

Abstract
UDP-Glucose 6-dehydrogenase (UGDH) produces UDP-α-D-glucuronic acid, the precursors for glycosaminoglycans (GAGs) and proteoglycans (PGs) of the extracellular matrix. Elevated GAG formation has been implicated in a variety of human diseases, including glioblastoma (GBM). In our previous study, we found that krÜppel-like factor 4 (KLF4) promotes GBM cell migration by binding to methylated DNA, mainly methylated CpGs (mCpG) and transactivating gene expression. We identified UDGH as one of the downstream targets of KLF4-mCpG binding activity. In this study, we show that KLF4 upregulates UGDH expression in a mCpG-dependent manner, and UGDH is required for KLF4 induced cell migration in vitro. UGDH knockdown decreases glycosaminoglycan (GAG) abundance in GBM cells, as well as cell proliferation and migration in vitro. In intracranial xenografts, reduced UGDH inhibits tumor growth and migration, accompanied by a decrease in the expression of extracellular matrix proteins such as tenascin C, brevican. Our studies demonstrate a novel DNA methylation-dependent UGDH upregulation by KLF4. Developing UGDH antagonists to decrease the synthesis of extracellular matrix components will be a useful strategy for GBM therapy.
Introduction
Glioblastoma (GBM, Grade IV glioma) is one of the most devastating forms of cancer and characterized by highly proliferative tumor growth and intensive tumor cell infiltration into normal brain tissues.1,2 An increased understanding of the molecular mechanisms underlying the aggressive behavior of tumor cells and the microenvironment in which they invade could provide insights into novel treatment strategies for this deadly disease. The extracellular matrix (ECM) is one of the critical components of the tumor microenvironment and provides essential biochemical and mechanical cues that direct cell growth, survival, migration and differentiation.3,4 Cell adhesion to the ECM permits growth factor-dependent activation of oncogenic signals, which promotes cell cycle progression and cell proliferation, while also functioning as either a barrier or a movement track to inhibit or promote cell migration.5 The ECM is mainly composed of fibrous proteins (e.g., collagen) and gel-like substance, such as glycosaminoglycans (GAGs), which are long polysaccharide chains with negative charges that attract water and soluble molecules including growth factors.6
GAGs are synthesized by an enzyme called UDP-glucose 6-dehydrogenase (UGDH). In our prior work, we found that krÜppel-like factor 4 (KLF4) binds to methylated CpGs (mCpG) in cis-regulatory elements, and activates gene expression, including UGDH, in GBM cells to exert phenotypic changes such as increased cell migration.7 DNA methylation at cis-regulatory regions, mostly occurring at the CpG dinucleotide sites, is linked to gene repression and more recently gene activation.7,8 Given the importance of extracellular matrix function, specifically GAGs, in GBM; and the fact that UGDH is implicated as a rate-limiting and essential step in GAG monosaccharide synthesis.9–11 the mCpG-dependent activation of UGDH prompts us to investigate the biological function of UGDH in GBM.
GAG formation is part of glucose metabolism: glucose is converted to glucose-1-phosphate then to UDP-glucose (UDP-Glu), an active form of glucose, which is further converted to UDP-glucuronic acid (UDP-GlcA). UDP-GlcA is the indispensable precursor for the synthesis of GAGs. The enzyme UDP-glucose 6-dehydrogenase (UGDH) catalyzes the biosynthetic oxidation of UDP-glucose to UDP-glucuronic acid,9,11 which are the building blocks of GAGs including hyaluronic acid and proteoglycans such as brevican, versican, aggregan etc. GAG synthesis pathways and key players are shown in (Figure 1).
Figure 1.
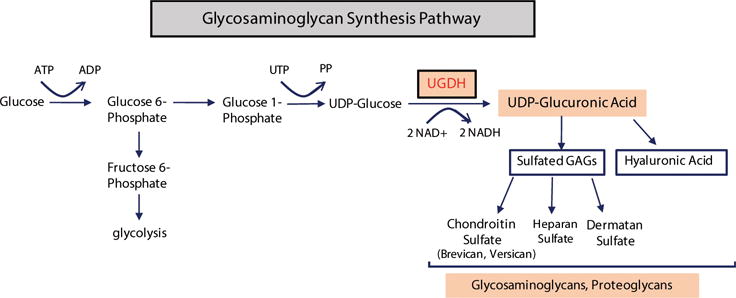
Schematic illustration of GAG synthesis pathway, different GAGs and UGDH function in GAG synthesis.
GAGs reside in the extracellular space providing structural support for cells, as well as promoting cell adhesion, motility, angiogenesis and wound healing.12,13 Elevated GAG formation is implicated in a variety of human diseases, including the progression of epithelium tumors, breast cancers and brain tumors.6,14 Although GAGs are shown to be implicated in tumor progression, decrease in the synthesis of GAG precursor UDP-glucuronic acid in GBM biology has not been investigated. In this current work, we investigated the methylation-dependent regulation of UGDH, as well as the biological function of UGDH in GBM cells. These findings identify UGDH as a potential therapeutic target for GBMs.
Materials and Methods
Reagents and Cell Cultures
All reagents were purchased from Sigma-Aldrich unless otherwise stated. Doxycline (Dox) was diluted to a concentration of 1μg/ml in cell culture medium as a working concentration. The human glioblastoma (GBM) cell lines U87 were originally purchased from ATCC (Manassas, VA). GBM neurosphere culture (HSR-GBM1A) were originally established by Vescovi and colleagues15 and further characterized by us.16–18 Both cells lines are free from mycoplasma and authenticated with short tandem repeat (STR) profiling by Johns Hopkins Genetic Resources Core facility using Promega GenePrint 10 system (Madison, WI). U87 cells were cultured in Minimum Essential Media (MEM, Thermo Fisher Scientific, Grand Island, NY) supplemented with sodium pyruvate (1%), sodium bicarbonate (2%), non-essential amino acid (1%) and 10% fetal calf serum (FCS, Gemini Bio-products, West Sacramento, CA). HSR-GBM1A (GBM1A) cells contain CD133+ GBM stem-like cells and form infiltrative orthotropic xenografts that have been extensively characterized by others and our group.19,20 GBM1A neurospheres were cultured in DMEM/F12 medium supplemented with epidermal growth factor (EGF) (Peprotech, Rocky Hill, NJ) and fibroblast growth factor (FGF) (Peprotech). Cells were incubated in a humidified incubator containing 5% CO2/95% air at 37°C, and passaged every 4-5 days.
Lentiviral Transduction
UGDH shRNA lentiviral particles were purchased from Dharmacon (Buckinghamshire, UK). Control (non-silencing) shRNA clone ID RHS4348, UGDH sh#1 clone ID V2LHS-171838 and UGDH sh#2 clone ID V3LHS-412961. GBM cells were transduced with virus for 48 hrs prior to puromycin selection (1 μg/ml) as previously described.21 Control cells were transduced with non-silencing control shRNA.
Quantitative real-time PCR
Total RNA was extracted using RNeasy Mini Kit (Qiagen, Mansfield, MA). After reverse transcription using MuLV reverse transcriptase (Applied Biosystems, Calsbad, CA) and Oligo(dT) primer, quantitative real-time PCR (qRT-PCR) was performed using SYBR Green PCR Mix (Applied Biosystems) and IQ5 detection system (Bio-Rad, Hercules, CA). Primer sequences are listed in Supplementary Table 1. Relative gene expression was normalized to 18S rRNA.
Immunoblot and Immunocytochemistry
Total cellular protein was extracted with radioimmunoprecipitation assay (RIPA) buffer containing protease and phosphatase inhibitors (Millipore, Billerica, MA) and sonicated for 15 seconds; the suspensions were centrifuged at 3,000 g for 10 minutes. SDS-PAGE was performed with 30-60 μg total proteins using 4% to 12% gradient Tris-glycine gels (LI-COR Biosciences, Lincoln, NE). Western blot analysis was performed using the Quantitative Western Blot System, with secondary antibodies labeled by IRDye infrared dyes (LI-COR Biosciences). Antibodies were purchased from: anti-KLF4 (Santa-Cruz, Dallas, TA); anti-UGDH, anti- RABGEF1, anti-NGEF, anti-Brevican (Abcam, San Francisco, CA); anti- PHLDB2 (Bethyl Labs, Montgomery, TX); anti-tenascin C (Millipore), anti- phospho-AKT, anti-total AKT, anti-cyclin B1, E1 and D1 (Cell signaling, Danvers, MA) and anti-β-actin.
For staining, GBM cells grown on chamber slides were fixed with 4% paraformaldehyde for 30 min at 4°C and permeabilized with PBS containing 0.1% Triton X-100 for 10 min. The cells were then incubated with primary antibodies at 4°C overnight and then incubated with appropriate corresponding secondary antibodies conjugated with alexa fluorescent 488 or cy3 for 1hr at room temperature. Slides were mounted with vectashield antifade solution containing DAPI (Vector Laboratories, Burlingame, CA) and observed under fluorescent microscopy. Immunofluorescent images were taken and analyzed using Axiovision software (Zeiss, Germany).
Chromatin immunoprecipitation (ChIP)-PCR
A commercial ChIP-grade anti-KLF4 antibody (H180; Santa Cruz) recognizing the N-terminal region of KLF4 was used for ChIP (DNA-binding domains of KLF4 are located to the C-terminus). Tet-on KLF4 WT and R458A GBM cells were treated with Dox for 48 hr followed by ChIP using the anti-KLF4 antibody and Dynabeads Protein A/G (Thermo Fisher Scientific) according to a protocol described previously.7,8 Primers targeting KLF4 binding sites were identified from previous ChIP-Seq analysis7 Primers targeting promoter regions lacking KLF4 binding sites were used as a negative control.
Assessment of CpG methylation status by bisulfite sequencing
Sanger bisulfite sequencing was performed as previously described.8 Purified genomic DNA from GBM cells were treated by EZ DNA Methylation-Direct Kit (Zymo Research, Irvine, CA). After bisulfite conversion, regions of interest were PCR-amplified using Taq polemerase. The primers used for bisulfite sequencing are listed in Supplemental Table 2. PCR products were cleaned up and cloned into a TA vector (Thermo Fisher Scientific). Individual clones were sequenced (Genewiz, Cambridge, MA) and aligned with the reference sequence.
Quantitative measurement of glycosaminoglycans (GAG)
The quantity of GAG was determined using the 1,9 dimethyl-methylene blue (DMMB) binding method with modifications as described by Barbosa et al.22 Briefly, the cells were digested with 0.2% papain in reaction buffer consisting of 0.1 M NaH2PO4, 5 mM Na2-EDTA, and 5 mM cysteine–HCl pH 6.0, at 60 °C for 6 h. Total cell lysates were concentrated and adjusted to a concentration of 0.1 mg protein in 50 μl before mixing with 200 μl of DMMB solution (40 mM glycine, 40 mM NaCl, 9.5 mM HCl, and 0.0016% DMMB, pH 3.0) in each well of a 96-well microtiter plate. The absorbance was immediately recorded at 525 nm using a plate reader (Perkin Elmer, Waltham, MA). Serial dilutions of shark chondroitin-6-sulphate, ranging from 5 to 25 μg/ml, were used to establish a standard curve, and duplicate wells with 50 μl of papain digestion buffer were used as blanks. Sample concentrations were calculated using linear regression of the standard curves.
Transwell migration assay
Cell migration assays were performed using transwell chambers as we previously described.21,23 The upper chamber medium consisted of either neurosphere cell culture medium without EGF/FGF or U87 cell media without FBS, and the lower chamber medium consisted of DMEM with 10% FBS. After 4-24 hours, cells that had migrated through the filter were fixed with Diff-Quick kit (Thermo Fisher Scientific). Cells on the upper side of the transwells were gently wiped off with Q-tips. Cells migrating through the filter were stained with 4′-6-Diamidino-2-phenylindole (DAPI). Migration was quantified by counting cells on five randomly selected fields per transwell in at least three independent experiments.23
Scratch Assay
GBM cells were grown under 10% FCS medium in 35 mm dishes until confluent. Several scratches were created using a 10 μl pipette tip through the confluent cells. Dishes were washed with PBS for 3 times and cells were grown in 0.1% FCS medium for 24-48 hr. Phase contrast pictures were taken at different time points. The width of the scratch was measured and areas of wound healing were measured and quantified using ImageJ.
Neurosphere formation assays
Viable cells (2 × 103/well or 2 × 104/well) were cultured in 48-well or 6-well plates, respectively. After 7-14 days, neurospheres were fixed in medium with 1% agarose, stained with 1% Wright stain solution and counted by computer-assisted morphometry (MCID software, Cambridge, UK) by measuring the number of neurospheres (>50 μm or >100 μm in diameter, as indicated) in three random fields per well.
Colony formation Assay
Anchorage-independent tumor cell proliferation was assessed by colony formation in soft agar. UGDH knockdown U87 cells or control-transduced cells were plated 10,000 cells per well in 0.5% agarose medium on top of regular medium containing 1% agarose and incubated for 2 weeks. The cells were stained blue with 1% Wright stain solution, and the number of colonies larger than 100 μm in diameter was determined by computer-assisted image analysis MCID.
Cell Cycle Analysis
Cell cycle was analyzed by flow cytometry on a FACSCalibur (Becton-Dickinson, Mountain View, CA).24 After plating in normal medium overnight, U87 cells were synchronized by changing into 0.1% FCS medium for 48 hrs followed by stimulation with 10% FCS for the indicated time points. To harvest, cells were trypsinized and dissociated by pipetting, fixed with 75% ethanol at 4 °C for 30 min. Cells were then incubated with DNase-free RNase at 37 °C for 30 min followed by propidium iodide (100 ng/ml) for 1 h at 37 °C. The percentage of cells at each cell-cycle phase (G1/G0, S and G2/M) was analyzed using CellQuest software (Becton-Dickinson).
Intracranial tumor implantation
All animal protocols were approved by the Johns Hopkins School of Medicine Animal Care and Use Committee. Each 6-8 week old female BALB/c strain immunodeficient (SCID) mouse received 10,000 viable neurosphere cells in 2 μL PBS by stereotactic injection to the right caudate/putamen (AP = 0 mm, ML = −2.5 mm, DV = −3.0 mm). After 7 weeks, mice were sacrificed and perfused with 4% paraformaldehyde; the brains were removed for histological analysis. Tumor sizes were quantified by measuring maximum tumor volume on hematoxylin and eosin–stained brain coronal sections using computer-assisted morphometry (MCID software) and then applying the formula Volume = (square root of maximum cross-sectional area).3,20 The primary antibodies used for immunofluorescent staining are the following: monoclonal anti-Ki67 (BD Biosciences, Franklin Lakes, NJ), anti-TNC (Millipore), anti-Brevican (Abcam), and anti-laminin (Millipore).
To observe tumor cell migration in vivo, we mixed one portion of red fluorescent protein (RFP)-labeled control non-silencing cells (50,000) with three portion of green fluorescent protein (GFP)-labeled UGDH knockdown cells (150,000), and injected the mixture into the same mouse brains (n=3). Animals were sacrificed after 4 weeks and brain sections were observed under fluorescence microscopy.
Large scale data analysis, statistical analysis
All the raw data for our large-scale RNA-sequencing and KLF4 chromatin immunoprecipitation-sequencing (ChIP-seq) have been deposited in GEO (GSE97632). The link is: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=wjydoqaqxzgnxwz&acc=GSE97632. The methods to generate this data have been described in Wan et al in details.7
Statistical analysis was performed using Prizm software (GraphPad, La Jolla, CA, www.graphpad.com). Post hoc tests included the Students T-test and Tukey multiple comparison tests as appropriate. All in vitro experiments reported here represent at least three independent replications. All data are represented as mean value ± standard error of mean (S.E.); significance was set at P < 0.05.
Results
KLF4-mCpG interactions activate genes involved in migration
In our previous studies, we firmly established that KLF4 could activate transcription of >100 genes via a non-canonical binding activity to methylated CpG (mCpG) to promote GBM cell migration.7 We observed that one third of such genes are involved in cytoskeletal organization, extracellular matrix formation and cell migration (Supplemental Fig. 1). To further interrogate the biological function of KLF4-mCpG binding activity, two previously characterized tet-on inducible human glioblastoma (GBM) U87 cell lines were used: one expressing KLF4 wild type (KLF4 WT) and the other KLF4 site-specific mutant (KLF4 R458A), which lacks the mCpG-dependent binding activity but retains KLF4 canonical binding activity.7 Real-time PCR (RT-PCR) and ChIP-PCR were utilized to examine the expression of selected KLF4 downstream target genes in GBM cells. Genes activated by KLF4 WT but not by KLF4 R458A would be recognized as a putative KLF4-mCpG target.
We selected the top 20 genes significantly upregulated by KLF4 WT, and associated with migration and cytoskeletal reorganization from our RNA-seq and gene ontology data,7 respectively. Gene expression was quantified in tet-on inducible U87 KLF4 WT cells +/− Dox, and tet-on inducible U87 KLF4 R458A cells +/− Dox (Figure 2A). Our results demonstrated that KLF4 WT and KLF4 R458A differently induced gene expression. Specifically, eleven of the twenty genes, such as neuronal guanine nucleotide exchange factor (NGEF), pleckstrin homology like domain family B member 2 (PHLDB2), RAB guanine nucleotide exchange factor (GEF) 1 (RABGEF1), and UDP-α-D-glucose 6-dehydrogenase (UGDH), were highly induced by KLF4 WT, but not by KLF4 R458A at the mRNA level (Figure 2A, asterisks), indicating that activation of these genes was dependent on mCpG-dependent binding of KLF4. To further confirm that the gene activation occurred via KLF4 binding to mCpGs in cis-regulatory regions, we performed bisulfite sequencing to examine the methylation status of the KLF4-binding regions of these genes based on our previous ChIP-seq data.7 We found that nine of the eleven KLF4 WT-upregulated genes were associated with highly methylated regions (Figure 2A, bold), and some examples are shown in Figure 2B.
Figure 2. KLF4-mCpG binding activity activates genes involved in GBM cytoskeletal organization and migration.
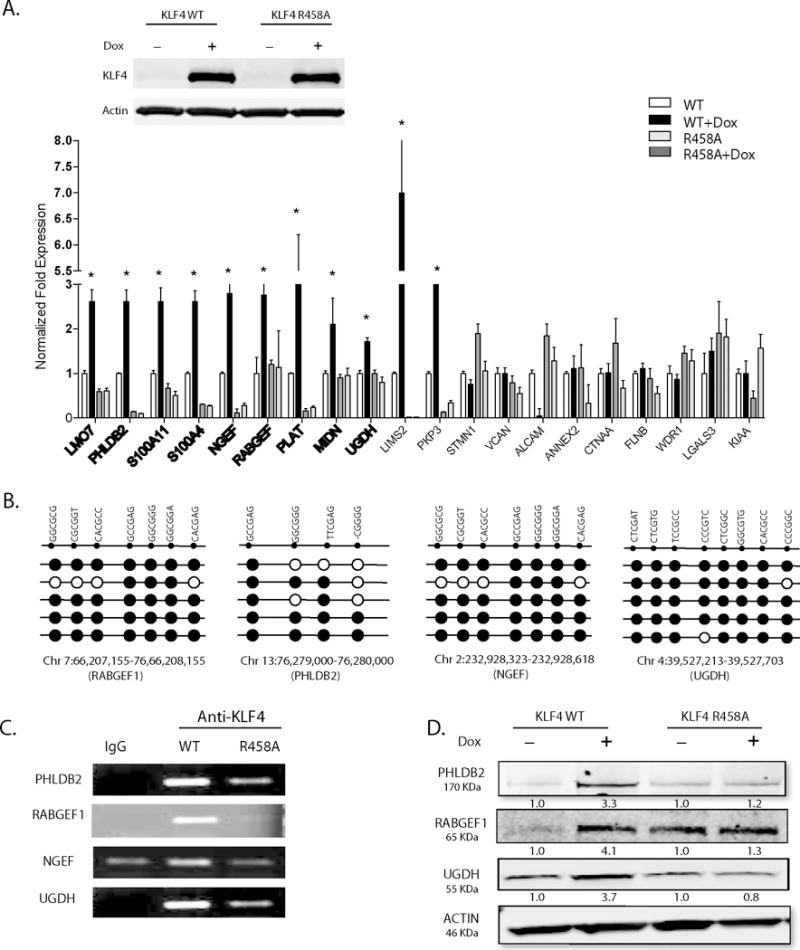
(A) Upper panel: Western blot analysis showed KLF4 WT or KLF4 R458A expression in tet-on stable U87 GBM cell lines upon doxycyline treatment (1 ug/ml, 48 hrs). Lower panel: Twenty putative KLF4-mCpG gene targets involved in cell migration pathway were picked from our previous RNA-seq studies. Real time-PCR (RT-PCR) revealed 11 of the 20 genes were significantly upregulated by KLF4 WT only, with no change in KLF4 R458A expressing cells (+Dox, 48 hr), confirming a mCpG-dependent gene activation mechanism (asterisks). Bold showed targets with methylated cis-regulatory elements in the gene. (B) Sanger bisulfite sequencing indicated DNA methylation in tested cis-regulatory regions of putative KLF4-mCpG targets. Examples of four genes showed highly methylated KLF4 binding regions of these genes. Each row represents one sequenced clone; each column represents one CpG site; filled circles stand for methylation. (C) Confirmation that KLF4 WT but not KLF4 R458R preferentially bound to the methylated cis-regulatory regions of selected genes. A KLF4 antibody was used to precipitate cross-linked genomic DNA from U87 cells expressing KLF4 WT or KLF4 R458A. Rabbit IgG was used to control for non-specific binding. De-crosslinked DNA samples were served as the input for ChIP-PCR. KLF4 binding to the selected regions was enriched in KLF4 WT expressing cells. (D) Western blot analysis indicating increased protein expression of the selected targets by KLF4 WT, but not by KLF4 R458A.
We next performed ChIP-PCR to examine binding activity of KLF4 WT and KLF4 R458A to the cis-regulatory regions of a subset of the nine genes, including PHLDB2, RABGEF1, NGEF and UGDH. An anti-KLF4 antibody that recognizes both KLF4 WT and KLF4 R458A was used to ChIP the chromatins in KLF4 WT and KLF4 R458A expressing cells 48-hrs post Dox induction.7 The resulting ChIP-PCR data demonstrated that KLF4 WT preferentially bound to the cis-regulatory regions of these upregulated genes, whereas KLF4 R458A showed much weaker or no binding signals to the same region (Figure 2C). In addition, KLF4 WT increased protein expression of PHLDB2, RABGEF1, and UGDH in comparison to KLF4 R458A (Figure 2D), corroborating that these genes could be functional KLF4-mCpG direct targets.
UGDH correlates with KLF4 expression in GBMs and is regulated by KLF4 via a DNA methylation-dependent mechanism
Next, we decided to perform detailed analysis on a single candidate to elucidate the mechanism underlying KLF4’s methylation dependent increase in GBM cell migration. We queried the REMBRANT database to assess gene expression in tumor and non-tumor specimens, and to determine whether selected genes correlate with KLF4 expression and patient survival in GBM. We chose UGDH as the model target because it was one of the three targets to have associations with GBM tumor progression. It is the least studied target in glioblastoma biology, and importantly, the only one to have consistent results across three cell lines we examined. We therefore focused on determining the biological function of UGDH in GBM as it is an important enzyme for the synthesis of extracellular matrix components. As shown in Figure 3A, UGDH was found to be upregulated in GBM samples when compared with normal brains. UGDH expression also moderately correlated with KLF4 expression in mesenchymal GBM samples (R= 0.32, P<0.001) (Figure 3B). Furthermore, we found that high expression of UGDH is associated with poor survival in mesenchymal GBM patients (Figure 3C, P<0.05, UGDH high n=38, median survival =10.4 months; UGDH low n=118, median survival =13.9 months).
Figure 3. UDGH correlates with KLF4 expression in GBM samples and is regulated via a mCpG dependent mechanism by KLF4.
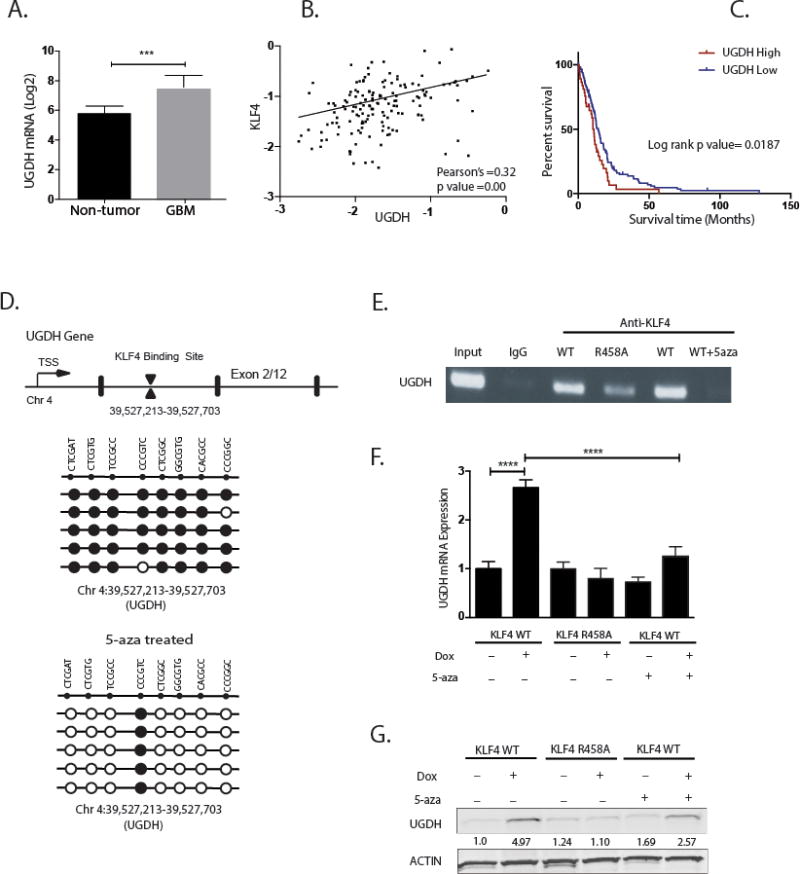
(A) UGDH gene expression intensity (mean) from REMBRANDT database. UGDH expression is significantly upregulated in GBM samples when compared with non-neoplastic brain samples (P<0.001). (B) Pearson’s correlation plots of UGDH and KLF4 expression in mesenchymal glioma patients of mesenchymal subtype. Plots show positive correlation in the expression of UGDH and KLF4 (R=0.32, P < 0.001). (C) Kaplan-Meier Survival Plots for mesenchymal glioma patients based on differential gene expression of UGDH (from REMBRANT database). The probability of survival is significantly lower in samples with high UGDH gene expression compared to samples with low expression in GBM mesenchymal subtype (P < 0.001). (D) Upper panel: schematic of UGDH gene structure showing KLF4 binding site on the first intron. Lower panels: sanger bisulfite sequencing of DNA methylation on KLF4 binding site of UGDH gene before and after treatment with DNA methyltransferase inhibitor 5-Aza. Cells pretreated with 5-aza showed 80% reduction in DNA methylation. (E) ChIP-PCR showed KLF4 WT preferentially bound to the methylated cis-regulatory region of UGDH; 5-aza treatment decreased KLF4 WT binding to UGDH. (F) RT-PCR showed UGDH mRNA was significantly induced by KLF4 WT only, which was abrogated by 5-aza. (G) Western blot showed induction of UGDH by KLF4 WT only. Consistent with ChIP-PCR and RT-PCR, 5-aza treatment partially blocked UGDH induction by KLF4 WT, in keeping with a methylation-dependent mechanism.
With evidence supporting UGDH was upregulated by KLF4 WT only, but not the KLF4 R458A, we determined whether UGDH expression is activated by KLF4 via a methylation-dependent mechanism. We tested the effect of 5-aza-2′-deoxycytidine (5-Aza), a potent inhibitor of DNA methyltransferase, on KLF4 binding to the UGDH cis-regulatory region, and on UGDH expression. Cells treated with 5-Aza (1μmol/L) for 2 weeks had 80% fewer methylated sites in UGDH cis-regulatory regions (Figure 3D). Consistent with the reduction of mCpG sites, we observed a complete loss of KLF4 WT binding to the UGDH cis-regulatory region in the presence of 5-Aza (Figure 3E). Furthermore, 5-Aza abrogated KLF4 WT-induced UGDH upregulation by nearly 60% at both the mRNA and protein levels (Figure 3F, G). These results demonstrate in part that KLF4 activates UGDH transcription in a DNA methylation-dependent manner.
UGDH knockdown decreases GAG abundance and cell migration
Given the importance of extracellular matrix on GBM progression and the relatively unknown functional significance of UGDH in GBM, we focused our efforts on determining the biological function of UGDH in GBM cells. We hypothesized that knocking down UGDH would inhibit tumor cell migration and proliferation, because UGDH catalyzes the reaction that generates the key precursor for glycosaminoglycans, building blocks for extracellular matrix components.11 Two GBM model systems, U87 cells and GBM neurosphere cells HSR-GBM1A (GBM1A), were used to conduct UGDH loss-of-function studies. Each cell line was transduced with lentivirus containing either one of the two distinct UGDH shRNAs (sh#1, sh#2). Both shRNAs induced a significant reduction (80%-85%) in UGDH protein in U87 (Figure 4A, left panel) and GBM1A cells (Figure 4A, right panel). We tested the level of the end products of UGDH, GAGs, by utilizing a polysaccharide binding dye 1,9 dimethyl-methylene blue (DMMB). There was a substantial reduction in the abundance of cell-associated GAGs in UGDH knockdown cells when compared to cells transduced with control shRNAs (e.g., 48% by sh#1, P<0.001; and 79% by sh#2 in U87 cells, P<0.001) (Figure 4B; left panel). Similar effects were observed in GBM 1A cells (Figure 4B; right panel). To examine the effect of UGDH knockdown on GBM cell migration, transwell and wound healing scratch migration assays were performed in 0.1% serum medium to minimize cell proliferation. Transwell assays 24 hrs after cell plating revealed that UGDH silencing significantly decreased the migratory ability of GBM cells (by ~50% in U87, P<0.01, and 30% in GBM1A cells, P<0.05) (Figure 4C and Supplemental Fig. 2A). UGDH knockdown also decreased cell motility in wound healing assays by 25% (P<0.05) when compared to cells transduced with control shRNAs after 24 hrs of scratch, and by 28% after 48 hrs (P<0.05) (Figure 4D and Supplemental Fig. 2B). Supplementing the culture medium with one of the GAGs, hyaluronic acid (HA, 100 ng/ml) rescued migration inhibition by UGDH shRNAs (Figure 4E), supporting the hypothesis that the effect of silencing UGDH on cell migration response is GAG-dependent. To substantiate the notion that the decreased cell migration in UGDH knockdown cells was not due to reduced cell growth under the given conditions and time points, we quantified the total cell number at 48 hrs under 0.1% FBS, and did not find any significant difference in cell growth (115,000 ± 3000 vs 113,000 ±2000 and Supplemental Fig. 3A).
Figure 4. UGDH knockdown decreases GAG abundance and cell migration.
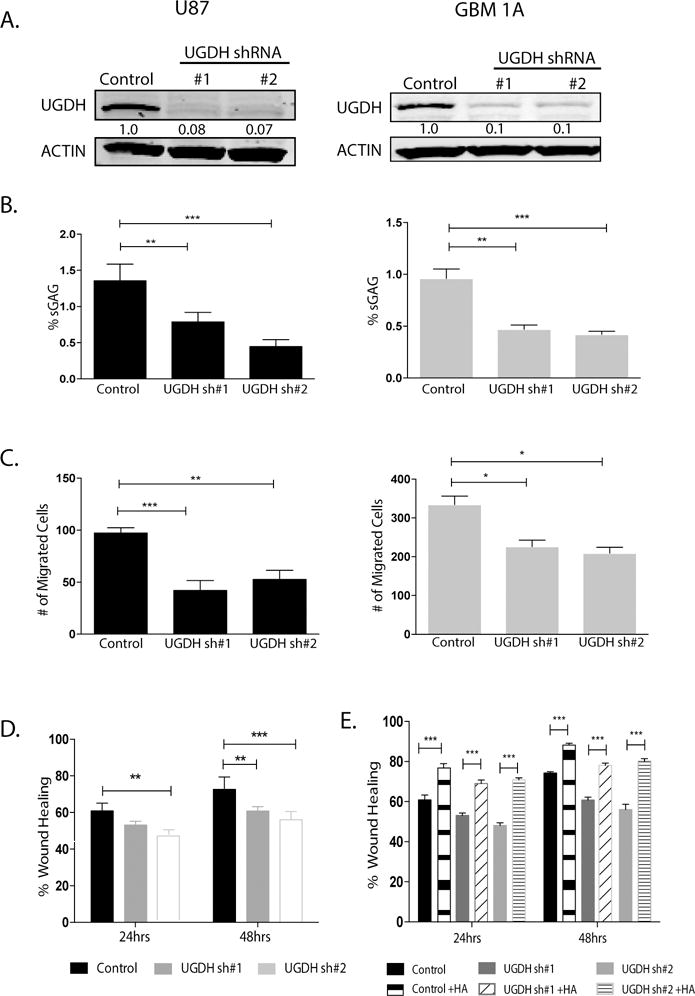
(A) U87 and HSR-GBM1A (GBM1A) cells were transduced with lentivirus coding for nonsilencing shRNA (Control), UGDH shRNA #1 or UGDH shRNA #2. UGDH knockdown was quantified by immunoblot analysis. Both UGDH shRNAs decreased UGDH protein level by more than 80%. (B) Sulfated GAG (sGAG) concentration was quantified by DMMB assay. UGDH knockdown showed significant reduction in GAG concentration in both U87 (Left panel) and GBM1A cells (right panel). (C) Transwell migration assays showed significant decrease in migration of UGDH knockdown U87 cells (left panel) and GBM1A cells (right panel). (D) Wound healing scratch assays showed UGDH knockdown decreased migration in U87 cells. (E). Exogenous HA rescued cell migration in scratch assays, in both U87 control and UGDH shRNA transduced cells.
UGDH knockdown decreases GBM cell proliferation and clonogenicity
Studies show that, in addition to a prominent role in migration and metastasis, GAGs can influence signal transduction, proliferation and differentiation.10 We hypothesized that reduction of UGDH could also influence cell proliferation. Cell counting demonstrated that UGDH knockdown inhibited cell proliferation by 45% and 20-50% after 9 days in U87 and GBM1A cultures, respectively (Figure 5A, P<0.001). Trypan blue staining demonstrated the reduced cell growth was not due to cell death as both control cells and UGDH knockdown cells had comparable percentage of non-viable cells (Supplemental Fig. 3B). Next, we examined the effects of UGDH knockdown on clonogenicity of U87 cells and the capacity for neurosphere formation in GBM1A cells. We found that compared to controls, reduction of UGDH dramatically impaired the ability of U87 cells to form colonies in soft agar (by 50-60%, P<0.001) (Figure 5B; left panel). We also observed a ~70% reduction in the neurosphere forming capacity of GBM neurosphere cells as compared with the controls (P<0.001) Figure 5B; right panel). Next, we analyzed cell cycle progression in U87 control and UGDH knockdown cells. Cell cycle was synchronized by incubating cells in 0.1% serum for 48 hrs, followed by stimulation with 10% serum for up to 32 hrs. Cell cycle progression was analyzed at 0, 8, 16, 24 and 32 hr post serum addition. At 0 h, both control and UGDH knockdown cells had ~80% of cells in G1/G0 phase. After replenishing serum for 32 hrs, 83% of UGDH knock down cells remained in G1/G0, in comparison to only 60% in control cells (P<0.001, Figure 5C, Supplementary Figure 4A). Thus, UGDH knockdown displayed a delay in G1/G0 to S phase transition. Analysis of cell cycle regulators by Western blot revealed a 65%-80% reduction in cyclin E and a 40-50% decrease in cyclin D1 in UGDH knockdown cells (Figure 5D), providing further evidence that UGDH loss-of-function promoted a delay in cell cycle progression, which led to decreased cell proliferation in GBM cells. Because UGDH is a rate limiting enzyme of producing GAGs, which can function as signaling molecules as well as a growth factor depot, we hypothesized that the decrease in proliferation of UGDH knockdown cells will be as a result of decrease in GAGs. To test this hypothesis, we performed rescuing experiments by adding HA (75ug/ml) to the UGDH knockdown cells and allowed them to grow for 9 days. We observed a significant rescue in the growth of UGDH knockdown cells on days 6 and 9 (Figure 5E). Furthermore, we surveyed a number of growth factor mediated signaling pathways. As shown in Figure 5F, phosphorylated AKT (p-AKT) levels were dramatically downregulated in UGDH knockdown cells compared with that in non-silencing control cells. Total AKT (t-AKT) levels were slightly upregulated in UGDH knockdown cells. The ratio of p-AKT versus t-AKT in UGDH knockdown cells were only 3-17% of that in non-silencing cells. This was consistent with our previously identified decrease in cyclin E1/D1, and a dramatic decrease in cell growth of UGDH knockdown cells.
Figure 5. UGDH Knockdown decreases GBM cell proliferation and clonogenicity.
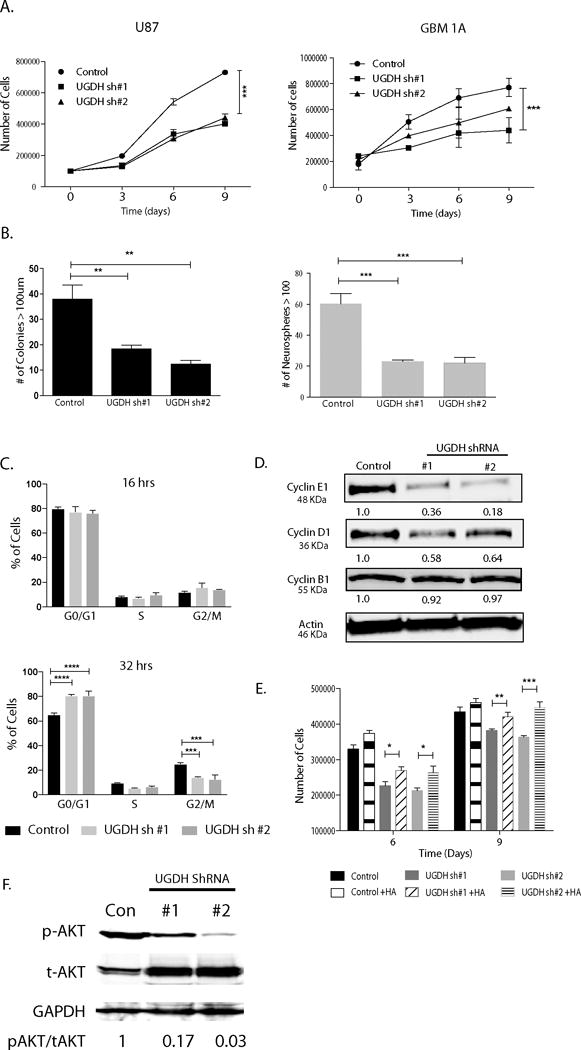
(A) Cell growth curve up to 9 days after plating. Trypsinized cells were stained with Trypan blue and both viable (unlabeled) cells were counted on the days indicated. (B) Colony formation assays showing significant decrease in anchorage independent clongenicity in UGDH knockdown U87 cells (right panel). For GBM neurosphere cells, equal numbers of viable GBM1A cells were plated and cultured for 14 days to allow neurosphere formation. Neurospheres (>100 μm diameter) were counted with MCID software. UGDH silencing inhibited neurosphere formation. (C) Cell cycle was synchronized. There was a delayed progression to S phase in U87 UGDH knockdown cells compared to controls after 32 hrs of serum replenishing. (D) UGDH silencing decreased cyclin D1 and E protein levels in GBM cells. (E). HA (75ug/ml) supplemented in U87 cells rescued the decreased cell growth effect observed in UGDH knockdown cells on days 6 and 9. (F) Phosphorylated AKT levels were significantly decreased in UGDH knockdown U87 cells compared to controls as evidenced by Western blots (*: P < 0.05; ** P<0.01; *** P<0.001).
UGDH knockdown reduces growth of GBM xenografts
The results that UGDH knockdown dramatically decreased GBM cell growth and migration in vitro prompted us to hypothesize that dramatic reduction of GAGs by UGDH knockdown would alter tumor growth in vivo. To examine the effect of UGDH silencing on orthotropic GBM growth, we employed GBM1A cells that form infiltrative orthotropic xenografts in immunodeficient (SCID) mice to examine the effect of UGDH silencing in vivo. GBM1A cells stably expressing either control shRNA or UGDH shRNA#1 were implanted into the brains of SCID mice and sacrificed 50 days after implantation according to our previous studies using this model.20 Coronal brain sections were stained for hematoxylin and eosin. Tumor size measurement demonstrated a ~65% reduction in tumor growth in mice implanted with UGDH shRNA transduced cells (~2.0 mm3) compared to control shRNA transduced cells (~6.0 mm3) (Figure 6A, P<0.001). The anti-tumor effects of UGDH inhibition could be explained in part by a ~25% inhibition of the tumor cell proliferation index measured by anti-Ki67 immunohistochemical staining (Figure 6B, P<0.001), and ~25% inhibition of the vascular density index measured by anti-laminin staining, respectively (Figure 6C, P<0.001).
Figure 6. UGDH knockdown inhibits GBM growth and migration in vivo.
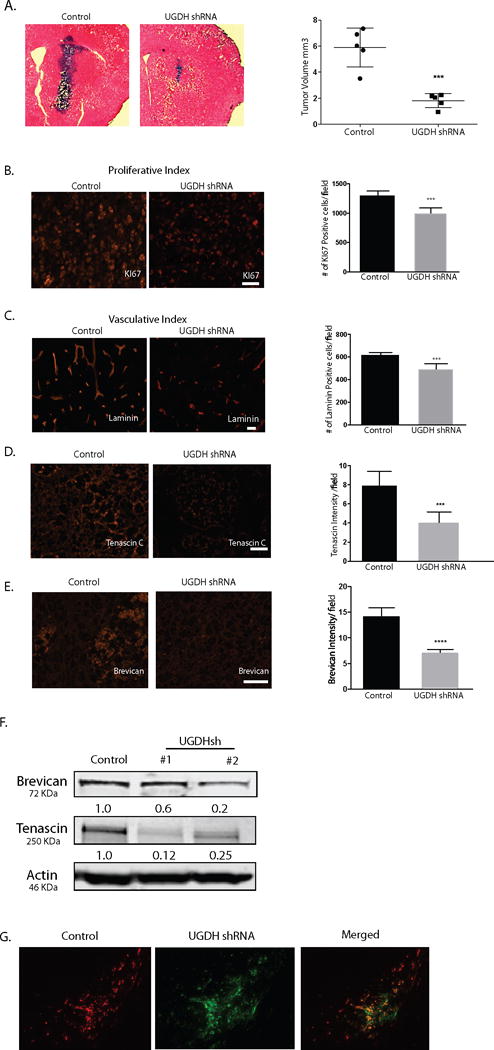
(A) Control shRNA or UGDH shRNA transduced GBM1A cells (100,000) were implanted by stereotactic injection to caudate/putamen of severe combined immunodeficiency mice (SCID). Animals were sacrificed 50 days after implantation. Hematoxylin and eosin-stained coronal brain sections (20 μm) obtained from animals showed dramatically decreased tumor size in UGDH knockdown groups (left panels, bar = 500 μm). Right panel: quantification of xenograft tumor volume shows that silencing UGDH repressed xenograft growth by more than 65% (6.4 in control vs. 2.3 in UGDH sh#1, P < 0.001). B. UGDH knockdown significantly inhibited tumor cell proliferation by 31% as evidenced by Ki67 staining. C. UGDH loss-of-function inhibited blood vessel density by 25% as evidenced by laminin staining. (D, E) Xenografts with UGDH knockdown decreased the abundance of key extracellular matrix components tenascin C and brevican in GBM1A xenografts. B to E: Bar = 20 μm. (F) Western blots showing decreased brevican and tenascin C protein in U87 UGDH knockdown cells in vitro. (G). A mixture of RFP-labeled control non-silencing cells (50,000) and GFP-labeled UGDH knockdown cells (150,000) was injected into the mouse brain. Dual-fluorescence experiment showed that UGDH knockdown cells (green) stayed in the tumor core (arrowheads), whereas the control cells (red) were preferentially located in the tumor periphery (arrows). Bar = 200 μm.
UGDH knockdown decreases expression of extracellular matrix proteins and decreased tumor cell migration in GBM xenografts
Given the evidence that UGDH knockdown inhibits GAG expression in vitro, we investigated the extracellular matrix components in UGDH knockdown xenografts, hypothesizing that GAG expression inhibited by UGDH knockdown would alter extracellular matrix formation and therefore impact tumor growth. Expression of brevican, a proteoglycan downstream of UGDH pathway and tenascin C, a glycoprotein that binds and connects proteoglycans in the ECM, was quantified by immunohistochemical staining of intracranial xenograft sections from control and UGDH knockdown tumor groups. Brevican and tenascin C staining was reduced by 40-50% in tumors harboring UGDH shRNA compared to control tumors as evidenced by Image J software quantification of staining intensity (Figure 6D, E, P<0.001). In vitro Western blot analysis of U87 and GBM1A cells further confirmed reduced brevican (50%) and tenascin C (~65%) expression in response to UGDH silencing (Figure 6F, P<0.001). These results demonstrate that UGDH knockdown decreases the expression of GAGs and other extracellular matrix components in vitro and in vivo.
To determine the effect of UGDH knockdown on tumor cell migration in animals, we employed the internally-controlled dual-fluorescence approach developed in our laboratory.23 Since the size of UGDH knockdown xenografts was only 1/3 of that of control, to accommodate the discrepancy of tumor cell proliferation rate that may complicate the interpretation of tumor cell migration, we mixed 50,000 RFP-labeled control non-silencing tumor cells with 150,000 GFP-labeled UGDH knockdown cells, and injected the mixture into the mouse brains (n=3). In all three animals, the dual-fluorescence approach clearly demonstrated that even with 3 fold more injected numbers, UGDH knockdown cells were less migratory in vivo, consistent with our in vitro studies. An example of the results was shown in Figure 6G. The core of the tumor was stained with DAPI and showed in Supplemental Fig. 5.
UGDH is required for induction of GBM cell migration by KLF4-mCpG interactions
Finally, we investigated the involvement of UGDH in cellular phenotype changes induced by KLF4-mCpG interactions. In tet-on U87 and GBM1A KLF4 WT cells, we knocked down UGDH expression with shRNAs to generate stable cells lines. Immunoblot analysis showed an ~80% to ~90% inhibition of UGDH in both U87 KLF4 WT and GBM1A KLF4 WT model systems (Figure 7A). KLF4 expression increased UGDH protein expression in the control shRNA transduced U87 and GBM1A cells as we have shown before, but failed to do so in the UGDH knockdown cell models (Figure 7A). A concomitant ~40-60% increase in GAGs in both U87 and GBM 1A cells was also observed after KLF4 WT expression was induced. However, UGDH knockdown abolished the increased GAG levels induced by KLF4 (Figure 7B). Next, we examined the effects of UGDH knockdown on KLF4-mCpG mediated GBM cell migration. KLF4 WT expression increased cell migration in transwell and wound healing assays, while UGDH knockdown reversed these changes induced by KLF4-mCpG interactions (Figure 7C, D). These results support that UGDH is required for cell migration mediated via KLF4 binding to methylated CpGs.
Figure 7. UGDH is required for KLF4-mCpG-dependent increase in GBM cell migration.
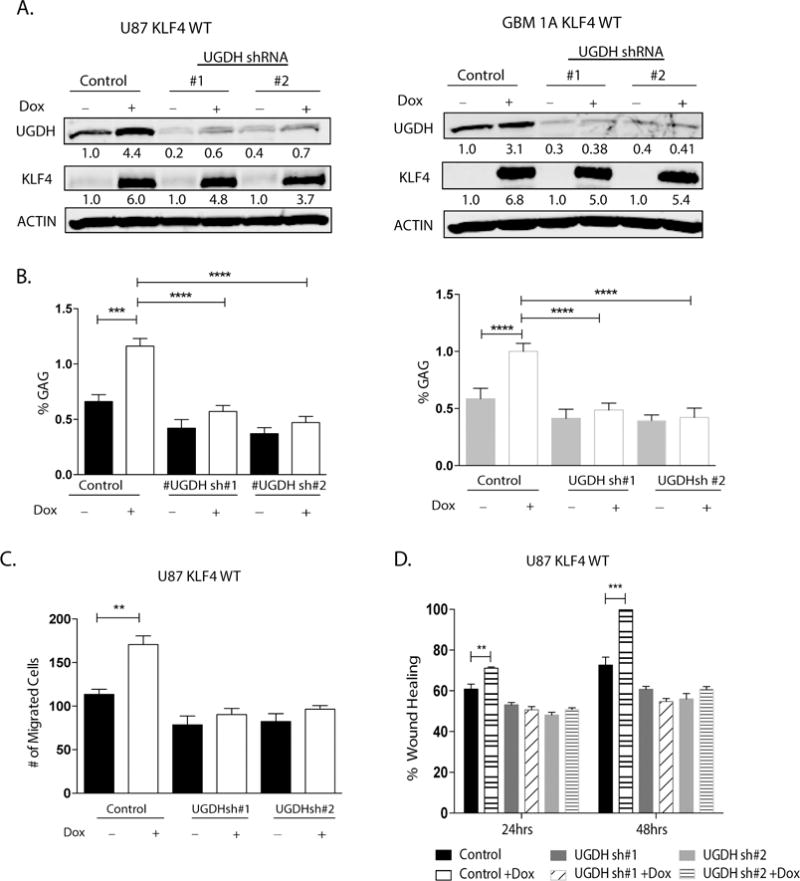
(A) U87 KLF4 WT and GBM1A KLF4 WT cells were transduced with lentivirus coding for nonsilencing shRNA (Control), UGDH shRNA #1 or UGDH shRNA #2. Cells were treated with Dox for 48 hrs before each analysis. Western blot showed UGDH knockdown significantly reverses KLF4 WT dependent induction of UGDH in both U87 and GBM1A cells. (B) Sulfated GAG (sGAG) concentration in U87 and GBM1A cells expressing KLF4 WT was determined by DMMB assay. UGDH knockdown significantly decreased the KLF4 dependent increase in GAG concentration (C) UGDH knockdown significantly reversed the KLF4-dependent increase in cell migration in transwell assays. Cell migration was evaluated 24 hrs later by counting DAPI-stained cells. Five fields per well were counted. (D) U87 KLF4 WT cells harboring UGDH shRNA were treated with Dox for 5 days till confluence. A scratch was made and cells were maintained in 0.1% FCS medium overnight. Microphotographs were taken 0 hr and 24 hrs after scratching. Bar = 100 um. UGDH knockdown inhibited the increased ability of KLF4 WT cells to migrate towards scratched area.
Discussion
Elevated glycosaminoglycan (GAG), major components in tumor microenvironment, has been shown to regulate multiple oncogenic pathways including tumor growth, invasion and migration.4,6,13 Consistent with these findings, several studies have demonstrated that inhibiting GAG synthesis diminishes tumor growth and metastasis.12,13,25 In our prior study, we found that krÜppel-like factor 4 (KLF4) promotes GBM cell migration by binding to methylated DNA (mCpG) and activating gene expression. In this study, we investigated a subset of KLF4-mCpG direct targets and focused on UDP-glucose dehydrogenase (UGDH), given its importance as an enzyme involved in the synthesis of the precursors for GAGs. Although UGDH is implicated as a rate-limiting and essential step in GAG monosaccharide synthesis9–11 the biological function of this enzyme in GBMs has not been explored. We demonstrate that UGDH is regulated via a methylation-dependent pathway; UGDH regulates cell migration and proliferation in vitro; silencing UGDH decreases levels of GAGs and some key components of the extracellular matrix in vitro and in vivo, and results in inhibition of GBM growth. A model of UGDH’s regulation and implication in GBM biology is proposed in Figure 8.
Figure 8.
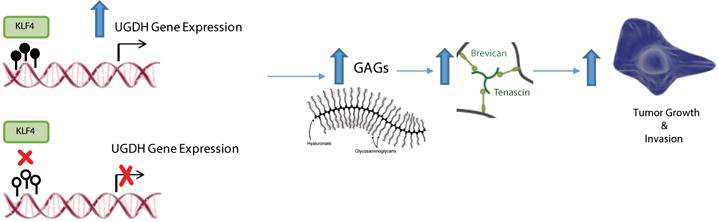
A model of UGDH’s regulation and implication in GBM biology.
Expression of proteoglycan brevican and glycoprotein tenascin C, both of which are overexpressed in primary brain tumors as well as in experimental models of glioma, have been implicated in GBM progression.21,25,26 In our study, cell migration and proliferation was rescued by exogenous HA in UGDH knockdown cells, supporting the notion that the biological function of UGDH occurs via GAG production. Although it is not surprising that silencing UGDH expression leads to decreased tumor progression, our work provides a direct link between GAGs and other extracellular matrix proteins including tenascin C, a glycoprotein with a short carbohydrate chain. Tenascin C is shown to be elevated in the extracellular matrix of malignant brain tumor models and mediates tumor progression.21 In our GBM models, we found that a reduction of GAGs facilitates decreased expression of tenascin C in vitro and in vivo, possibly via a post-translational mechanism. It has been reported that in human brains, the most abundant gel-like, long charge GAG, hyaluronic acid, serves as the backbone for the connection of other proteoglycans such as the brevican, which are further linked by tenascin C. It is conceivable that a decrease in glycosaminoglycan expression would disrupt the formation of these complex networks, resulting in a decreased deposit of tenascin C. The exact mechanism through which silencing UGDH induces a decrease in tenascin C requires further detailed studies.
Whereas silencing UGDH dramatically decreased tumor cell migration and proliferation in vitro, our in vivo UGDH knockdown xenografts showed a dramatic decrease in tumor growth, mainly via decreased tumor cell proliferation. Unlike our previously published studies on tenascin C21 in which the tumor/normal brain interface demonstrated well-demarcated borders in TNC knockdown orthotropic xenografts, our UGDH knockdown models did not support a role for UGDH knockdown in tumor cell infiltration in vivo. This could be due to a moderate decrease in extracellular matrix components, such as brevican and tenascin C, in UGDH knockdown xenografts, as opposed to a complete elimination of these components in TNC knock down xenografts.21
DNA methylation, mainly at the C5 of the CpG dinucleotides, is present throughout the genome and more than 70% of CpGs are methylated in GBM.27,28 In fact, DNA hypermethylation is among the most commonly investigated epigenetic alterations in GBMs.28 An increased understanding of how abnormal DNA methylation patterns contribute to transcription factor binding ultimately leads to downstream gene transcription is important for developing effective epigenetic-related therapies against GBM tumor progression. In this work, the anti-proliferative and pro-migratory role of UGDH were shown in vitro; a DNA methylation-dependent mechanism for UGDH regulation was identified, and modulation of GBM cell migration via KLF4-mCpG interactions was demonstrated. While little is known about the epigenetic regulation of UGDH expression, this study shows for the first time that KLF4 upregulates UGDH expression via a methylation-dependent manner and also increases GAGs in GBM cells. In our previous studies, KLF4 in U87 cells did not affect cell proliferation in vitro.7 Similarly, in GBM1A cells, KLF4 did not affect GBM1A neurosphere proliferation in vitro (data not shown). Therefore, although UGDH knockdown had a profound effect on GBM cell growth inhibition in vitro and in vivo, our current models were not suitable to test whether UGDH was involved in KLF4-mediated gliomagenesis and tumor growth. We do found that KLF4 affects GBM cell differentiation, which will be followed up in the future.
Finally, considering the importance of the enzyme UGDH in GAG synthesis and the critical role of GAG in tumor growth, we provide evidence supporting the notion that UGDH could be a potential therapeutic target to treat GBM malignancy. Future studies with small molecular inhibitors specific for UDGH would test if UGDH could serve as a therapeutic target alone or in combination with other therapies for GBMs.
Supplementary Material
Acknowledgments
We thank Dr. H. Lopez-Bertoni for technical expertise. This work was supported by grants from NIH R01NS091165 (S.X.), NIH R01 NS096754 (J.L.), NS076759 (J.L.), NIH GM111514 (H.Z.), NIH R01 GM111514 (H.Z.), NIH R33CA186790 (H.Z.), NIH U54 HG006434 (H.Z.), NIH U24 CA160036 (H.Z.), Ford Foundation pre-doctoral fellowship program (O.O.) and NIH T32 GM007445 (O.O.), United Negro College Fund/Merck Science Initiative and Burroughs Wellcome Fund to (C.R.G.).
Footnotes
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
The authors indicate no potential conflicts of interest.
References
- 1.Adamson C, Kanu OO, Mehta AI, Di C, Lin N, Mattox AK, et al. Glioblastoma multiforme: a review of where we have been and where we are going. Expert opinion on investigational drugs. 2009;18:1061–1083. doi: 10.1517/13543780903052764. [DOI] [PubMed] [Google Scholar]
- 2.Batash R, Asna N, Schaffer P, Francis N, Schaffer M. Glioblastoma Multiforme, Diagnosis and Treatment; Recent Literature Review. Current medicinal chemistry. 2017 doi: 10.2174/0929867324666170516123206. [DOI] [PubMed] [Google Scholar]
- 3.Marzenna Wiranowska, Mumtaz V, Rojiani DAG. Extracellular Matrix Microenvironment in Glioma Progression, Glioma - Exploring Its Biology and Practical Relevance. InTech. 2011 [Google Scholar]
- 4.Venning FA, Wullkopf L, Erler JT. Targeting ECM Disrupts Cancer Progression. Front Oncol. 2015;5:224. doi: 10.3389/fonc.2015.00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. The Journal of cell biology. 2012;196:395–406. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Afratis N, Gialeli C, Nikitovic D, Tsegenidis T, Karousou E, Theocharis AD, et al. Glycosaminoglycans: key players in cancer cell biology and treatment. The FEBS journal. 2012;279:1177–1197. doi: 10.1111/j.1742-4658.2012.08529.x. [DOI] [PubMed] [Google Scholar]
- 7.Wan J, Su Y, Song Q, Tung B, Oyinlade O, Liu S, et al. Methylated cis-regulatory elements mediate KLF4-denpendent gene transactivation and cell migration. eLife. 2017;6:e20068. doi: 10.7554/eLife.20068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu S, Wan J, Su Y, Song Q, Zeng Y, Nguyen HN, et al. DNA methylation presents distinct binding sites for human transcription factors. eLife. 2013;2:e00726. doi: 10.7554/eLife.00726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Egger S, Chaikuad A, Kavanagh KL, Oppermann U, Nidetzky B. UDP-glucose dehydrogenase: structure and function of a potential drug target. Biochem Soc Trans. 2010;38:1378–1385. doi: 10.1042/BST0381378. [DOI] [PubMed] [Google Scholar]
- 10.Clarkin CE, Allen S, Kuiper NJ, Wheeler BT, Wheeler-Jones CP, Pitsillides AA. Regulation of UDP-glucose dehydrogenase is sufficient to modulate hyaluronan production and release, control sulfated GAG synthesis, and promote chondrogenesis. J Cell Physiol. 2011;226:749–761. doi: 10.1002/jcp.22393. [DOI] [PubMed] [Google Scholar]
- 11.Wen Y, Li J, Wang L, Tie K, Magdalou J, Chen L, et al. UDP-glucose dehydrogenase modulates proteoglycan synthesis in articular chondrocytes: its possible involvement and regulation in osteoarthritis. Arthritis Research & Therapy. 2014;16:484. doi: 10.1186/s13075-014-0484-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Toole BP. Hyaluronan: from extracellular glue to pericellular cue. Nat Rev Cancer. 2004;4:528–539. doi: 10.1038/nrc1391. [DOI] [PubMed] [Google Scholar]
- 13.Park JB, Kwak H-J, Lee S-H. Role of hyaluronan in glioma invasion. Cell Adhesion & Migration. 2008;2:202–207. doi: 10.4161/cam.2.3.6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu P, Takai K, Weaver VM, Werb Z. Extracellular Matrix Degradation and Remodeling in Development and Disease. Cold Spring Harbor perspectives in biology. 2011;3 doi: 10.1101/cshperspect.a005058a005058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer research. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 16.Yu F, Li J, Chen H, Fu J, Ray S, Huang S, et al. Kruppel-like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene. 2011;30:2161–2172. doi: 10.1038/onc.2010.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tilghman J, Wu H, Sang Y, Shi X, Guerrero-Cazares H, Quinones-Hinojosa A, et al. HMMR maintains the stemness and tumorigenicity of glioblastoma stem-like cells. Cancer research. 2014;74:3168–3179. doi: 10.1158/0008-5472.CAN-13-2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ying M, Tilghman J, Wei Y, Guerrero-Cazares H, Quinones-Hinojosa A, Ji H, et al. Kruppel-like factor-9 (KLF9) inhibits glioblastoma stemness through global transcription repression and integrin alpha6 inhibition. The Journal of biological chemistry. 2014;289:32742–32756. doi: 10.1074/jbc.M114.588988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu Y, Richard JP, Wang SD, Rath P, Laterra J, Xia S. Regulation of glioblastoma multiforme stem-like cells by inhibitor of DNA binding proteins and oligodendroglial lineage-associated transcription factors. Cancer science. 2012;103:1028–1037. doi: 10.1111/j.1349-7006.2012.02260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rath P, Lal B, Ajala O, Li Y, Xia S, Kim J, et al. In Vivo c-Met Pathway Inhibition Depletes Human Glioma Xenografts of Tumor-Propagating Stem-Like Cells. Translational oncology. 2013;6:104–111. doi: 10.1593/tlo.13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xia S, Lal B, Tung B, Wang S, Goodwin CR, Laterra J. Tumor microenvironment tenascin-C promotes glioblastoma invasion and negatively regulates tumor proliferation. Neuro-oncology. 2016;18:507–517. doi: 10.1093/neuonc/nov171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barbosa I, Garcia S, Barbier-Chassefière V, Caruelle J-P, Martelly I, Papy-García D. Improved and simple micro assay for sulfated glycosaminoglycans quantification in biological extracts and its use in skin and muscle tissue studies. Glycobiology. 2003;13:647–653. doi: 10.1093/glycob/cwg082. [DOI] [PubMed] [Google Scholar]
- 23.Wang SD, Rath P, Lal B, Richard JP, Li Y, Goodwin CR, et al. EphB2 receptor controls proliferation/migration dichotomy of glioblastoma by interacting with focal adhesion kinase. Oncogene. 2012;31:5132–5143. doi: 10.1038/onc.2012.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reznik TE, Sang Y, Ma Y, Abounader R, Rosen EM, Xia S, et al. Transcription-dependent epidermal growth factor receptor activation by hepatocyte growth factor. Molecular cancer research : MCR. 2008;6:139–150. doi: 10.1158/1541-7786.MCR-07-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wade A, Robinson AE, Engler JR, Petritsch C, James CD, Phillips JJ. Proteoglycans and their roles in brain cancer. The FEBS journal. 2013;280:2399–2417. doi: 10.1111/febs.12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nutt CL, Zerillo CA, Kelly GM, Hockfield S. Brain enriched hyaluronan binding (BEHAB)/brevican increases aggressiveness of CNS-1 gliomas in Lewis rats. Cancer Res. 2001;61:7056–7059. [PubMed] [Google Scholar]
- 27.Craig JM, Bickmore WA. The distribution of CpG islands in mammalian chromosomes. Nat Genet. 1994;7:376–382. doi: 10.1038/ng0794-376. [DOI] [PubMed] [Google Scholar]
- 28.Medvedeva YA, Khamis AM, Kulakovskiy IV, Ba-Alawi W, Bhuyan MSI, Kawaji H, et al. Effects of cytosine methylation on transcription factor binding sites. BMC Genomics. 2014;15:119. doi: 10.1186/1471-2164-15-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.