

Abstract
Serratia marcescens, a member of the Enterobacteriaceae family, was long thought to be a non-pathogenic bacterium prevalent in environmental habitats. Together with other members of this genus, it has emerged in recent years as an opportunistic nosocomial pathogen causing various types of infections. One important feature of pathogens belonging to this genus is their intrinsic and acquired resistance to a variety of antibiotic families, including β-lactam, aminoglycosides, quinolones and polypeptide antibiotics. The aim of this study was to elucidate which genes participate in the intrinsic and acquired antibiotic resistance of this genus in order to determine the Serratia genus resistome. We performed phylogenomic and comparative genomic analyses using 32 Serratia spp. genomes deposited in the NCBI GenBank from strains isolated from different ecological niches and different lifestyles. S. marcescens strain SmUNAM836, which was previously isolated from a Mexican adult with obstructive pulmonary disease, was included in this study. The results show that most of the antibiotic resistance genes (ARGs) were found on the chromosome, and to a lesser degree, on plasmids and transposons acquired through horizontal gene transfer. Four strains contained the gyrA point mutation in codon Ser83 that confers quinolone resistance. Pathogenic and environmental isolates presented a high number of ARGs, especially genes associated with efflux systems. Pathogenic strains, specifically nosocomial strains, presented more acquired resistance genes than environmental isolates. We may conclude that the environment provides a natural reservoir for antibiotic resistance, which has been underestimated in the medical field.
Keywords: resistome, genus Serratia, antibiotics resistance genes, intrinsic resistance, acquired resistance
Introduction
Human infections caused by members of the genus Serratia, most commonly by Serratia marcescens, were not well identified until the latter half of the 20th Century, probably due to the taxonomic difficulty in describing the species. S. marcescens and to a lesser extent, other members of this genus, are now considered opportunistic nosocomial pathogens causing an array of infections including meningitis, sepsis, urinary tract infections, skin infections, bloodstream and respiratory infections, as well as being important ocular pathogens (Engelhart et al., 2003; Shanks et al., 2013; Wu et al., 2013; Gupta et al., 2014). Serratia species harbor several virulence factors including hemolysins (ShlAB), Quorum-Sensing proteins (LuxI-R), biofilm development proteins (BsmB) (only seen in S. marcescens), phospholipases (PhlA), peptidases (Clp), metalloproteases, chitinases (ChiABC), siderophores and hemophores (HasA), the lipopolysaccharide LPS, and motility and adherence factors such as flagella and fimbriae (Mahlen, 2011). S. marcescens is among the 10 most recovered pathogens in hospitals worldwide (Mahlen, 2011; Bertrand and Dowzicky, 2012) and has been cultured from a variety of sources including disinfectants, pressure transducers, bronchoscopes, multi-dose medication vials, contaminated antiseptic solutions, fentanyl-containing fluids, contaminated MgSO4 and contaminated saline syringes among others (Sunenshine et al., 2007; Liu et al., 2011; Chiang et al., 2013; Merkier et al., 2013; Šiširak and Hukić, 2013; Liou et al., 2014; Hervé et al., 2015; Dawczynski et al., 2016; Morillo et al., 2016; Vetter et al., 2016). Historically, outbreaks of S. marcescens have been reported since 1950 and have been considered nosocomial in origin (Mahlen, 2011). Recent S. marcescens outbreaks have been reported mostly in North America and Europe, probably due to more efficient surveillance systems in those regions. The majority of the outbreaks occurred in neonatal ICUs, cardiac surgical ICUs, orthopedic clinics and dialysis units. There have been some S. liquefaciens outbreak reports in past years in various countries (Grohskopf et al., 2001). Pathogenic strains from other Serratia species have been isolated from individual patients were not associated with any epidemic outbreak, as seen in the case of a S. rubideae isolated from the sputum of a patient in China (Yao et al., 2016). There is a large amount of published data about Serratia epidemiology and resistance patterns among human populations worldwide. The S. marcescens strains recovered in UCIs from 6 different geographic regions (Africa, Asia, the Asia-Pacific Rim, Europe, Latin America and North America) show a similar resistance/susceptibility pattern in all regions: resistance to all penicillins and susceptibility to all carbapenems. However, Latin American strains show a higher percentage of resistance to all antibiotic tested (Bertrand and Dowzicky, 2012).
Many members of the Serratia genus contain genes related to antimicrobial resistance, which confer resistance to β-lactam, aminoglycosides, quinolones, macrolides and polypeptide antimicrobials. Intrinsic resistance in microorganisms is conferred by antibiotic resistance genes (ARGs), including genes associated with efflux pumps, which are present on the chromosome and shared by members of the same species or genus. Acquired resistance is conferred by the gains of novel resistance genes via horizontal gene transfer (HGT) or by mutations of particular chromosomal genes. Generally, horizontally transferred resistance genes are located on mobile genetic elements, such as plasmids, integrons, transposons or genomic islands (Blair et al., 2014; Hu et al., 2015), and can be defined as any segment of DNA that can translocate from one part of the genome to another or, between genomes (Van Hoek et al., 2011).
The aim of this study was to identify the genes responsible for intrinsic and acquired multidrug-resistance (resistome) of the genus Serratia using 32 Serratia spp. genomes deposited in the NCBI database belonging to strains isolated from different ecological niches from 19 countries and 4 continents. We also included the S. marcescens SmUNAM836 strain, which was previously sequenced by our group.
Materials and Methods
Bacterial Strains and Genomes
Comparative genomic analysis was carried out using 32 whole, sequenced Serratia genomes retrieved from NCBI’s GenBank and RefSeq repositories (Supplementary Table S1), including the S. marcescens SmUNAM836 strain, which was sequenced at the Yale Center for Genome Analysis (YCGA) and assembled and annotated by our group (Sandner-Miranda et al., 2016).
Computing Conservative Consensus Core- and Pan-Genomes
High stringency homologous gene clusters were computed with the GET_HOMOLOGUES (Contreras-Moreira and Vinuesa, 2013) software package by imposing a minimum of 90% query coverage on the all-against-all BLASTP results and performing a PFAM-domain scanning on each sequence to ensure that all homologous gene clusters contain the same domain composition and order. Clustering was performed with the BDBH (-e -D), COGtriangles (-G -t 0 -D) and OMCL (-M -t 0 -D) algorithms implemented in GET_HOMOLOGUES (Contreras-Moreira and Vinuesa, 2013) and the indicated parameters. A consensus core-genome was computed with the aid of the compare_clusters.pl script (-t 34) from the clusters generated by each of the tree algorithms, as detailed elsewhere (Vinuesa and Contreras-Moreira, 2015). Similarly, a consensus pan-genome was computed from the COGtriangles and OMCL clusters using the ‘-m -t 0’ parameters, to generate the pan-genome matrix reporting clusters of all sizes.
Estimating a Robust Maximum-Likelihood Core-Genome Phylogeny for the Genus Serratia
A core-genome phylogeny was estimated under the maximum-likelihood (ML) optimality criterion using the consensus core-genome clusters computed by GET_HOMOLOGUES (Contreras-Moreira and Vinuesa, 2013) as described in the previous section and passing them to the GET_PHYLOMARKERS (Vinuesa et al., 2018, in revision) software suite, which was run in default mode (-R 1 -t DNA). The latter is freely available on GitHub1. Briefly, the GET_PHYLOMARKERS pipeline was used to select core-genome loci with optimal phylogenetic attributes, namely those passing the phi recombination test (Bruen et al., 2006), producing tree topologies and branch-lengths not significantly deviating (kdetrees test) from the expected distribution of these parameters under the multispecies coalescent (Weyenberg et al., 2014) and displaying average branch support values >0.7 (see Vinuesa et al., 2018 for the details). The clustal–omega (Sievers et al., 2011) codon alignments passing these filters were concatenated and a ML phylogeny estimated with IQ-TREE 1.6.1 (Nguyen et al., 2015) using the best fitting model and selecting the phylogeny with the highest likelihood score from those found among independent searches.
Computing Pairwise Core-Genome Average Nucleotide Identity Values From OMCL Clusters (cgANIb-OMCL)
Pairwise cgANIb values were computed from the BLASTN alignments identified by OMCL as belonging to the core-genome with the aid of the get_homologues.pl script, run with the ‘-A -a CDS’ parameters. The resulting cgANIb-OMCL matrix was then displayed as an ordered, bi-dimensional heatmap with the aid of the plot_matrix_heatmap.sh script, distributed with the GET_HOMOLOGUES package.
Phylogeny of Serratia spp. Based on Its Pan-Genome
A parsimony pan-genome phylogeny for the 32 Serratia genomes was estimated from the consensus pan-genome matrix of presence/absence data for homologous clusters created with the aid of the compare_clusters.pl script from the GET_HOMOLOGUES (Contreras-Moreira and Vinuesa, 2013) package, run with the -m -t 0 -T parameters, which calls PARS from the PHYLIP suite. The total number of resistance determinants and the number of acquired and intrinsic resistance genes, including the number of efflux pump genes identified in each genome, are indicated on the tree.
Identification of Antimicrobial Resistance Genes in the Serratia Genus
The whole genome for all strains was analyzed using BLASTN to identify ARGs (Camacho et al., 2009). Searches were also made against locally maintained versions of the CARD (a rigorously curated collection of known resistance determinants) (McArthur et al., 2013) and ResFinder (Zankari et al., 2012) databases. Mutations of genes associated with resistance were searched with BioEdit 7.2.5 (Hall, 1999) and MEGA7 softwares (Kumar et al., 2008). We aligned the 32 Serratia whole genomes using Mauve 2.4.0 (Darling et al., 2010) in order to enable identification of the chromosomal genes associated with intrinsic antibiotic resistance by sequence homology. Mutations of genes gyrA and gyrB (resistance to quinolones), murA (resistance to fosfomycin) and folP (resistance to sulfamethoxazole) were assessed for acquired resistance using BioEdit 7.2.5. In addition, each genome was screened for additional and strain-exclusive ARGs. In order to identify acquired genes on plasmids or integrons, we used BLASTN (Altschul et al., 1990), Mauve 2.4.0. and ISfinder (Siguier et al., 2006).
Relationship Between the Isolation Source of Each Serratia Species and Its Resistance Genes Content
We performed a Principal Coordinate Analysis (PCoA) (Gower, 1998) in order to visualize similarities and dissimilarities among the 32 Serratia spp. resistomes and their relationship with their ecology. We grouped the isolation sources of the strains into three main categories: pathogenic, environmental and symbiotic strains. The environmental strains were divided into three sub-groups: environmental strains associated with soil and plants, environmental strains isolated from water and environmental strains isolated from food sources. Analysis was performed for each Serratia strain based on antibiotic resistance gene content.
Results
A Robust and Highly Resolved Core-Genome Phylogeny for the Genus Serratia
A highly stringent consensus core-genome of 396 genes was computed for the 32 Serratia spp. genomes and two Yersinia spp. genomes, used as outgroup sequences (Supplementary Figure S1). We used the GET_PHYLOMARKERS pipeline to compute a ML core-genome phylogeny from the concatenated supermatrix of 264 top-scoring codon alignments passing the sequential filters imposed by the pipeline. Figure 1 shows the ML tree estimated under the best-fit GTR+F+ASC+R4 model (lnL score: -1793389.569) and rooted at the branch subtending the clade grouping the two Yersinia genomes. The tree is highly resolved, as indicated by the approximate Bayesian and UFBootstrap values computed for each bipartition by IQ-TREE. The two most basal branches of the ingroup clade correspond to environmental (ATCC39006) and endosymbiotic (S. symbiotica) organisms. The latter has a strongly reduced genome, containing only 672 CDSs, being also atypical due to its low G+C content, which is only ∼ 29%. Seven lineages were resolved further inside the tree, which are consistent with the species-level classification of the genome sequences (numbered branches in Figure 1). However, several taxonomic inconsistencies were identified in the S. marcescens clade, which tightly groups S. nematodiphila DSM 21420 and S. ureilytica Lr5/4 nested within it. In addition, the species-tree presented in Figure 1 strongly supports the classification of strain AS12 as a member of S. plymuthica, strains FS14, SCBI and YD25 as members of S. marcescens and strain Serratia sp. FGI94 as S. rubidea. These reclassifications were fully supported by average core-genome identity values (cgANIb) computed from the pairwise BLASTN alignments used by OMCL to cluster the core-genome loci. In all cases these strains had a cgANIb value >98% when compared to the closest named species (Supplementary Figure S2), as indicated in the previous sentence and shown on the ML species-tree depicted in Figure 1.
FIGURE 1.

Core-genome phylogeny estimated using the GET_PHYLOMARKERS pipeline to compute a maximum-likelihood (ML) tree from the concatenated supermatrix of 264 top-scoring codon alignments.
Phylogenetic Structure of Serratia spp. Pan-Genome and the ARGs Number
The parsimony pan-genome tree was derived from a consensus presence/absence matrix of homologous genes based on the clustering algorithms OMCL and COGtriangles. The Venn diagram shows the number of gene clusters of the 32 Serratia spp. pan-genome (12,347 clusters) (Supplementary Figure S3) that were used to generate the pan-genome matrix and the phylogeny.
The pan-genome tree shows five main groups (Figure 2). The results of the phylogenetic analysis placed all the S. marcescens strains in group A, together with the misclassified S. ureilytica, an environmental strain from a geothermal spring and S. nematodiphila DSM 21420, a nematode symbiont and an insect pathogen and 3 uncharacterized strains: Serratia sp. YD25, Serratia sp. FS14 and Serratia sp. SCB1. All these strains should be re-classified as S. marcescens, based on the evidence gained from the core-genome phylogeny (Figure 1) and the cgANIb data (Supplementary Figure S2). Most of the acquired ARGs and efflux pump genes are found in strains of this group. Group B consists of two sub-groups: one with the strains belonging to all S. plymuthica strains; and the other with the S. liquefaciens and S. proteamaculans strains, which are all environmental strains, with the exception of S. liquefaciens HUMV-21 and S. liquefaciens FDAARGOS 125, which are both nosocomial strains. Group C comprises only two strains, Serratia sp. FGI94, a fungus symbiont and S. rubidaea 1122 isolated from a patient in China. The former has a cgANIb value of 99.2 when compared to the latter, and hence strain FGI94 should be classified as S. rubidea. S. fonticola GS2 and S. fonticola DSM4576 are placed in group D, both environmental strains. Finally, group E, holds 3 distantly related strains: Chania multitudinisentens isolated from the soil of an ex-landfill site; Serratia sp. ATCC 39006 isolated from water; and the co-obligate aphid symbiont, S. symbiotica ‘Cinara Cedri’. The genomes of these 3 strains harbor the fewest ARGs on their chromosomes. Their cgANIb values are <85% with respect to the other species in the main Serratia spp. cluster, and therefore their classification as members of the former genus is questionable.
FIGURE 2.

Parsimony pan-genome tree for 32 Serratia genomes derived from presence/absence of homolog genes in a consensus pan-genome matrix computed by the COGtriangles y OrthoMCL clustering algorithms. This phylogeny was the most parsimonious tree found in a tree search made with PARS from the PHYLIP suite. The tree has a total length of 20,453 steps. Capital letters A-E indicate the phylogenetic groups.
Phylogenetically, S. marcescens SmUNAM836 and S. marcescens SM39 are closely related with both genomes sharing the same intrinsic ARGs and efflux pump genes (Iguchi et al., 2014) but different acquired ARGs.
Identification of Antimicrobial Resistance Genes in the 32 Serratia spp. Genomes
Using the CARD database, we looked for ARGs reported to be associated with enterobacteria and more specifically with Serratia spp., which were identified on the genomes of the Serratia spp. strains using BLASTN. We then aligned the 32 genomes using Mauve 2.4.0 and performed BLASTN to look for the percentage of identity and coverage between the different ARGs. The sequence alignments showed that those genes that appeared to be homologous in Mauve, shared more than 70% of identity between them when performing BLASTN; this, together with the use of global multiple sequence alignments, makes our identification of ARGs more robust (Pearson, 2013). Strain specific ARGs and efflux pump components were detected running a blast search against the CARD database using the following parameters: coverage = 95/90% and identity = 70/50%, respectively.
Frequency of ARGs According to Each Class of Antibiotic in the Serratia spp. Resistome
The general frequency of ARGs found in the 32 Serratia spp. genomes (efflux pumps not included in this analysis) according to the class of antibiotic was: 0.13% for macrolides; 0.54% for sulfonamides; 3.69% for chloramphenicol; 3.83% for phosphonic antimicrobials; 3.96% for quinolones; 4.1% for aminocoumarin antibiotics (novobiocin); 7.11% associated with aminoglycoside resistance; 20.38% for β-lactam, 56.22% for antimicrobial polypeptides (Figure 3). It is not surprising that the highest frequency of ARGs corresponded to the genes that confer resistance to polypeptide antibiotics since S. marcescens and the whole genus has a natural resistance to polymyxin B (Olaitan et al., 2014). The second highest frequency in our sample corresponded to β-lactam resistance genes. This fact was expected due to the high number of β-lactamases acquired through HGT in plasmids or transposons found in this genus. The importance of β-lactam resistance genes is reinforced with the fact that up to 1,000 genes related to β-lactam resistance have been described and classified in recent years, among them many new alleles and new genes found in remote geographical niches (Allen et al., 2009; Bush and Jacoby, 2010). In our analysis, the fact that the β-lactamases acquired on plasmids were found exclusively in nosocomial bacteria, except for blaACT-28, which was identified in an environmental strain associated with plants, is a noteworthy observation (Supplementary Table S3). HDT has been shown to play a major role in the transmission and evolution of β-lactam resistance genes in pathogenic and environmental enteric bacteria (Davies and Davies, 2010; Iredell et al., 2016).
FIGURE 3.
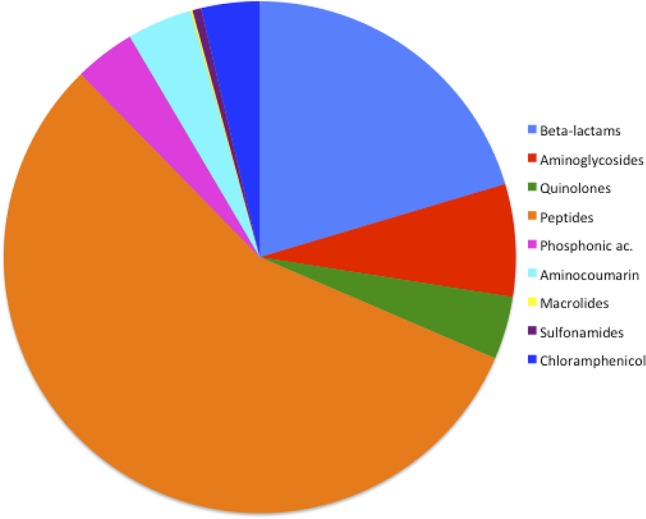
Frequency of ARGs according to the antibiotic class in Serratia spp. resistome.
Total Antibiotic Resistance Genes
A total of 123 different classes of genes associated with intrinsic and acquired antibiotic resistance were found among the 32 Serratia species (detailed information is shown in Supplementary Table S2). These ARGs include genes that encode modifying enzymes, antibiotic hydrolysis enzymes, efflux pumps, porines, regulatory proteins, genes with mutations that confer antibiotic resistance, and alleles of some genes and efflux pumps. From the 123 total resistance genes: 33 are intrinsic resistance genes; 16 are acquired resistance genes [13 on plasmids (Supplementary Table S3), 2 on transposons, and the gene gyrA with the Ser83 mutation that confers quinolone resistance] (Figure 4); and 74 are genes associated with efflux systems, from which 3 were localized on plasmids (Figure 5).
FIGURE 4.

Number of intrinsic and acquired ARGs identified in the 32 Serratia genomes (efflux pump genes not included). Intrinsic ARGs in orange color, acquired in red, and acquired by mutation in green.
FIGURE 5.

Box plot showing the number of genes associated with the five families of efflux pumps identified in the 32 Serratia genomes: multidrug and toxic-compound extrusion family (MATE), the small multidrug resistance family (SMR), the major facilitate superfamily (MFS), the resistance nodulation division family (RND), and the ATP binding cassette superfamily (ABC).
Intrinsic Resistance Genes
Intrinsic resistance comprises all of the inherent properties provided by the characteristics of a particular microorganism that limit the action of antimicrobials (Fernández and Hancock, 2012). Most of the genes related to antimicrobial resistance are encoded on the chromosome of the Serratia genus. In most genomes, we identified the Ambler class C β-lactamase gene ampC, which confers resistance to all penicillins, as well as to 3rd generation cephalosporins and aztreonam when overexpressed (Paterson and Bonomo, 2005), in addition, we found all its regulatory genes: ampD, ampE, ampG, and ampR (MacDougall, 2011). A class A β-lactamase gene blaCTX-M and class C β-lactamase gene blaACT-29 were found only in the two S. fonticola genomes. Of the 14 ARGs associated with β-lactam resistance, 10 were found in S. marcescens CAV1492, a nosocomial strain (Supplementary Table S2).
Three aminoglycoside resistance genes, the aminoglycoside phosphotransferase gene aph(3″), which confers resistance to streptomycin, and the aminoglycoside acetyltransferase gene aac(6′)-Ic to amikacin and tobramycin (Shaw et al., 1992) were present on most of the chromosomes studied. We found the aminoglycoside nucleotidyltransferase gene aadA16 in only 4 genomes from environmental strains. S. symbiotica, and C. multitudinisentens RB-25 did not present any of these genes.
We identified the qnr gene that confers quinolone resistance in the chromosome of 25 strains. Six different alleles of this gene were identified: allele qnrB15 in the S. phymutica group, allele qnrB23 in the S. marcescens and S. nematodiphila group, qnrB31 also in the S. marcescens group, qnrB32 in the S. liquefaciens and S. proteamaculans group, qnrB37 in the S. marcescens and S. ureilytica (reclassified as S. marcescens) group, and qnrB57 in the rest of the S. phymuthica strains. Due to the fact that this gene is unique to the Serratia spp. chromosome, it could be the possible source of Qnr determinants for the plasmid-encoded Qnr in other species of bacteria (Velasco et al., 2009).
Serratia spp. is intrinsically resistant to polymyxin B. Polymyxin B resistance in this genus can be explained by the presence of the arnBCADTEF operon, the two-component systems pmrAB/phoPQ and the regulatory gene mgrB in all strains, except for S. symbiotica ‘Cinara Cedri’. Activation of the two-component systems is set off by environmental stimuli that result in an overexpression of LPS-modifying genes (Lin et al., 2014; Olaitan et al., 2014) and leading to polymyxin B and colistin resistance.
Besides the above mentioned antibiotics, the genus Serratia is intrinsically resistant to other classes of antibiotics: 31 out of 32 strains have the bacA gene on their chromosomes, conferring resistance to bacitracin, another polypeptide antibiotic. In addition, 30 strains harbor the gene hslJ and 28 strains the gene fosA, which confer novobiocin and fosfomycin resistance, respectively. The chloramphenicol acetyltransferase cat gene, which confers resistance to chloramphenicol is also present in 27 strains (Potrykus and Wegrzyn, 2001; Schwarz et al., 2004). Only 12 strains, mostly environmental, possess the vancomycin resistance gene vanX on its chromosome (detailed information is shown in Supplementary Table S2).
Intrinsic Resistance Conferred by Efflux Pumps
Active drug extrusion outside of the bacterial cell is one of the most common mechanisms associated with resistance. We identified 74 different genes related to the 5 efflux pump families: the multidrug and toxic-compound extrusion family (MATE), the small multidrug resistance family (SMR), the major facilitator superfamily (MFS), the resistance nodulation division family (RND), and the ATP binding cassette superfamily (ABC) (Supplementary Table S2).
Some of these efflux pumps have been described previously in the Serratia genus and have homolog in other bacteria: the SsmE efflux pump of the SMR family (conferring resistance to ethidium bromide) is homologous to the E. coli EmrE (Minato et al., 2008); SmfY of the MFS family (conferring resistance to norfloxacin, acriflavine and ethidium bromide) homologous to QacA of Staphylococcus aureus (Shahcheraghi et al., 2007); SdeXY (conferring resistance to erythromycin, tetracycline, norfloxacin, benzalkonium chloride, ethidium bromide, acriflavine, and rhodamine); SdeAB (conferring resistance to fluoroquinolones) and SdeCDE (conferring resistance to novobiocin) all belonging to the RND family and having a high degree of homology with the AcrAB-TolC, OqxAB and MdtABC efflux pumps of E. coli respectively (Chen et al., 2003; Kumar and Worobec, 2005; Begic and Worobec, 2008); and finally, SmdAB (conferring resistance to norfloxacin, tetracycline, and tetraphenylphosphonium chloride) of the ABC superfamily, an homolog of MdlAB of E. coli (Matsuo et al., 2008).
Other homologous genes of efflux systems from different bacteria that confer resistance to several kinds of antibiotics and antiseptics were identified on the 32 genomes: 2 alleles of the aminoglycoside efflux pump AcrD from the RND family (Rosenberg et al., 2000); 2 quinolone efflux pumps, DinF and MdtK, from the MATE family (Brown et al., 2007); SugE, a SMR family efflux pump which confers resistance to quaternary ammonium compounds (Chung and Saier, 2002); 9 different MSF family efflux pumps, among them two alleles of EmrAB conferring resistance to thiolactomycin (Furukawa et al., 1993), Bcr to bacitracin (Bernard et al., 2005), Fsr, an homolog of E. coli RosA, which confers fosmidomycin resistance, and MdfA, which confers resistance to chloramphenicol (Heng et al., 2015). The efflux pump MacAB from the ABC superfamily, which confers resistance to macrolides (Lu and Zgurskaya, 2012; Fitzpatrick et al., 2017) was also identified on the chromosomes of the Serratia spp. genomes, and the efflux pump TetA from the MFS family conferring resistance to tetracycline (Aldema et al., 1996). In addition, 16 genes belonging to 8 efflux systems had alleles on different chromosomal loci, including copies on genomic islands, for example, the aminoglycoside efflux protein AcrD, a transporter belonging to the resistance-nodulation-division family (RND family), which has three different alleles (acrD, acrD′, and acrD″) distributed among the 32 Serratia genomes. The nucleotidic identity percentage between acrD and acrD′ is 67.2%, while it is 60.9% between acrD and acrD″ and 62.6% between acrD′ and acrD″. Two of the nosocomial strains, SmUNAM836 and Sm39, were the only strains that presented all three alleles.
The majority of the genes associated with efflux pumps of the 32 genomes belong to the RND efflux family (Figure 5). Most of these genes are shared by all the strains, except for S. symbiotica ‘Cinara Cedri’ that only contains 5 of the reported 74 efflux pumps genes. It is not surprising that this strain harbors very few ARGs and efflux pumps because of its reductive genome evolution related to its lifestyle inside the stable and protected niche of the host cell of the aphid Cinara cedri (Lamelas et al., 2011; Dutta and Paul, 2012). The highest number of efflux pump genes (57) is present in the Mexican nosocomial strain S. marcescens SmUNAM836 sequenced by our group (Sandner-Miranda et al., 2016). Pathogenic strains, including nosocomial strains, show an average number of 50.1 efflux-associated genes, whereas the environmental strains show an average of 44.1 efflux-associated genes and the symbiotic strains only 33.5 efflux-associated genes (Supplementary Table S2).
Acquired Resistance Genes on Plasmids, Transposons, and Integrons
In order to identify genes associated with acquired antibiotic resistance, we screened all the plasmids of the 32 Serratia spp. strains. A total of 16 plasmids are reported in the NCBI database for this strain collection (Supplementary Table S3). We found 13 plasmid-borne ARGs, 7 β-lactam resistance genes, 3 aminoglycoside resistance genes encoding modifying enzymes, one macrolide resistance gene, one sulfonamide resistance gene, one trimethoprim resistance gene and additionally 3 efflux pump genes associated with resistance to quaternary ammonium compounds, disinfectants and macrolides also on plasmids.
The β-lactam resistance genes class A blaTEM-1, blaKPC-2 and blaSV H-30, class B blaIMP-1, class C blaACT-28 and blaCMY -8, and class D blaOXA-9 were identified in plasmids of nosocomial strains, with the exception of blaACT-28, a class C β-lactamase found on plasmid pSF001 of S. fonticola GS2, a strain that is associated with a plant. Looking at the nosocomial strains S. marcescens SmUNAM836 and S. marcescens CAV1429, its plasmids (pSmUNAM836 and pKPC_CAV1492 respectively) harbor class A blaTEM-1 β-lactamase, which is classified as a broad spectrum β-lactamase (Bush and Jacoby, 2010) and confers resistance to all penicillins and β-lactamase inhibitors. The strain S. marcescens CAV1429 also carries two copies of the class A β-lactamase, namely blaKPC-2, a carbapenemase (Wang et al., 2014), and the blaOXA-9 gene that encodes an oxacillinase-carbenicillinase (Bojorquez et al., 1998) on plasmid pKPC_CAV1492. The blaSV H-30 gene, which encodes an ESBL that confers resistance to penicillins and cephalosporines (Bradford, 2001), was located on plasmid pCAV1492-73 of strain S. marcescens CAV1492. The imipenem metallo-β-lactamase gene, blaIMP-1, and the cephalosporin resistance gene, blaCMY-8, were only found on the plasmid pSMC1 of the nosocomial strain S. marcescens Sm39.
The aminoglycoside resistance genes aac(6′)-1b and aadA1, encoding modifying enzymes tobramycin and amikacin 6′ acetyltransferase (Rather et al., 1992) and a streptomycin 3″-adenylyltransferase were found on plasmid pKPC_CAV1492 of the nosocomial strain S. marcescens CAV1429. Gene aadA2, encoding another streptomycin 3″-adenylyltransferase was found on plasmid pSMC1 of the nosocomial strain S. marcescens Sm39. Strain S. marcescens Sm39 harbors two copies of the sulfonamide resistance gene sul1 on plasmid pSMC1.
Three efflux pump genes, qacE, qacH, and msrE, were found on plasmids of the nosocomial strain S. marcescens CAV1492. Two copies of gene qacE were found on plasmid pSMC1 of S. marcescens SM39. These efflux pumps confer resistance to quaternary ammonium compounds, disinfectants and macrolides, respectively.
As previously reported, it is common to find various ARGs on the same mobile element. Such is the case of the genes sul1, qacE and some β-lactamases, which are found together in the clinical class 1 integron (Wyrsch et al., 2016). In this study we found two plasmids harboring this integron, plasmid pSMC1 of the nosocomial strain S. marcescens Sm39 and plasmid pKPC_CAV1492 of the nosocomial strain S. marcescens CAV1492. The class 1 integron is associated with various families of transposons, the Tn3 transposon present in plasmid pKPC_CAV1492 (Deng et al., 2015) and the Tn91 transposon present in plasmid pSMC1. Other plasmids harboring heavy metal resistance are also present in some of the Serratia spp. genomes (mercury resistance genes in plasmid pCAV1492-73 and pSMC1from the nosocomial strains S. marcescens CAV1492 and S. marcescens Sm39, respectively, and copper resistance genes in plasmid pCAV1492-199 also from strain S. marcescens CAV1492).
The aminoglycoside phosphotransferase gene, aph(3′), was only found on S. rubidaea 1122 and Serratia sp. FGI94 chromosomes, associated with a transposon. Gene vanW, which confers resistance to vancomycin, was found on a transposon of the environmental strain S. fonticola DSM4576. These mobile elements have not been previously classified and were inferred in this analysis on the basis of being unique sequences harboring transposases (Supplementary Table S2).
Many other genes encoding transposases and integrases were found on all Serratia spp. chromosomes, but neither associated with ARGs.
Efflux Pumps Acquired on the Genomic Islands of Serratia spp.
Three efflux pumps were identified on different genomic islands of some of the S. marcescens strains using IslandViewer 4 (Bertelli et al., 2017). These genomic islands have not been characterized before. Two of them, MexGHI (Aendekerk et al., 2005) and MexPQ-OprE (Mima et al., 2005), which confer resistance to norfloxacin and macrolides, respectively, showed 82 and 84% homology to the P. aeruginosa efflux pumps, indicating a probable horizontal transfer event between these bacteria. The third efflux pump was an allele of etsABC that encodes an E. coli efflux pump of a putative ABC transport system contained within the pAPEC-O2-ColV plasmid, that confers resistance to macrolides (Johnson et al., 2006). These efflux pumps are also present in other Serratia species and most probably on genomic islands as well (Supplementary Table S2).
Resistance Acquired by Gene Mutations
The gene gyrA mutation at codon Ser83 was identified in three nosocomial strains, namely S. marcescens SmUNAM836, S. marcescens Sm39, and S. marcescens CAV1492, and also on S. symbiotica ‘Cinara Cedri,’ an aphid symbiont. Resistance may arise due to point mutations that result in amino acid substitutions within the topoisomerase genes (gyrA, gyrB, parC, parE), with or without a decreased expression of outer membrane porins and overexpression of multidrug efflux pumps (Hopkins et al., 2005). S. marcescens SmUNAM836 and S. marcescens CAV1492 had an isoleucine residue instead of the expected serine at position 83 of GyrA while the strain S. marcescens Sm39 had an arginine residue. S. symbiotica, which is a co-obligate symbiont, had a threonine in this position. These mutations are likely to induce a local conformation change of the A subunit of the DNA gyrase, modifying the binding affinity of the quinolones for this enzyme (Yoshida et al., 1990; Weigel et al., 1998). Resistance to one type of quinolone will confer resistance to all (Webber and Piddock, 2001). Neither of the strains had the mutation on gene folP that confers resistance to sulfamethoxazole. The folP single C→T transition resulting in a Pro→Ser substitution at amino acid position 64 is absent in all genomes (Vedantam et al., 1998). Amino acid substitutions (Asp369Asn and Leu370Ile) in MurA are a major factor in fosfomycin resistance, but neither of the strains showed these mutations (Takahata et al., 2010).
The Serratia spp. Resistome
We classified the resistome according to the niche or lifestyle of the bacteria. We divided the sample of 32 Serratia spp. strains into three lifestyles: pathogens (nosocomial and animal pathogens), environmental (bacteria associated with plant, soil, water, or food) and symbiotic bacteria (Figure 6). The diagram shows that pathogenic (brown) and environmental strains (blue) do not show a significant different median number of total ARGs, intrinsic and acquired, but symbiotic bacteria (green) do show the smallest number. Regarding efflux pump genes, pathogenic bacteria harbor more of these genes than environmental and symbiotic bacteria. We can appreciate that the difference among resistance gene content between nosocomial/pathogenic and environmental strains is not significant. We believe that this is due to the environment providing a natural reservoir for ARGs.
FIGURE 6.

Box plot showing the relationship between the number of resistance determinants and the lifestyle of the strains. There are 3 main lifestyle groups, nosocomial/pathogen [P (brown)], environmental/free living [E (blue)] and environmental/symbiotic [S (green)]. P, E, and S represent the total resistance determinants. P.1, E.1, and S.1 are the total efflux pump genes. P.2, E.2, and S.2 represent the acquired resistance genes and P.3, E.3, and S.3 the intrinsic AGRs.
In this comparative genomic study, we report that the Serratia spp. resistome consists of genes shared by the majority of the strains and genes exclusive of niche. With the analysis of 32 Serratia spp. genomes, we report a resistome for the Serratia genus totaling 123 ARGs, 49 intrinsic and acquired ARGs, and 74 efflux pump-associated genes. Of the 123 ARGs, 80 are present in one or more strains from each niche, 7 are shared by pathogens and environmental strains only, 2 are shared by pathogens and symbiotic strains, and one is shared by environmental and symbiotic strains. 18 ARGs were found exclusively in pathogenic strains, 14 in environmental strains and one in symbiotic strains (Figure 7).
FIGURE 7.
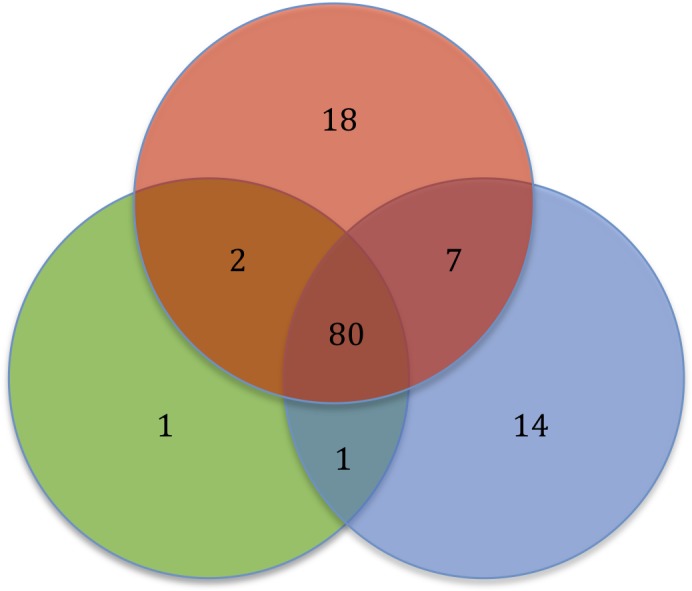
Diagram showing the Serratia spp. resistome based on the bacterial niche. Pathogens (red), environmental (blue), and symbiotic (green).
The polymyxin resistance operon and the genes catA, bacA, fosA and hslJ, which confer resistance to chloramphenicol, bacitracin, fosfomycin and novobiocin, respectively, are among the shared ARGs. In addition, the aminoglycoside acelyltransferase gene, aac(6′)-Ic, the aminoglycoside phosphotransferase gene, aph(3″), and the quinolone resistance gene, qnr, are shared by the majority of the strains, regardless of their lifestyles. The class C β-lactamase gene, ampC, and all the regulatory genes were also found in most of the strain chromosomes. Most of the strains share most of the genes associated with efflux systems.
The 18 ARGs exclusive of pathogens are genes located on plasmids, mainly β-lactamase coding genes, with the exception of the alleles of the efflux pumps EmrAB and AcrD, which are located on the chromosome (Supplementary Table S2). The 14 ARGs exclusive of environmental strains are genes found on the chromosome, all of intrinsic resistance except for the vancomycin resistance gene vanW found on a transposon. Only one ARG was found to be exclusive of a symbiotic strain, an allele of the efflux pump AcrF that confers resistance to ciprofloxacin. Five efflux pumps genes, mexP, mexQ, opmE and triC, from the RND family conferring resistance to macrolides, fluoroquinolones and triclosan, respectively, and the gene mdsC, which is part of the efflux pump MdsAB (absent in this sample), are shared by pathogen and environmental strains. The aminoglycoside phosphotransferase gene aph(3′) and the mutation of gyrA, which confers resistance to quinolones, were located in pathogens and symbiotic strains. Only the streptogramin efflux pump gene, vgaC, is shared by environmental and symbiotic strains and is absent in pathogens.
Similarities and Dissimilarities Between the Serratia spp. Resistomes
The intrinsic and acquired ARGs, and the efflux pump-associated genes, which were identified in the 10 Serratia species and in the 4 non-characterized strains, were grouped according to the lifestyle of each strain in a matrix of similarity/dissimilarity. A PCoA analysis was performed to visualize the similarities of the Serratia spp. resistomes according to the 3 previously mentioned ecological niches. The environmental strains were sub-divided into strains associated with soil and plants, strains isolated from water and strains isolated from food. The plot of Euclidean distances shows that the resistomes of nosocomial and pathogenic strains are similar to those isolated from the natural environment based on the number of ARGs (Figure 8). The most dissimilar resistome was from the co-obligate aphid symbiont, followed by the resistome of strain Serratia sp. ATCC 39006 isolated from salt marsh water and a questionable member of the genus Serratia, the resistome of the fungus garden symbiont Serratia sp. FGI94 (reclassified in this work as S. rubidea) and the resistome of C. multitudinisentens RB-25 isolated from soil. Nosocomial strains with the highest number of acquired ARGs, S. marcescens Sm39 and S. marcescens CAV1429, also showed dissimilarity with respect to the others. It is not surprising that nosocomial strains present the highest number of acquired ARGs as they are subject to a strong selection pressure due to frequent antibiotic use in hospitals.
FIGURE 8.

Principal Coordinate Analysis (PCoA) plot depicting Euclidean distances between resistomes of nosocomial strains (red), animal pathogens (orange), environmental associated with soil and plants (green), environmental isolated from water (blue), environmental isolated from food (purple), and environmental symbiotic bacteria (yellow) calculated using ARGs counts. The blue and green square represents a group of many strains isolated from water and soil/plants, the triangle represents de pathogen/symbiotic strain S. nematodiphila.
It would be interesting to include a new set of Serratia isolates to be collected from animal and environment sources that could confirm the selection pressure of the indiscriminate use of antibiotics in the hospital environment on nosocomial strains and address unsolved issues on the zoonotic origin of antimicrobial resistance genes (Cloeckaert et al., 2017).
Discussion
The microbial pan-genome is the cumulative number of different genes found within genomes of a particular taxonomic rank, usually within a species, though this can be extended to higher levels, such as a genus (Tettelin et al., 2008). It contains the core genes, common to all strains of the study, the accessory genome containing genes present between two and n–1 strains, and the unique or singleton genes present only in a single strain. Inside the pan-genome, we can study different features, such as the resistome (Rouli et al., 2015). The pan-genome size, and whether it is open or closed, depends in part on bacterial lifestyle. Large and open pan-genomes are associated with bacterial species that live within communities and that are prone to horizontal gene exchange. However, the more genomes used to predict the pan-genome, the larger its predicted size, due to the contribution of rare genes (Tettelin et al., 2008). Here, we report an observed pan-genome of 12,347 clusters of genes for a collection of 32 strains of the genus Serratia, which we consider a relatively large pan-genome based on the small sample size of this genus. It is also considered to be relatively large when compared to the pan-genome of 5,398 clusters for 50 Streptococcus genomes from 14 species (Contreras-Moreira and Vinuesa, 2013), and around 10,000 family genes for the Salmonella genus (Jacobsen et al., 2011). On the other hand, for the genus Vibrio, there is a report of a pan-genome consisting of 26,504 genes for 43 different species (Thompson et al., 2009), which is a very large pan-genome and reflects the high diversity of this genus. For the small genome of genus Mycoplasma there is an estimate of a pan-genome consisting of 8,000 genes, which is very large if we consider the small genome size (0.5 and 1.4 Mb) of this genus; this huge pan-genome size may be the reflection of their diverse lifestyles (Liu et al., 2012). Large pan-genomes indicate that these genomes have a considerable amount of unique sequences, such as mobile elements, genomic islands, transposons or prophages.
The phylogenetic analysis based on the pan-genome of the 32 Serratia spp. is perfectly supported by our maximum likelihood core-genome phylogeny. All S. marcescens strains group together with the uncharacterized strains Serratia sp. YD25, Serratia sp. FS14 and Serratia sp. SCB1, suggesting that they should be reclassified as members of the S. marcescens species. This is clearly supported by our cgANIb estimates which are >98% in all cases. It is worth noting that S. nematodiphila DSM 21420 clusters in the same subgroup as S. marcescens Db11 and Serratia sp. SCB1, which are both insect pathogens. This fact can be explained due to the dual quality of S. nematodiphila, which is a nematode symbiont, but a pathogen of the insect that is parasitized by the nematode (Kwak et al., 2015). The evidence presented in this study from the core-genome phylogeny and cgANIb analyses strongly suggest that strains DSM 21420 and SCB1 should be reclassified as S. marcescens. S. phymuthica, S. liquefaciens and S. proteamaculans strains are placed in the sister group to S. marcescens strains. Two of the S. liquefaciens strains are nosocomial pathogens. This grouping is congruent with previous studies (Abebe-Akele et al., 2015; Li et al., 2015). Serratia sp. FGI94, which is a fungus symbiont strain and S. rubidaea 1122 isolated from a patient in China are placed together in group C. The evidence from the core-genome phylogeny and cgANIb data clearly indicate that FGI94 belongs to the S. rubidaea species and that it is wrongly classified. Both have similar chromosome size and harbor the aminoglycoside phosphotransferase gene aph(3′) on a transposon. The most distantly related strains are C. multitudinisentens [recently reclassified as C. multitudinisentens (Ee et al., 2016)] isolated from the soil of an ex-landfill site, Serratia sp. ATCC 39006 isolated from water and the co-obligate aphid symbiont S. symbiotica ‘Cinara Cedri’. The genomes of these 3 strains harbor the fewest ARGs on their chromosomes. Based on their <85% cgANIb values when compared to the other Serratia genomes, their classification in this genus should be carefully revised.
Bacterial antimicrobial resistance occurs by one or a combination of different mechanisms: a reduction in antibiotic passage through the bacterial outer membrane preventing access to the target; modification of antibiotic targets by modifying enzymes; antibiotic hydrolysis; and increased transport of the antibiotic out of the cell by efflux pumps (Shaikh et al., 2015; Munita and Arias, 2016). The modulation of resistance to certain antibiotics depends also on the activation of regulatory genes, mutations of specific genes, intrinsic differences in the structure of the outer membrane, such as porin alterations that reduce the entry of antimicrobials, such as carbapenems (Gupta et al., 2011), or due to the acquisition of novel resistance genes by HGT (Blair et al., 2014). It is important to recognize that the concept of antimicrobial resistance is a phenomenon with many layers of complexity. Due to this complexity, a resistance genotype does not necessarily produce a resistant phenotype all the time; an example of this is illustrated with the aac (6′)-Ic gene. The aac(6′)-Ic gene was cloned from S. marcescens (Shaw et al., 1992) and DNA hybridization analysis demonstrated that all S. marcescens strains carried the aac(6′)-Ic gene, however, not all presented the AAC(6′)-Ic resistance profile (Shaw et al., 1993). In this comparative study, we looked for all classes of genes involved in the different antibiotic resistance mechanisms mentioned above, including regulatory genes (detailed information is shown in Supplementary Table S2).
The resistome of Serratia spp. is composed mainly by intrinsic resistance genes (structural), mostly by genes encoding efflux pumps systems. These pumps, besides conferring resistance to antibiotics, have other important physiological roles and therefore, have greater clinical relevance than is usually attributed to them (Piddock, 2006). These groups of bacteria harbor a “natural” resistance to most of the antibiotics, such as the β-lactam family, due to the presence of the β-lactamase AmpC (Jacoby, 2009), to polypeptide antibiotics through the operon arn, and to quinolones by having the gene qnr. This resistance to a wide spectrum of antimicrobials makes it difficult to select an appropriate treatment for Serratia spp. infections. Surprisingly, the Serratia genus lacks resistance genes for trimethoprim and sulfonamides (with the exception of S. marcescens CAV1492 which has acquired the trimethoprim and sulfonamide resistance genes dfr5 and sul1 on one of its plasmids, and S. marcescens Sm39 which harbors the sulfonamide gene sul1 on plasmid pSMC1). Serratia genomes also lack the folP mutation in position 64 that results in a Pro→Ser aminoacid substitution and that confers sulfonamide resistance (Vedantam et al., 1998), which makes this combination of antibiotics a good choice in the treatment of bacterial infections caused by this genus. One evolutionary hypothesis for the lack of these resistance genes in this genus could be based on the antibiotic resistance cost. Resistance is often associated with reduced bacterial fitness and the reduction in antibiotic use will benefit the fitter susceptible bacteria (Andersson and Hughes, 2010). The combination of these antimicrobials has been suspended in many countries relaxing the selection pressure on these genes and leading to gene loss. An alternative hypothesis could be that this genus is intrinsically susceptible to trimethoprim-sulfamethoxazole and that they have never carried these genes on their chromosomes. The presence of these ARGs on the plasmids of two nosocomial S. marcescens strains can be explained by HGT events and by genetic co-selection, where the use of one antibiotic will exert selection pressure on all the ARGs that are on the same mobile element.
Our results show that most of the resistomes of pathogenic and environmental bacteria of the genus Serratia are very similar in the number of ARGs shared, being the content of horizontally transferred genes what determine the differences with the nosocomial strains. To date there are around 350 S. marcescens genomes in the NCBI database, and more than 450 for the whole genus, but only 10.8% of these genomes are assembled and annotated completely. We decided to work with complete genomes retrieved from NCBI’s GenBank and RefSeq repositories to avoid biases introduced by highly fragmented genomes in phylogenomic inference and comparative pan-genomic. This can lead to a potential limitation of our results; nevertheless, we presume that the results obtained here are a suitable approximation for the resistome of this bacterial genus.
The nosocomial strains with the highest number of acquired ARGs are S. marcescens CAV1429 and Sm39 with 14 and 8 genes on their plasmids, respectively, mostly β-lactamases. We also found that both pathogenic and environmental strains share genes with high nucleotide identities confirming what has been previously observed, in that the environmental isolates from soil (D’Costa et al., 2006), plants and water represent a natural reservoir for ARGs (Riesenfeld et al., 2004). While ARGs of environmental isolates may originally have had different functions aside from conferring resistance to antibiotics produced by other competing bacteria, these genes have now been acquired as resistance genes in pathogenic bacteria via HGT (Berglund, 2015). There are several other known factors that promote resistance in susceptible bacteria: selection pressure placed on susceptible microbes through the use of therapeutic agents, over-prescription, self-medication, treatment non-compliance, use of antibiotics in food-producing animals and in agriculture in general (Knobler, 2003), and an increase in antimicrobial residues found in the environment, most particularly in water (Harris et al., 2013). It is important to raise awareness concerning the excessive use of antibiotics in agricultural, poultry and livestock industries by creating specialized surveillance institutions that coordinate the health, food and environmental sectors, enabling the identification of the many routes for both dissemination and acquisition of ARGs from agricultural and environmental microbial communities to human pathogens, preventing nosocomial outbreaks. It is also crucial to identify the intrinsic resistance profile of a given microorganism or species in order to select the most suitable antimicrobial treatment not forgetting to link the laboratory-based phenotype antibiogram to the genomic data to understand which ARGs are in circulation and which represent a threat to the medical community (McArthur and Wright, 2015). Due to the fact that the Serratia species have a high number of efflux systems, no matter how many times a new antibiotic molecule is generated, resistance will persist. Therefore, new strategies for generating effective treatments have to take into account other targets, for example, inhibiting the expression of regulatory genes of the efflux pumps.
Whole genome sequencing and metagenomics are opening the door to rapid genotype-based resistance diagnosis (Bertelli and Greub, 2013; Wyrsch et al., 2016) that in turn are helping in the rapid detection and effective treatment of bacterial infections.
Conclusion
Awareness of the increasing problem related to antibiotic resistance has been a great concern in public health in recent years. Combating this problem requires an understanding of the mechanisms, evolution and spread of ARGs and the manufacture of new drugs that can circumvent resistance. Members of the Serratia genus are gram-negative bacteria of the Enterobacteriacea family that have been isolated from various ecological niches, such as soil, water and hospitals. S. marcescens has emerged recently as an opportunistic pathogen associated mostly with nosocomial infections. Serratia spp. infections pose a major health problem due to their high multidrug resistance, mainly because of the high number of efflux pump genes present in their genomes. This makes it difficult to choose suitable treatments. Knowing the genomic composition of Serratia spp. and the genes that confer resistance, will allow timely treatments to be determined and avoid those antibiotics to which the bacterium has a natural or acquired resistance.
Author Contributions
LS-M performed the experiments, did the phylogenetic analysis, analyzed the data, and wrote the paper. PV performed the bioinformatic analysis. AC the final approval of the version of the manuscript to be published. RM-E conceived and designed the experiments, contributed in writing the paper, and gave the final approval for the publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by El Programa de Doctorado en Ciencias Biomédicas, Universidad Nacional Autónoma de México (UNAM) of which LS-M is a doctoral student. We are also grateful to Gabriela Delgado for her helpful discussions, to José Luis Méndez for his support in the laboratory essays and to Luis Fernando Espinosa for his technical support.
Funding. This research was funded by DGAPA-PAPIIT Grant numbers IN213816 and IN211814 from Universidad Nacional Autónoma de México and by CONACYT Grant numbers 100343 and 179133.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00828/full#supplementary-material
Venn diagram of the consensus core-genome of the 32 Serratia spp. genomes generated by COG, OMCL, and BDBH algorithms.
Pairwise core-genome average nucleotide identity values from OMCL clusters (cgANIb-OMCL). The colors in the heat map represent pairwise ANI values, with a gradient from light yellow (high identity) to red (low identity).
Venn analysis of the pan-genome of the 32 Serratia spp. genomes generated by COG and OMCL algorithms.
List of the 32 Serratia spp. genomes used in this study, source of isolation, geographic region, continent, NCBI genome reference number, and genome size.
Total number of ARGs and efflux pumps identified in the 32 Serratia genomes. Genes found on the chromosome in white color, genes found on plasmids in orange color, genes found on transposons in blue color, genes found on probable genomic islands in purple color, regulatory genes in pink color and genes with mutations that confer resistance in green color.
Plasmids and acquired ARGs of the 32 Serratia spp. genomes. β-lactam resistance genes: blue, aminoglycoside resistance genes: red, macrolide resistance gene: yellow, sulfamide/trimethoprim resistance gene: pink, quaternary ammonium compounds: green.
References
- Abebe-Akele F., Tisa L. S., Cooper V. S., Hatcher P. J., Abebe E., Thomas W. K. (2015). Genome sequence and comparative analysis of a putative entomopathogenic Serratia isolated from Caenorhabditis briggsae. 16:531. 10.1186/s12864-015-1697-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aendekerk S., Diggle S. P., Song Z., Høiby N., Cornelis P., Williams P., et al. (2005). The MexGHI-OpmD multidrug efflux pump controls growth, antibiotic susceptibility and virulence in Pseudomonas aeruginosa via 4-quinolone-dependent cell-to-cell communication. 151 1113–1125. 10.1099/mic.0.27631-0 [DOI] [PubMed] [Google Scholar]
- Aldema M. L., McMurry L. M., Walmsley A. R., Levy S. B. (1996). Purification of the Tn10-specified tetracycline efflux antiporter TetA in a native state as a polyhistidine fusion protein. 19 187–195. 10.1046/j.1365-2958.1996.359886.x [DOI] [PubMed] [Google Scholar]
- Allen H. K., Moe L. A., Rodbumrer J., Gaarder A., Handelsman J. (2009). Functional metagenomics reveals diverse β-lactamases in a remote Alaskan soil. 3 243–251. 10.1038/ismej.2008.86 [DOI] [PubMed] [Google Scholar]
- Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. (1990). Basic local alignment search tool. 215 403–410. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- Andersson D. I., Hughes D. (2010). Antibiotic resistance and its costs: it is possible to reverse resistance? 8 260–271. 10.1038/nrmicro2319 [DOI] [PubMed] [Google Scholar]
- Begic S., Worobec E. (2008). Characterization of the Serratia marcescens SdeCDE multidrug efflux pump studied via gene knockout mutagenesis. 54 411–416. 10.1139/w08-019 [DOI] [PubMed] [Google Scholar]
- Berglund B. (2015). Environmental dissemination of antibiotic resistance genes and correlation to anthropogenic contamination with antibiotics. 5:28564. 10.3402/iee.v5.28564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard R., El Ghachi M., Mengin-Lecreulx D., Chippaux M., Denizot F. (2005). BcrC from Bacillus subtilis acts as an undecaprenyl pyrophosphate phosphatase in bacitracin resistance. 280 28852–28857. 10.1074/jbc.M413750200 [DOI] [PubMed] [Google Scholar]
- Bertelli C., Greub G. (2013). Rapid bacterial genome sequencing: methods and applications in clinical microbiology. 19 803–813. 10.1111/1469-0691.12217 [DOI] [PubMed] [Google Scholar]
- Bertelli C., Laird M. R., Williams K. P., Simon Fraser University Research Computing Group Lau B. Y., Hoad G. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. 45 W30–W35. 10.1093/nar/gkx343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand X., Dowzicky M. J. (2012). Antimicrobial susceptibility among Gram-negative isolates collected from Intensive Care Units in North America, Europe, the Asia-Pacific Rim, Latin America, the Middle East, and Africa between 2004 and 2009 as part of the tigecycline evaluation and surveillance trial. 34 124–137. 10.1016/j.clinthera.2011.11.023 [DOI] [PubMed] [Google Scholar]
- Blair J. M. A., Webber M. A., Baylay A. J., Ogbolu D. O., Piddock L. J. V. (2014). Molecular mechanisms of antibiotic resistance. 13 42–51. 10.1038/nrmicro3380 [DOI] [PubMed] [Google Scholar]
- Bojorquez D., Belei M., Delira S. F., Sholly S., Mead J., Tolmasky M. E. (1998). Characterization of OXA-9, a beta-lactamase encoded by the multiresistance transposon Tn1331. 44 483–491. [PubMed] [Google Scholar]
- Bradford P. A. (2001). Extended-Spectrum β-Lactamases in the 21st Century: characterization, epidemiology, and detection of this important resistance threat. 14 933–951. 10.1128/CMR.14.4.933-951.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D. G., Swanson J. K., Allen C. (2007). Two host-induced Ralstonia solanacearum genes, acrA and dinF, encode multidrug efflux pumps and contribute to bacterial wilt virulence. 73 2777–2786. 10.1128/AEM.00984-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruen T. C., Philippe H., Bryant D. (2006). A simple and robust statistical test for detecting the presence of recombination. 172 2665–2681. 10.1534/genetics.105.048975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush K., Jacoby G. A. (2010). Updated functional classification of β-lactamases. 54 969–976. 10.1128/AAC.01009-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., et al. (2009). BLAST+: architecture and applications. 10:421. 10.1186/1471-2105-10-421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Kuroda T., Huda M. N., Mizushima T., Tsuchiya T. (2003). An RND-type multidrug efflux pump SdeXY from Serratia marcescens. 52 176–179. 10.1093/jac/dkg308 [DOI] [PubMed] [Google Scholar]
- Chiang P. C., Wu T. L., Kuo A. J., Huang Y. C., Chung T. Y., Lin C. S., et al. (2013). Outbreak of Serratia marcescens postsurgical bloodstream infection due to contaminated intravenous pain control fluids. 17 e718–e722. 10.1016/j.ijid.2013.02.012 [DOI] [PubMed] [Google Scholar]
- Chung Y. J., Saier M. H. (2002). Overexpression of the Escherichia coli sugE gene confers resistance to a narrow range of quaternary ammonium compounds. 184 2543–2545. 10.1128/JB.184.9.2543-2545.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloeckaert A., Zygmunt M. S., Doublet B. (2017). Editorial: genetics of acquired antimicrobial resistance in animal and zoonotic pathogens. 8:2428. 10.3389/fmicb.2017.02428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras-Moreira B., Vinuesa P. (2013). GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. 79 7696–7701. 10.1128/AEM.02411-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling A. E., Mau B., Perna N. T. (2010). Progressive Mauve: multiple genome alignment with gene gain, loss and rearrangement. 5:e11147. 10.1371/journal.pone.0011147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J., Davies D. (2010). Origins and evolution of antibiotic resistance. 74 417–433. 10.1128/MMBR.00016-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawczynski K., Proquitté H., Roedel J., Edel B., Pfeifer Y., Hoye H., et al. (2016). Intensified colonisation screening according to the recommendations of the German Commission for Hospital Hygiene and Infectious Diseases Prevention (KRINKO): identification and containment of a Serratia marcescens outbreak in the neonatal Intensive Care Unit, Jena, Germany, 2013–2014. 44 739–746. 10.1007/s15010-016-0922-y [DOI] [PubMed] [Google Scholar]
- D’Costa V. M., McGrann K. M., Hughes D. W., Wright G. D. (2006). Sampling the antibiotic resistome. 311 374–377. 10.1126/science.1120800 [DOI] [PubMed] [Google Scholar]
- Deng Y., Bao X., Ji L., Chen L., Liu J., Miao J., et al. (2015). Resistance integrons: class 1 2 and 3 integrons. 14:45. 10.1186/s12941-015-0100-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta C., Paul S. (2012). Microbial lifestyle and genome signatures. 13 153–162. 10.2174/138920212799860698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ee R., Lim Y.-L., Yin W.-F., See-Too W.-S., Roberts R. J., Chan K.-G. (2016). Novel methyltransferase recognition motif identified in Chania multitudinisentens RB-25T gen. nov., sp. nov. 7:1362 10.3389/fmicb.2016.01362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhart S., Saborowski F., Krakau M., Scherholz-Schlösser G., Heyer I., Exner M. (2003). Severe Serratia liquefaciens sepsis following vitamin C infusion treatment by a naturopathic practitioner. 41 3986–3988. 10.1128/JCM.41.8.3986-3988.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández L., Hancock R. E. W. (2012). Adaptive and mutational resistance: role of porins and efflux pumps in drug resistance. 25 661–681. 10.1128/CMR.00043-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick A. W. P., Llabrés S., Neuberger A., Blaza J. N., Bai X.-C., Okada U., et al. (2017). Structure of the MacAB–TolC ABC-type tripartite multidrug efflux pump. 2:17070. 10.1038/nmicrobiol.2017.70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa H., Tsay J. T., Jackowski S., Takamura Y., Rock C. O. (1993). Thiolactomycin resistance in Escherichia coli is associated with the multidrug resistance efflux pump encoded by emrAB. 175 3723–3729. 10.1128/jb.175.12.3723-3729.1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gower J. (1998). “Principal coordinates analysis,” in , eds Armitage P., Coulton T. (Hoboken, NJ: John Wiley and Sons, Inc; ), 5 3514–3518. [Google Scholar]
- Grohskopf L. A., Roth V. R., Feikin D. R., Arduino M. J., Carson L. A., Tokars J. I., et al. (2001). Serratia liquefaciens bloodstream infections from contamination of epoetin alfa at a hemodialysis center. 3 1491–1497. 10.1056/NEJM200105173442001 [DOI] [PubMed] [Google Scholar]
- Gupta N., Hocevar S. N., Moulton-Meissner H. A., Stevens K. M., McIntyre M. G., Jensen B., et al. (2014). Outbreak of Serratia marcescens bloodstream infections in patients receiving parenteral nutrition prepared by a compounding pharmacy. 59 1–8. 10.1093/cid/ciu218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta N., Limbago B. M., Patel J. B., Kallen A. J. (2011). Carbapenem-Resistant Enterobacteriaceae: epidemiology and prevention. 53 60–67. 10.1093/cid/cir202 [DOI] [PubMed] [Google Scholar]
- Hall T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. 41 95–98. [Google Scholar]
- Harris S., Cormican M., Cummins E. (2013). Risk ranking of antimicrobials in the aquatic environment from human consumption: an Irish case study. 19 1264–1284. 10.1080/10807039.2012.713826 [DOI] [Google Scholar]
- Heng J., Zhao Y., Liu M., Liu Y., Fan J., Wang X., et al. (2015). Substrate-bound structure of the E. coli multidrug resistance transporter MdfA. 25 1060–1073. 10.1038/cr.2015.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hervé B., Chomali M., Gutiérrez C., Luna M., Rivas J., Blamey R., et al. (2015). Outbreak due to Serratia marcescens associated with intrinsic contamination of aqueous chlorhexidine. 32 517–522. 10.4067/S0716-10182015000600004 [DOI] [PubMed] [Google Scholar]
- Hopkins K. L., Davies R. H., Threlfall E. J. (2005). Mechanisms of quinolone resistance in Escherichia coli and Salmonella: recent developments. 25 358–373. 10.1016/j.ijantimicag.2005.02.006 [DOI] [PubMed] [Google Scholar]
- Hu Y., Zhu Y., Ma Y., Liu F., Lu N., Yang X., et al. (2015). Genomic insights into intrinsic and acquired drug resistance mechanisms in Achromobacter xylosoxidans. 59 1152–1161. 10.1128/AAC.04260-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iguchi A., Nagaya Y., Pradel E., Ooka T., Ogura Y., Katsura K., et al. (2014). Genome evolution and plasticity of Serratia marcescens, an important multidrug-resistant nosocomial pathogen. 6 2096–2110. 10.1093/gbe/evu160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iredell J., Brown J., Tagg K. (2016). Antibiotic resistance in Enterobacteriaceae: mechanisms and clinical implications. 352:h6420. 10.1136/bmj.h6420 [DOI] [PubMed] [Google Scholar]
- Jacobsen A., Hendriksen R. S., Aaresturp F. M., Ussery D. W., Friis C. (2011). The Salmonella enterica pan-genome. 62 487–504. 10.1007/s00248-011-9880-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacoby G. A. (2009). AmpC?β-lactamases Clin. 22 161–182. 10.1128/CMR.00036-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson T. J., Siek K. E., Johnson S. J., Nolan L. K. (2006). DNA sequence of a ColV plasmid and prevalence of selected plasmid-encoded virulence genes among avian Escherichia coli strains. 188 745–758. 10.1128/JB.188.2.745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knobler S. L. (2003). “Forum on emerging infections. Factors contributing to the emergence of resistance,” in , eds Knobler S. L., Lemon S. M., Najafi M., et al. (Washington DC, National Academies Press, US; ), 5. 10.17226/10651 [DOI] [Google Scholar]
- Kumar A., Worobec E. A. (2005). Cloning, sequencing, and characterization of the SdeAB multidrug efflux pump of Serratia marcescens. 49 1495–1501. 10.1128/AAC.49.4.1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Nei M., Dudley J., Tamura K. (2008). MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. 9 299–306. 10.1093/bib/bbn017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak Y., Khan A. R., Shin J. H. (2015). Genome sequence of Serratia nematodiphila DSM 21420T, a symbiotic bacterium from entomopathogenic nematode. 193 1–2. 10.1016/j.jbiotec.2014.11.002 [DOI] [PubMed] [Google Scholar]
- Lamelas A., Gosalbes M. J., Manzano-Marín A., Peretó J., Moya A., Latorre A. (2011). Serratia symbiotica from the Aphid Cinara cedri: a missing link from facultative to obligate insect endosymbiont. 7:e1002357. 10.1371/journal.pgen.1002357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P., Kwok A. H. Y., Jiang J., Ran T., Xu D., Wang W., et al. (2015). Comparative genome analyses of Serratia marcescens FS14 reveals its high antagonistic potential. 10:e0123061. 10.1371/journal.pone.0123061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q. Y., Tsai Y. L., Liu M. C., Lin W. C., Hsueh P. R., Liaw S. J. (2014). Serratia marcescens arn, a PhoP-regulated locus necessary for polymyxin B resistance. 58 5181–5190. 10.1128/AAC.00013-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou B. H., Duh R. W., Lin Y. T., Lauderdale T. L. Y. Taiwan Surveillance of Antimicrobial Resistance (TSAR) Hospitals (2014). A multicenter surveillance of antimicrobial resistance in Serratia marcescens in Taiwan. 47 387–393. 10.1016/j.jmii.2013.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D., Zhang L. P., Huang S. F., Wang Z., Chen P., Wang H., et al. (2011). Outbreak of Serratia marcescens infection due to contamination of multiple-dose vial of heparin-saline solution used to flush deep venous catheters or peripheral trocars. 77 175–176. 10.1016/j.jhin.2010.10.003 [DOI] [PubMed] [Google Scholar]
- Liu W., Fang L., Li M., Li S., Guo S., Luo R., et al. (2012). Comparative genomics of Mycoplasma: analysis of conserved essential genes and diversity of the pan-genome. 7:e35698. 10.1371/journal.pone.0035698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S., Zgurskaya H. I. (2012). Role of ATP binding and hydrolysis in assembly of MacAB–TolC macrolide transporter. 86 1132–1143. 10.1111/mmi.12046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDougall C. (2011). Beyond susceptible and resistant, Part I: treatment of infections due to Gram-negative organisms with inducible β-lactamases. 16 23–30. 10.1056/NEJMra1313875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahlen S. D. (2011). Serratia infections: from military experiments to current practice. 24 755–791. 10.1128/CMR.00017-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo T., Chen J., Minato Y., Ogawa W., Mizushima T., Kuroda T., et al. (2008). SmdAB, a heterodimeric ABC-type multidrug efflux pump, in Serratia marcescens. 190 648–654. 10.1128/JB.01513-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur A. G., Waglechner N., Nizam F., Yan A., Azad M. A., Baylay A. J., et al. (2013). The comprehensive antibiotic resistance database. 57 3348–3357. 10.1128/AAC.00419-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur A. G., Wright G. D. (2015). Bioinformatics of antimicrobial resistance in the age of molecular epidemiology. 27 45–50. 10.1016/j.mib.2015.07.004 [DOI] [PubMed] [Google Scholar]
- Merkier A. K., Rodríguez M. C., Togneri A., Brengi S., Osuna C., Pichel M., et al. (2013). Outbreak of a cluster with epidemic behavior due to Serratia marcescens after colistin administration in a hospital setting. 51 2295–2302. 10.1128/JCM.03280-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mima T., Sekiya H., Mizushima T., Kuroda T., Tsuchiya T. (2005). Gene cloning and properties of the RND-type multidrug efflux pumps MexPQ-OpmE and MexMN-OprM from Pseudomonas aeruginosa. 49 999–1002. 10.1111/j.1348-0421.2005.tb03696.x [DOI] [PubMed] [Google Scholar]
- Minato Y., Shahcheraghi F., Ogawa W., Kuroda T., Tsuchiya T. (2008). Functional gene cloning and characterization of the SsmE multidrug efflux pump from Serratia marcescens. 31 516–519. 10.1248/bpb.31.516 [DOI] [PubMed] [Google Scholar]
- Morillo Á., González V., Aguayo J., Carreño C., Torres M. J., Jarana D., et al. (2016). A six-month Serratia marcescens outbreak in a neonatal Intensive Care Unit. 34 645–651. 10.1016/j.eimc.2016.01.006 [DOI] [PubMed] [Google Scholar]
- Munita J. M., Arias C. A. (2016). Mechanisms of antibiotic resistance. 4 1–37. 10.1128/microbiolspec.VMBF-0016-2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L.-T., Schmidt H. A., von Haeseler A., Minh B. Q. (2015). IQ-TREE: a fast and effective Stochastic algorithm for estimating Maximum-Likelihood phylogenies. 32 268–274. 10.1093/molbev/msu300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olaitan A. O., Morand S., Rolain J. M. (2014). Mechanisms of polymyxin resistance: acquired and intrinsic resistance in bacteria. 5:643 10.3389/fmicb.2014.00643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson D. L., Bonomo R. A. (2005). Extended-Spectrum Beta-lactamases: a clinical update. 18 657–686. 10.1128/CMR.18.4.657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson W. R. (2013). An introduction to sequence similarity (“homology”) searching. 42 3.1.1–3.1.8. 10.1002/0471250953.bi0301s42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piddock L. (2006). Multidrug-resistance efflux pumps? not just for resistance. 4 629–636. 10.1038/nrmicro1464 [DOI] [PubMed] [Google Scholar]
- Potrykus J., Wegrzyn G. (2001). Chloramphenicol-sensitive Escherichia coli strain expressing the chloramphenicol acetyltransferase (cat) gene. Antimicrob. Agents Chemother. 45 3610–3612. 10.1128/AAC.45.12.3610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rather P. N., Munayyer H., Mann P. A., Hare R. S., Miller G. H., Shawt K. J. (1992). Genetic analysis of bacterial acetyltransferases: identification of amino acids determining the specificities of the aminoglycoside 6’-N-acetyltransferase lb and Ila proteins. 174 3196–3203. 10.1128/jb.174.10.3196-3203.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riesenfeld C. S., Goodman R. M., Handelsman J. (2004). Uncultured soil bacteria are a reservoir of new antibiotic resistance genes. 6 981–989. 10.1111/j.1462-2920.2004.00664.x [DOI] [PubMed] [Google Scholar]
- Rosenberg E. Y., Ma D., Nikaido H. (2000). AcrD of Escherichia coli is an aminoglycoside efflux pump. 182 1754–1756. 10.1128/JB.182.6.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouli L., Merhej V., Fournier P.-E., Raoult D. (2015). The bacterial pangenome as a new tool for analyzing pathogenic bacteria. 7 72–85. 10.1016/j.nmni.2015.06.00 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandner-Miranda L., Vinuesa P., Soberón-Chávez G., Morales-Espinosa R. (2016). Complete genome sequence of Serratia marcescens SmUNAM836, a nonpigmented multidrug-resistant strain isolated from a Mexican patient with obstructive pulmonary disease. 4:e01417–15. 10.1128/genomeA.01417-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz S., Kehrenberg C., Doublet B., Cloeckaert A. (2004). Molecular basis of bacterial resistance to chloramphenicol and florfenicol. 28 519–542. 10.1016/j.femsre.2004.04.001 [DOI] [PubMed] [Google Scholar]
- Shahcheraghi F., Minato Y., Chen J., Mizushima T., Ogawa W., Kuroda T., et al. (2007). Molecular cloning and characterization of a multidrug efflux pump, SmfY, from Serratia marcescens. 30 798–800. 10.1248/bpb.30.798 [DOI] [PubMed] [Google Scholar]
- Shaikh S., Fatima J., Shakil S., Rizvi S. M. D., Kamal M. A. (2015). Antibiotic resistance and extended spectrum beta-lactamase: types, epidemiology and treatment. 22 90–101. 10.1016/j.sjbs.2014.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanks R. M. Q., Davra V. R., Romanowski E. G., Brothers K. M., Stella N. A., Godboley D., et al. (2013). An eye to a kill: using predatory bacteria to control Gram-negative pathogens associated with ocular infections. 8:e66723. 10.1371/journal.pone.0066723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw K. J., Rather P. N., Hare R. S., Miller G. H. (1993). Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. 57 138–163. 10.1016/j.abb.2004.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw K. J., Rather P. N., Sabatelli F. J., Mann P., Munayyer H., Mierzwa R., et al. (1992). Characterization of the chromosomal aac(6’)-Ic gene from Serratia marcescens. 36 1447–1455. 10.1128/aac.36.7.1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. 7:539. 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siguier P., Perochon J., Lestrade L., Mahillon J., Chandler M. (2006). ISfinder: the reference centre for bacterial insertion sequences. 34 32–36. 10.1093/nar/gkj014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šiširak M., Hukić M. (2013). An outbreak of multidrug-resistant Serratia marcescens: the importance of continuous monitoring of nosocomial infections. 42 25–31. 10.5644/ama2006-124.67 [DOI] [PubMed] [Google Scholar]
- Sunenshine R. H., Tan E. T., Terashita D. M., Jensen B. J., Kacica M. A., Sickbert-Bennett E. E., et al. (2007). A multistate outbreak of Serratia marcescens bloodstream infection associated with contaminated intravenous magnesium sulfate from a compounding pharmacy. 45 527–533. 10.1086/520664 [DOI] [PubMed] [Google Scholar]
- Takahata S., Ida T., Hiraishi T., Sakakibara S. (2010). Molecular mechanisms of fosfomycin resistance in clinical isolates of Escherichia coli. 35 333–337. 10.1016/j.ijantimicag.2009.11.011 [DOI] [PubMed] [Google Scholar]
- Tettelin H., Riley D., Cattuto C., Medini D. (2008). Comparative genomics: the bacterial pan-genome. 12 472–477. 10.1016/j.mib.2008.09.006 [DOI] [PubMed] [Google Scholar]
- Thompson C. C., Vicente A. C., Souza R. C., Vasconcelos A. T., Vesth T., Alves N., Jr., et al. (2009). Genomic taxonomy of vibrios. 9:258. 10.1186/1471-2148-9-258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hoek A. H., Mevius D., Guerra B., Mullany P., Roberts A. P., Aarts H. J. M. (2011). Acquired antibiotic resistance genes: an overview. 2:203 10.3389/fmicb.2011.00203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedantam G., Guay G. G., Austria N. E., Doktor S. Z., Nichols B. P. (1998). Characterization of mutations contributing to sulfathiazole resistance in Escherichia coli. 42 88–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco C., Rodríguez-Martínez J. M., Briales A., Díaz de Alba P., Calvo J., Pascual A. (2009). Smaqnr, a new chromosome-encoded quinolone resistance determinant in Serratia marcescens. 65 239–242. 10.1093/jac/dkp424 [DOI] [PubMed] [Google Scholar]
- Vetter L., Schuepfer G., Kuster S. P., Rossi M. (2016). A hospital-wide outbreak of Serratia marcescens, and Ishikawa’s “Fishbone” analysis to support outbreak control. 25 1–7. 10.1097/QMH.0000000000000078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinuesa P., Ochoa-Sánchez L. E., Contreras-Moreira B. (2018). GET_PHYLOMARKERS, a software package to select optimal orthologous clusters for phylogenomics and inferring pan-genome phylogenies, used for a critical geno-taxonomic revision of the genus Stenotrophomonas. 9:771 10.3389/fmicb.2018.00771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinuesa P., Contreras-Moreira B. (2015). “Robust identification of orthologues and paralogues for microbial pan-genomics using GET_HOMOLOGUES: a case study of pIncA/C Plasmids,” in , eds Mengoni A., Galardini M., Fondi M. M. (New York, NY: Humana Press; ). [DOI] [PubMed] [Google Scholar]
- Wang D., Hou W., Chen J., Mou Y., Yang L. J., Yang L. Q., et al. (2014). Characterization of the blaKPC-2 and blaKPC-3 genes and the novel blaKPC-15 gene in Klebsiella pneumonia. 63 981–987. 10.1099/jmm.0.073841-0 [DOI] [PubMed] [Google Scholar]
- Webber M., Piddock L. J. (2001). Quinolone resistance in Escherichia coli. 32 275–284. 10.1051/vetres:2001124 [DOI] [PubMed] [Google Scholar]
- Weigel L. M., Steward C. D., Tenover F. C. (1998). gyrA mutations associated with fluoroquinolone resistance in eight species of Enterobacteriaceae. 42 2661–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyenberg G., Huggins P. M., Schardl C. L., Howe D. K., Yoshida R. (2014). kdetrees: non-parametric estimation of phylogenetic tree distributions. 30 2280–2287. 10.1093/bioinformatics/btu258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y. M., Hsu P. C., Yang C. C., Chang H. J., Ye J. J., Huang C. T., et al. (2013). Serratia marcescens meningitis: epidemiology, prognostic factors and treatment outcomes. 46 259–265. 10.1016/j.jmii.2012.07.006 [DOI] [PubMed] [Google Scholar]
- Wyrsch E. R., Chowdhury P. R., Chapman T. A., Charles I. G., Hammond J. M., Djordjevic S. P. (2016). Genomic microbial epidemiology is needed to comprehend the global problem of antibiotic resistance and to improve pathogen diagnosis. 7:843. 10.3389/fmicb.2016.00843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X., Sun Q., Liu W., Yin X., Pei G., Wang Y., et al. (2016). Complete genome sequence of Serratia rubidaea isolated in China. 4:e00283–16 10.1128/genomeA.00283-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H., Bogaki M., Nakamura M., Nakamura S. (1990). Quinolone resistance-determining region in the DNA gyrase gyrA gene of Escherichia coli. 34 1271–1272. 10.1128/AAC.34.6.1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zankari E., Hasman H., Cosentino S., Vestergaard M., Rasmussen S., Lund O., et al. (2012). Identification of acquired antimicrobial resistance genes. 67 2640–2644. 10.1093/jac/dks261 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Venn diagram of the consensus core-genome of the 32 Serratia spp. genomes generated by COG, OMCL, and BDBH algorithms.
Pairwise core-genome average nucleotide identity values from OMCL clusters (cgANIb-OMCL). The colors in the heat map represent pairwise ANI values, with a gradient from light yellow (high identity) to red (low identity).
Venn analysis of the pan-genome of the 32 Serratia spp. genomes generated by COG and OMCL algorithms.
List of the 32 Serratia spp. genomes used in this study, source of isolation, geographic region, continent, NCBI genome reference number, and genome size.
Total number of ARGs and efflux pumps identified in the 32 Serratia genomes. Genes found on the chromosome in white color, genes found on plasmids in orange color, genes found on transposons in blue color, genes found on probable genomic islands in purple color, regulatory genes in pink color and genes with mutations that confer resistance in green color.
Plasmids and acquired ARGs of the 32 Serratia spp. genomes. β-lactam resistance genes: blue, aminoglycoside resistance genes: red, macrolide resistance gene: yellow, sulfamide/trimethoprim resistance gene: pink, quaternary ammonium compounds: green.