

Abstract
This study investigates the role of substrate stiffness on non-viral transfection of human adipose-derived stem cells (hASCs) with the aim to maximize hASCs expression of vascular endothelial growth factor (VEGF). The results confirm the direct effect of substrate stiffness in regulating cytoskeletal remodeling and corresponding plasmid internalization.
Graphical abstract
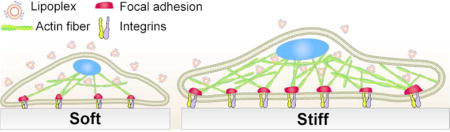
Growth factors play a key role for stem cell-based regenerative therapies1, 2. Stem cells produce a wide variety of these bioactive proteins, which control fundamental processes such as cell proliferation, differentiation, and homeostasis3. For instance, human adipose-derived stem cells (hASCs) express vascular endothelial growth factor A (VEGF-A) as one of the regulator of their proliferation and differentiation. As demonstrated in a recent study, neutralization of VEGF-A activity by exposing hASCs to monoclonal anti-VEGF antibody caused a significant reduction in cell proliferation. Aside from its mitogenic effects in hASCs, VEGF signaling cascade can also enhance hASCs’ differentiation into adipogenic, chondrogenic, and osteogenic lineages.4 VEGF is also a potent chemoattractant signal for bone marrow derived mesenchymal stem cells. Several studies have reported the active role of this growth factor in mediating cell-cell interaction and promoting the recruitment of undifferentiated stem cells to the fracture area in calvarial defects.5 These examples demonstrate the therapeutic potential of stem cells for tissue regeneration through the design of novel strategies for controlling the expression of angiogenic growth factors.
This goal can be achieved following a variety of strategies. One of these consists of culturing stem cells under stressed conditions such as hypoxia and serum deprivation, or by fabricating cell spheroids.6, 7 The composition and concentration of secreted angiogenic growth factors vary according to the selected approach. For example, culturing hASCs spheroids in serum-free media is a well-established method to obtain a higher expression of pro-angiogenic growth factors when compared to conventional in vitro 2D cell culture.8 Aside from modifying the cell culture conditions, more precise control can be achieved by nanocarrier-based gene delivery to obtain a selective angiogenic protein expression.9
Gene delivery to stem cells is carried out using both viral and non-viral nanocarriers, which are designed to improve the internalization of plasmid DNA vectors encoding pro-angiogenic genes.10, 11 Viral carriers, such as adenoviruses, lentivirus and retrovirus yield a high transfection efficiency because the transduction of viral DNA is facilitated by the viral envelopes surrounding their capsids. However, several safety concerns limit their applicability in gene therapy including biosafety, immunogenicity and possibility of mutagenesis.12 On the contrary, non-viral gene nanocarriers such as cationic polymers, lipids, and peptide conjugates provide a safer alternative to viral carriers given their low toxicity, lack of pathogenicity, versatile surface chemistry, and controllable size.13, 14 These set of unique advantages makes this type of nanocarriers a suitable candidate to ensure enhanced DNA delivery with limited cytotoxic effects. The selected carrier can have also a positive impact on transfection efficiency by promoting transmembrane diffusion of the genetic material, circumventing lysosomal degradation of the complex, and enhancing intracellular translocation to the nucleus.15 For all these reasons, the selection of an appropriate nanocarrier directly influences the final outcome of gene-based therapies aimed to regulate transient growth factor expression in stem cells.
Aside from these carrier properties, internalization of plasmid DNA nanocomplexes is also dependent on the elasticity of the cellular microenvironment, which controls the cell membrane tension and the cytoskeletal remodeling.16 Plasmid DNA nanocomplexes are internalized through several pathways including macropinocytosis, caveolin-mediated endocytosis,17 and clathrin-mediated endocytosis18, 19 which in turn are regulated by the polymerization of actin stress fibers in the cytoskeleton.20 Therefore, understanding the influence of substrate stiffness in modifying the intracellular organization of actin stress fibers is beneficial in optimizing transfection efficiency in non-viral gene delivery. Specifically, culturing pre-osteoblasts on stiff substrates, which mimic the elasticity of hard tissues, has been found to enhance non-viral transfection efficiency.21
Based on this concept, we evaluated the role of substrate stiffness as a parameter in gene transfection of hASCs with the goal of maximizing the transient expression of VEFG-A. hASCs were chosen as a model stem cell line due to their high proliferative potential, ease of harvest, and inherent ability to produce angiogenic growth factors. hASCs were purchased from RoosterBio and harvested from human lipoaspirate of female donors (31–45 years old). Our hypothesis is that transfection efficiency and corresponding growth factor production of hASCs can be enhanced by varying the physical properties of the substrate. Overall, our findings can be beneficial towards the design of scaffolds with suitable stiffness for optimal transfection efficiency. This point makes our study relevant in the field of gene delivery since it investigates the importance of substrate elasticity as primary modifier of cytoskeletal rearrangement to control the process of endocytosis of genetic material. Specifically, this study will provide useful insights regarding the role of actin fiber stress as one of the main regulators of plasmid DNA internalization.
As the first step of our investigation, we studied the effect of gelatin-coated silicone hydrogel stiffness on the morphology and cytoskeletal arrangement of hASCs. Soft (0.5 kPa) and stiff (32 kPa) substrates were chosen to resemble the stiffness of adipose and bone tissues, respectively.22 hASCs displayed a diverse morphology on the different substrates as indicated by immunofluorescent staining (Figure 1A and Figure S1). Cells seeded on the stiffer hydrogels showed an elongated morphology characterized by a larger surface cell area (4173 μm2) with respect to the hASCs on the softer hydrogels (2195 μm2) (Figure 1B). Paxillin staining was carried out to investigate the differences in cell focal adhesion expression.23 A greater focal adhesion area was observed for the hASCs seeded on the stiff hydrogels, and this result is congruent with previous reports that investigated paxillin expression in other cells lines (Figure 1C).24 Aside from a change in the focal adhesion area, a higher number of actin stress fibers was quantified in the hASCs on the 32 kPa hydrogels (Figure 1D). Similar results were found also in a study comparing the percentage of actin stress fibers in human fibroblasts seeded on poly(dimethylsiloxane) (PDMS) and polyacrylamide hydrogels presenting stiffness values ranging from 5 kPa to 2 MPa. Fibroblasts seeded on the stiffer matrix presented a more aligned distribution of F-actin and a higher cell area.
Fig. 1.
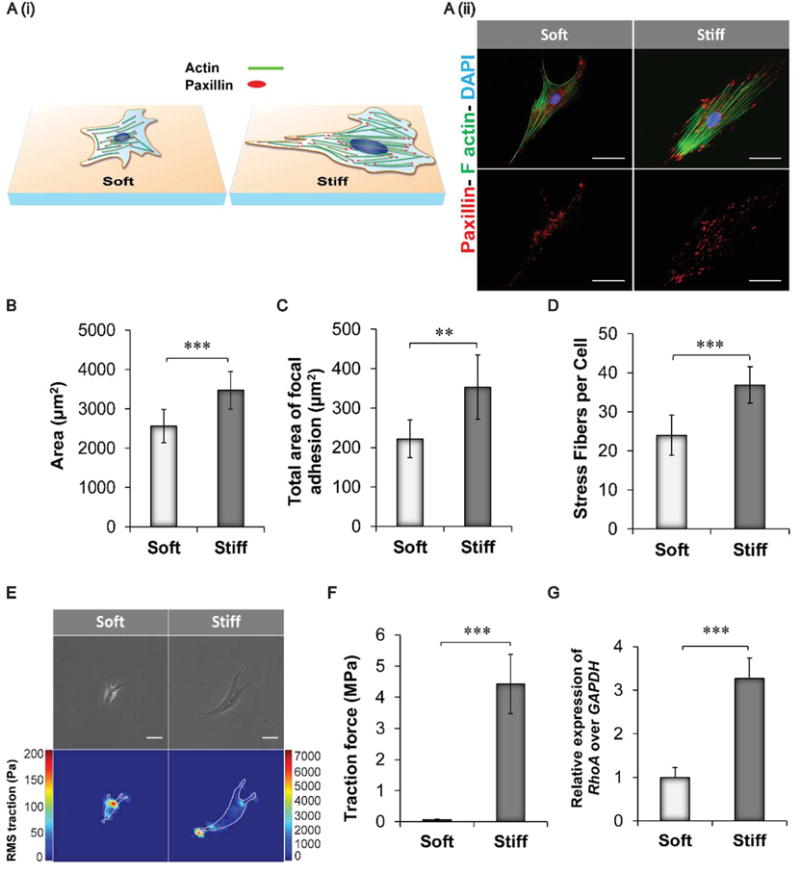
Influence of substrate stiffness on hASC morphology and cytoskeletal remodeling. A) (i) Schematic representing the main changes in cytoskeletal composition and focal adhesion distribution observed by culturing hASCs on soft and stiff hydrogels. Cells cultured on softer substrates displayed reduced cell surface area, lower total area of focal adhesions, and reduced formation of actin filaments. (ii) Immunofluorescence staining of hASCs seeded on soft (0.5 kPa) and stiff (32 kPa) silicone hydrogels pre-coated with a solution of gelatin 1% w/v. Cells were stained with Alexa Fluor 488 Phalloidin to visualize actin stress fibers (green) and with Diamidino-2-phenylindole dilactate (DAPI) to stain the nuclei (blue). In addition, paxillin staining (red) was carried out to identify the area of focal adhesion. Scale bar = 50 μm. B) Quantification of cell area of hASCs cultured on the two different substrates. Results are reported as mean ± standard deviation (n=10) . C) Quantification of the total area of focal adhesions expressed by hASCs on the different substrates. Results are calculated based on the red fluorescence intensity obtained from the immunofluorescent staining for paxillin expression (n=7). D) Actin stress fiber quantification obtained by counting the number of actin stress fiber on each cell at 40X magnification. Results are reported as mean ± standard deviation (n=9). E) Phase contrast pictures and traction map images of hASCs cultured on soft and stiff silicone hydrogels. Scale bar = 80 μm. F) Root-mean-square (RMS) traction values of hASCs indicated an increase in cell traction on the stiffer substrate (n=7). G) RhoA gene expression in hASCs seeded on the different substrates after 24 hours. Results were normalized based on the gene expression of cells cultured on the softer substrates (0.5 kPa) n=3. Results are reported as mean ± standard deviation * = p < 0.05, ** = p < 0.01 and *** = p < 0.001.
Furthermore, the average local stress magnitude exerted by the cells on the substrates was calculated based on constrained Fourier transform traction (FTTM). Results confirmed that hASCs seeded on stiffer hydrogels displayed a higher traction force with an increase in the mean value from 0.5 MPa to 5.7 MPa (Figures 1E–F). This difference can be attributed to the increase in tension within the networks of actin fibers in the cell’s cytoskeleton, and furthermore, the cells on stiffer substrates exhibited a reduced cytoskeletal mobility. This change in mobility is also evident by the increase in measured traction forces.25, 26 These findings were further confirmed by qPCR analysis. Specifically, we evaluated the expression of RhoA gene, which regulates the assembly of both contractile actomyosin filaments and focal adhesion complexes. RhoA was found to be upregulated in the 32 kPa group with a 2.1-fold increase compared to the softer hydrogel group (Figure 1G).27
After studying the effects of substrate stiffness on cell activity and cytoskeletal remodeling, we proceeded to evaluate the effects of substrate stiffness on the efficiency of gene delivery to hASCs. Following our previous observations, we hypothesized that an increase in the number of actin stress fibers will promote the internalization of the Lipofectamine-DNA complexes (lipoplexes) and therefore increase the transfection efficiency (Figure 2A). The Lipofectamine used in this study has a mean diameter of 346.96 ± 34.42 nm and a positive zeta potential value of 15.65 ± 0.89 as measured by dynamic light scattering (DLS) analysis. Rhodamine-tagged plasmids were electrostatically complexed with Lipofectamine and DLS analysis confirmed the formation of the complex (Figure 2B). For instance, the lipoplexes displayed an increase in the mean diameter value as well as a change in the zeta potential measurement from positive to negative values. Similar results were found in other reports investigating the formation of complex between plasmid DNA and cationic liposomes28. The lipoplexes were used to transfect hASCs seeded on the soft and stiff silicone hydrogels. The process of internalization was evaluated one hour post-transfection, then cells were imaged to visualize the quantity of plasmid internalized (Figure 2C). Quantification of the fluorescence intensity of the rhodamine-tagged plasmid revealed a higher presence of plasmid DNA in the hASCs cultured on the 32 kPa hydrogels. A three-fold increase in the fluorescent intensity was detected in the more rigid hydrogels with respect to the softer substrates (Figure 2D). This trend was further confirmed by qPCR analysis to determine the quantity of plasmid that was internalized into the cells (Figure 2E).29 The amount of plasmid internalized was measured based on a calibration curve obtained by qPCR analysis of different concentration of GFP plasmid (0.9 ng to 30 ng) in the cell lysates (Figure S2). As a proof of concept, we also transfected hASCs with a green fluorescent protein (GFP) plasmid using Lipofectamine. hASCs seeded on the stiff substrates displayed a higher GFP expression as confirmed by FACS analysis and fluorescent imaging of GFP positive fluorescent cells (Figure S3). Our findings suggest that the differences in cell morphology and cytoskeletal remodeling play a fundamental role in influencing the cell’s ability to uptake the lipoplexes. These results are in agreement with other reports studying the effects of matrix elasticity as the main mediator of cellular endocytosis in other cell lines. For instance, fibroblasts, myoblasts, and bone marrow stromal cells displayed higher internalization of fluorescently labeled DNA when seeded on the stiffer fibronectin-conjugated poly(ethylene glycol) diacrylate hydrogels (670 kPa) with respect to cells seeded on softer substrates.30
Fig. 2.
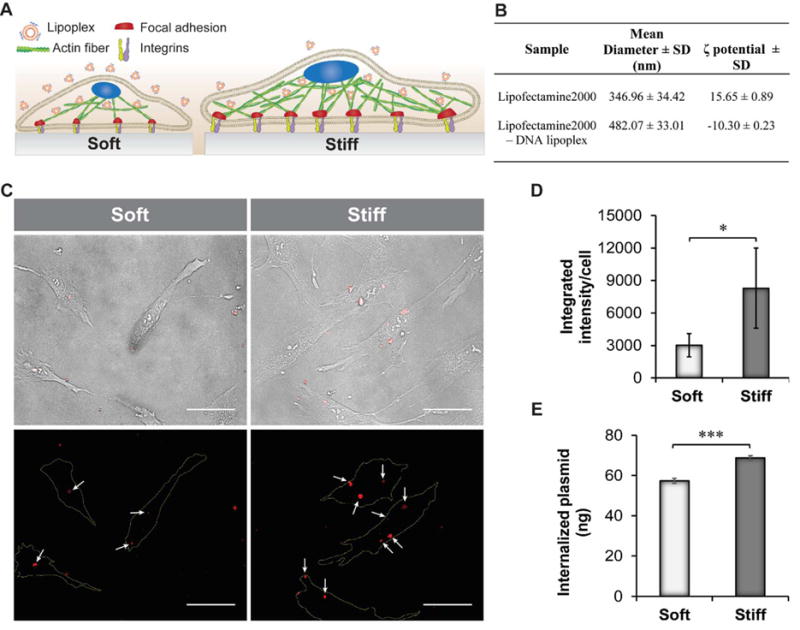
Effect of substrate stiffness on nanolipoplexes internalization. A) Schematic indicating the internalization efficiency of lipoplexes on hASCs adhering to substrates with different stiffness. Cells seeded on 32 kPa substrate internalized a higher quantity of plasmid DNA. B) Mean diameter and ζ-potential results obtained by DLS analysis of Lipofectamine and lipoplexes. C) Bright field images of hASCs seeded on soft and stiff substrates show the presence of internalized red-fluorescent lipoplexes. Scale bar = 50 μm. Corresponding fluorescent images displaying the red fluorescent complexes (white arrows). Cell boundaries are shown with a yellow line in the fluorescent images. Scale bar = 50 μm. D) Quantification of the fluorescence intensity of the rhodamine-tagged plasmid performed using image analysis (n=5). E) Quantification of plasmid internalization in hASCs seeded on soft and stiff substrates by qPCR analysis. Results are reported as mean ± standard deviation (n=4). * = p < 0.05, ** = p < 0.01 and *** = p < 0.001.
As the next step, we investigated the expression of the genes involved in endocytosis to define the role of actin stress fibers in regulating the internalization of the lipoplexes. We studied the gene expression of several key proteins directly involved in both caveolin and clathrin-mediated endocytosis pathways, since both uptake mechanisms are commonly activated in the presence of Lipofectamine complexes.31 The genes and primers used as well as the functions of each gene have been reported in the supplementary info (Table S1, Table S2, Supporting Information). The gene expression profile of hASCs was evaluated 1 hour and 4 hour post-transfection with lipoplexes. These two different times points were selected based on previous studies investigating the trafficking processes of the lipoplexes within the cytosol. Lipoplexes commonly bind with the cell membrane followed by association with actin fibers within the first hour upon transfection. Subsequently, lipoplexes can migrate to the perinuclear region through active transport using microtubules within the next four hours as shown in other reports (Figure 3A).32
Fig. 3.
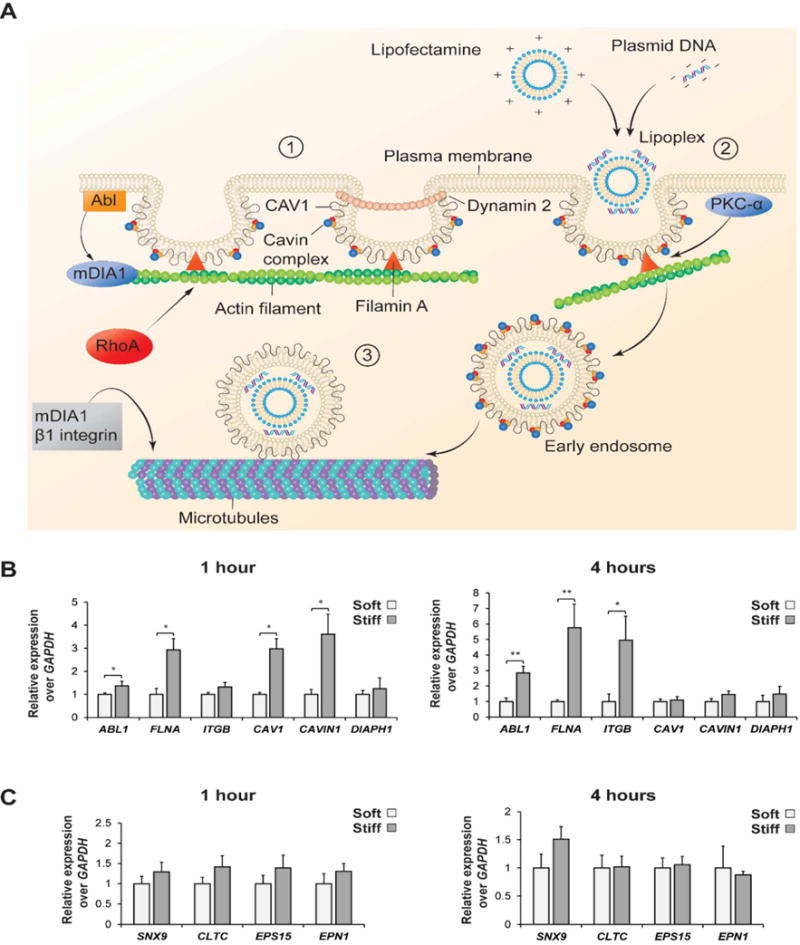
qPCR analysis of caveolin and clathrin genes regulating the process of endocytosis of nano-lipoplexes. A) Schematic illustrating the role of main proteins regulating the caveolin-mediated endocytosis. This process is divided into three steps: (1) Dynamic of caveolae at the plasma membrane, (2) Caveolae internalization by actin fibers stress and (3) endocytotic trafficking by microtubules. B) Fold increase expression of the main genes involved in caveolin endocytosis pathways after 1 and 4 hours of lipoplexes internalization. C) Fold increase expression of the main genes regulating clathrin endocytosis pathways after 1 and 4 hours of lipoplexes internalization. Results are normalized based on the gene expression of hASCs cultured on the soft substrate (0.5 kPa) (n=3). *= p < 0.05, ** = p < 0.01.
According to the qPCR results, caveolin-mediated genes were upregulated in the cells cultured on the 32 kPa substrate (Figure 3B), which confirmed the role of substrate stiffness as an indirect regulator of endocytosis. Specifically, Filamin A (FLNA) and Abl tyrosine kinases-1 (ABL1) genes were more expressed in hASCs cultured on the stiffer silicone hydrogels. Both genes are essential regulators of caveolae formation and migration. Filamin A is a protein that participates in the later migration of caveolae membrane and regulates the interaction between caveolin 1 (CAV1) and actin stress fibers. A depletion of FLNA gene is commonly associated with an alteration in the caveolae dynamic and a reduction in the caveolae formation. Additionally, Abl tyrosine kinase-1 controls the polymerization of actin stress fibers and regulates actin interaction with CAV 1. CAV1 and CAVIN1 genes were instead only upregulated after 4 hours, indicating a later expression of these proteins essential for caveolae formation. The higher upregulation of all the selected genes is justified by the increased presence of actin fibers in hASCs cultured on the stiffer hydrogels. Finally, β1 Integrin ITGB1 was upregulated after 1 hour of transfection in the 32 kPa group, which is indicative of a stabilization of microtubules that are involved in the cell trafficking of caveolae and recycling of CAV1 to the cellular membrane.17, 33 Overall, the upregulation of all the analyzed genes can be explained by the greater presence of well-aligned actin fibers in the hASCs seeded on the stiffer substrates.
On the contrary, no significant differences were found among clathrin-mediated genes in the two tested groups (Figure 3C). A possible explanation is that actin only plays a primary role in caveolin-mediated pathway, while its contribution to clathrin-mediated endocytosis is reported to be secondary. For instance, actin is not necessary to promote endocytosis of transferrin, which is a common ligand used to investigate clathrin-mediated endocytosis.
Another explanation for the predominance of a caveolin-mediated pathway is that the size of the lipoplexes used in this study was larger than 200 nm (482.07 ± 33.01 nm) as evaluated by DLS analysis. Clathrin-mediated endocytosis becomes the predominant mechanism of internalization only with nanomaterials characterized by a diameter less than 200 nm.34
Finally, we evaluated how the changes in plasmid internalization affected the cell’s ability to transiently express angiogenic growth factor. hASCs seeded on the different substrates were transfected with plasmid DNA encoding for the human VEGF-A gene. While hASCs possess a natural ability to produce angiogenic growth factors, this inherent capability can be augmented by using gene delivery and controlling the physical properties of the substrate on which hASCs are seeded.35 Higher gene transfection efficiency observed in hASCs cultured on the stiffer substrates resulted in a greater production of VEGF, which was evaluated by human umbilical vein endothelial cells (HUVEC) migration assay and ELISA quantification.36 HUVEC migration was monitored over time and a larger migration area was evident after 24 hours in the group exposed to VEGF produced by hASCs cultured on the 32 kPa hydrogels (Figure 4A–B). Finally, a four-fold increase in VEGF concentration was found in the media of the 32 kPa sample compared to the 0.5 kPa group after 72 hours (Figure 4C).
Fig. 4.
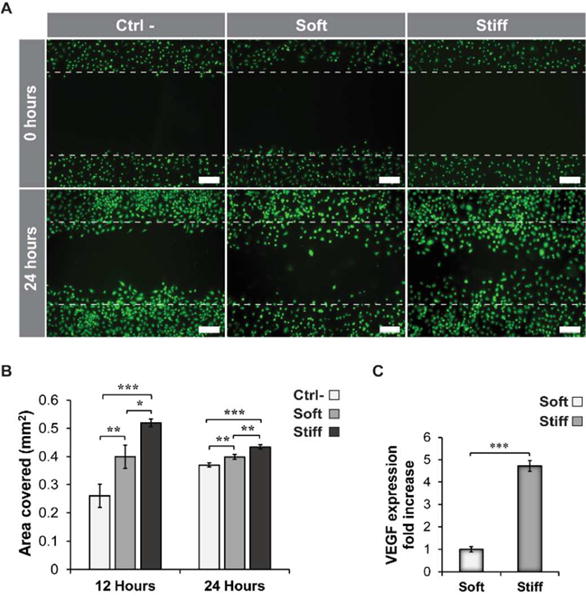
Modulation of VEGF expression of hASCs transfected on silicone hydrogels with different stiffness. A) Fluorescence images of HUVECs stained with calcein and seeded on a regular tissue culture-treated 24 well plate. Scratches were made in the 24 well plate at time zero on HUVEC monolayer. Cells were allowed to migrate into the void area for 24 hours. The conditioned media in the soft and stiff group was supplemented with VEGF secreted by hASCs after 72 hours post-transfection. No VEGF and recombinant human fibroblast growth factor-B (rhFGF-B) were present in the media of the control group (Ctrl-). Scale bar = 200μm. B) Quantification of the surface area covered by HUVECs after proliferation through the original scratches at 12 and 24 hours. The area covered was calculated based on the original area of the scratches at time zero (n=3). C) Quantification of VEGF expression by ELISA after 72 hours post-transfection. Results are normalized based on the VEGF expression of hASCs cultured on the soft substrate (0.5 kPa) (n=3).*= p < 0.05, ** = p < 0.01, *** = p < 0.001.
Conclusions
Overall, this study highlights the importance of substrate stiffness as an essential regulator of cellular cytoskeletal remodelling and internalization of exogenous genes in hASCs. Specifically, the role of actin stress fibers in the process of endocytosis of lipoplexes has been confirmed defining its direct contribution in plasmid internalization. Thus, hASCs ability to produce angiogenic growth factors can be augmented by transfecting them in a stiffer substrate and this strategy can be applied to enhance their angiogenic potential. Finally, our findings could be potentially translated to other stem cell types, although proper selection of the optimal substrate stiffness is an essential parameter that needs to be accurately evaluated.
Supplementary Material
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Conflicts of interest
There are no conflicts to declare.
References
- 1.Rodrigues M, Griffith LG, Wells A. Stem Cell Research & Therapy. 2010;1:32. doi: 10.1186/scrt32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pacelli S, Acosta F, Chakravarti AR, Samanta SG, Whitlow J, Modaresi S, Ahmed RPH, Rajasingh J, Paul A. Acta Biomaterialia. 2017;58:479–491. doi: 10.1016/j.actbio.2017.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pacelli S, Basu S, Whitlow J, Chakravarti AR, Acosta F, Varshney A, Modaresi S, Berkland C, Paul A. Advanced Drug Delivery Reviews. 2017 doi: 10.1016/j.addr.2017.07.011. DOI: http://dx.doi.org/10.1016/j.addr.2017.07.011. [DOI] [PMC free article] [PubMed]
- 4.Chen G, Shi X, Sun C, Li M, Zhou Q, Zhang C, Huang J, Qiu Y, Wen X, Zhang Y, Zhang Y, Yang S, Lu L, Zhang J, Yuan Q, Lu J, Xu G, Xue Y, Jin Z, Jiang C, Ying M, Liu X. PLoS One. 2013;8:e73673. doi: 10.1371/journal.pone.0073673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou Y, Huang R, Fan W, Prasadam I, Crawford R, Xiao Y. Journal of Tissue Engineering and Regenerative Medicine. 2017:n/a–n/a. doi: 10.1002/term.2327. [DOI] [PubMed] [Google Scholar]
- 6.Van Pham P, Vu NB, Phan NK. Biomedical Research and Therapy. 2016;3 [Google Scholar]
- 7.Waters R, Pacelli S, Maloney R, Medhi I, Ahmed RP, Paul A. Nanoscale. 2016;8:7371–7376. doi: 10.1039/c5nr07806g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petrenko Y, Syková E, Kubinová Š. Stem Cell Research & Therapy. 2017;8:94. doi: 10.1186/s13287-017-0558-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dall’Era JE, Meacham RB, Mills JN, Koul S, Carlsen SN, Myers JB, Koul HK. Int J Impot Res. 2008;20:307–314. doi: 10.1038/ijir.2008.1. [DOI] [PubMed] [Google Scholar]
- 10.Schönmeyr BH, Soares M, Avraham T, Clavin NW, Gewalli F, Mehrara BJ. Tissue Engineering Part A. 2010;16:653–662. doi: 10.1089/ten.tea.2009.0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nikol S. Cardiovasc Res. 2007;73:443–445. doi: 10.1016/j.cardiores.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 12.Seow Y, Wood MJ. Molecular Therapy. 17:767–777. doi: 10.1038/mt.2009.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Medina-Kauwe LK, Xie J, Hamm-Alvarez S. Gene Ther. 2005;12:1734–1751. doi: 10.1038/sj.gt.3302592. [DOI] [PubMed] [Google Scholar]
- 14.Ramamoorth M, Narvekar A. J Clin Diagn Res. 2015;9:GE01-06. doi: 10.7860/JCDR/2015/10443.5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adler AF, Leong KW. Nano today. 2010;5:553–569. doi: 10.1016/j.nantod.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Missirlis D. PLoS One. 2014;9:e96548. doi: 10.1371/journal.pone.0096548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Echarri A, Del Pozo MA. Journal of Cell Science. 2015;128:2747–2758. doi: 10.1242/jcs.153940. [DOI] [PubMed] [Google Scholar]
- 18.Yao LH, Rao Y, Bang C, Kurilova S, Varga K, Wang CY, Weller BD, Cho W, Cheng J, Gong LW. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33:15793–15798. doi: 10.1523/JNEUROSCI.2171-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McMahon HT, Boucrot E. Nature reviews Molecular cell biology. 2011;12:517–533. doi: 10.1038/nrm3151. [DOI] [PubMed] [Google Scholar]
- 20.Smythe E, Ayscough KR. Journal of Cell Science. 2006;119:4589–4598. doi: 10.1242/jcs.03247. [DOI] [PubMed] [Google Scholar]
- 21.Kong HJ, Liu J, Riddle K, Matsumoto T, Leach K, Mooney DJ. Nature materials. 2005;4:460–464. doi: 10.1038/nmat1392. [DOI] [PubMed] [Google Scholar]
- 22.Vertelov G, Gutierrez E, Lee SA, Ronan E, Groisman A, Tkachenko E. Sci Rep. 2016;6:33411. doi: 10.1038/srep33411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pacelli S, Maloney R, Chakravarti AR, Whitlow J, Basu S, Modaresi S, Gehrke S, Paul A. Sci Rep. 2017;7:6577. doi: 10.1038/s41598-017-06028-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prager-Khoutorsky M, Lichtenstein A, Krishnan R, Rajendran K, Mayo A, Kam Z, Geiger B, Bershadsky AD. Nat Cell Biol. 2011;13:1457–1465. doi: 10.1038/ncb2370. [DOI] [PubMed] [Google Scholar]
- 25.Ehrlicher AJ, Krishnan R, Guo M, Bidan CM, Weitz DA, Pollak MR. Proc Natl Acad Sci U S A. 2015;112:6619–6624. doi: 10.1073/pnas.1505652112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Butler JP, Tolić-Nørrelykke IM, Fabry B, Fredberg JJ. American Journal of Physiology - Cell Physiology. 2002;282:C595. doi: 10.1152/ajpcell.00270.2001. [DOI] [PubMed] [Google Scholar]
- 27.Chi X, Wang S, Huang Y, Stamnes M, Chen JL. Int J Mol Sci. 2013;14:7089–7108. doi: 10.3390/ijms14047089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Son KK, Patel DH, Tkach D, Park A. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2000;1466:11–15. doi: 10.1016/s0005-2736(00)00176-0. [DOI] [PubMed] [Google Scholar]
- 29.Carapuca E, Azzoni AR, Prazeres DM, Monteiro GA, Mergulhao FJ. Mol Biotechnol. 2007;37:120–126. doi: 10.1007/s12033-007-0007-3. [DOI] [PubMed] [Google Scholar]
- 30.Chu C, Kong H. Acta Biomater. 2012;8:2612–2619. doi: 10.1016/j.actbio.2012.04.014. [DOI] [PubMed] [Google Scholar]
- 31.Cui S, Wang B, Zhao Y, Chen H, Ding H, Zhi D, Zhang S. Biotechnol Lett. 2014;36:1–7. doi: 10.1007/s10529-013-1325-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cardarelli F, Digiacomo L, Marchini C, Amici A, Salomone F, Fiume G, Rossetta A, Gratton E, Pozzi D, Caracciolo G. 2016;6:25879. doi: 10.1038/srep25879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parton RG, del Pozo MA. Nature reviews Molecular cell biology. 2013;14:98–112. doi: 10.1038/nrm3512. [DOI] [PubMed] [Google Scholar]
- 34.Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Biochem J. 2004;377:159–169. doi: 10.1042/BJ20031253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yarak S, Okamoto OK. An Bras Dermatol. 2010;85:647–656. doi: 10.1590/s0365-05962010000500008. [DOI] [PubMed] [Google Scholar]
- 36.Paul A, Hasan A, Kindi HA, Gaharwar AK, Rao VT, Nikkhah M, Shin SR, Krafft D, Dokmeci MR, Shum-Tim D, Khademhosseini A. ACS Nano. 2014;8:8050–8062. doi: 10.1021/nn5020787. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.