF1, CSF1R, CD68, and CD163 (Fig. 2E), but not with the melanoma-specific marker genes MIA (melanoma inhibitory activity) and TYR (tyrosinase), which we analyzed as “housekeeping” controls (fig. S2). Together, these results strongly argue that the abundance of CD8+ T cells positively correlates with TAM infiltration in human cutaneous melanoma.
Tumor cells are a source of CSF1 in human melanoma
We then asked whether melanoma cells could be a source of CSF1 in the tumors. We performed multiplexed immunofluorescence staining on histological sections of eight melanoma specimens (table S1). Melanoma cells were identified by their expression of the S100 protein, which is the most sensitive diagnostic marker for melanoma cells (47). We found that about 80% of the S100+ melanoma cells expressed CSF1 (Fig. 3, A and B). We then reassessed CSF1 expression in relation to CD8+ T cell abundance and found that it was higher in tumor regions with high than low CD8+ T cell density (fig. S3, A to C), which confirmed the IHC data (Fig. 2, C and D).
Fig. 3. CSF1 is expressed in human melanoma.
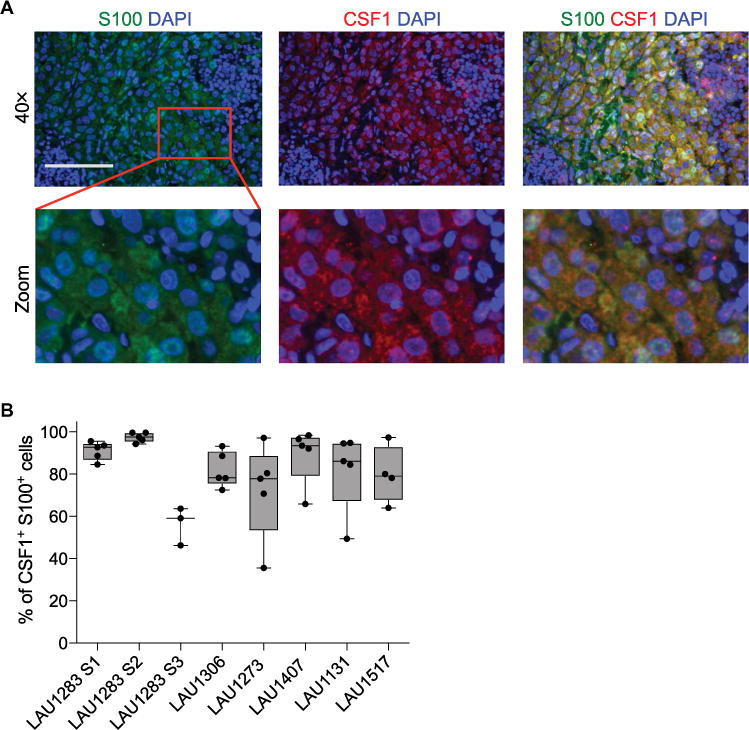
(A) Representative multiplexed fluorescence staining images of tumor tissue from one melanoma patient (LAU1283) stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue) and antibodies against S100 (green) or CSF1 (red). The red square in the upper panel indicates a region of interest that has been magnified in the lower panels. Scale bar, 100 μm. (B) Percentage of S100+ melanoma cells that are also CSF1+ in primary melanomas (n = 4) and melanoma metastases (n = 4) from the patients listed in table S1. Each data point represents the average (means ± SEM) of three to five images per tumor area for each patient. S1 to S3 indicate three individual specimens from patient LAU1283 (details are provided in table S1).
Melanoma-specific CTLs trigger CSF1 production by melanoma cells
To evaluate whether melanoma cells constitutively secrete CSF1, we tested four low-passage human melanoma cell lines for their ability to produce CSF1 (Fig. 4A). Although CSF1 was low or undetectable in medium conditioned by untreated melanoma cell cultures, it was induced by coculturing the melanoma cell lines with melanoma-specific cytotoxic CD8+ T lymphocyte (CTL) clones (Fig. 4B) (48). The CTLs were specific for human leukocyte antigen–A2 (HLA-A2)/MelanA/MART-1 (hereafter MelanA), and all four melanoma cell lines were positive for both HLA-A2 and MelanA (48, 49). Two of the four CTL-melanoma cocultures were autologous (T1185B and T1015A), whereas the other two (Me275 and Me290) were matched for HLA-A2. Notably, HLA-A2–restricted CTLs with irrelevant specificity (yellow fever virus–specific CTLs) did not trigger CSF1 secretion by melanoma cells.
Fig. 4. CSF1 secretion by melanoma cells is induced by CTL-derived cytokines.
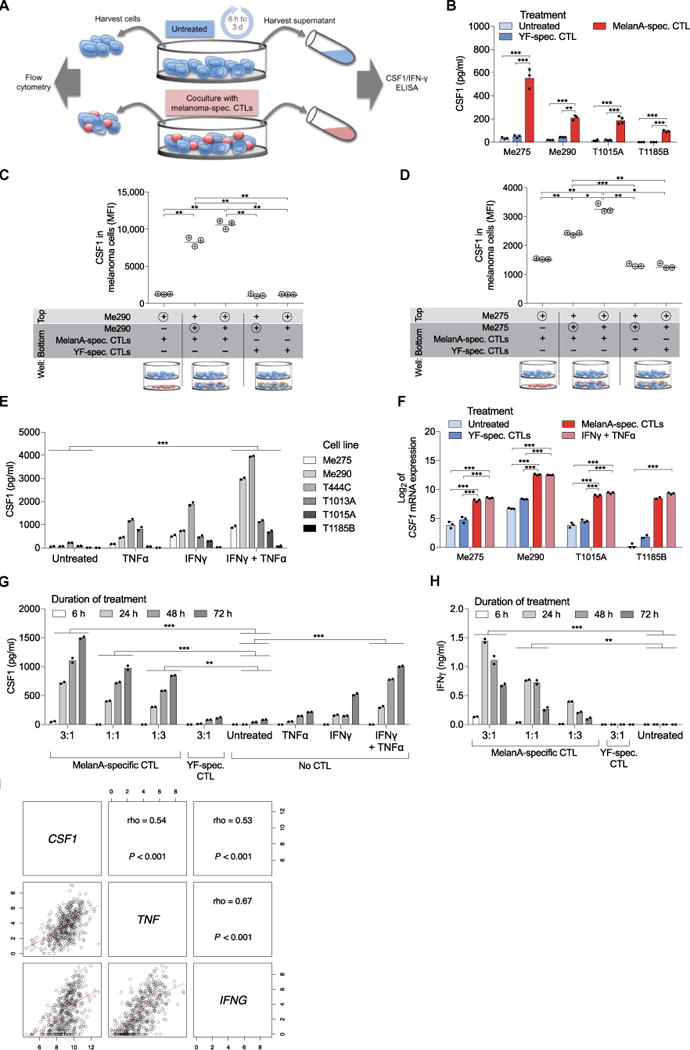
(A) Experimental setup of the coculture study analyzed in (B) and (F) to (H). Cocultures were established at a 1:1 CTL/melanoma cell ratio, unless stated otherwise. (B) Co-culture experiments. CSF1 protein concentration in supernatants of melanoma cell lines after 24 hours of culture alone or in the presence of MelanA- or yellow fever virus (YF)–specific CTLs, quantified by ELISA. Four melanoma cell lines are shown. Data are means ± SD; three independent cell cultures per cell line are shown, except for T1015A, of which four independent cell cultures were analyzed for melanoma cells cultured alone or with MelanA-specific CTLs. (C and D) Transwell experiments. CSF1 expression was analyzed by flow cytometry in the melanoma cells indicated by the symbol ⊕ after 48 hours of culture with the indicated CTLs (n = 3). Pore size of the filter was 0.4 μm, allowing soluble factors to pass but not cell migration between the top and bottom chamber. MFI, median fluorescence intensity. (E) CSF1 concentration in supernatant of six different melanoma cell lines after 48 hours of culture alone, in the presence of IFNγ, TNFα, or both, quantified by ELISA. Data represent the mean of two independent cell cultures per cell line. (F) Log2-transformed, normalized ex pression value of CSF1 mRNA in melanoma cells after 24 hours of the indicated treatment, quantified by NanoString. Data are means ± SD, except for T1185B, of which two independent cell cultures were analyzed for melanoma cells cocultured with MelanA- or YF-specific CTLs. (G and H) Concentration of CSF1 (G) or IFNγ (H) determined by ELISA in supernatants of Me275 melanoma cells cultured as indicated for 6 to 72 hours. Data represent the mean of two independent cell cultures. (I) Matrix of scatter-plots showing correlations between the expression of CSF1, IFNG, and TNF genes in the SKCM metastatic cohort (n = 369) of TCGA (46). Spearman’s correlation coefficients are indicated, and the red lines show the local regression (LOESS) fit. P, P value; n, number of samples; rho, Spearman’s correlation coefficient. Statistical analysis by one-way analysis of variance (ANOVA) (B to D and F) or two-way ANOVA (E, G, and H) with correction for multiple comparisons by post hoc Tukey’s test (B to H) and Geisser-Greenhouse correction (C and D). ***P < 0.001; **P < 0.01; *P < 0.05.
We then analyzed intracellular CSF1 expression in the Me275 melanoma cell line by flow cytometry. Intracellular CSF1 increased in the melanoma cells after encounter with melanoma-specific CTLs (fig. S4A). Intracellular CSF1 was also increased in CTLs cocultured with antigen-expressing melanoma cells (fig. S4A). Together, these data support the notion that melanoma-specific CD8+ T cells stimulate CSF1 expression in human melanoma.
To assess whether the induction of CSF1 secretion was dependent on direct contacts between CTLs and melanoma cells, we performed distinct cell culture assays. Two different melanoma cell lines (Me290 and Me275) up-regulated CSF1 expression in the presence of activated MelanA-specific CTLs, both in direct contact (coculture) and in a transwell assay enabling the passage of soluble factors but not cells (Fig. 4, C and D). Nonspecific CTLs failed to induce CSF1 in either cell culture assay. Thus, CTLs that have been activated through antigen-specific interaction can induce CSF1 expression in neighboring melanoma cells without the requirement of further direct cell contacts.
We then asked whether CTL-derived soluble factors induce CSF1 production in melanoma cells. To this aim, we cultured human melanoma cell lines in the presence of combinations of IFNγ and TNFα, which are cytokines prominently secreted by activated CTLs (50). The two cytokines induced CSF1 secretion from the melanoma cell lines in the absence of melanoma-specific CTLs (Fig. 4E). Although CSF1 was induced by either IFNγ or TNFα alone, the combination of the two cytokines had the strongest effects. Fifteen additional melanoma cell lines were tested and found to secrete only low amounts of CSF1 in the absence of cytokines, but stimulation with IFNγ and TNFα strongly increased CSF1 secretion in all cell lines (fig. S4B and table S2). In addition, blockade of IFNγ and TNFα during coculture attenuated the expression and secretion of CSF1 by melanoma cells (fig. S4, C and D). CSF1 mRNA expression was strongly increased in all four examined cell lines within 24 hours of coculture with melanoma-specific CTLs or stimulation with IFNγ and TNFα, but not in the presence of CTLs with irrelevant specificity (Fig. 4F), confirming the rapid and strong transcriptional up-regulation of CSF1 in melanoma cells. A 72-hour kinetic analysis of melanoma–T cell cocultures revealed a peak of IFNγ concentration at 24 hours, which coincided with the onset of CSF1 protein in the cell culture supernatant (Fig. 4, G and H). Finally, to explore the potential occurrence of autocrine signaling, we analyzed CSF1R expression in melanoma cells either unstimulated or exposed to IFNγ and TNFα but found no detectable CSF1R expression on melanoma cells (fig. S5). Together, these data identify IFNγ and TNFα as CTL-derived factors that induce CSF1 expression in melanoma cells.
We next examined the expression of CSF1, IFNG, and TNF in the metastatic cohort of TCGA melanoma data set (46). We found significant correlations (P < 0.001) between the expression of the three genes in cutaneous melanoma samples (Fig. 4I). The finding that CTL-derived cytokines promote CSF1 secretion is consistent with our previous study showing that, upon encountering melanoma-specific CTLs, melanoma cells activate a number of immune cell genes (49), some of which have been implicated in resistance to immunotherapy (51–54). Motivated by these earlier findings, we analyzed the expression of additional factors that may support immunosuppressive macrophage recruitment and found that melanoma cells also up-regulated TGFB, IL10, VEGFA, and VEGFC upon coculture with MelanA-specific CTLs or after exposure to IFNγ and TNFα (fig. S6).
Markers of CD8+ T cells and TAMs correlate with response to PD1 blockade in human melanoma
To further explore the associations between CTLs and TAMs in human melanoma, we analyzed the expression of CD8A and a compendium of macrophage-specific genes, previously identified in metastatic melanomas through single-cell RNA sequencing (RNA-seq) (55), in the SKCM metastatic cohort of TCGA (46). Compared to tumors with low CD8A expression, tumors with high CD8A expression were highly enriched for the macrophage signature (mean fold change > 2; P < 0.001) (Fig. 5A and table S3).
Fig. 5. Markers of CD8+ T cells and TAMs correlate with response to PD1 blockade in human melanoma.
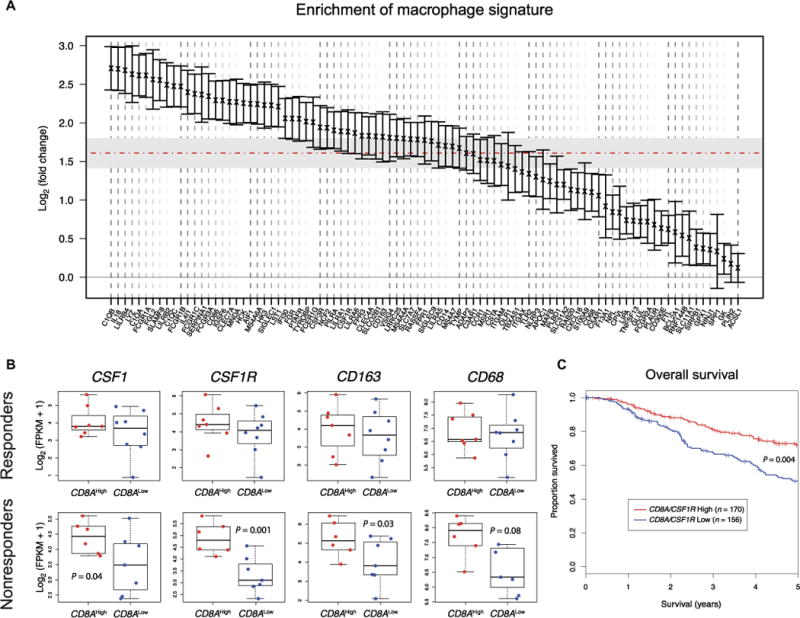
(A) Enrichment of melanoma-derived macrophage-specific transcriptional signature from (55) in CD8AHigh versus CD8ALow melanomas of the SKCM metastatic cohort (n = 369) (46). Genes are ranked based on mean fold change expression. Error bars indicate 95% confidence interval for each gene. The horizontal dashed line indicates mean fold change of the macrophage signature. Gray region indicates 95% confidence interval of the overall mean fold change. (B) CSF1, CSF1R, CD163, and CD68 expression in responder (n = 15) and nonresponder (n = 13) melanomas to anti-PD1 therapy, obtained from (56). Stratification as CD8AHigh or CD8ALow was performed using the median expression. Statistical analysis by Student’s t test. FPKM, fragments per kilobase of transcript per million mapped reads. (C) Kaplan-Meier estimate of survival in the SKCM metastatic cohort (n = 326, for which survival data were available) of TCGA (46), stratified as (CD8A/CSF1R) ratio high (n = 170) or (CD8A/CSF1R) ratio low (n = 156), using median ratio as cutoff value. The CD8A/CSF1R ratio denotes the numeric difference between log-transformed expression data. P value was obtained using the log-rank test and adjusted for tumor stage.
To explore the clinical significance of the CTL-TAM interplay, we examined the expression of CSF1, CSF1R, CD163, and CD68 in pretreatment tumor biopsies of metastatic melanoma patients stratified into responders and nonresponders after anti-PD1 therapy (56). The overall expression of the selected genes was not different between responders and nonresponders (fig. S7). However, further stratifying tumors based on CD8A expression revealed that, unlike the responders, the nonresponders showed an association between high CD8A expression and high expression of CSF1, CSF1R, and CD163 (Fig. 5B). To evaluate the impact of CTL and TAM infiltrates on melanoma prognosis, we analyzed CD8A and CSF1R expression and overall patient survival in the SKCM metastatic cohort (46). We found that a high CD8A/CSF1R ratio—a surrogate for the relative abundance of CD8+ T cells over TAMs—correlated with improved overall survival (P < 0.01; Fig. 5C). We obtained similar results by using alternative markers for both CD8+ T cells (GZMA and PRF1) and TAMs (CD68 and CD163) (fig. S8), which indicates that the observed correlations are cell type–specific and valid beyond single markers. Collectively, these findings support the notion that CTLs may drive the recruitment of immunosuppressive macrophages to human melanoma. Moreover, they suggest that TAM recruitment by activated CTLs may contribute to limit a potentially more robust antitumor immune response in the patients.
CSF1R inhibition enhances the therapeutic efficacy of PD1 blockade in BRAFV600E-driven transplant melanoma models
The results described above prompted us to investigate whether TAMs may limit melanoma response to immune checkpoint blockade. We used ovalbumin (OVA)–expressing SM1 cells (SM1-OVA), a mutant BRAFV600E-driven mouse melanoma cell line (57) that secretes high amounts of CSF1 (fig. S9). When inoculated in syngeneic C57Bl/6 mice, SM1 cells establish macrophage-rich tumors (27). We found that combined CSF1R and PD1 blockade improved tumor growth inhibition compared to the single agents (Fig. 6, A to C). Dual CSF1R and PD1 blockade induced complete regression of all tumors by 17 days after tumor challenge. Notably, single CSF1R blockade only slightly delayed tumor growth, whereas single PD1 blockade induced tumor regression in several, but not all, mice and with delayed kinetics compared to the combined treatment.
Fig. 6. CSF1R blockade enhances the therapeutic efficacy of anti-PD1 treatment in BRAFV600E-driven transplant melanoma models.
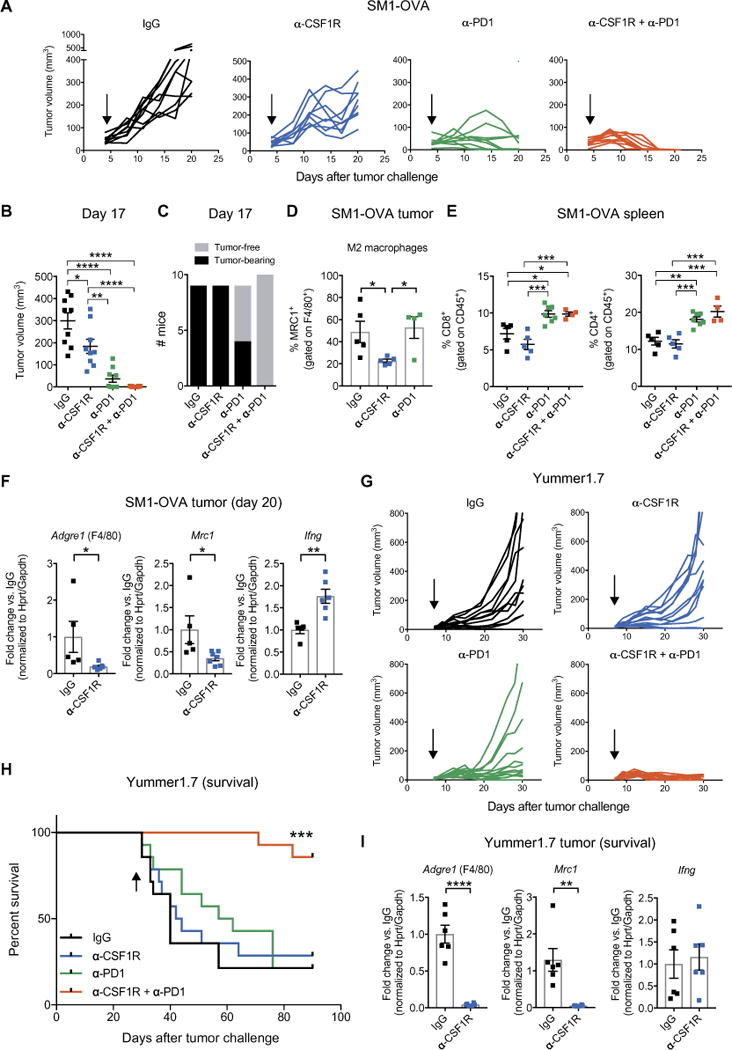
(A) Tumor volumes of subcutaneous SM1-OVA melanomas treated as indicated. IgG (n = 9), α-CSF1R (n = 9), α-PD1 (n = 9), and α-CSF1R + α-PD1 (n = 10). Arrows indicate start of treatment. (B) SM1-OVA tumor volumes (means ± SEM), measured at day 17 after tumor inoculation. Each dot represents one tumor. Statistical analysis by one-way ANOVA with Tukey’s correction for multiple comparisons. (C) Number of tumor-free and SM1-OVA tumor-bearing mice on day 17 after tumor inoculation. (D) Percentage of MRC1+ (M2-like) TAMs (means ± SEM) at day 20 after tumor inoculation, determined by flow cytometry of whole tumor-derived cells. Each dot represents one tumor. Flow cytometric analysis was performed on tumors selected for having a comparable size (only the smaller tumors in the IgG and CSF1R were analyzed) from the experiment shown in (A). IgG (n = 5), α-CSF1R (n = 5), and α-PD1 (n = 4). Note that tumors in the combination group could not be analyzed because they had fully regressed by day 20. Statistical analysis by one-way ANOVA with Fisher’s least significant difference (LSD) test. (E) Percentage of splenic CD8+ or CD4+ T cells (means ± SEM) determined by flow cytometry. IgG (n = 5), α-CSF1R (n = 5), α-PD1 (n = 7), and α-CSF1R + α-PD1 (n = 4). Flow cyto-metric analysis was performed on spleen from mice also used in (D). For the α-PD1 group, three additional spleens from mice whose tumors had regressed were used to exclude differences between tumor-free and tumor-bearing mice. Each dot represents one spleen. Statistical analysis by one-way ANOVA with Tukey’s correction for multiple comparisons. (F) qPCR analysis of Adgre1 (F4/80), Mrc1 (CD206), and Ifng from whole tumor lysates. Data indicate mean fold change values ± SEM over the reference sample (IgG) after normalization to the average of Hprt and Gapdh housekeeping genes. IgG (n = 5) and α-CSF1R (n = 6 to 7). Each dot represents one tumor; qPCR analysis was performed on tumors with similar size from the experiment shown in (A). Statistical analysis by Student’s t test. (G) Tumor volumes of subcutaneous Yummer1.7 melanomas treated as indicated. IgG (n = 14), α-CSF1R (n = 14), α-PD1 (n = 14), and α-CSF1R + α-PD1 (n = 14). Arrows indicate start of treatment. (H) Survival of mice bearing Yummer1.7 melanomas; the mice were euthanized when the tumors reached a volume of 1000 mm3. The arrow indicates the last treatment. Statistical analysis by log-rank test. (I) qPCR analysis of Adgre1 (F4/80), Mrc1 (CD206) and Ifng expression in lysates of Yummer1.7 melanomas treated as indicated and analyzed at termination (between day 30 and day 42 after tumor challenge). Data indicate mean fold change values ± SEM over the reference sample (IgG) after normalization to the average of Hprt and Gapdh housekeeping genes. For qPCR, we analyzed tumors of the first six mice that reached the termination endpoint (1000 mm3) in the experiment shown in (G). IgG (n = 5 to 6) and α-CSF1R (n = 6). Each dot represents one tumor. Statistical analysis by Student’s t test. ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05.
Flow cytometric analysis of the tumors showed a significant reduction of MRC1+ (M2-like) TAMs (9) after anti-CSF1R treatment (Fig. 6D) and a significant increase of both CD4+ and CD8+ T cells in the spleens of mice treated with anti-PD1 or combined anti-PD1 and anti-CSF1R antibodies (Fig. 6E). Accordingly, quantitative polymerase chain reaction (qPCR) analysis of tumor samples revealed decreased expression of Mrc1 (CD206) and Adgre1 (F4/80), indicative of macrophage elimination, and elevated expression of Ifng, after CSF1R blockade (Fig. 6F). Six mice in the anti-PD1 plus anti-CSF1R treatment cohort were monitored for six additional weeks after the termination of the treatment and remained tumor-free.
We then used another transplant melanoma model, Yummer1.7, which does not express the highly immunogenic OVA protein but has been subject to subsequent rounds of ultraviolet irradiation to induce de novo mutations (58) that better recapitulate the high mutational load of human melanoma (46). Yummer1.7 cells express CSF1 constitutively (fig. S9). Anti-PD1 treatment inhibited Yummer1.7 tumor growth in most of the mice (Fig. 6G) but did not extend survival after therapy withdrawal (Fig. 6H). Similar to findings in the SM1-OVA model, CSF1R blockade had no growth-inhibitory activity in the Yummer1.7 model, although both M2-like MRC1+ TAMs and total F4/80+ TAMs were markedly decreased upon treatment (Fig. 6I). Remarkably, the combination of anti-CSF1R and anti-PD1 antibodies fully eradicated most of the tumors and greatly extended mouse survival after therapy, indicating strongly additive therapeutic effects of the two antibodies.
Finally, we generated a transgenic melanoma model, called iBIP2 (see Supplementary Materials and Methods), which is driven by tamoxifen- and doxycycline-inducible Cdkn2a and Pten deletion and BRAFV600 expression in skin melanocytes. iBIP2 transgenic mice were obtained by refining the existing iBIP melanoma model, which carries constitutively deleted Cdkn2a alleles, floxed Pten alleles, and inducible BRAFV600 expression (59). At variance with results obtained with transplant tumor models, combined CSF1R and PD1 blockade did not inhibit melanoma growth in iBIP2 transgenic mice (Fig. 7A). Notably, anti-CSF1R antibodies failed to deplete protumorigenic macrophages in the transgenic melanomas, as shown by both flow cytometry (Fig. 7B) and qPCR analysis of macrophage-specific genes (Fig. 7C). These findings suggest that transgenic iBIP2 melanomas are refractory to TAM elimination and/or repolarization by anti-CSF1R antibodies, which may explain the lack of therapeutic responses.
Fig. 7. Differences in the tumor microenvironment between transplant and transgenic BRAFV600E-driven melanoma models may underlie refractoriness of iBIP2 tumors to immunotherapy.
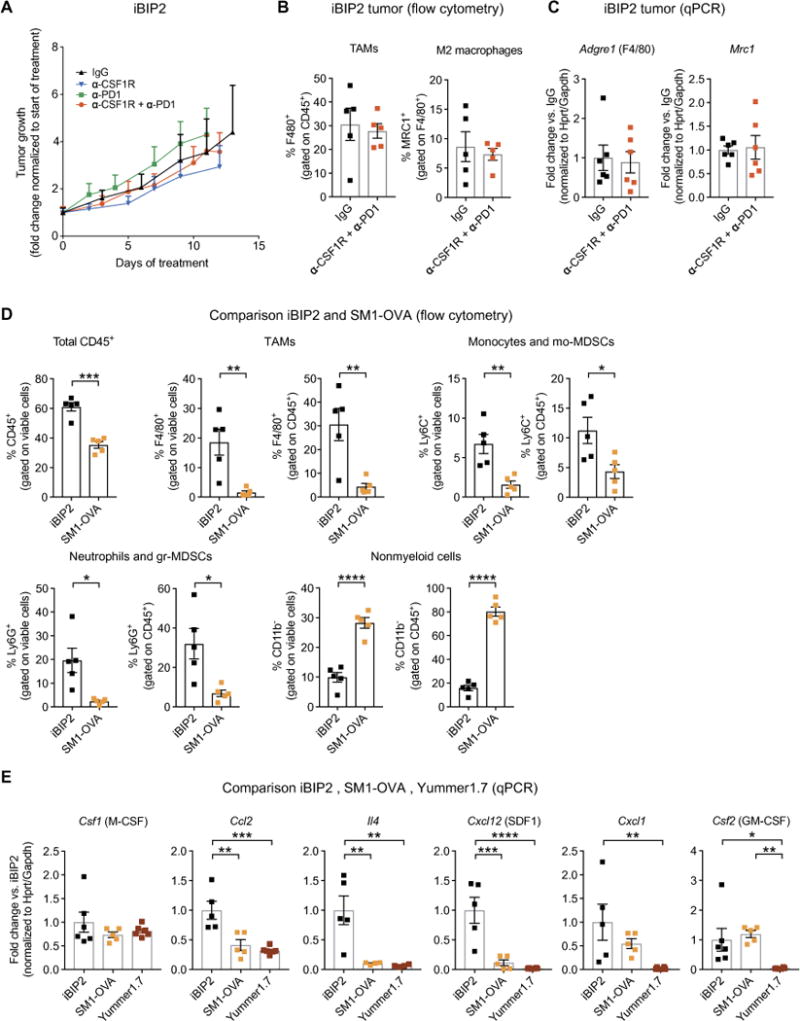
(A) Mean change of tumor volume (±SEM; versus tumor volume at treatment start) in iBIP2 mice treated as indicated. IgG (n = 5), α-CSF1R (n = 7), α-PD1 (n = 8), and α-CSF1R + α-PD1 (n = 18). Statistical analysis by linear mixed modeling, showing no statistically significant interaction effect between time and treatment as predictors of tumor volume fold change. (B) Percentage of F4/80+ and MRC1+ (M2-like) TAMs (means ± SEM) in iBIP2 tumors treated as indicated and analyzed by flow cytometry. IgG (n = 5) and α-CSF1R + α-PD1 (n = 5). Each dot represents one tumor; the analysis was performed on tumors with similar size from the experiment shown in (A). Statistical analysis by Student’s t test. No statistically significant differences were observed. (C) qPCR analysis of Adgre1 (F4/80) and Mrc1 (CD206) in lysates of iBIP2 tumors treated as indicated. Data represent mean fold change values ± SEM over the reference sample (IgG) after normalization to the average of Hprt and Gapdh housekeeping genes. IgG (n = 6) and α-CSF1R + α-PD1 (n = 6). Each dot represents one tumor; the analysis was performed on tumors with similar size from the experiment shown in (A). Statistical analysis by Student’s t test. No statistically significant differences were observed. (D) Percentage of CD45+ hematopoietic cells, F4/80+ TAMs, Ly6C+ monocytes and mo-MDSCs, Ly6G+ neutrophils and gr-MDSCs, and CD11b− non-myeloid cells (means ± SEM) in iBIP2 and SM1-OVA tumors treated with control IgGs and analyzed by flow cytometry. iBIP2 (n = 5) and SM1-OVA (n = 5). Statistical analysis by Student’s t test. (E) qPCR analysis of Csf1, Ccl2, Il4, Cxcl12 (SDF1), Cxcl1, and Csf2 [granulocyte-macrophage CSF (GM-CSF)] in lysates of iBIP2, SM1-OVA, and Yummer1.7 tumors treated with control IgGs. Data represent mean fold change ± SEM over the reference sample (IgG control-treated iBIP2 tumors) after normalization to the average of Hprt and Gapdh housekeeping genes. iBIP2 (n = 5 to 6), SM1-OVA (n = 4 to 5), and Yummer1.7 (n = 4 to 6). Statistical analysis by one-way ANOVA with Fisher’s LSD test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Flow cytometric analysis of immunoglobulin G (IgG) control-treated iBIP2 and SM1-OVA tumors showed that iBIP2 tumors contained more abundant CD45+ hematopoietic cell infiltrates (Fig. 7D). Myeloid cells encompassing F4/80+ TAMs, Ly6C+ monocytes/monocytic myeloid-derived suppressor cells (mo-MDSCs), and Ly6G+ neutrophils/granulocytic MDSCs (gr-MDSCs) were all more abundant in iBIP2 compared to SM1-OVA tumors. Conversely, nonmyeloid (CD11b−) cells, which comprise lymphocytes and natural killer cells, were less abundant in the iBIP2 tumors. qPCR analysis of chemokine and cytokine genes involved in myeloid cell recruitment and activation (Fig. 7E) showed greatly enhanced expression of Ccl2, Il4, and Cxcl12 in iBIP2 compared to both SM1-OVA and Yummer1.7 tumors. It is possible that the cytokine networks active in iBIP2 melanomas may sustain tumor refractoriness to anti-CSF1R–mediated macrophage elimination and drive resistance to combined CSF1R and PD1 blockade.
DISCUSSION
This study suggests that adaptive CSF1 secretion upon exposure to T cell–derived cytokines may act detrimentally to recruit TAMs and consequently hamper antitumor immune responses. We observed elevated CSF1 in patients with advanced malignant melanoma, and CSF1 expression correlated with disease progression. Histological analysis of biopsies of primary and metastatic melanomas, as well as transcriptomic analysis of metastatic melanomas from TCGA (46), revealed a positive correlation between CD8A and CSF1 or the macrophage markers CSF1R and CD163. High CD8A was associated with higher CSF1R, CD68, and CD163 gene expression in pretreatment melanomas from patients who did not respond to anti-PD1 therapy (56). Accordingly, IFNγ and TNFα secreted by activated tumor-specific T cells induced melanoma cells to secrete CSF1 in vitro. Finally, mouse experiments using transplant melanoma models showed that combined PD1 and CSF1R blockade could eradicate the tumors.
Tumor resistance to targeted therapies is frequently due to selection of resistant cancer cell clones. Although this principle has been broadly demonstrated for therapies targeting oncogenes (60), the mechanisms of resistance to immunotherapies are diverse and not yet fully understood. Resistance can also develop in the absence of therapeutic pressure, for example, through evolutionarily conserved mechanisms that slow down or halt T cell responses after acute immune responses to avoid overwhelming tissue destruction (49). These conserved mechanisms have been referred to as adaptive immune resistance (40, 51–53). The best-studied example of adaptive resistance is the well-known immune checkpoint PDL1-PD1 axis (54). PDL1 is one of the ligands for the immune checkpoint inhibitory molecule PD1, which is expressed on activated T cells. The induction of PDL1 on cancer cells and tumor-associated stromal cells was shown to be directly mediated by IFNγ signaling (51), one of the main cytokines secreted by activated CD8+ T and CD4+ helper 1 T cells. IFNγ can thus exert opposing roles in tumors. On the one hand, it ensues from the activation of T cells and can counteract tumor progression. On the other hand, it can indirectly suppress activated T cells through the up-regulation of immune inhibitors in the tumor microenvironment, such as PDL1 or indoleamine-pyrrole 2,3-dioxygenase (IDO), through a negative feedback mechanism (61, 62). Accordingly, the treatment of cancer patients with IFNγ has not yielded positive results (63–65).
Our patient data suggest that CSF1 induction in melanoma represents a conserved, adaptive resistance mechanism triggered by activated (IFNγ- and TNFα-secreting) T cells, which correlates with disease progression. In line with our findings, Varney et al. (66) previously reported augmented CSF1 mRNA in melanoma thicker than 0.75 mm. CSF1 is known to play a key role in shaping the tumor myeloid cell compartment toward immunosuppression by inducing the differentiation and accumulation of M2-like TAMs and MDSCs (3–5, 31). For example, infiltrating CD68+ or CD163+ TAMs correlated with poor outcome in solid tumors, such as breast and pancreatic cancers (67–69). Although CSF1R blockade provided limited therapeutic benefits, if any, in most of the cancer types tested so far (22, 34–36), preclinical mouse studies of breast, lung, and pancreatic tumors, and melanoma, suggest that its combination with other cancer immunotherapies, such as adoptive T cell transfer and checkpoint blockade (27, 29, 70), might improve clinical responses by relieving cancer-associated immunosuppression (71). Here, combined PD1 and CSF1R blockade induced complete tumor regressions in two transplant BRAFV600E-driven melanoma models (SM1-OVA and Yummer1.7). In both models, the anti-CSF1R antibody AFS98 (72) efficiently depleted TAMs. Conversely, dual CSF1R and PD1 blockade did not impair tumor growth in an inducible, BRAFV600E-driven transgenic melanoma model. In this experimental setup, antibody-mediated CSF1R blockade by the CSF1R antibody 2G2 (9, 31) neither eliminated TAMs nor altered their polarization, which may explain the lack of therapeutic benefit. Notably, 2G2 is a potent TAM-depleting agent, as shown previously in both transplant and transgenic cancer models (9, 31, 73). Failure of 2G2 to deplete TAMs in the iBIP2 model was associated with higher pretreatment expression of Ccl2, Il4, and Cxcl12, as well as infiltration by monocytic and granulocytic myeloid cells, compared to both SM1-OVA and Yummer1.7 models. Notably, elevated interleukin-4 (IL-4) expression may sustain TAM survival under emactuzumab (an anti-human CSF1R antibody), as shown in a recent clinical study (74). Furthermore, both CCL2 and CXCL12 are potent monocyte chemoattractants (1, 3, 6–8), which may enhance monocyte-derived TAM turnover in the presence of CSF1R inhibition.
Earlier studies have shown that the combination of a CSF1R small-molecule inhibitor (PLX3397) and adoptive T cell transfer prolonged mouse survival compared to the single treatments in the BRAFV600E-driven SM1 and SM1-OVA mouse models in a T cell–dependent manner (27). In another study, melanoma-bearing mice were treated with a BRAF inhibitor (PLX4720), a CSF1R inhibitor (PLX3397), and anti-PD1 or anti-PDL1 antibodies (75). The triple combination delayed tumor growth compared to dual BRAF and CSF1R inhibition, although the growth delay achieved by the combination of PD1 or PDL1 blockade and the CSF1R inhibitor was smaller than in our study. Differences in the therapeutic agents and dosage regimens may underlie the differences between earlier works and our study (27, 75). We used CSF1R antibodies (clones AFS98 and 2G2) that are specific for the ligand-binding domain of CSF1R and do not bind other tyrosine kinase receptors (31, 72). Conversely, PLX3397 inhibits several kinases, including CSF1R [IC50 (median inhibitory concentration), 0.02 μM], cKIT (IC50, 0.01 μM), and FLT3 (IC50, 0.16 μM), so it may have broader effects than CSF1R antibodies on the immune tumor microenvironment (67). cKIT (CD117) is expressed by immature progenitor cells (72, 76), so PLX3397 may intervene at more upstream stages of myelopoiesis than AFS98. PLX3397 also suppresses extramedullary hematopoiesis by inhibiting the expansion of splenic myeloid precursors, granulocytes, monocytes, and macrophages, as shown in mice (77). Different modes of CSF1R inhibition, for example, by monoclonal antibodies versus kinase inhibitors with a broader spectrum of molecular targets, may induce tumor responses that vary with the cancer type and patient population (78).
Tumor responses to immunotherapy are generally heterogeneous and can be influenced by various parameters, including tumor type and the extent of immune cell infiltration, among other factors (79). The CTL-TAM molecular cross-talk identified in our study may help to predict clinical responses to PD1 or PDL1 blockade. Several genomic and transcriptomic markers of response or resistance to PD1/PDL1 blockade have been proposed (41, 56, 80–85). Our analysis of the Hugo et al. data set (56) revealed that, unlike responders to anti-PD1 therapy, nonresponders showed increased expression of CSF1, CSF1R, CD163, and CD68 in CD8AHigh tumors compared to CD8ALow tumors, pointing to higher protumoral macrophage infiltrates in the nonresponders. This is in line with previous studies showing a higher proximity of CD68+ myeloid cells to CD8+ T cells in nonresponders after PD1 blockade (84). Therefore, combined macrophage elimination and immune checkpoint blockade may improve clinical outcomes in melanoma patients who do not respond to immune checkpoint blockade and, possibly, other T cell–based therapies.
Our study also has limitations that should be addressed with further work. Although we focused on CSF1, additional factors, such as transforming growth factor β (TGFB), IL-10, vascular endothelial growth factor A (VEGFA), and VEGFC, may be induced by CD8+ T cells (49) and contribute to recruit or polarize immunosuppressive macrophages in melanoma. Therefore, the relative contribution of CSF1 versus other factors to recruiting or activating immunosuppressive TAMs remains to be studied. Another unresolved issue is the discrepancy between therapeutic responses in transplant and transgenic melanoma models. It is currently unknown whether transgenic iBIP2 melanomas recapitulate more accurately than transplant melanoma models the cellular and molecular milieu of human melanoma. It is also possible that distinct patient subsets present biological features in their tumors that approximate either melanoma model. Ongoing clinical trials, such as those referenced in table S4, are expected to provide useful information on the potential of combined CSF1R and immune checkpoint blockade in patients with solid cancers. Several clinical trials involve patients enrolled for combination therapies of CSF1R kinase inhibitors (PLX3397, ARRY-382, and BLZ945) and anti-PD1 or anti-PDL1 antibodies (NCT02777710, NCT02452424, NCT02880371, and NCT02829723). In further trials, patients with solid tumors are being treated with anti-CSF1R antibodies (RG7155, FPA008, and AMG820) in combination with PD1, PDL1, or CTLA4 blockade (NCT02323191, NCT02526017, NCT02713529, and NCT02718911). Notably, an anti-CSF1 antibody is also being tested in combination with PD1 blockade (NCT02807844). On the basis of current evidence, our data would encourage the stratification of patients for treatment with CSF1R inhibitors with PD1 or PDL1 blockade based on T cell infiltration, IFNγ-regulated gene signatures, and abundance of M2-like TAMs in the tumor microenvironment.
MATERIALS AND METHODS
Study design
The patients analyzed in this study were diagnosed with cutaneous melanoma and gave informed consent before study inclusion. Patient studies and human sample collection were performed according to protocols (188/12 and 400/11) approved by the Institutional Review and Privacy Board of the University Hospital of Lausanne (CHUV, Lausanne, Switzerland) and the Human Research Ethics Committee of the Canton de Vaud (Switzerland). The patient characteristics are described in table S1.
Studies involving large and independent experimental cohorts of mice were performed once. Mice with transplant or transgenic melanoma were randomized to the various experimental cohorts by excluding mice that lacked palpable tumors at the time of enrollment. The treatment of mice carrying transplant tumors was begun when the tumors had reached a size of about 50 mm3, whereas treatment of iBIP2 mice was begun when the tumors had reached a size of 50 to 100 mm3. All mouse cohorts received antibody injections at the dosage regimens indicated in Supplementary Materials and Methods. The investigators were not blinded when they administered therapeutic antibodies or while assessing the results of the experiments. All procedures involving transplant tumor models were performed according to a protocol approved by the Veterinary Authorities of the Canton Vaud according to the Swiss Law (protocol 3049). All procedures involving iBIP2 transgenic mice were performed according to a protocol approved by the Veterinary Authorities of the Canton Vaud according to the Swiss Law (protocol 3100).
Statistical analysis
Information on the study outline, sample size, and statistical analysis is shown in the main text, figures, and figure legends. For the analysis of human samples, we used the nonparametric unpaired Mann-Whitney U test to assess statistical significance of differences between two data sets. Statistical analysis by one-way ANOVA or two-way ANOVA, both with correction for multiple comparisons, was used to compare data sets involving more than two experimental groups. In the experiments involving cell cultures, replicates indicate independent cell cultures. For IHC of human melanoma samples, correlation coefficients were computed using the Spearman’s rank-order correlation. For the analysis of published RNA-seq data sets of human melanoma, correlation between selected genes was assessed using the Spearman’s rank-order correlation.
For the analysis of preclinical experiments involving mouse models (tumor volumes, quantification of immune cell infiltrates, etc.), we used either unpaired Student’s t test or one-way ANOVA with correction for multiple comparisons for assessing statistics involving two or more than two experimental groups, respectively. The statistical analysis of mouse survival data was performed by using the log-rank (Mantel-Cox) test. For the comparison of tumor growth curves in the iBIP2 model, statistical analysis was performed using a linear mixed modeling approach. Briefly, we approximated each mouse’s tumor growth curve by a linear fit. By treating mouse-specific deviations as random, we estimated a population mean for each treatment group. To test whether population means were different between treatment groups, we compared a full model including an interaction term between the categorical variable representing treatment groups and the linear fit representing tumor growth to a null model without such interaction term. On rare occasions, outliers at endpoint were excluded by using the ROUT method to identify outliers (provided in GraphPad Prism). In some cases, selected samples were excluded from specific analyses (for example, flow cytometry) because of technical flaws during sample processing or data acquisition.
Data show the mean values ± SD or SEM, as indicated in the figure legends. In all statistical analyses, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. All numerical values (primary raw data when n < 20) are reported in table S5.
Supplementary Material
Fig. S1. Selection of regions with high or low density of tumor-infiltrating CD8+ T cells.
Fig. S2. Correlations between genes expressed in the SKCM metastatic cohort.
Fig. S3. CSF1+ cell density in high and low CD8+ cell-infiltrated tumor regions.
Fig. S4. CSF1 expression by melanoma cell lines and CTLs.
Fig. S5. Absence of CSF1R on the surface of untreated or cytokine-treated melanoma cell lines.
Fig. S6. Gene expression in human melanoma cells exposed to MelanA-specific CTLs or IFNγ and TNFα.
Fig. S7. Expression of macrophage genes in a transcriptomic data set of human melanoma treated with anti-PD1 therapy.
Fig. S8. Impact of CTL and TAM infiltrates on melanoma prognosis.
Fig. S9. Concentration of CSF1 in cell culture medium conditioned by mouse melanoma cells.
Table S1. Melanoma patients, clinical parameters, and specimens analyzed in this study.
Table S2. Origin of the melanoma cell lines used in this study.
Table S3. List of melanoma-derived macrophage signature genes.
Table S4. A selection of currently active clinical trials of CSF1 or CSF1R blockade in combination with immune checkpoint blockade (ClinicalTrials.gov; 18 October 2017).
Table S5. Primary raw data shown in the figures.
Acknowledgments
We thank A. Ribas, S. Kaech, M. Bosenberg, V. Du, K. Park, and D. Rimoldi for the melanoma cell lines and excellent documentation; D. Hanahan for his support in developing the refined iBIP2 mouse model; S. Leuba, K. Fortis, and E. Pavlidou for tissue processing and technical support with IHC; S. Winkler and A. Wilson for help with flow cytometry; O. Michielin, M. Matter, B. Martins Moura, L. Trueb, P. Combe, L. Leyvraz, L. Cagnon, S. Abed Maillard, P. Marcos Mondéjar, K. Ellefsen, and K. de Jonge for clinical and research contributions; S. Rusakiewicz and P. Baumgaertner for expert experimental guidance; N. Montandon for help with ELISA; L. Pradel for the CSF1R flow cytometry protocol; C. Soneson for the analysis of NanoString data; A. Guichard for help with qPCR analysis; and D. Barras for statistical analyses in Fig. 4.
Funding: This work was supported by grants from the Swiss Institute for Experimental Cancer Research Foundation (to N.J.N.), Emma Muschamp Foundation (to N.J.N.), Swiss Cancer Research (3507-08-2014 to D.E.S.), Swiss National Science Foundation (CRSII3-160708 to D.E.S. and 31003A-165963 to M.D.P.), Leenaards Foundation (to M.D.P. and G.C.), SwissTransMed (KIP 18 to D.E.S. and K.H.), Alfred and Annemarie von Sick (to D.E.S.), Cancer Research Institute (to D.E.S.), and Roche (to M.D.P.).
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/10/436/eaan3311/DC1
Materials and Methods
Author contributions: Conception and design: N.J.N., M.S., N.B., S.N., W.D., M.D.P., and D.E.S.; development of methodology: N.J.N., M.S., N.B., S.N., W.D., N.W., S.H., C.H.R., S.A.F.M., M.D.P., and D.E.S.; acquisition of data: N.J.N., M.S., N.B., S.N., N.W., L.T., I.K., C.M., K.I., and K.H.; analysis and interpretation of data: N.J.N., M.S., N.B., S.N., N.W., L.T., C.M., H.M.-E.H., K.I., K.H., S.A.F.M., M.D.P., and D.E.S.; writing, review, and revision of manuscript: N.J.N., M.S., N.B., S.N., W.D., L.K., G.C., N.W., S.A.F.M., M.D.P., and D.E.S.; technical and material support: E.D., S.H., and C.H.R.; study supervision: P.G.F., M.D.P., and D.E.S.
Competing interests: C.H.R., I.K., and S.H. are employees of Roche Diagnostics GmbH. C.H.R. and S.H. are inventors of granted and pending patent applications relating to RG7155, and stockholders of F. Hoffmann–La Roche. Roche is investigating combinations of the CSF1R antibody emactuzumab and immune checkpoint inhibitors. C.H.R. is inventor on patent applications (WO2011107553A1 and WO2013132044A1) filed by Hoffmann–La Roche that cover CSF1R blockade and its combination with immune checkpoint inhibitors for cancer treatment. The other authors declare that they have no competing interests.
Data and materials availability: The anti-CSF1R 2G2 monoclonal antibody was provided by Roche under conditions expressed in sponsored research agreements between Roche and École Polytechnique Federale de Lausanne (M.D.P.). Interested researchers may contact Roche for requests concerning 2G2.
REFERENCES AND NOTES
- 1.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat Immunol. 2010;11:889–896. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 2.Erdag G, Schaefer JT, Smolkin ME, Deacon DH, Shea SM, Dengel LT, Patterson JW, Slingluff CL., Jr Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res. 2012;72:1070–1080. doi: 10.1158/0008-5472.CAN-11-3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balkwill FR, Mantovani A. Cancer-related inflammation: Common themes and therapeutic opportunities. Semin Cancer Biol. 2012;22:33–40. doi: 10.1016/j.semcancer.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 5.Mantovani A, Sica A. Macrophages, innate immunity and cancer: Balance, tolerance, and diversity. Curr Opin Immunol. 2010;22:231–237. doi: 10.1016/j.coi.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 6.Noy R, Pollard JW. Tumor-associated macrophages: From mechanisms to therapy. Immunity. 2014;41:49–61. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell. 2013;23:277–286. doi: 10.1016/j.ccr.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 8.Lewis CE, Harney AS, Pollard JW. The multifaceted role of perivascular macrophages in tumors. Cancer Cell. 2016;30:18–25. doi: 10.1016/j.ccell.2016.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baer C, Squadrito ML, Laoui D, Thompson D, Hansen SK, Kiialainen A, Hoves S, Ries CH, Ooi CH, De Palma M. Suppression of microRNA activity amplifies IFN-γ-induced macrophage activation and promotes anti-tumour immunity. Nat Cell Biol. 2016;18:790–802. doi: 10.1038/ncb3371. [DOI] [PubMed] [Google Scholar]
- 10.Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, Setty M, Leslie CS, Oei Y, Pedraza A, Zhang J, Brennan CW, Sutton JC, Holland EC, Daniel D, Joyce JA. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19:1264–1272. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, Delaney A, Jones SJ, Iqbal J, Weisenburger DD, Bast MA, Rosenwald A, Muller-Hermelink HK, Rimsza LM, Campo E, Delabie J, Braziel RM, Cook JR, Tubbs RR, Jaffe ES, Lenz G, Connors JM, Staudt LM, Chan WC, Gascoyne RD. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N Engl J Med. 2010;362:875–885. doi: 10.1056/NEJMoa0905680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang M, He Y, Sun X, Li Q, Wang W, Zhao A, Di W. A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J Ovarian Res. 2014;7:19. doi: 10.1186/1757-2215-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang QW, Liu L, Gong CY, Shi HS, Zeng YH, Wang XZ, Zhao YW, Wei YQ. Prognostic significance of tumor-associated macrophages in solid tumor: A meta-analysis of the literature. PLOS ONE. 2012;7:e50946. doi: 10.1371/journal.pone.0050946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Senovilla L, Vacchelli E, Galon J, Adjemian S, Eggermont A, Fridman WH, Sautès-Fridman C, Ma Y, Tartour E, Zitvogel L, Kroemer G, Galluzzi L. Trial watch: Prognostic and predictive value of the immune infiltrate in cancer. Oncoimmunology. 2012;1:1323–1343. doi: 10.4161/onci.22009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hillen F, Baeten CIM, van de Winkel A, Creytens D, van der Schaft DWJ, Winnepenninckx V, Griffioen AW. Leukocyte infiltration and tumor cell plasticity are parameters of aggressiveness in primary cutaneous melanoma. Cancer Immunol Immunother. 2008;57:97–106. doi: 10.1007/s00262-007-0353-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Storr SJ, Safuan S, Mitra A, Elliott F, Walker C, Vasko MJ, Ho B, Cook M, Mohammed RAA, Patel PM, Ellis IO, Newton-Bishop JA, Martin SG. Objective assessment of blood and lymphatic vessel invasion and association with macrophage infiltration in cutaneous melanoma. Mod Pathol. 2012;25:493–504. doi: 10.1038/modpathol.2011.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kiss J, Tímár J, Somlai B, Gilde K, Fejõs Z, Gaudi I, Ladányi A. Association of microvessel density with infiltrating cells in human cutaneous malignant melanoma. Pathol Oncol Res. 2007;13:21–31. doi: 10.1007/BF02893437. [DOI] [PubMed] [Google Scholar]
- 18.Jensen TO, Schmidt H, Møller HJ, Hoyer M, Maniecki MB, Sjoegren P, Christensen IJ, Steiniche T. Macrophage markers in serum and tumor have prognostic impact in American Joint Committee on Cancer stage I/II melanoma. J Clin Oncol. 2009;27:3330–3337. doi: 10.1200/JCO.2008.19.9919. [DOI] [PubMed] [Google Scholar]
- 19.Varney ML, Johansson SL, Singh RK. Tumour-associated macrophage infiltration, neovascularization and aggressiveness in malignant melanoma: Role of monocyte chemotactic protein-1 and vascular endothelial growth factor-A. Melanoma Res. 2005;15:417–425. doi: 10.1097/00008390-200510000-00010. [DOI] [PubMed] [Google Scholar]
- 20.Piras F, Colombari R, Minerba L, Murtas D, Floris C, Maxia C, Corbu A, Perra MT, Sirigu P. The predictive value of CD8, CD4, CD68, and human leukocyte antigen-D-related cells in the prognosis of cutaneous malignant melanoma with vertical growth phase. Cancer. 2005;104:1246–1254. doi: 10.1002/cncr.21283. [DOI] [PubMed] [Google Scholar]
- 21.Hume DA, MacDonald KPA. Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling. Blood. 2012;119:1810–1820. doi: 10.1182/blood-2011-09-379214. [DOI] [PubMed] [Google Scholar]
- 22.Ries CH, Hoves S, Cannarile MA, Rüttinger D. CSF-1/CSF-1R targeting agents in clinical development for cancer therapy. Curr Opin Pharmacol. 2015;23:45–51. doi: 10.1016/j.coph.2015.05.008. [DOI] [PubMed] [Google Scholar]
- 23.Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2008;9:239–252. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forget MA, Voorhees JL, Cole SL, Dakhlallah D, Patterson IL, Gross AC, Moldovan L, Mo X, Evans R, Marsh CB, Eubank TD. Macrophage colony-stimulating factor augments Tie2-expressing monocyte differentiation, angiogenic function, and recruitment in a mouse model of breast cancer. PLOS ONE. 2014;9:e98623. doi: 10.1371/journal.pone.0098623. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Chambers SK, Kacinski BM, Ivins CM, Carcangiu ML. Overexpression of epithelial macrophage colony-stimulating factor (CSF-1) and CSF-1 receptor: A poor prognostic factor in epithelial ovarian cancer, contrasted with a protective effect of stromal CSF-1. Clin Cancer Res. 1997;3:999–1007. [PubMed] [Google Scholar]
- 26.Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol. 2009;9:259–270. doi: 10.1038/nri2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mok S, Koya RC, Tsui C, Xu J, Robert L, Wu L, Graeber TG, West BL, Bollag G, Ribas A. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res. 2014;74:153–161. doi: 10.1158/0008-5472.CAN-13-1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sluijter M, van der Sluis TC, van der Velden PA, Versluis M, West BL, van der Burg SH, van Hall T. Inhibition of CSF-1R supports T-cell mediated melanoma therapy. PLOS ONE. 2014;9:e104230. doi: 10.1371/journal.pone.0104230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, Wang-Gillam A, Goedegebuure SP, Linehan DC, DeNardo DG. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74:5057–5069. doi: 10.1158/0008-5472.CAN-13-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patwardhan PP, Surriga O, Beckman MJ, de Stanchina E, Dematteo RP, Tap WD, Schwartz GK. Sustained inhibition of receptor tyrosine kinases and macrophage depletion by PLX3397 and rapamycin as a potential new approach for the treatment of MPNSTs. Clin Cancer Res. 2014;20:3146–3158. doi: 10.1158/1078-0432.CCR-13-2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, Rey-Giraud F, Pradel LP, Feuerhake F, Klaman I, Jones T, Jucknischke U, Scheiblich S, Kaluza K, Gorr IH, Walz A, Abiraj K, Cassier PA, Sica A, Gomez-Roca C, de Visser KE, Italiano A, Le Tourneau C, Delord JP, Levitsky H, Blay JY, Rüttinger D. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014;25:846–859. doi: 10.1016/j.ccr.2014.05.016. [DOI] [PubMed] [Google Scholar]
- 32.Tap WD, Wainberg ZA, Anthony SP, Ibrahim PN, Zhang C, Healey JH, Chmielowski B, Staddon AP, Cohn AL, Shapiro GI, Keedy VL, Singh AS, Puzanov I, Kwak EL, Wagner AJ, Von Hoff DD, Weiss GJ, Ramanathan RK, Zhang J, Habets G, Zhang Y, Burton EA, Visor G, Sanftner L, Severson P, Nguyen H, Kim MJ, Marimuthu A, Tsang G, Shellooe R, Gee C, West BL, Hirth P, Nolop K, van de Rijn M, Hsu HH, Peterfy C, Lin PS, Tong-Starksen S, Bollag G. Structure-guided blockade of CSF1R kinase in tenosynovial giant-cell tumor. N Engl J Med. 2015;373:428–437. doi: 10.1056/NEJMoa1411366. [DOI] [PubMed] [Google Scholar]
- 33.Cassier PA, Italiano A, Gomez-Roca CA, Le Tourneau C, Toulmonde M, Cannarile MA, Ries C, Brillouet A, Müller C, Jegg AM, Bröske AM, Dembowski M, Bray-French K, Freilinger C, Meneses-Lorente G, Baehner M, Harding R, Ratnayake J, Abiraj K, Gass N, Noh K, Christen RD, Ukarma L, Bompas E, Delord JP, Blay JY, Rüttinger D. CSF1R inhibition with emactuzumab in locally advanced diffuse-type tenosynovial giant cell tumours of the soft tissue: A dose-escalation and dose-expansion phase 1 study. Lancet Oncol. 2015;16:949–956. doi: 10.1016/S1470-2045(15)00132-1. [DOI] [PubMed] [Google Scholar]
- 34.Moskowitz CH, Younes A, de Vos S, Bociek RG, Gordon LI, Witzig TE, Gascoyne RD, West B, Nolop K, Steidl C. CSF1R inhibition by PLX3397 in patients with relapsed or refractory Hodgkin lymphoma: Results from a phase 2 single agent clinical trial. Blood. 2012;120:1638. [Google Scholar]
- 35.von Tresckow B, Morschhauser F, Ribrag V, Topp MS, Chien C, Seetharam S, Aquino R, Kotoulek S, de Boer CJ, Engert A. An open-label, multicenter, phase I/II study of JNJ-40346527, a CSF-1R inhibitor, in patients with relapsed or refractory Hodgkin lymphoma. Clin Cancer Res. 2015;21:1843–1850. doi: 10.1158/1078-0432.CCR-14-1845. [DOI] [PubMed] [Google Scholar]
- 36.Butowski N, Colman H, De Groot JF, Omuro AM, Nayak L, Wen PY, Cloughesy TF, Marimuthu A, Haidar S, Perry A, Huse J, Phillips J, West BL, Nolop KB, Hsu HH, Ligon KL, Molinaro AM, Prados M. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro Oncol. 2016;18:557–564. doi: 10.1093/neuonc/nov245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–461. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abril-Rodriguez G, Ribas A. SnapShot: Immune checkpoint inhibitors. Cancer Cell. 2017;31:848–848.e1. doi: 10.1016/j.ccell.2017.05.010. [DOI] [PubMed] [Google Scholar]
- 39.Atkins MB, Clark JI, Quinn DI. Immune checkpoint inhibitors in advanced renal cell carcinoma: Experience to date and future directions. Ann Oncol. 2017;28:1484–1494. doi: 10.1093/annonc/mdx151. [DOI] [PubMed] [Google Scholar]
- 40.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, Seja E, Cherry G, Gutierrez AJ, Grogan TR, Mateus C, Tomasic G, Glaspy JA, Emerson RO, Robins H, Pierce RH, Elashoff DA, Robert C, Ribas A. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16:275–287. doi: 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Callahan MK, Postow MA, Wolchok JD. Targeting T cell co-receptors for cancer therapy. Immunity. 2016;44:1069–1078. doi: 10.1016/j.immuni.2016.04.023. [DOI] [PubMed] [Google Scholar]
- 43.Satriano JA, Hora K, Shan Z, Stanley ER, Mori T, Schlondorff D. Regulation of monocyte chemoattractant protein-1 and macrophage colony-stimulating factor-1 by IFN-gamma, tumor necrosis factor-alpha, IgG aggregates, and cAMP in mouse mesangial cells. J Immunol. 1993;150:1971–1978. [PubMed] [Google Scholar]
- 44.Gershenwald JE, Scolyer RA, Hess KR, Sondak VK, Long GV, Ross MI, Lazar AJ, Faries MB, Kirkwood JM, McArthur GA, Haydu LE, Eggermont AMM, Flaherty KT, Balch CM, Thompson JF, American Joint Committee on Cancer Melanoma Expert Panel and the International Melanoma Database and Discovery Platform Melanoma staging: Evidence-based changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J Clin. 2017;67:472–492. doi: 10.3322/caac.21409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med. 1999;189:1363–1372. doi: 10.1084/jem.189.9.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell. 2015;161:1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Salama I, Malone PS, Mihaimeed F, Jones JL. A review of the S100 proteins in cancer. Eur J Surg Oncol. 2008;34:357–364. doi: 10.1016/j.ejso.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 48.Neubert NJ, Soneson C, Barras D, Baumgaertner P, Rimoldi D, Delorenzi M, Fuertes Marraco SA, Speiser DE. A well-controlled experimental system to study interactions of cytotoxic T lymphocytes with tumor cells. Front Immunol. 2016;7:326. doi: 10.3389/fimmu.2016.00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Neubert NJ, Tillé L, Barras D, Soneson C, Baumgaertner P, Rimoldi D, Gfeller D, Delorenzi M, Fuertes Marraco SA, Speiser DE. Broad and conserved immune regulation by genetically heterogeneous melanoma cells. Cancer Res. 2017;77:1623–1636. doi: 10.1158/0008-5472.CAN-16-2680. [DOI] [PubMed] [Google Scholar]
- 50.Hamann D, Baars P, Rep M, Hooibrink B, KerkhofGarde SR, Klein MR, vanLier R. Phenotypic and functional separation of memory and effector human CD8+ T cells. J Exp Med. 1997;186:1407–1418. doi: 10.1084/jem.186.9.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ribas A. Adaptive immune resistance: How cancer protects from immune attack. Cancer Discov. 2015;5:915–919. doi: 10.1158/2159-8290.CD-15-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, Chen S, Klein AP, Pardoll DM, Topalian SL, Chen L. Colocalization of inflammatory response with B7-H1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4:127ra37. doi: 10.1126/scitranslmed.3003689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ribas A, Hu-Lieskovan S. What does PD-L1 positive or negative mean? J Exp Med. 2016;213:2835–2840. doi: 10.1084/jem.20161462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tirosh I, Izar B, Prakadan SM, Wadsworth MH, II, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, Fallahi-Sichani M, Dutton-Regester K, Lin J-R, Cohen O, Shah P, Lu D, Genshaft AS, Hughes TK, Ziegler CGK, Kazer SW, Gaillard A, Kolb KE, Villani A-C, Johannessen CM, Andreev AY, Van Allen EM, Bertagnolli M, Sorger PK, Sullivan RJ, Flaherty KT, Frederick DT, Jane-Valbuena J, Yoon CH, Rozenblatt-Rosen O, Shalek AK, Regev A, Garraway LA. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, Seja E, Lomeli S, Kong X, Kelley MC, Sosman JA, Johnson DB, Ribas A, Lo RS. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Koya RC, Mok S, Otte N, Blacketor KJ, Comin-Anduix B, Tumeh PC, Minasyan A, Graham NA, Graeber TG, Chodon T, Ribas A. BRAF inhibitor vemurafenib improves the antitumor activity of adoptive cell immunotherapy. Cancer Res. 2012;72:3928–3937. doi: 10.1158/0008-5472.CAN-11-2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang J, Perry CJ, Meeth K, Thakral D, Damsky W, Micevic G, Kaech S, Blenman K, Bosenberg M. UV-induced somatic mutations elicit a functional T cell response in the YUMMER1.7 mouse melanoma model. Pigment Cell Melanoma Res. 2017;30:428–435. doi: 10.1111/pcmr.12591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kwong LN, Boland GM, Frederick DT, Helms TL, Akid AT, Miller JP, Jiang S, Cooper ZA, Song X, Seth S, Kamara J, Protopopov A, Mills GB, Flaherty KT, Wargo JA, Chin L. Co-clinical assessment identifies patterns of BRAF inhibitor resistance in melanoma. J Clin Invest. 2015;125:1459–1470. doi: 10.1172/JCI78954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, Place CS, Taylor-Weiner A, Whittaker S, Kryukov GV, Hodis E, Rosenberg M, McKenna A, Cibulskis K, Farlow D, Zimmer L, Hillen U, Gutzmer R, Goldinger SM, Ugurel S, Gogas HJ, Egberts F, Berking C, Trefzer U, Loquai C, Weide BN, Hassel JC, Gabriel SB, Carter SL, Getz G, Garraway LA, Schadendorf D, Dermatologic Cooperative Oncology Group of Germany (DeCOG) The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014;4:94–109. doi: 10.1158/2159-8290.CD-13-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zaidi MR, Merlino G. The two faces of interferon-γ in cancer. Clin Cancer Res. 2011;17:6118–6124. doi: 10.1158/1078-0432.CCR-11-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol. 2006;6:836–848. doi: 10.1038/nri1961. [DOI] [PubMed] [Google Scholar]
- 63.Alberts DS, Marth C, Alvarez RD, Johnson G, Bidzinski M, Kardatzke DR, Bradford WZ, Loutit J, Kirn DH, Clouser MC, Markman M, GRACES Clinical Trial Consortium Randomized phase 3 trial of interferon gamma-1b plus standard carboplatin/paclitaxel versus carboplatin/paclitaxel alone for first-line treatment of advanced ovarian and primary peritoneal carcinomas: Results from a prospectively designed analysis of progression-free survival. Gynecol Oncol. 2008;109:174–181. doi: 10.1016/j.ygyno.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 64.Mauldin IS, Wages NA, Stowman AM, Wang E, Smolkin ME, Olson WC, Deacon DH, Smith KT, Galeassi NV, Bullock KAC, Dengel LT, Marincola FM, Petroni GR, Mullins DW, Slingluff CL. Intratumoral interferon-gamma increases chemokine production but fails to increase T cell infiltration of human melanoma metastases. Cancer Immunol Immunother. 2016;65:1189–1199. doi: 10.1007/s00262-016-1881-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Meyskens FL, Jr, Kopecky KJ, Taylor CW, Noyes RD, Tuthill RJ, Hersh EM, Feun LG, Doroshow JH, Flaherty LE, Sondak VK. Randomized trial of adjuvant human interferon gamma versus observation in high-risk cutaneous melanoma: A Southwest Oncology Group study. J Natl Cancer Inst. 1995;87:1710–1713. doi: 10.1093/jnci/87.22.1710. [DOI] [PubMed] [Google Scholar]
- 66.Varney ML, Olsen KJ, Mosley RL, Singh RK. Paracrine regulation of vascular endothelial growth factor-A expression during macrophage-melanoma cell interaction: Role of monocyte chemotactic protein-1 and macrophage colony-stimulating factor. J Interferon Cytokine Res. 2005;25:674–683. doi: 10.1089/jir.2005.25.674. [DOI] [PubMed] [Google Scholar]
- 67.DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SA, Rugo HS, Hwang ES, Jirström K, West BL, Coussens LM. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1:54–67. doi: 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kurahara H, Shinchi H, Mataki Y, Maemura K, Noma H, Kubo F, Sakoda M, Ueno S, Natsugoe S, Takao S. Significance of M2-polarized tumor-associated macrophage in pancreatic cancer. J Surg Res. 2011;167:e211–e219. doi: 10.1016/j.jss.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 69.Shabo I, Stal O, Olsson H, Doré S, Svanvik J. Breast cancer expression of CD163, a macrophage scavenger receptor, is related to early distant recurrence and reduced patient survival. Int J Cancer. 2008;123:780–786. doi: 10.1002/ijc.23527. [DOI] [PubMed] [Google Scholar]
- 70.Holmgaard RB, Brachfeld A, Gasmi B, Jones DR, Mattar M, Doman T, Murphy M, Schaer D, Wolchok JD, Merghoub T. Timing of CSF-1/CSF-1R signaling blockade is critical to improving responses to CTLA-4 based immunotherapy. Oncoimmunology. 2016;5:e1151595. doi: 10.1080/2162402X.2016.1151595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.MacDonald KPA, Palmer JS, Cronau S, Seppanen E, Olver S, Raffelt NC, Kuns R, Pettit AR, Clouston A, Wainwright B, Branstetter D, Smith J, Paxton RJ, Cerretti DP, Bonham L, Hill GR, Hume DA. An antibody against the colony–stimulating factor 1 receptor depletes the resident subset of monocytes and tissue- and tumor-associated macrophages but does not inhibit inflammation. Blood. 2010;116:3955–3963. doi: 10.1182/blood-2010-02-266296. [DOI] [PubMed] [Google Scholar]
- 72.Sudo T, Nishikawa S, Ogawa M, Kataoka H, Ohno N, Izawa A, Hayashi SI, Nishikawa SI. Functional hierarchy of c-kit and c-fms in intramarrow production of CFU-M. Oncogene. 1995;11:2469–2476. [PubMed] [Google Scholar]
- 73.Hoves S, Ooi CH, Wolter C, Sade H, Bissinger S, Schmittnaegel M, Ast O, Giusti AM, Wartha K, Runza V, Xu W, Kienast Y, Cannarile MA, Levitsky H, Romagnoli S, De Palma M, Rüttinger D, Ries CH. Rapid activation of tumor-associated macrophages boosts preexisting tumor immunity. J Exp Med. 2018;215:859–876. doi: 10.1084/jem.20171440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pradel LP, Ooi CH, Romagnoli S, Cannarile MA, Sade H, Rüttinger D, Ries CH. Macrophage susceptibility to emactuzumab (RG7155) treatment. Mol Cancer Ther. 2016;15:3077–3086. doi: 10.1158/1535-7163.MCT-16-0157. [DOI] [PubMed] [Google Scholar]
- 75.Ngiow SF, Meeth KM, Stannard K, Barkauskas DS, Bollag G, Bosenberg M, Smyth MJ. Co-inhibition of colony stimulating factor-1 receptor and BRAF oncogene in mouse models of BRAFV600E melanoma. Oncoimmunology. 2016;5:e1089381. doi: 10.1080/2162402X.2015.1089381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Richards DM, Hettinger J, Feuerer M. Monocytes and macrophages in cancer: Development and functions. Cancer Microenviron. 2013;6:179–191. doi: 10.1007/s12307-012-0123-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chitu V, Nacu V, Charles JF, Henne WM, McMahon HT, Nandi S, Ketchum H, Harris R, Nakamura MC, Stanley ER. PSTPIP2 deficiency in mice causes osteopenia and increased differentiation of multipotent myeloid precursors into osteoclasts. Blood. 2012;120:3126–3135. doi: 10.1182/blood-2012-04-425595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stanley ER, Chitu V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harb Perspect Biol. 2014;6:a021857. doi: 10.1101/cshperspect.a021857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Anderson KG, Stromnes IM, Greenberg PD. Obstacles posed by the tumor microenvironment to T cell activity: A case for synergistic therapies. Cancer Cell. 2017;31:311–325. doi: 10.1016/j.ccell.2017.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ascierto ML, McMiller TL, Berger AE, Danilova L, Anders RA, Netto GJ, Xu H, Pritchard TS, Fan J, Cheadle C, Cope L, Drake CG, Pardoll DM, Taube JM, Topalian SL. The intratumoral balance between metabolic and immunologic gene expression is associated with anti–PD-1 response in patients with renal cell carcinoma. Cancer Immunol Res. 2016;4:726–733. doi: 10.1158/2326-6066.CIR-16-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, Saco J, Homet Moreno B, Mezzadra R, Chmielowski B, Ruchalski K, Shintaku IP, Sanchez PJ, Puig-Saus C, Cherry G, Seja E, Kong X, Pang J, Berent-Maoz B, Comin-Anduix B, Graeber TG, Tumeh PC, Schumacher TNM, Lo RS, Ribas A. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375:819–829. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, Lieeping C, Pardoll DM, Topalian SL, Anders RA. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014;20:5064–5074. doi: 10.1158/1078-0432.CCR-13-3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Roh W, Chen PL, Reuben A, Spencer CN, Prieto PA, Miller JP, Gopalakrishnan V, Wang F, Cooper ZA, Reddy SM, Gumbs C, Little L, Chang Q, Chen WS, Wani K, De Macedo MP, Chen E, Austin-Breneman JL, Jiang H, Roszik J, Tetzlaff MT, Davies MA, Gershenwald JE, Tawbi H, Lazar AJ, Hwu P, Hwu WJ, Diab A, Glitza IC, Patel SP, Woodman SE, Amaria RN, Prieto VG, Hu J, Sharma P, Allison JP, Chin L, Zhang J, Wargo JA, Futreal PA. Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci Transl Med. 2017;9:eaah3560. doi: 10.1126/scitranslmed.aah3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen PL, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, Miller JP, Bassett RL, Gopalakrishnan V, Wani K, De Macedo MP, Austin-Breneman JL, Jiang H, Chang Q, Reddy SM, Chen WS, Tetzlaff MT, Broaddus RJ, Davies MA, Gershenwald JE, Haydu L, Lazar AJ, Patel SP, Hwu P, Hwu WJ, Diab A, Glitza IC, Woodman SE, Vence LM, Wistuba II, Amaria RN, Kwong LN, Prieto V, Davis RE, Ma W, Overwijk WW, Sharpe AH, Hu J, Futreal PA, Blando J, Sharma P, Allison JP, Chin L, Wargo JA. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. 2016;6:827–837. doi: 10.1158/2159-8290.CD-15-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Charoentong P, Finotello F, Angelova M, Mayer C, Efremova M, Rieder D, Hackl H, Trajanoski Z. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 2017;18:248–262. doi: 10.1016/j.celrep.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 86.Speiser DE, Baumgaertner P, Voelter V, Devevre E, Barbey C, Rufer N, Romero P. Unmodified self antigen triggers human CD8 T cells with stronger tumor reactivity than altered antigen. Proc Natl Acad Sci USA. 2008;105:3849–3854. doi: 10.1073/pnas.0800080105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Goel VK, Ibrahim N, Jiang G, Singhal M, Fee S, Flotte T, Westmoreland S, Haluska FS, Hinds PW, Haluska FG. Melanocytic nevus-like hyperplasia and melanoma in transgenic BRAFV600E mice. Oncogene. 2009;28:2289–2298. doi: 10.1038/onc.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yamazaki T, Akiba H, Koyanagi A, Azuma M, Yagita H, Okumura K. Blockade of B7-H1 on macrophages suppresses CD4+ T cell proliferation by augmenting IFN-γ-induced nitric oxide production. J Immunol. 2005;175:1586–1592. doi: 10.4049/jimmunol.175.3.1586. [DOI] [PubMed] [Google Scholar]
- 89.Stack EC, Wang C, Roman KA, Hoyt CC. Multiplexed immunohistochemistry, imaging, and quantitation: A review, with an assessment of Tyramide signal amplification, multispectral imaging and multiplex analysis. Methods. 2014;70:46–58. doi: 10.1016/j.ymeth.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 90.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yaari G, Bolen CR, Thakar J, Kleinstein SH. Quantitative set analysis for gene expression: A method to quantify gene set differential expression including gene-gene correlations. Nucleic Acids Res. 2013;41:e170. doi: 10.1093/nar/gkt660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Therneau TM, Grambsch PM. Modeling Survival Data: Extending the Cox Model. Springer Science & Business Media; 2013. [Google Scholar]
- 94.Therneau TM. A package for survival analysis in S Version 2.38. 2015 https://cran.r-project.org/package=survival.
- 95.R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2016. www.R-project.org/ [Google Scholar]
- 96.Nikolaev SI, Rimoldi D, Iseli C, Valsesia A, Robyr D, Gehrig C, Harshman K, Guipponi M, Bukach O, Zoete V, Michielin O, Muehlethaler K, Speiser D, Beckmann JS, Xenarios I, Halazonetis TD, Jongeneel CV, Stevenson BJ, Antonarakis SE. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat Genet. 2012;44:133–139. doi: 10.1038/ng.1026. [DOI] [PubMed] [Google Scholar]
- 97.Valsesia A, Rimoldi D, Martinet D, Ibberson M, Benaglio P, Quadroni M, Waridel P, Gaillard M, Pidoux M, Rapin B, Rivolta C, Xenarios I, Simpson AJG, Antonarakis SE, Beckmann JS, Jongeneel CV, Iseli C, Stevenson BJ. Network-guided analysis of genes with altered somatic copy number and gene expression reveals pathways commonly perturbed in metastatic melanoma. PLOS ONE. 2011;6:e18369. doi: 10.1371/journal.pone.0018369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Berard F, Blanco P, Davoust J, Neidhart-Berard EM, Nouri-Shirazi M, Taquet N, Rimoldi D, Cerottini JC, Banchereau J, Palucka AK. Cross-priming of naive CD8 T cells against melanoma antigens using dendritic cells loaded with killed allogeneic melanoma cells. J Exp Med. 2000;192:1535–1544. doi: 10.1084/jem.192.11.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rimoldi D, Rubio-Godoy V, Dutoit V, Liénard D, Salvi S, Guillaume P, Speiser D, Stockert E, Spagnoli G, Servis C, Cerottini JC, Lejeune F, Romero P, Valmori D. Efficient simultaneous presentation of NY-ESO-1/LAGE-1 primary and nonprimary open reading frame-derived CTL epitopes in melanoma. J Immunol. 2000;165:7253–7261. doi: 10.4049/jimmunol.165.12.7253. [DOI] [PubMed] [Google Scholar]
- 100.Berger Y, Bernasconi CC, Juillerat-Jeanneret L. Targeting the endothelin axis in human melanoma: Combination of endothelin receptor antagonism and alkylating agents. Exp Biol Med. 2006;231:1111–1119. [PubMed] [Google Scholar]
- 101.Held G, Neumann F, Sturm C, Kaestner L, Dauth N, de Bruijn DR, Renner C, Lipp P, Pfreundschuh M. Differential presentation of tumor antigen-derived epitopes by MHC-class I and antigen-positive tumor cells. Int J Cancer. 2008;123:1841–1847. doi: 10.1002/ijc.23728. [DOI] [PubMed] [Google Scholar]
- 102.Huber F, Lang HP, Backmann N, Rimoldi D, Gerber C. Direct detection of a BRAF mutation in total RNA from melanoma cells using cantilever arrays. Nat Nanotechnol. 2013;8:125–129. doi: 10.1038/nnano.2012.263. [DOI] [PubMed] [Google Scholar]
- 103.Caballero OL, Zhao Q, Rimoldi D, Stevenson BJ, Svobodová S, Devalle S, Röhrig UF, Pagotto A, Michielin O, Speiser D, Wolchok JD, Liu C, Pejovic T, Odunsi K, Brasseur F, Van den Eynde BJ, Old LJ, Lu X, Cebon J, Strausberg RL, Simpson AJ. Frequent MAGE mutations in human melanoma. PLOS ONE. 2010;5:e12773. doi: 10.1371/journal.pone.0012773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Karageorgis A, Claron M, Jugé R, Aspord C, Thoreau F, Leloup C, Kucharczak J, Plumas J, Henry M, Hurbin A, Verdié P, Martinez J, Subra G, Dumy P, Boturyn D, Aouacheria A, Coll JL. Systemic delivery of tumor-targeted bax-derived membrane-active peptides for the treatment of melanoma tumors in a humanized SCID mouse model. Mol Ther. 2017;25:534–546. doi: 10.1016/j.ymthe.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lissina A, Briceno O, Afonso G, Larsen M, Gostick E, Price DA, Mallone R, Appay V. Priming of qualitatively superior human effector CD8+ T cells using TLR8 ligand combined with FLT3 ligand. J Immunol. 2016;196:256–263. doi: 10.4049/jimmunol.1501140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Speiser DE, Baumgaertner P, Barbey C, Rubio-Godoy V, Moulin A, Corthesy P, Devevre E, Dietrich PY, Rimoldi D, Liénard D, Cerottini JC, Romero P, Rufer N. A novel approach to characterize clonality and differentiation of human melanoma-specific T cell responses: Spontaneous priming and efficient boosting by vaccination. J Immunol. 2006;177:1338–1348. doi: 10.4049/jimmunol.177.2.1338. [DOI] [PubMed] [Google Scholar]
- 107.Reed DS, Romero P, Rimoldi D, Cerottini JC, Schaack J, Jongeneel CV. Construction and characterization of a recombinant adenovirus directing expression of the MAGE-1 tumor-specific antigen. Int J Cancer. 1997;72:1045–1055. doi: 10.1002/(sici)1097-0215(19970917)72:6<1045::aid-ijc20>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 108.Valmori D, Liénard D, Waanders G, Rimoldi D, Cerottini JC, Romero P. Analysis of MAGE-3-specific cytolytic T lymphocytes in human leukocyte antigen-A2 melanoma patients. Cancer Res. 1997;57:735–741. [PubMed] [Google Scholar]
- 109.Bioley G, Jandus C, Tuyaerts S, Rimoldi D, Kwok WW, Speiser DE, Tiercy JM, Thielemans K, Cerottini JC, Romero P. Melan-A/MART-1-specific CD4 T cells in melanoma patients: Identification of new epitopes and ex vivo visualization of specific T cells by MHC class II tetramers. J Immunol. 2006;177:6769–6779. doi: 10.4049/jimmunol.177.10.6769. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Selection of regions with high or low density of tumor-infiltrating CD8+ T cells.
Fig. S2. Correlations between genes expressed in the SKCM metastatic cohort.
Fig. S3. CSF1+ cell density in high and low CD8+ cell-infiltrated tumor regions.
Fig. S4. CSF1 expression by melanoma cell lines and CTLs.
Fig. S5. Absence of CSF1R on the surface of untreated or cytokine-treated melanoma cell lines.
Fig. S6. Gene expression in human melanoma cells exposed to MelanA-specific CTLs or IFNγ and TNFα.
Fig. S7. Expression of macrophage genes in a transcriptomic data set of human melanoma treated with anti-PD1 therapy.
Fig. S8. Impact of CTL and TAM infiltrates on melanoma prognosis.
Fig. S9. Concentration of CSF1 in cell culture medium conditioned by mouse melanoma cells.
Table S1. Melanoma patients, clinical parameters, and specimens analyzed in this study.
Table S2. Origin of the melanoma cell lines used in this study.
Table S3. List of melanoma-derived macrophage signature genes.
Table S4. A selection of currently active clinical trials of CSF1 or CSF1R blockade in combination with immune checkpoint blockade (ClinicalTrials.gov; 18 October 2017).
Table S5. Primary raw data shown in the figures.