

Abstract
Respiratory syncytial virus (RSV) and pneumonia virus of mice (PVM) are viruses of the family Paramyxoviridae, subfamily pneumovirus, which cause clinically important respiratory infections in humans and rodents, respectively. The respiratory epithelial target cells respond to viral infection with specific alterations in gene expression, including production of chemoattractant cytokines, adhesion molecules, elements that are related to the apoptosis response, and others that remain incompletely understood. Here we review our current understanding of these mucosal responses and discuss several genomic approaches, including differential display reverse transcription-polymerase chain reaction (PCR) and gene array strategies, that will permit us to unravel the nature of these responses in a more complete and systematic manner.
Keywords: bronchiolitis, gene expression, pneumonia virus of mice, pneumovirus, respiratory syncytial virus
Introduction
RSV and PVM are viruses of the family Paramyxoviridae, subfamily pneumovirus; they are enveloped, single-stranded, nonsegmented RNA viruses that can cause intense viral bronchiolitis in humans and mice, respectively. In its most severe form, the lower respiratory tract infection caused by pneumoviruses is associated with the development of peribronchiolar infiltrates that are accompanied by submucosal edema and bronchorrhea, and ultimately leads to bronchiolar obstruction and compromised oxygen transfer. As the infection is confined to the respiratory epithelium, the responses of these cells are clearly of primary importance in determining the nature and extent of the resulting inflammatory process.
Most of our understanding of responses to pneumovirus infection has emerged from studies of RSV infection of human epithelial target cells in vitro; a list of genes and/or gene products produced by epithelial cells in response to RSV infection in vitro is provided in Table 1. At the cellular level, epithelial cells initially respond to RSV infection by reducing their ciliary beat frequency. Production and release of chemoattractant cytokines (chemokines) can be observed as early as 12 h after infection, leading to the recruitment of specific leukocyte subsets to the lung tissue. RSV-infected epithelial cells become resistant to tumor necrosis factor (TNF)-α-induced apoptosis, but later fuse to form giant-cell syncytia and die by cellular necrosis. We review the molecular bases (to the extent that they are understood) of these specific responses, and discuss several novel strategies that may permit us to study the responses to RSV and PVM infection in a more coherent and systematic manner.
Table 1.
Alterations in gene expression in response to RSV infection
| Group | Examples |
| Cell-surface | ICAM-1 (CD54) |
| molecules | CD18 |
| Vascular cell adhesion molecule (VCAM) | |
| Major histocompatibility complex (MHC) I and II | |
| CD14 | |
| CD15 | |
| Fas | |
| Muscarinic receptors | |
| β2-adrenergic receptors | |
| Lewisx antigen | |
| Chemokines and | IL-8 |
| cytokines | IL-11 |
| IL-6 | |
| Soluble TNF receptor | |
| RANTES | |
| Monocyte chemoattractant protein (MCP)-1 | |
| MIP-1α | |
| Granulocyte-macrophage colony-stimulating | |
| factor (GM-CSF) | |
| IL-1β | |
| IFN-β | |
| Interleukin-1α | |
| Transcription | NF-κB |
| factors | NF-IL-6 |
| Others | Inducible nitric oxide synthase (iNOS) |
| Vascular endothelial growth factor (VEGF)/VPF | |
| Endothelin-1 | |
| 5-Lipoxygenase | |
| Leukotrienes | |
| IEX-1L | |
| Cytokeratin-17 |
Increased expression of the these genes and/or gene products has been observed in response to RSV infection of target cells in vitro. VPF, vascular permeability factor.
Ciliostasis
Tristram et al [1] observed that explanted respiratory epithelial cells slow their ciliary beat frequency almost immediately after exposure to RSV, with complete ciliostasis seen as early as 6 h after the initial infection. The molecular basis of ciliostasis remains completely unknown.
Production and release of chemokines
The chemokines and cytokines with production and release that has been associated with RSV infection of human epithelial cells are listed in Table 1. Much of this work was also recently reviewed elsewhere [2,3]. We focus here on the three chemokines whose molecular mechanisms and physiologic implications are best understood.
Interleukin-8
The earliest reports on this subject described production of the neutrophil chemoattractant IL-8 from tissue culture supernatants from RSV-infected cells [4,5,6] and in nasal secretions from patients with viral rhinitis [7]. IL-8 has since been detected in lower airway secretions from patients with severe RSV bronchiolitis [8], and the neutrophil influx observed in response to this infection is probably due, at least in part, to the activity of this chemokine.
At the cellular level IL-8 production can be observed in response to inactivated RSV virions, whereas IL-8 production in response to active infection was inhibited by ribavarin, amiloride, and antioxidants [9,10]. Several groups have demonstrated activation of the transcription factor nuclear factor-κB (NF-κB) in response to RSV infection, and NF-κB is recognized for its central role in eliciting the production of IL-8 [9,11,12]. The transcription factor NF-IL-6 is also produced in response to RSV infection [13], and participates in a co-operative manner with NF-κB in the regulation of IL-8 gene expression [11], although later studies suggest that activator protein-1 may function preferentially in this role [14]. Interestingly, the NF-κB regulator IκBα, which functions by inhibiting NF-κB activation in response to TNF-α, was produced with different kinetics and does not promote a reversal of NF-κB activation in response to RSV infection as it does in response to TNF-α [15]. Most recently, Casola et al [16] demonstrated that the IL-8 promoter contains independent response elements, with nucleotides –162 to –132 representing a unique RSV response element that is distinct from elements necessary for IL-8 production in response to TNF-α. This concept of a stimulus-specific response will probably make an important contribution toward our understanding of how pneumoviruses promote transcription of unique and specific sets of independent gene products.
Regulated upon activation, normal T-cell expressed and secreted
The pleiotropic chemokine regulated upon activation, normal T-cell expressed and secreted (RANTES) has also been detected in supernatants from RSV-infected epithelial cells in culture [17,18], as well as in upper and lower airway secretions from patients infected with this virus [7,8]. RANTES acts as a chemoattractant for eosinophils and monocytes in vitro, although its role in vivo is somewhat less clear. Similar to IL-8, RANTES can be produced in vitro in response to inactivated virions [8], and involves NF-κB activation, binding, and nuclear translocation [19]. However, Koga et al [20] demonstrated that stabilization of RANTES mRNA, a response to RSV infection mediated in part by nucleotides 11–389 of the RANTES gene, is probably the primary mechanism underlying increased production and secretion of RANTES protein. Further studies will determine whether a similar mechanism is also in place for IL-8 and other RSV-mediated responses.
Macrophage inflammatory protein-1α
Several groups have recently shown that macrophage inflammatory protein (MIP)-1α is released from RSV-infected cells in culture [7,21]; MIP-1α was also detected in upper and lower airway secretions from RSV-infected patients [7,8]. Interestingly, of the three aforementioned chemokines, MIP-1α is the one that is most closely correlated with the presence of eosinophil degranulation products; this, together with data from our PVM model of pneumovirus infection [22], has suggested to us that MIP-1α plays a pivotal role in eosinophil recruitment in response to primary pneumovirus infection. Interestingly, production of MIP-1α in cell culture requires active viral replication [8], which suggests that this response may proceed by a mechanism that is completely distinct from that which elicits production of RANTES and IL-8. However, no reports to date have addressed the molecular mechanism that underlies the RSV-mediated MIP-1α response.
Cell-surface adhesion molecules
A list of cell-surface molecules that have been reported as expressed in response to RSV infection is shown in Table 1. We focus here on the expression of intercellular adhesion molecule (ICAM)-1 (CD54) and the leukocyte integrin CD18.
Intercellular adhesion molecule-1
Increased expression of this cell-surface adhesion protein was observed in both respiratory epithelial cell lines [23,24] and in human nasal epithelial cells [25] in response to infection with RSV in vitro. Chini et al [26] demonstrated that the expression of ICAM-1 mRNA, similar to IL-8 and RANTES, was dependent on an intact NF-κB site in the gene promoter, and demonstrated a role for the consensus binding site for the factor CCAAT/ enhancer-binding protein. Stark et al [27] demonstrated that ICAM-1 and CD18 expressed in response to RSV serve to enhance neutrophil and eosinophil binding to epithelial cells.
CD18
CD18 is a polypeptide of the integrin family that functions in mediating cell–cell interactions. Several groups have observed expression of CD18 on epithelial cells in response to RSV infection [27,28], with CD18 shown to enhance the degranulation of eosinophils in this specific setting [28]. Of particular interest are the recent findings relating expression of CD18 (along with CD14) to earlier literature on bacterial superinfections in the setting of viral infections. Earlier studies [29,30] reported enhanced binding of bacteria to respiratory epithelial cells that were infected with RSV, findings that had clinical implications relating to acute bacterial otitis media in infants.
Two more recent studies addressed the question of binding sites. Saadi et al [31] determined that two strains of the pathogen Bordetella pertussis bound more efficiently to RSV-infected cells, and that the binding was reduced upon pretreatment of the cells with anti-CD14 or anti-CD18 antibodies. Similarly, Raza et al [32] reported that both CD14 and CD18 on RSV-infected epithelial cells contributed to the binding of nonpilate Neisseria meningitidis. In vivo testing is required before the clinical significance of these intriguing findings can be appreciated.
Protection from programmed cell death
RSV-infected epithelial cells in culture do not show features that are suggestive of apoptosis (ie no evidence of membrane blebbing, fragmentation of chromosomal DNA, or characteristic changes in nuclear morphology). Takeuchi et al [33] showed that, although RSV-infected epithelial cells express a number of apoptosis-associated genes, including interferon regulatory factor-1, IL-1β-converting enzyme and caspase 3, they do not undergo formal apoptosis.
As part of our attempts to understand mucosal responses in a more systematic manner (see below), we discovered that RSV-infected epithelial cells express the recently described antiapoptosis gene IEX-1L [34]. In our studies, we found that expression of IEX-1L is a response to active virus; no gene expression was observed in response to irradiated, replication-incompetent virus. Moreover, expression of IEX-1L is not observed in response to adenoviral infection, suggesting that expression of this gene is not a universal response to cellular perturbation, or indeed to all viral infections. Functionally, we also demonstrated that RSV infection protects epithelial cells from TNF-α-induced apoptosis, an effect that is temporally associated with the expression of IEX-1L.
Apoptosis is generally considered to be a highly efficient self-defense mechanism employed by host target cells, because it permits the infected host to dispose of viral proteins and nucleic acids on a single-cell basis without inducing an inflammatory response. It is thus not surprising that many viruses have evolved strategies to circumvent this response. Of interest, Krilov et al [35] demonstrated that monocytes and cord blood mononuclear cells are similarly protected from apoptosis when infected with RSV.
Although virus-induced protection from apoptosis appears advantageous to the virus alone, another interpretation may be considered. Because respiratory epithelial cells are now recognized as a major source of leukocyte chemoattractants, and because leukocyte recruitment to the lung has been associated with enhanced viral clearance and prolonged survival in pneumovirus infection [22], the ability to maintain chemoattractant production from viable cells may ultimately benefit the host organism as well.
Syncytia formation
In tissue culture, RSV-infection is characterized by the formation of giant-cell syncytia. The mechanisms for the formation of these fused masses of cells depend in part on the expression of the RSV-specific fusion (F) protein on the surface of infected host cells, and in part on virus-mediated changes in cytoskeletal architecture. It is important to note that RSV-induced changes in cytoskeletal architecture are not restricted to cell lines grown in vitro, as giant-cell syncytia have also been found in pathologic lung specimens obtained from both humans and animals that were infected with RSV.
Again, as part of our systematic study of gene expression in response to pneumovirus infection, we found that human respiratory epithelial cells respond to RSV infection with increased expression of the cytoskeletal protein cytokeratin-17 [36]. Cytokeratin-17 is a 46-kDa cytoskeletal protein that belongs to the class I acidic cytokeratin family. In the lung, expression of cytokeratin-17 is normally restricted to basal epithelial cells of the larynx, trachea, and bronchi. In RSV-infected cells, we found expression of Ck-17 predominantly at sites of syncytia formation, and thus provided the first description of a unique component of these pathognomonic structures at the molecular level. Similar to what has been reported for the production of IL-8, expression of Ck-17 is dependent on the activity of the transcription factor NF-κB, and future studies will determine the role of the NF-κB consensus site (–200 to –208 of the cytokeratin-17 promoter) in mediating this response.
A novel model of inflammatory viral bronchiolitis: pneumonia virus of mice
To date, efforts to study pneumovirus-induced alterations in gene expression have relied heavily on in vitro models of virus-infected cells and cell lines. The intrinsic value of characterizing the genes identified in this artificial system is by definition limited, and the clinical and physiologic significance of any findings must ultimately be tested in vivo. To some extent, the study of gene products in clinical specimens is possible, but this approach is limited, cumbersome, and dictated by sample availability. It is clear that an appropriate animal model of inflammatory bronchiolitis is required to characterize the alterations in gene expression discovered using the available in vitro models. Although RSV has been used for the study of specific allergic reactions to viral antigens, it is not a natural pathogen of mice, and intranasal inoculation of virus at high titer results in, at best, a minimal primary infection with a correspondingly minimal inflammatory response. In order to study gene expression in response to primary pneumovirus infection in vivo, we developed a novel mouse model of inflammatory bronchiolitis using the natural rodent pneumovirus pathogen and closest phylogenetic relative of RSV [37] — PVM. We presented our initial findings on PVM infection in mice in three recent publications [22,38,39]. A summary of these findings is presented in Table 2 and Fig. 1.
Table 2.
Features of RSV infection of humans, RSV in mice, and PVM infection in mice: a comparison
| Features | Humans infected with RSV | Mice infected with PVM | Mice infected with RSV |
| Inoculum required | Very low (presumed) | <30 pfu | >0.5–2 million pfu |
| Pulmonary inflammation | Granulocytic bronchiolitis | Granulocytic bronchiolitis | Mild mononuclear cell infiltrate |
| Eosinophil recruitment | Yes | Yes (8–12%); see arrows in panel D, Fig. 1 | No |
| Progression to ARDS | Yes | Yes | No |
| Percent with respiratory failure | 10% | 60% (low dose); 100% (high dose) | 0% |
| Weight loss | Not reported, but likely | 30% | 12–15% |
| Peak lung viral titers | 106 pfu/ml nasal secretion | 106–108 pfu/g lung | Less than inoculum |
ARDS, acute respiratory distress syndrome.
Figure 1.
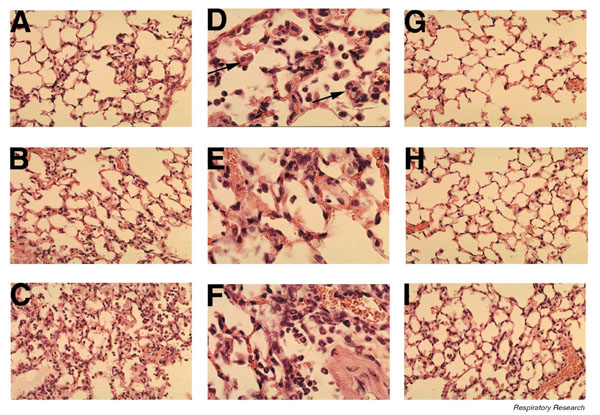
Pulmonary histopathology during pneumovirus infection. Lungs from mice infected with PVM (60 pfu) for (A, D) 3 days, (B, E) 5 days, and (C, F) 7 days. Images at lower magnification (A, B, and C × 400) show the extent of inflammatory changes, whereas the higher power images (D, E, and F ×1000) demonstrate that the cells are predominantly granulocytes. The arrows in panel D highlight the presence of eosinophils. Lung histology from animals infected with RSV (1 million pfu) for (G) 3, (H) 5, and (I) 7 days (× 400). Note the paucity of acute inflammatory changes.
To begin, we described the cellular and biochemical pathology observed in response to PVM infection in mice [38]. We found that infection could be established with as few as 30 plaque-forming units (pfu) of PVM in the inoculum, with infection resulting in significant morbidity and mortality, and viral recoveries in the order of 108 pfu/g lung tissue. We also noted inflammatory bronchiolitis as among the immediate responses to this infection, with bronchoalveolar lavage fluid containing virtually 100% neutrophils and eosinophils obtained as early as 3 days after inoculation. Furthermore, we found that infection was accompanied by the production of the proinflammatory chemokine MIP-1α, which was previously shown by Cook et al [40] to be an important component of the inflammatory response to the orthomyxovirus influenza virus.
We also described the role played by MIP-1α in the pathogenesis of PVM-induced bronchiolitis [22]. Specifically, we explored the responses of gene-deleted mice to infection with PVM, and found no inflammatory response in mice deficient in MIP-1α expression (MIP-1α-/-) and only minimal virus-induced inflammation in mice that lacked the major MIP-1α receptor on granulocytes chemokine receptor (CCR)1 (CCR1-/-). Although the inflammatory response is often considered to be unnecessary and indeed detrimental, we demonstrated that the absence of granulocytic inflammation was associated with enhanced recovery of infectious virions, as well as with accelerated mortality. These results suggest that the MIP-1α/CCR1-mediated acute inflammatory response protects mice by delaying the lethal sequelae of viral infection.
Our most recent report on this subject [39] presents a direct comparison between the responses of mice to challenge with PVM and RSV. Although RSV is not a natural pathogen of mice, it has been used extensively in mouse models of human infection because a limited, or 'semipermissive' infection can be established via intranasal inoculation of virus at very high titers. In this regard, we found (as have others) that RSV infection did not result in any measurable degree of morbidity, and that viral recovery was significantly lower than that found in the inoculum; these results suggested that there was no significant viral replication in mouse lung tissue. We further demonstrated that the inflammatory response to RSV challenge was minimal, as few leukocytes were recruited to the lungs (Fig. 1).
Taken together, our results suggest that infection of mice with PVM provides a superior model for the study of acute inflammatory bronchiolitis in response to pneumovirus infection in vivo. The advantages of this model include the following: clinical parameters — morbidity and mortality — that can be measured clearly and specifically; clear evidence of viral replication in lung tissue, with incremental recoveries that, at peak, are in excess of 108 pfu/g in response to as few as 30 pfu in the inoculum; and a dramatic granulocytic response that is modulated at least in part by the proinflammatory chemokine MIP-1α and its receptor CCR1.
Genomic approaches to alterations in gene expression
Traditionally, analysis of gene expression through measurement of steady-state levels of individual mRNAs could be conducted only one gene at a time using northern blotting, dot blots, or quantitative reverse transcription-PCR. Differential display, serial analysis of gene expression, and total gene analysis offer great promise, because they are multiplex technologies that provide simultaneous analysis of multiple mRNAs isolated under conditions of interest via PCR amplification techniques. DNA hybridization arrays are theoretically the most efficient of the gene expression analysis techniques. Although many skeptics have described these genome-based approaches as expensive, nonhypothesis-driven 'fishing expeditions', we view them as broad-based screening techniques that will enable us to identify patterns of gene expression that can then be subjected to careful characterization and analysis.
Differential display
Differential display is a semiquantitative, reverse transcription-PCR-based technique that is used to compare mRNAs from two or more conditions of interest. Both increased and decreased expression of specific amplicons will be evident — an obvious advantage to this approach. Total RNA can be isolated from virus infected versus uninfected cells or mouse lungs both before and during infection, and differential display is performed using degenerate T11(XY) anchoring primers and random upstream oligomers, as described elsewhere [34,36]. The resulting PCR products are separated by electrophoresis, and the gel is dried and exposed to film. An example of our results comparing cDNA amplicons from RNA extracted from RSV-infected epithelial cells daily for 4 days is shown in Fig. 2. Differentially expressed bands are cut from the gel, eluted and reamplified using the same primers that generated the original signal, and northern blots generated from RNA extracted from pneumovirus-infected cells or tissue over time and probed with the differentially expressed amplicons serve to confirm differential expression of the identified sequence.
Figure 2.
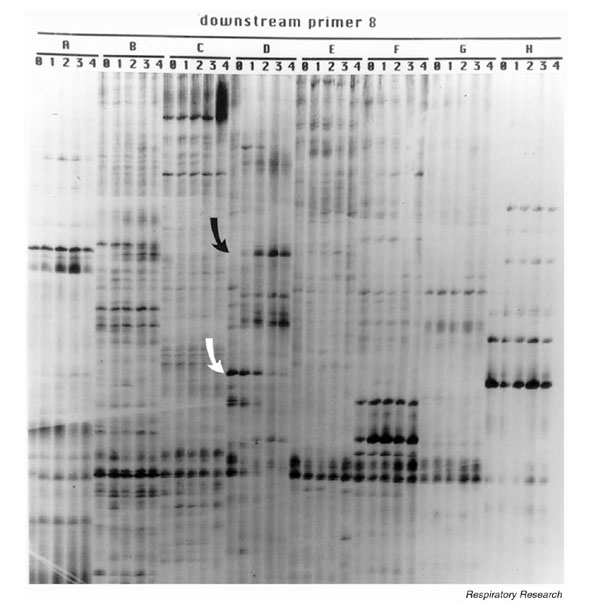
Display of amplicons generated from RNA extracted from RSV-infected cells at daily intervals following infection (days 0–4) using a single anchoring primer, T11GC (downstream primer 8) and (A-H) eight random 10mers. Two differentially expressed sequences are highlighted by arrows (the black arrow shows an upregulated amplicon, and the white arrow highlights a downregulated amplicon). Several other differentially expressed signals are also seen.
The DNA sequences of the newly identified differentially expressed amplicons are compared with sequences present in the GenBank database. Viral sequences are expected to be upregulated over time and can be identified immediately, because the entire genomes of both PVM and RSV are present in GenBank. In cases in which the amplicon represents a newly discovered gene, potential open-reading frames are compared with sequences that are present in the Swiss protein database; motifs that share homologies with known proteins represent important clues to the identity of the differentially expressed gene.
With the help of differential display, we have identified and characterized several genes that are upregulated in RSV-infected respiratory epithelial cells. Two specific examples of genes that were found to be induced during RSV infection, and later characterized as playing independent roles in the pathophysiology of RSV infection, are the antiapoptosis gene IEX-1L [34] and the gene that encodes the cytoskeletal protein cytokeratin-17 [36].
Unlike DNA viruses, which are known to encode virus-specific antiapoptosis genes, RSV — an RNA virus with a small (approximately 15.2 kb) viral genome — was shown to alter host cell expression of the apoptosis inhibitor IEX-1L. After demonstrating that IEX-1L mRNA was present at sevenfold higher concentrations in RSV-infected respiratory epithelial cells when compared with uninfected cells, we concluded that this cellular response protected against TNF-α-induced programmed cell death during viral infection. Further efforts to determine which of the 11 RSV proteins participate in the transactivation of the IEX-1L gene (either directly or indirectly) are ongoing.
A second example of a gene that is specifically upregulated in RSV-infected respiratory epithelium, as identified by differential display, is that which encodes cytokeratin-17 [36]. Upon characterizing the molecular events that are important for cytokeratin-17 induction, we demonstrated a link to an NF-κB signaling pathway. Above, we discussed the importance of this transcription factor in the regulation of proinflammatory cytokine gene expression, and because of this involvement we were not surprised to discover its role in virus-induced cytokeratin-17 gene regulation. Perhaps the most interesting observation made during these experiments was the in situ localization of cytokeratin-17 protein to areas of cytopathic syncytia formation, suggesting a role for this cytoskeletal protein in their formation. Of note, we observed a dramatic decrease in RSV replication and in syncytia formation when we blocked cytokeratin-17 expression, suggesting that blocking syncytia formation, at least in part, impairs the direct cell–cell spread and productive replication of virus.
Gene arrays
Although there are several companies that market these systems and components, the cytokine gene macroarray systems recently developed by R&D Systems (Sigma Genosys®; Minneapolis, MN, USA) and Clontech (Atlas®; Palo Alto, CA, USA) represent some of the newer opportunities available that have a focus on gene products that are known to be involved in inflammation. These arrays consist of different cDNAs printed as PCR products onto charged nylon membranes. An example of our experience with the Sigma Genosys array is shown in Fig. 3. For this example, total RNA was extracted from RSV-infected HEp-2 cells and uninfected controls at day3 after infection. Three micrograms of total RNA was used in a cDNA synthesis reaction, using a proprietary mixture of 378 primer pairs and trace amounts of 32P-radiolabeled dCTP. The resulting radiolabeled products were hybridized to the macroarrays overnight at 65°C, and then washed and exposed to film. The arrow highlights one of the most obviously upregulated sequences from this experiment, which was identified as the gene encoding human MIP-1α. The physiologic importance of MIP-1α upregulation during human RSV infection and during rodent PVM bronchiolitis has already been described.
Figure 3.
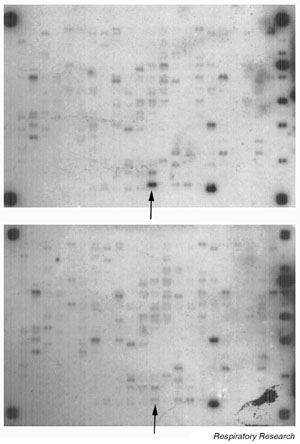
Cytokine macroarray probed with radiolabelled cDNA generated from total RNA extracted from epithelial cells 48 h after RSV infection (upper panel) or 48 h after exposure to conditioned medium (lower panel). Signal intensity of each gene under each condition is compared. The arrow highlights the signal for human MIP-1α present at 12-fold higher concentration in infected epithelial cells compared with the uninfected controls.
Microarrays can be differentiated from macroarrays in several ways. Among these differences, the microarray matrix is a glass or plastic slide, probes are labeled with fluorescent dye rather than via radioisotopes, and, most significantly, microarrays generally include a larger number and a higher density of imbedded sequences than do macroarrays. Although this may seem to be highly appealing at first, the massive amounts of data generated by microarray technology poses new challenges with respect to data normalization, management, and development of mathematical models to assist in data interpretation.
Conclusion
The pneumoviruses RSV and PVM enter respiratory epithelial cells via a receptor-mediated event. During host-cell attachment and internalization, the target cell begins to alter its gene expression, which, among other events, involves the transcriptional upregulation of cytokine and chemokine genes. As RSV replication progresses, additional changes in cellular gene expression can be observed, including induction of the potent antiapoptosis gene IEX-1L and increased expression of the otherwise quiescent gene that encodes cytokeratin-17. What we know regarding the physiologic importance of these genes and their gene products has been described, but there is more to be learned. As the available technologies evolve, we can continue to capitalize on the use of genomic approaches as large-scale screening tools to identify genes that play important roles in the pathophysiology of pneumovirus infection. These elegant and simple tools will provide us with the means for thorough and systematic exploration of gene expression, both in the established in vitro cell culture models and in our new murine model of acute pneumovirus infection.
Abbreviations
CCR = chemokine receptor; ICAM = intercellular adhesion molecule; MIP = macrophage inflammatory protein; NF-κB = nuclear factor-κB; PCR = polymerase chain reaction; pfu = plaque-forming units; PVM = pneumonia virus of mice; RANTES = regulated upon activation, normal T-cell expressed and secreted; RSV = respiratory syncytial virus; TNF = tumor necrosis factor.
Acknowledgments
Acknowledgements
We thank Dr Andrew Easton for his assistance with the PVM model. We thank Drs Leonard Weiner, Harry Malech, and John Gallin for their support for the ongoing research in our laboratories. This work was funded in part by an American Heart Association Scientist Development Grant to JBD, the Sinsheimer Foundation and the Children's Miracle Network of Central New York.
References
- Tristram DA, Hicks W, Jr, Hard R. Respiratory syncytial virus and human bronchial epithelium. Arch Otolaryngol. 1998;124:777–783. doi: 10.1001/archotol.124.7.777. [DOI] [PubMed] [Google Scholar]
- Crowe JE., Jr Host responses to respiratory virus infection and immunization. Curr Top Microbiol Immunol. 1999;236:191–214. doi: 10.1007/978-3-642-59951-4_10. [DOI] [PubMed] [Google Scholar]
- Garofalo RP, Haeberle H. Epithelial regulation of innate immunity to respiratory syncytial virus. Am J Respir Cell Mol Biol. 2000;23:581–585. doi: 10.1165/ajrcmb.23.5.f204. [DOI] [PubMed] [Google Scholar]
- Becker S, Koren HS, Henke DC. Interleukin-8 expression in normal nasal epithelium and its modulation by infection with respiratory syncytial virus and cytokines tumor necrosis factor, interleukin-1, and interleukin-6. Am J Respir Cell Mol Biol. 1993;8:20–27. doi: 10.1165/ajrcmb/8.1.20. [DOI] [PubMed] [Google Scholar]
- Fiedler MA, Wernke-Dollries K, Stark JM. Respiratory syncytial virus increases IL-8 gene expression and protein release in A549 cells. Am J Physiol. 1995;269:L865–L872. doi: 10.1152/ajplung.1995.269.6.L865. [DOI] [PubMed] [Google Scholar]
- Arnold R, Humbert B, Werchau H, Gallati H, Konig W. Interleukin-8, interleukin-6, and soluble tumour necrosis factor receptor type I release from a human pulmonary epithelial cell line (A549) exposed to respiratory syncytial virus. Immunology. 1994;82:126–133. [PMC free article] [PubMed] [Google Scholar]
- Bonville CA, Rosenberg HF, Domachowske JB. Macrophage inflammatory protein-1α and RANTES are present in nasal secretions during ongoing upper respiratory tract infection. Pediatr Allergy Immunol. 1999;10:39–44. doi: 10.1034/j.1399-3038.1999.101005.x. [DOI] [PubMed] [Google Scholar]
- Harrison AM, Bonville CA, Rosenberg HF, Domachowske JB. Respiratory syncytial virus-induced chemokine expression in the lower airways: eosinophil recruitment and degranulation. Am J Respir Crit Care Med. 1999;159:1918–1924. doi: 10.1164/ajrccm.159.6.9805083. [DOI] [PubMed] [Google Scholar]
- Fiedler MA, Wernke-Dollries K, Stark JM. Inhibiton of viral replication reverses respiratory syncytial virus-induced NF-κB activation and interleukin-8 gene expression in A549 cells. J Virol. 1996;70:9079–9082. doi: 10.1128/jvi.70.12.9079-9082.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde JG, Monick MM, Hunninghake GW. Oxidant tone regulates IL-8 production in epithelium infected with respiratory syncytial virus. Am J Respir Cell Mol Biol. 1995;13:237–244. doi: 10.1165/ajrcmb.13.2.7626291. [DOI] [PubMed] [Google Scholar]
- Mastronarde JG, He B, Monick MM, Mukaida N, Matsushima K, Hunninghake GW. Induction of interlukin (IL)-8 gene expression by respiratory syncytial virus involved activation of nuclear factor (NF)-κB and NF-IL-6. J Infect Dis. 1996;174:262–267. doi: 10.1093/infdis/174.2.262. [DOI] [PubMed] [Google Scholar]
- Garofalo R, Sabry M, Jamaluddin M, Yu RK, Casola A, Ogra PL, Brasier AR. Transcriptional activation of the interleukin-8 gene by respiratory syncytial virus infection in alveolar epithelial cells: nuclear translocation of the RelA transcription factor as a mechanism producing airway mucosal inflammation. J Virol. 1996;70:8773–8781. doi: 10.1128/jvi.70.12.8773-8781.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamaluddin M, Garofalo R, Ogra PL, Brasier AR. Inducible translational regulation of the NF-IL6 transcription factor by respiratory syncytial virus infection in pulmonary epithelial cells. J Virol. 1996;70:1554–1563. doi: 10.1128/jvi.70.3.1554-1563.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde JG, Monick MM, Mukaida N, Matsushima K, Hunninghake GW. Activator protein-1 is the preferred transcription factor for cooperative interaction with nuclear factor-κB in respiratory syncytial virus-induced interleukin-8 gene expression in airway epithelium. J Infect Dis. 1998;177:1275–1281. doi: 10.1086/515279. [DOI] [PubMed] [Google Scholar]
- Fiedler MA, Wernke-Dollries K. Incomplete regulation of NF-κB by IκBα during respiratory syncytial virus infection in A549 cells. J Virol. 1999;73:4502–4507. doi: 10.1128/jvi.73.5.4502-4507.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casola A, Garofalo RP, Jamaluddin M, Vlahopoulos S, Brasier AR. Requirement of a novel upstream response element in respiratory syncytial virus-induced IL-8 gene expression. J Immunol. 2000;164:5944–5951. doi: 10.4049/jimmunol.164.11.5944. [DOI] [PubMed] [Google Scholar]
- Saito T, Deskin RW, Casola A, Haeberle H, Olszewska B, Ernst PB, Alam R, Ogra PL, Garofalo R. Respiratory syncytial virus induces selective production of the chemokine RANTES by upper airway epithelial cells. J Infect Dis. 1997;175:497–504. doi: 10.1093/infdis/175.3.497. [DOI] [PubMed] [Google Scholar]
- Becker S, Reed W, Henderson FW, Noah TL. RSV infection of human airway epithelial cells causes production of the beta-chemokine RANTES. Am J Physiol. 1997;272:L512–L520. doi: 10.1152/ajplung.1997.272.3.L512. [DOI] [PubMed] [Google Scholar]
- Thomas LH, Friedland JS, Sharland M, Becker S. Respiratory syncytial virus-induced RANTES production from human bronchial epithelial cells is dependent on nuclear factor-κB nuclear binding and is inhibited by adenovirus-mediated expression of inhibitor of κBα. J Immunol. 1998;161:1007–1016. [PubMed] [Google Scholar]
- Koga T, Sardina E, Tidwell RM, Pelletier M, Look DC, Hotzman MJ. Virus-inducible expression of a host chemokine gene relies on replication-linked mRNA stabilization. Proc Natl Acad Sci USA. 1999;96:5680–5685. doi: 10.1073/pnas.96.10.5680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olszewska-Pazdrak B, Casola A, Saito T, Alam R, Crowe SE, Mei F, Ogra PL, Garofalo RP. Cell-specific expression of RANTES, MCP-1, and MIP-1α by lower airway epithelial cells and eosinophils infected with respiratory syncytial virus. J Virol. 1998;72:4756–4764. doi: 10.1128/jvi.72.6.4756-4764.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domachowske JB, Bonville CA, Gao JL, Murphy PM, Easton AJ, Rosenberg HF. The chemokine MIP-1α and its receptor CCR1 control pulmonary inflammation and anti-viral host defense in paramyxovirus infection. J Immunol. 2000;165:2677–2682. doi: 10.4049/jimmunol.165.5.2677. [DOI] [PubMed] [Google Scholar]
- Wang SZ, Hallsworth PG, Dowling KD, Alpers JH, Bowden JJ, Forsyth KD. Adhesion molecule expression on epithelial cells infected with respiratory syncytial virus. Eur Respir J. 2000;15:358–366. doi: 10.1183/09031936.00.15235800. [DOI] [PubMed] [Google Scholar]
- Arnold R, Konig W. ICAM-1 expression and low-molecular-weight G-protein activation of human bronchial epithelial cells (A549) infected with RSV. J Leukoc Biol. 1996;60:766–771. doi: 10.1002/jlb.60.6.766. [DOI] [PubMed] [Google Scholar]
- Matsuzaki Z, Okamoto Y, Sarashina N, Ito E, Togawa K, Saito I. Induction of intercellular adhesion molecule-1 in human nasal epithelial cells during respiratory syncytial virus infection. Immunology. 1996;88:565–568. doi: 10.1046/j.1365-2567.1996.d01-687.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chini BA, Fiedler MA, Milligan L, Hopkins T, Stark JM. Essential roles of NF-κB and C/EBP in the regulation of intercellular adhesion molecule-1 after respiratory syncytial virus infection of human respiratory epithelial cell cultures. J Virol. 1998;72:1623–1626. doi: 10.1128/jvi.72.2.1623-1626.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark JM, Godding V, Sedgwick JB, Busse WW. Respiratory syncytial virus infection enhances neutrophil and eosinophil adhesion to cultured respiratory epithelial cells. Roles of CD18 and intercellular adhesion molecule-1. J Immunol. 1996;156:4774–4782. [PubMed] [Google Scholar]
- Olszewska-Pazdrak B, Pazdrak K, Ogra PL, Garofalo RP. Respiratory syncytial virus-infected pulmonary epithelial cells induced eosinophil degranulation by a CD18-mediated mechanism. J Immunol. 1998;160:4889–4895. [PubMed] [Google Scholar]
- Raza MW, Ogilvie MM, Blackwell CC, Stewart J, Elton RA, Weir DM. Effect of respiratory syncytial virus infection on binding of Neisseria meningitidis and Haemophilus influenzae type b to a human epithelial cell line (HEp-2). Epidemiol Infect. 1993;110:339–347. doi: 10.1017/s095026880006828x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel J, Faden H, Sharma S, Ogra PL. Effect of respiratory syncytial virus on adherence, colonization and immunity of non-typable Haemophilus influenzae: implications for otitis media. Int J Pediatr Otorhinolaryngol. 1992;23:15–23. doi: 10.1016/0165-5876(92)90075-z. [DOI] [PubMed] [Google Scholar]
- Saadi AT, Blackwell CC, Essery SD, Raza MW, El Ahmer OR, MacKenzie DA, James VS, Weir DM, Ogilvie MM, Elton RA, Busuttil A, Keeling JW. Developmental and environmental factors that enhance binding of Bordetella pertussis to human epithelial cells in relation to sudden infant death syndrome (SIDS). FEMS Immunol Med Microbiol. 1996;16:51–59. doi: 10.1016/S0928-8244(96)00079-X. [DOI] [PubMed] [Google Scholar]
- Raza MW, El Ahmer OR, Ogilvie MM, Blackwell CC, Saadi AT, Elton RA, Weir DM. Infection with respiratory syncytial virus enhances expression of native receptors for non-pilate Neisseria meningitidis on HEp-2 cells. FEMS Immunol Med Microbiol. 1999;23:115–124. doi: 10.1016/S0928-8244(98)00125-4. [DOI] [PubMed] [Google Scholar]
- Takeuchi R, Tsutsumi H, Osaki M, Haseyama K, Mizue N, Chiba S. Respiratory syncytial virus infection of human alveolar epithelial cells enhances interferon regulatory factor 1 and interleukin-1β-converting enzyme gene expression but does not cause apoptosis. J Virol. 1998;72:4498–4502. doi: 10.1128/jvi.72.5.4498-4502.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domachowske JB, Bonville CA, Mortelliti AJ, Colella CB, Kim U, Rosenberg HF. Respiratory syncytial virus infection induces expression of the anti-apoptosis gene IEX-1L in human respiratory epithelial cells. J Infect Dis. 2000;181:824–830. doi: 10.1086/315319. [DOI] [PubMed] [Google Scholar]
- Krilov LR, McClosky TW, Harkness SH, Pahwa VS. Responses of monocytes in peripheral blood mononuclear leukocytes (PBMLs) exposed to respiratory syncytial virus (RSV) in vitro [abstract]. Pediatr Res. 1998;43:868A. [Google Scholar]
- Domachowske JB, Bonville CA, Rosenberg HF. Cytokeratin 17 is expressed in cells infected with respiratory syncytial virus via NF-κB activations and is associated with the formation of cytopathic syncytia. J Infect Dis. 2000;182:1022–1028. doi: 10.1086/315841. [DOI] [PubMed] [Google Scholar]
- Marriott AC, Easton AJ. Reverse genetics of the Paramyxoviridae. Adv Virus Res. 1999;53:321–340. [PubMed] [Google Scholar]
- Domachowske JB, Bonville CA, Dyer KD, Easton AJ, Rosenberg HF. Pulmonary eosinophilia and production of MIP-1α are prominent responses to infection with pneumonia virus of mice. Cell Immunol. 2000;200:98–104. doi: 10.1006/cimm.2000.1620. [DOI] [PubMed] [Google Scholar]
- Domachowske JB, Bonville CA, Gao JL, Murphy PM, Easton AJ, Rosenberg HF. MIP-1α is produced but it does not control pulmonary inflammation in response to respiratory syncytial virus infection in mice. Cell Immunol. 2001. [DOI] [PubMed]
- Cook DN, Beck MA, Coffman TM, Kirby SL, Sheridan JF, Pragnell IB, Smithies O. Requirement of MIP-1 alpha for an inflammatory response to viral infection. Science. 1995;269:1583–1585. doi: 10.1126/science.7667639. [DOI] [PubMed] [Google Scholar]