

Abstract
Early growth response 3 (Egr3) is an immediate early gene (IEG) that is regulated downstream of a cascade of genes associated with risk for psychiatric disorders, and dysfunction of Egr3 itself has been implicated in schizophrenia, bipolar disorder, and depression. As an activity-dependent transcription factor, EGR3 is poised to regulate the neuronal expression of target genes in response to environmental events. In the current study, we sought to identify a downstream target of EGR3 with the goal of further elucidating genes in this biological pathway relevant for psychiatric illness risk. We used electroconvulsive stimulation (ECS) to induce high-level expression of IEGs in the brain, and conducted expression microarray to identify genes differentially regulated in the hippocampus of Egr3-deficient (-/-) mice compared to their wildtype (WT) littermates. Our results replicated previous work showing that ECS induces high-level expression of the brain-derived neurotrophic factor (Bdnf) in the hippocampus of WT mice. However, we found that this induction is absent in Egr3-/- mice. Quantitative real-time PCR (qRT-PCR) validated the microarray results (performed in males) and replicated the findings in two separate cohorts of female mice. Follow-up studies of activity-dependent Bdnf exons demonstrated that ECS-induced expression of both exons IV and VI requires Egr3. In situ hybridization demonstrated high-level cellular expression of Bdnf in the hippocampal dentate gyrus following ECS in WT, but not Egr3-/-, mice. Bdnf promoter analysis revealed eight putative EGR3 binding sites in the Bdnf promoter, suggesting a mechanism through which EGR3 may directly regulate Bdnf gene expression. These findings do not appear to result from a defect in the development of hippocampal neurons in Egr3-/- mice, as cell counts in tissue sections stained with anti-NeuN antibodies, a neuron-specific marker, did not differ between Egr3-/- and WT mice. In addition, Sholl analysis and counts of dendritic spines in golgi-stained hippocampal sections revealed no difference in dendritic morphology or synaptic spine density in Egr3-/-, compared to WT, mice. These findings indicate that Egr3 is required for ECS-induced expression of Bdnf in the hippocampus and suggest that Bdnf may be a downstream gene in our previously identified biologically pathway for psychiatric illness susceptibility.
Keywords: electroconvulsive therapy, immediate early genes, early growth response 3, brain-derived neurotrophic factor, schizophrenia, psychosis treatment
Introduction
The risk to develop neuropsychiatric illnesses is determined by both genetic and environmental factors. We have hypothesized that immediate early gene (IEG) transcription factors are poised to modulate this dual contribution, as they are rapidly activated in the brain in response to environmental stimuli and, in turn, influence numerous neurobiological processes that are dysfunctional in the brains of patients with mental illness (Gallitano-Mendel et al., 2007, 2008; Huentelman et al., 2015). These include memory formation and synaptic plasticity (Gallitano-Mendel et al., 2007; Forbes et al., 2009), myelination (Jessen and Mirsky, 2002; Davis et al., 2003; Dwork et al., 2007), vascularization (Mechtcheriakova et al., 1999; Fahmy et al., 2003; Hanson and Gottesman, 2005), and growth factor response (Moises et al., 2002; Fahmy and Khachigian, 2007; Zakharyan et al., 2014). Our prior work has focused on investigating the functions of the IEG transcription factor early growth response 3 (Egr3), as it is regulated downstream of numerous proteins that are associated with risk for neuropsychiatric disorders (Gallitano-Mendel et al., 2007, 2008; Huentelman et al., 2015). We have previously reported that Egr3-deficient (-/-) mice have behavioral abnormalities consistent with animal models of mental illness, including schizophrenia and bipolar disorder, that are rescued by the antipsychotic medications used to treat these disorders (Gallitano-Mendel et al., 2007, 2008). EGR3 has been associated with risk for schizophrenia in Japanese, Korean, Han Chinese, and US populations of European descent (Yamada et al., 2007; Kim et al., 2010; Zhang et al., 2012; Huentelman et al., 2015) and levels of EGR3 are reduced in the brains of schizophrenia patients (Mexal et al., 2005; Yamada et al., 2007). Recently, EGR3 was identified as a master regulator in a network of genes differentially expressed in the postmortem brains of bipolar disorder patients, compared with controls, in two independent cohorts (Pfaffenseller et al., 2016). In addition, although EGR3 was not identified within one of the 108 loci found to be associated with schizophrenia risk in the Psychiatric Genomics Consortium genome-wide association study (GWAS), numerous genes encoding proteins that regulate or are regulated by EGR3 do map to these schizophrenia-associated loci (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014; Marballi and Gallitano, 2018).
The few confirmed downstream target genes of EGR3 have also been implicated in neuropsychiatric disorders. These include glutamic acid decarboxylase A4 (GABRA4; Roberts et al., 2005, 2006), and activity-regulated cytoskeleton associated protein (Arc-Arg3.1; Li et al., 2005). In addition, our prior studies identified a specific deficit in GluN2B-containing N-methyl D-aspartate receptors (NMDARs) in the hippocampus of Egr3-deficient (-/-) mice, indicating a requirement of Egr3 for the normal function of this receptor (Gallitano-Mendel et al., 2007). Hypofunction of NMDARs is one of leading hypotheses for the etiology of schizophrenia (Javitt and Zukin, 1991; Olney et al., 1999). Indeed, GRIN2A, the gene encoding the NMDAR subunit GluN2A, is also located at one of the 108 schizophrenia-associated loci (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014).
Based on these prior studies, we hypothesized that other EGR3 target genes in the brain may be critical contributors to neuropsychiatric disorders. Since neuronal expression of IEGs is activity dependent, we used electroconvulsive seizure (ECS) to maximally activate IEGs, and compared hippocampal gene expression between mice lacking Egr3 and their wildtype (WT) littermates. This experimental approach is particularly relevant as ECS is an experimental model of electroconvulsive therapy (ECT), which remains one of the most effective treatments for severe mood and psychotic disorders. However, the mechanisms underlying the efficacy of ECT remain elusive.
Here we show that induction of brain-derived neurotrophic factor (Bdnf), a critical neurotrophin involved in a wide range of neurobiological functions, is significantly diminished or absent in the hippocampus of mice lacking Egr3. Dysfunction of BDNF has been implicated in numerous neuropsychiatric disorders (Autry and Monteggia, 2012), including bipolar disorder and schizophrenia (Durany et al., 2001). Like Egr3, Bdnf is also induced in the hippocampus following ECS, and this expression is associated with the effectiveness of antidepressant treatments in reversing mood disorder-like phenotypes in rodents (Altar et al., 2003; Adachi et al., 2008; Inta et al., 2013).
Materials and Methods
Mice
Previously generated Egr3-/- mice (Tourtellotte and Milbrandt, 1998) were backcrossed to C57BL/6 mice for greater than 20 generations. Studies were conducted on homozygous adult littermate progeny of heterozygote matings, and assigned as “matched pairs” at the time of weaning. Matched pairs were exposed to identical conditions for all studies. The term WT refers to +/+ littermates of Egr3-/- mice generated from these crossings. Male mice ages 6–12 months (n = 4) were utilized for the microarray and follow-up quantitative real-time PCR (qRT-PCR) study. Results were validated in two separate cohorts of female Egr3 mice. The first cohort included older adult females, ages 12–15 months, from the same C57BL/6 background (n = 4–5 per group). A second cohort of females, age 3.5–6 months (n = 4–5 per group), had a mixed background resulting from crossing the above Egr3 C57BL/6 background mice to a GENSAT reporter line in a mixed FVB/N and Swiss Webster background. Since these animals contain a bacterial artificial chromosome expressing EGFP, in addition to a mixed background, results were analyzed separately from other groups.
Animals were housed on a 12 h light/dark schedule with ad libitum access to food and water. All studies were performed in accordance with the University of Arizona, Institutional Animal Care and Use Committee (IACUC). This study was carried out in accordance with the recommendations of IACUC guidelines, IACUC. The protocol was approved by IACUC.
Electroconvulsive Stimulation and Tissue Collection
To anesthetize the corneas of all animals, 0.5% proparacaine hydrochloride ophthalmic solution (Akorn, Inc., Lake Forest, IL, United States) was applied 5 min prior to electroconvulsive stimulation (ECS). Male mice utilized in the microarray study underwent ECS without general anesthesia. Female mice used in the replication studies were anesthetized with isoflurane (VetOne, Boise, ID, United States) administered in an enclosed chamber at a flow rate of 0.5 mL/min in oxygen. Animals were removed from the chamber after 2 min of full anesthetization, transferred to room air to recover to a level of light anesthesia, and then administered electrical stimulation of 260 A for 1 ms duration and a pulsewidth of 0.3 mm, 1 ms. (Ugo Basile, Varese, Italy) via orbital electrodes. Mice were observed to undergo seizure, and were placed in their home cage to recover for one hour prior to sacrifice. Control animals remained in their home cages undisturbed.
Tissue Collection and RNA Isolation
Animals were sacrificed via isoflurane overdose, followed by decapitation. The brains were removed, rinsed in sterile ice-cold phosphate buffered saline (PBS), and hemisected along the central sulcus into right and left hemispheres for further studies to quantify both the expression of mRNA with qRT-PCR and in situ hybridization, respectively.
Hippocampal tissue was dissected and immediately placed in RNAlater (Ambion, Waltham, MA, United States). Tissue was transferred to 1.5-mL Eppendorf tubes, immediately placed on dry ice, and transferred to -80°C until further use. For male cohort microarray and follow-up qRT-PCR, RNA was isolated using TRIzol reagent (Life Technologies, Carlsbad, CA, United States) followed by phenol/chloroform extraction, chloroform extraction, and isopropanol precipitation, per the manufacturer’s protocol. RNA was resuspended in RNAse-free water and quantitated by spectrophotometry. RNA quality and concentration was validated by Agilent Bioanalyzer 2100 prior to microarray analysis and reverse transcription for qRT-PCR. An aliquot of the RNA samples was sent to the Microarray Resource Center, Yale/NIH Neuroscience Microarray Center (New Haven, CT, United States) for analysis using an Illumina Mouse WG6 v3.0 expression beadchip microarray. For female cohorts, RNA isolation was performed using TRI reagent (Sigma-Aldrich, St. Louis, MO, United States) and MagMaxTM Total RNA isolation kit (Ambion, Waltham, MA, United States) according to the manufacturer’s protocol, and quantified using the NanoDrop ND-1000 spectrophotometer (Thermo Scientific, Waltham, MA, United States).
Microarray Procedure and Analysis
Data analysis and quality control was initially performed using Gene Pattern (Reich et al., 2006), with normalization using the cubic spline method using the following settings (FDR < 0.05) to determine significantly differentially expressed genes between the WT and Egr3-/- groups 1 h following ECS. This was the timepoint at which we expected the most enrichment for EGR3 transcription factor targets, as Egr3 is maximally induced by ECS (O’Donovan et al., 1998). A parallel analysis using the Illumina Genome studio 2010 software was used to determine differentially expressed genes by comparing the same two groups using the following settings: background subtraction, quantile normalization, p < 0.05 (T-test). Finally, a list of common differentially expressed genes using both programs was generated. Complete microarray results will be published separately.
qRT-PCR
For qRT-PCR studies, mRNA was reverse transcribed into cDNA using a standard protocol (Maple et al., 2015), and used as a template for qRT-PCR using FastStart SYBR Green Master mix (Roche Applied Science, Indianapolis, IN, United States) on a 7500 Fast Real-Time PCR machine (ThermoFisher Scientific, Waltham, MA, United States). Each sample was amplified in triplicate for both Bdnf and the housekeeping gene phosphoglycerate kinase 1 (Pgk1). Pgk1 was selected for use as a housekeeping gene as one of the least changed genes across experimental groups in the microarray data and validated by qRT-PCR in all cohorts.
General Bdnf primers were used to assess overall levels of total Bdnf mRNA (Bdnf F, R) (Tallaksen-Greene et al., 2014). These primers target a region common to all 12 transcripts of Bdnf as revealed by NCBI primer blast analysis. For follow-up exon-specific studies, we focused on Bdnf exons IV and VI as removal of these exons in vivo significantly reduces BDNF protein levels in the mouse hippocampus (Maynard et al., 2016). In addition, Bdnf transcripts containing exon IV and VI are highly induced in vivo following ECS in the mouse frontal cortex (Martinowich et al., 2011). Exon specific primers for Bdnf exons IV and VI (Zheng and Wang, 2009) were used in our study. Fold changes in gene expression and data were plotted using the 2-ΔCT method (Le et al., 2013). Primer sequences used were as follows, Bdnf F: TGG CCC TGC GGA GGC TAA GT; Bdnf R: AGG GTG CTT CCG AGC CTT CCT; Pgk1 F: TGT TAG CGC AAG ATT CAG CTA GTG; Pgk1 R: CAG ACA AAT CCT GAT GCA GTA AAG AC; Bdnf exon IV F: CTC CGC CAT GCA ATT TCC AC; Bdnf exon VI F: GTG ACA ACA ATG TGA CTC CAC; and Bdnf exon IV/VI R: GCC TTC ATG CAA CCG AAG TA.
Prediction of EGR3 Binding Sites in Bdnf Gene Regulatory Regions
Bioinformatic identification of putative EGR binding sites in the promoter of the mouse Bdnf gene was carried out using the TFBIND1 (Tsunoda and Takagi, 1999) website that utilizes the transcription factor database TRANSFAC R. 3.3. Briefly, the nucleotide sequence 4000 bp upstream of the start ATG in the mouse Bdnf gene was exported from the UCSC genome browser2 using genome assembly GRCm38/mm10 and used to query the TFBIND site. This generated an output that showed all the putative binding sites for the EGR3 TF.
Golgi-Cox Preparation
Tissue was treated using the Golgi-Cox method, as previously described (Gibb and Kolb, 1998; Gallitano et al., 2016). Briefly, 10 animals (5 Egr3-/- and 5 WT) were decapitated under isoflurane and the brains were rapidly extracted. The brains were rinsed in 0.9% saline and immersed in Golgi-Cox solution for at least 14 days followed by 30% sucrose for at least 3 days. The solution was made in the laboratory from ingredients purchased from Sigma-Aldrich (St. Louis, MO, United States). Brains were then sectioned on a Vibratome at 200 μm and mounted on gelatin-coated slides.
Golgi-Cox Imaging and Analysis
Ten fully impregnated cells were selected in each region, from the hippocampal Cornu Ammonis (CA) regions CA1 and CA3, and the dentate gyrus (DG) of each animal, and were imaged in a z-stack throughout the thickness of the section using a brightfield microscope equipped with a digital camera (AmScope, Irvine, CA, United States). Using ImageJ (NIH) or Metamorph (Molecular Devices) software, several measures of dendritic arborization were obtained (Figure 3). The Sholl technique (Sholl, 1953) involves overlaying the neuron with a series of concentric circles (20 μm apart) and recording the number of dendritic processes intersecting each circle to a maximal distance of 380 μm. In addition, branching order was calculated for all cells. The primary dendrites originating from the soma are assigned a branch order of one, while dendritic processes originating from that dendrite are second-order branches, and each subsequent bifurcation is assigned a progressively higher branch order (van Pelt et al., 1986). Dendritic spine density and morphology were analyzed at 100x under oil immersion. Dendritic spines were counted in a sample of two random 20-μm-long segments in several distinct regions of each cell according to the classification of Lorente de No (1934). For CA1 and CA3, these regions were: the basal tuft (stratum oriens), the mid-point of the apical tuft (near the distal edge of the stratum radiatum), and the distal tip of the apical tuft (stratum lacunosum moleculare). In the DG, dendritic spines were counted in the inner molecular layer (IML), middle molecular layer (MML), and outer molecular layer (OML) as defined by dividing the length of the molecular layer into three equal parts.
FIGURE 3.
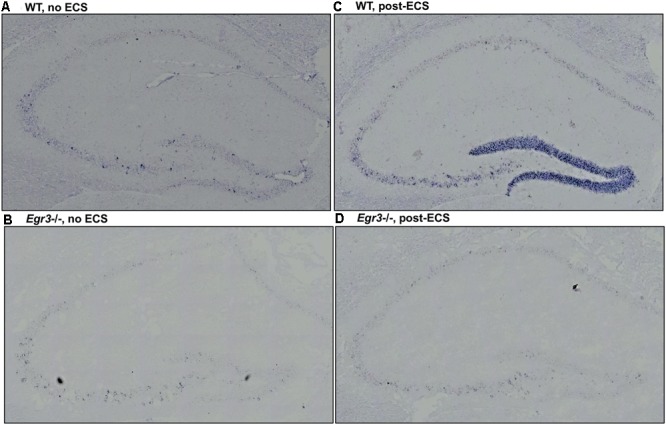
Bdnf induction in the hippocampal dentate gyrus requires Egr3. Representative tissue sections demonstrating in situ hybridization labeling of Bdnf mRNA expressing cells in the hippocampus of WT and Egr3–/– mice at baseline (A,B) and 1 h following ECS (C,D). (A) Bdnf is expressed sparsely in CA3, CA2, and the DG of WT mice at baseline. (C) ECS induces high-level Bdnf expression in the DG, and increased expression in CA3, CA2, and CA1 of WT mice. (B) Bdnf expression in Egr3–/– mice is limited to rare cells in the CA3 region of the hippocampus, with no clear expression above background in the DG, CA2, or CA1, at baseline. (D) ECS produces little to no increase in Bdnf expression in the DG, CA3, CA2, and CA1 regions of Egr3–/– mice.
Immunohistochemistry
Animals were sacrificed by isoflurane euthanasia and were perfused with PBS followed by fixation with 4% paraformaldehyde (PFA). Brains were harvested and were post-fixed in 4% PFA for 24 h, rinsed in Tris-buffered saline (TBS, pH 7.4), transferred into 30% sucrose, and stored at 4°C until saturated, rinsed, and stored in TBS at 4°C. Tissue was sectioned coronally at a thickness of 20 μm on a sliding microtome and stored until further processing in a cryopreservative solution.
Immunohistochemistry
Tissue was rinsed in TBS, 3x × 5 min. Endogenous peroxidase was quenched with 1% H2O2 × 10 min in a buffer/detergent solution (0.4% Triton-X in TBS). The reaction was stopped by rinsing the tissue 3x × 5 min in TBS. Tissue was blocked with 4% normal goat serum (Sigma-Aldrich, St. Louis, MO, United States) prepared in 0.4% Triton-X in TBS at room temperature (RT) for 2 h. Polyclonal rabbit primary antibody raised against neuronal nuclei, NeuN (1:1000, Millipore, Cat #ABN78) was prepared in 4% normal goat serum in 0.4% Triton-X in TBS and was incubated at RT for 1 h with a 24-h incubation at 4°C. A secondary biotinylated antibody raised against rabbit (1:1000, Vector Labs, #BA-1000) was prepared in 4% normal goat serum in 0.4% TBS-TX and was incubated with the tissue for 1 h at RT. Avidin–biotin complex (ABC; ThermoFisher Scientific, Waltham, MA, United States) was prepared at a concentration of 1:1000 in TBS. Tissue was incubated in ABC for 1 h at RT. Detection of the antigen was performed with 3,3′-diaminobenzidine (DAB) kit (ImmPACT DAB Peroxidase Substrate, Cat No. SK-4105, Vector Laboratories, Burlingame, CA, United States). Tissue sections were imaged with a Zeiss Imager M2 microscope using a 40x objective, photographed using an Axiocam 506 camera, and tiled together using Zen 2012 software (Zeiss Microscopy, Oberkochen, Germany).
Neurons were counted from defined anatomical regions of CA1 and CA3 pyramidal neurons, as well as both the suprapyramidal (DGsp) and infrapyramidal (DGip) blades of the DG. For each brain, neurons were counted from four sections spanning a total rostral–caudal distance of 200 μm including, and caudal to, coordinates: interaural, 1.74 mm; bregma, 2.06 mm (Paxinos and Franklin, 2001). The number of neurons within each analyzed region of the hippocampus was estimated using Abercrombie’s (1946) correction.
In Situ Hybridization
Tissue Preparation
Tissue was flash frozen in -40°C methylbutane and was sectioned coronally at 10 μm thickness with a cryostat (Leika, Buffalo Grove, IL, United States), mounted on super-charged slides and stored at -80°C. Tissue sections from matched experimental pairs of animals were mounted on a single slide to control for possible staining differences (Egr3-/- control, Egr3-/- ECS, WT control, and WT ECS).
Probe Generation
Mouse BDNF DNA plasmid (accession #X55573) was kindly donated by Dr. Stanley Watson, University of Michigan, Ann Arbor. The DNA sequence was validated by comparison to Bdnf reference sequence using FinchTV software (Geospiza, Inc., Seattle, WA, United States). Sense and antisense strands were generated by in vitro transcription with the AMBION Maxiscript T7 kit (Ambion, Waltham, MA, United States), labeled with digoxigenin RNA labeling mix (Roche, Basel, Switzerland), and purified with mini columns according to the manufacturer’s protocol. (Roche, Basel, Switzerland).
In Situ Hybridization
Tissue was fixed in 4% PFA at 4°C for 5 min., followed by a wash in 2x SSC for 2 min at RT. Tissue was acetylated in acetic anhydride (0.625%) for 10 min at RT, rinsed in sterilized water, and delipidized with a 1:1 mixture of acetone and methanol for 5 min. Tissue was rinsed in 2x SSC for 5 min, before application of the probe (300 ng) prepared in hybridization buffer (Sigma, St. Louis, MO, United States). The probe was applied directly to the mounted tissue (250 μL/slide) and was cover-slipped (FisherBrand, Fisher Scientific). Tissue was hybridized in a humid, sealed chamber with a mixture of 2x SSC and formamide overnight at 56°C. Stringency washes were performed on Day 2 to remove any non-specific binding (protocol available on request). Endogenous peroxidase was quenched with 2% hydrogen peroxide, and tissue was permeabilized with 1x SSC-Tween followed by rinsing in 0.1 M TBS (pH 7.4). The tissue was blocked with a mixture of 0.5% TSA blocking buffer (PerkinElmer, Waltham, MA, United States) and 5% normal sheep serum prepared in 0.1 M TBS. Anti-digoxigenin (Roche, Basel, Switzerland; 1:400 dilution in blocking buffer) was applied to the slides at RT for 2 h. Slides were washed in 0.1 M TBS-Tween 4x × 15 min at RT and the antibody was detected through alkaline phosphatase development, SIGMAFAST BCIP/NBT tablets (Sigma, St. Louis, MO, United States). Slides were imaged with bright-field microscopy (Axiovision, Zeiss, Oberkochen, Germany) at a magnification of 40x.
Statistical Analysis
All statistical analyses for qRT-PCR and in situ data were carried out using graphpad prism. In all cases, two-way analysis of variance (ANOVA) was performed followed by Tukey’s post hoc test, corrected for multiple comparisons, and significance was set at p < 0.05. Golgi-Cox impregnated granule cells and pyramidal cells were analyzed in independent ANOVAs. Sholl data were analyzed by repeated-measures ANOVA, while branching order and spine density measures used general factorial ANOVA, with genotype and hippocampal region (i.e., either CA1, CA3a/b, and CA3c or DGsp and DGip) as factors. The neuron density (number of neurons per unit area) was compared between WT and Egr3-/- brains using a 2 × 5 ANOVA with genotype and hippocampal region (i.e., CA1, CA3a/b, CA3c, DGsp, and DGip) as factors.
Results
Activity-Dependent Hippocampal Bdnf Expression Depends on Egr3
To identify genes regulated by Egr3 in the mouse hippocampus, we compared the complement of genes expressed in Egr3-/- mice to that of WT mice. Since Egr3 is an activity-dependent IEG, and thus expressed at low levels in the absence of a stimulus, we used ECS to maximally activate IEG expression in both Egr3-/- and WT mice, and compared levels of induced genes to those at baseline. We conducted an expression microarray to screen for genes that were differentially expressed in response to seizure in the hippocampus of male WT, compared to Egr3-/-, mice.
Results of the microarray showed 65 genes to be differentially expressed between WT and Egr3-/- mice following ECS. Of these, 40 genes were increased (greater than 1.5-fold), while 13 were decreased, in WT mice following ECS compared to Egr3-/- mice following ECS. Twelve genes were minimally changed across both groups (1–1.4-fold). Bdnf was the only growth factor among these 65 putative EGR3-dependent, ECS-induced genes. Complete results of the microarray will be published separately.
Bdnf was of particular interest to us, as it has been shown to contribute to the therapeutic effects of antidepressant treatments, including ECS, in rodent models (Altar et al., 2003; Adachi et al., 2008; Inta et al., 2013). Results of the expression microarray demonstrated that, in WT mice, ECS produced a 2.5-fold increase in Bdnf mRNA levels, measured 1 h following seizure, compared to baseline unstimulated controls. This induction is dependent upon Egr3, as Egr3-/- mice did not show a change in Bdnf mRNA expression following seizure (Figure 1A). A two-way ANOVA revealed a significant interaction of genotype and treatment [F(1,12) = 12.65, p = 0.004]. Post hoc analysis showed that WT mice had significantly higher levels of Bdnf induction after ECS compared to the untreated WT (p < 0.001) mice, while Egr3-/- mice did not show a significant increase in Bdnf expression following ECS compared to baseline. In addition, there was a significant difference in Bdnf levels between WT and Egr3-/- mice following ECS (p < 0.01).
FIGURE 1.
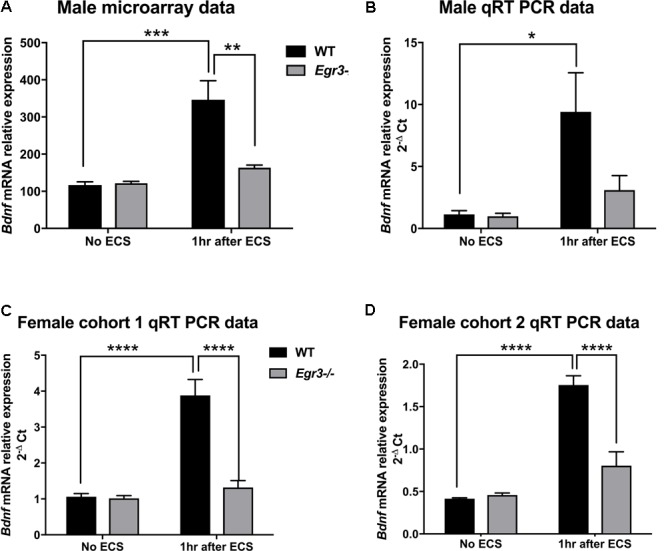
Electroconvulsive seizure (ECS)-induced hippocampal Bdnf expression is Egr3-dependent. Expression microarray analysis (A), and validation using qRT-PCR (B–D), of hippocampal Bdnf expression from WT, and Egr3–/–, mice at baseline (no ECS) and 1 h after ECS. Two-way ANOVAs showed significant interaction of genotype and ECS treatment on Bdnf expression in (A) male microarray data (p = 0.004) and qRT-PCR data in (C) female cohort 1 (p = 0.0005) and (D) female cohort 2 (p = 0.0003), and (B) significant effect of treatment in original male cohort (p = 0.0098; n = 4–5 animals/group; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001 controlled for multiple comparisons).
To validate these findings, we performed qRT-PCR from the RNA samples that were used for the expression microarray (Figure 1B). ECS induced a greater than six-fold increase in Bdnf expression in WT mice (compared to non-stimulated control), while it failed to induce a significant increase in Bdnf expression in the Egr3-/- mice. These results validate the microarray data. A main effect of treatment was observed using two-way ANOVA [F(1,12) = 9.393, p = 0.0098] and post hoc analysis demonstrated that ECS-treated WT mice had significantly higher levels of Bdnf after ECS compared to the untreated WT (p < 0.05). ECS did not result in a significant change in Bdnf expression in Egr3-/- mice (p = 0.8133).
To determine whether the requirement of Egr3 for activity-dependent induction of Bdnf is sex specific, we conducted ECS in female Egr3-/- and WT mice. Figure 1C shows that, as in male mice, ECS induced a significant increase (3.8-fold) in Bdnf mRNA in WT female mice, but did not result in a statistically significant increase in Egr3-/- mice. Cohort 1 (Figure 1C) showed a significant interaction of genotype and treatment [F(1,14) = 20.52, p = 0.0005] and post hoc analysis revealed significant elevations in Bdnf levels post ECS in the WT group compared to WT controls (p < 0.0001) and Egr3-/- ECS-treated mice (p < 0.0001), respectively.
To determine whether the effect was also seen in younger animals, we used a second cohort of female mice. Figure 1D shows a similar result to that seen in adult male, and older female, C57BL/6 mice, that ECS produces a fourfold increase in Bdnf expression in WT mice that is dependent upon Egr3. Two-way ANOVA demonstrated a significant interaction of genotype and treatment [F(1,13) = 23.85, p = 0.0003]. Post hoc analysis revealed significant elevations in Bdnf post ECS in the WT group compared to WT controls (p < 0.0001) and to Egr3-/- ECS-treated mice (p < 0.0001). No significant effect of ECS on Bdnf induction was observed in Egr3-/- mice in either group. These data suggest that Bdnf induction by ECS is dependent, at least in part, on Egr3.
ECS-Induced Exon IV and Exon VI Bdnf Expression Is Egr3 Dependent
Given that the Bdnf gene has multiple splice variants, we next examined whether Egr3 is required for ECS-induced expression of specific splice variants. This was of particular interest as ECS induces specific exon variants in the mouse brain in vivo (Martinowich et al., 2011). Martinowich et al. (2011) showed that mRNAs containing exon IV and exon VI are highly upregulated following ECS compared to untreated mice, and mice that lack exon IV fail to show induction of six splice variants following ECS in vivo (Martinowich et al., 2011). In addition, both exon IV and exon VI containing mRNAs maximally contribute to total BDNF protein levels in the mouse hippocampus (Maynard et al., 2016). Since exons IV and VI are required for expression of the remaining BDNF exons, we decided to evaluate levels of transcripts containing these two critical exons in WT and Egr3-/- mice following ECS compared with untreated controls.
Quantitative real-time PCR using exon specific primers showed that WT mice demonstrated a 10-fold increase in Bdnf exon IV expression, 1 h post-ECS that was absent in Egr3-/- mice (Figure 2A). Two-way ANOVA revealed a significant interaction of genotype and treatment [F(1,12) = 12.52, p = 0.0041]. Post hoc analysis indicated that WT mice had significantly higher levels of Bdnf exon IV mRNA induction after ECS compared to the untreated WT (p < 0.001) mice. Egr3-/- mice did not show a significant increase in Bdnf exon IV expression following ECS compared to baseline, but showed decreased Bdnf exon IV expression compared to WT mice following ECS (p < 0.01). Similar results were seen in both female cohorts. Female cohort 1 (Figure 2B) WT mice exhibited a sevenfold induction in Bdnf exon IV post ECS compared to baseline. Two-way ANOVA showed a significant interaction of genotype and treatment [F(1,14) = 14.46, p = 0.0019]. Post hoc analysis demonstrated that WT mice had a significantly greater induction of Bdnf exon IV following ECS compared to baseline (p < 0.0001) and compared to Egr3-/- mice post ECS (p < 0.001). WT mice from the second female cohort demonstrated a ninefold induction in Bdnf exon IV post ECS compared to non-stimulated controls (Figure 2C). Two-way ANOVA revealed a significant interaction of genotype and treatment [F(1,13) = 30.24, p = 0.0001]. Post hoc analysis revealed WT mice showed a significant induction of Bdnf exon IV post ECS compared to baseline (p < 0.0001) and Egr3-/- mice that underwent ECS (p < 0.0001).
FIGURE 2.
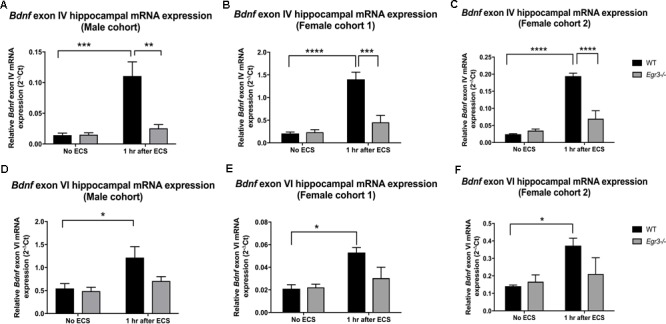
Electroconvulsive seizure (ECS)-mediated hippocampal induction of Bdnf exon IV and VI requires Egr3. Exon IV (A–C) and VI (D–F) hippocampal Bdnf expression determined using qRT PCR from WT, and Egr3–/–, mice at baseline (no ECS) and 1 h after ECS. Two-way ANOVAs showed significant interactions of genotype and ECS treatment on Bdnf exon IV expression in (A) original male cohort data (p = 0.0041), (B) female cohort 1 (p = 0.0019), and (C) female cohort 2 (p = 0.0001). Two-way ANOVAs showed significant effect of treatment on Bdnf exon VI expression in original (D) male cohort (p = 0.0099), (E) female cohort 1 (p = 0.0064), and (F) female cohort 2 (p = 0.0237; n = 4–5 animals/group; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001 controlled for multiple comparisons).
In comparison, Bdnf exon VI mRNA was induced 2.2-fold in male WT mice following ECS compared to baseline that was absent in Egr3-/- mice (Figure 2D). Two-way ANOVA showed a significant effect of treatment [F(1,12) = 9.361, p = 0.0099] and post hoc analysis revealed a significant induction of Bdnf exon VI in WT mice that underwent ECS vs. non-stimulated WT mice (p < 0.05). These data were replicated in both female cohorts. WT mice showed a 2.24- (Figure 2E) and a 2.64-fold (Figure 2F) induction post ECS compared to baseline, in the first and second female cohorts, respectively, that was absent in Egr3-/- mice. Two-way ANOVA revealed a significant effect of treatment in both female cohorts; female cohort 1 [F(1,14) = 10.26, p = 0.0064] and female cohort 2 [F(1,13) = 6.557, p = 0.0237]. Post hoc analysis revealed that significant induction of Bdnf exon VI in WT mice that underwent ECS compared to baseline (p < 0.05) in both female cohorts, while this effect was not observed in Egr3-/- mice.
ECS-Induced Bdnf Expression in Dentate Gyrus Requires Egr3
To identify the location of Bdnf expression in the hippocampus following ECS, and to determine in which regions it is dependent on EGR3, we conducted in situ hybridization to detect expression of Bdnf mRNA in hippocampal tissue sections. In situ hybridization histochemistry was performed on tissue sections from female Egr3-/- and WT mice at baseline, and 1-h post ECS. In WT mice, ECS resulted in a strong induction of Bdnf mRNA in the DG of the hippocampus 1 h after ECS (Figure 3C) compared with baseline, unstimulated controls (Figure 3A). Expression of Bdnf in the hippocampus of WT mice at baseline is found in sparsely distributed individual cells in the DG as well as CA regions 2 and 3 (Figure 3A). One hour following ECS, high level Bdnf expression is evident uniformly throughout the dorsal and ventral blades of the DG (Figure 3C). There is no evident increase in the individual cellular pattern of labeling in CA2 and CA3 (Figure 3C). In Egr3-/- mice ECS did not result in increased expression of Bdnf (Figure 3D) compared to baseline (Figure 3B), and neither condition demonstrated strong labeling of Bdnf in the DG, or in CA1, CA2, or CA3 regions (Figures 3B,D).
EGR3 Binding Sites Are Present in Bdnf Regulatory Regions
If Egr3 is required to directly regulate expression of Bdnf, then EGR3 binding sites should be present in the promoter of the Bdnf gene. The genomic sequence 4000 bp upstream of the Bdnf transcription start site contains eight high-probability putative binding sites for EGR3 (Figure 4 and Table 1), with a cutoff value of 0.74 designated by TFBIND that calculates transcription factor-specific cutoffs using an algorithm (Tsunoda and Takagi, 1999). These sites represent a potential mechanism by which EGR3 could regulate expression of the Bdnf gene. Further studies will be necessary to determine whether EGR3 binds to these sites in vivo, and whether this binding may be functional.
FIGURE 4.
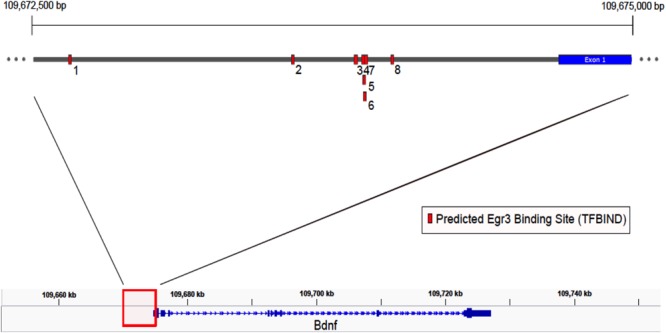
Map of predicted EGR3 binding sites in Bdnf upstream region. EGR3 binding sites identified in Table 1 are indicated in red. Positions are shown relative to exon 1 of the mouse Bdnf gene as identified in the NCBI Refseq database. Numbers under each binding site correspond to numbered rows in Table 1.
Table 1.
EGR3 binding sites identified in the Bdnf promoter using TFBIND.
| ID∗1 | Score∗2 | Position∗3 | Strand∗4 | Consensus sequence∗5 | Identified sequence∗6 |
|---|---|---|---|---|---|
| V$EGR3_01 | 0.807500 | 1947 | (-) | NTGCGTGGGCGK | CCTCCCACGTCA |
| V$EGR3_01 | 0.753542 | 2881 | (-) | NTGCGTGGGCGK | ACTCGCACTCAT |
| V$EGR3_01 | 0.741042 | 3144 | (-) | NTGCGTGGGCGK | CCGCATCCGCCT |
| V$EGR3_01 | 0.901667 | 3175 | (-) | NTGCGTGGGCGK | ACGCCCGCGCAC |
| V$EGR3_01 | 0.748958 | 3179 | (-) | NTGCGTGGGCGK | CCGCGCACACGC |
| V$EGR3_01 | 0.771042 | 3181 | (-) | NTGCGTGGGCGK | GCGCACACGCGC |
| V$EGR3_01 | 0.805417 | 3187 | (-) | NTGCGTGGGCGK | ACGCGCACACAC |
| V$EGR3_01 | 0.777083 | 3296 | (-) | NTGCGTGGGCGK | CAGCCTGCGCAG |
∗1ID: transcription factor matrix ID (from TRANSFAC R.3.4). V: vertebrate. ∗2Score: degree of similarity between input sequence and registered sequence for the transcription factor binding sites at the position shown in the position column (ranging from 0 to 1, representing low–high degree of similarity). ∗3Position: location of putative EGR3 binding site in the 4000 nucleotides upstream of Bdnf transcription start site. ∗4Strand: location of EGR3 consensus sequences on + vs. - strand of DNA. ∗5Consensus sequence (fixed) of the transcription factor binding sites. K = G or T; N = any base pair. ∗6Putative EGR3 binding sequence identified by TFBIND.
Hippocampal Neuronal Numbers Are Not Affected by Developmental Absence of Egr3
Egr3-/- mice lack expression of the functional gene throughout development and life. It is therefore possible that the absence of induction of Bdnf in the hippocampus following ECS in these animals may be a consequence of abnormal development, or survival, of hippocampal neurons in the absence of Egr3. To determine whether this may be the case, we conducted detailed regional cell counts of anti-NeuN antibody immunolabeled neurons in the hippocampus of matched pairs of adult Egr3-/- and WT littermate mice. WT and Egr3-/- brains display a comparable distribution and density of neurons (Figure 5). As expected (van Strien et al., 2009), a significant difference was observed on the basis of hippocampal region [F(4,50) = 9.13, p < 0.001], with the DG showing the tightest cell packing density, while the CA3 regions were the least densely packed. However, there was no difference in the number of neurons in any hippocampal regions between Egr3-/- and WT control mice. No significant main effect of genotype [F(1,50) = 1.19, p = 0.28] or interaction between genotype and hippocampal region [F(4,50) = 0.10, p = 0.98] was observed.
FIGURE 5.
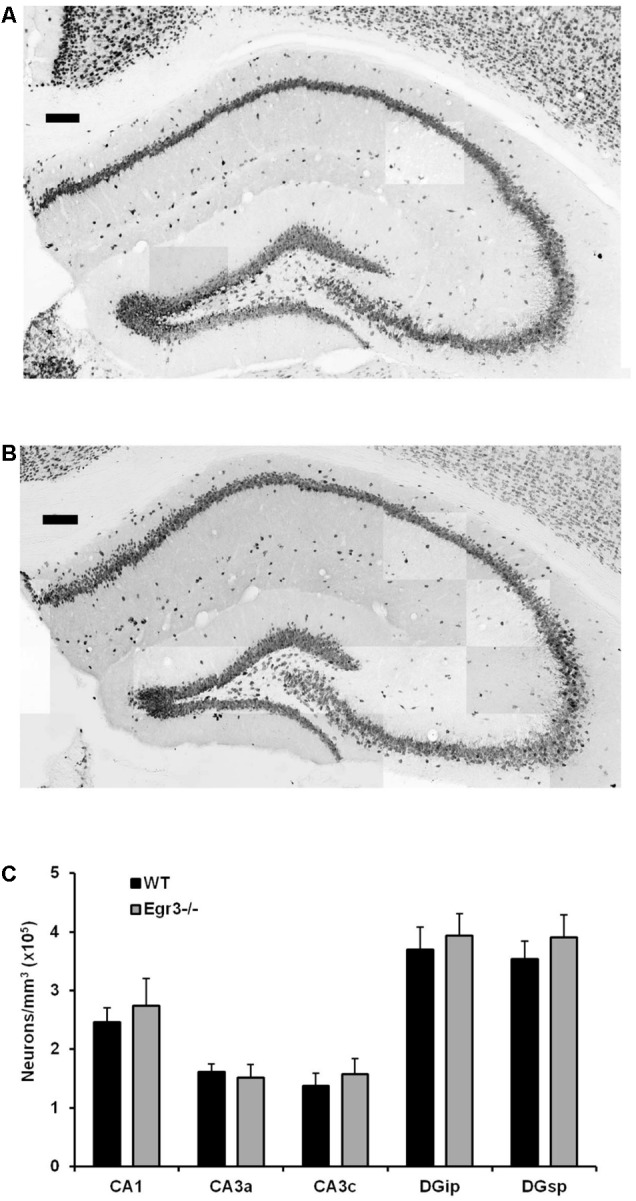
Egr3 is not required for development of normal numbers of hippocampal neurons. Sample montaged photomicrographs from the hippocampus of anti-NeuN stained (A) WT and (B) Egr3–/– mice show no obvious signs of changes in gross morphology (scale bar = 100 μm). Consistent with this observation, (C) quantification of NeuN+ cells (n = 6 animals/genotype) demonstrated no significant differences in neuron density in any hippocampal region between WT (black) and Egr3–/– (gray) mice.
Dendritic Complexity Remains Normal in Egr3-/- Mice
To further characterize how the absence of functional Egr3 may alter the structure of hippocampal neurons, we conducted detailed analyses of cells impregnated with Golgi-Cox solution in adult WT and Egr3-/- littermate mice (Figures 6A,B). In pyramidal cells (Table 2), no significant differences were observed in the Sholl analysis [F(1,24) = 2.71, p = 0.11] or branching order [F(1,24) = 0.81, p = 0.38] on the basis of genotype. Moreover, no significant main effect of region was observed on Sholl analysis [F(2,24) = 1.32, p = 0.29], or branching order [F(2,24) = 0.68, p = 0.51], and no interaction between genotype and region was observed (p > 0.05 in all cases). Similarly, granule cells (Table 3) showed no significant differences in the Sholl analysis [F(1,16) = 0.11, p = 0.75) or branching order [F(1,16) = 1.678, p = 0.21] on the basis of genotype. A significant main effect of region was observed on Sholl analysis [F(1,16) = 20.52, p < 0.001], indicating that granule cells in the DGsp are significantly larger and more complex than granule cells from the DGip, consistent with previous observations (Gallitano et al., 2016), although branching order was not significantly different [F(1,16) = 3.15, p = 0.09]. No interaction between genotype and region was observed (p > 0.05 in all cases).
FIGURE 6.
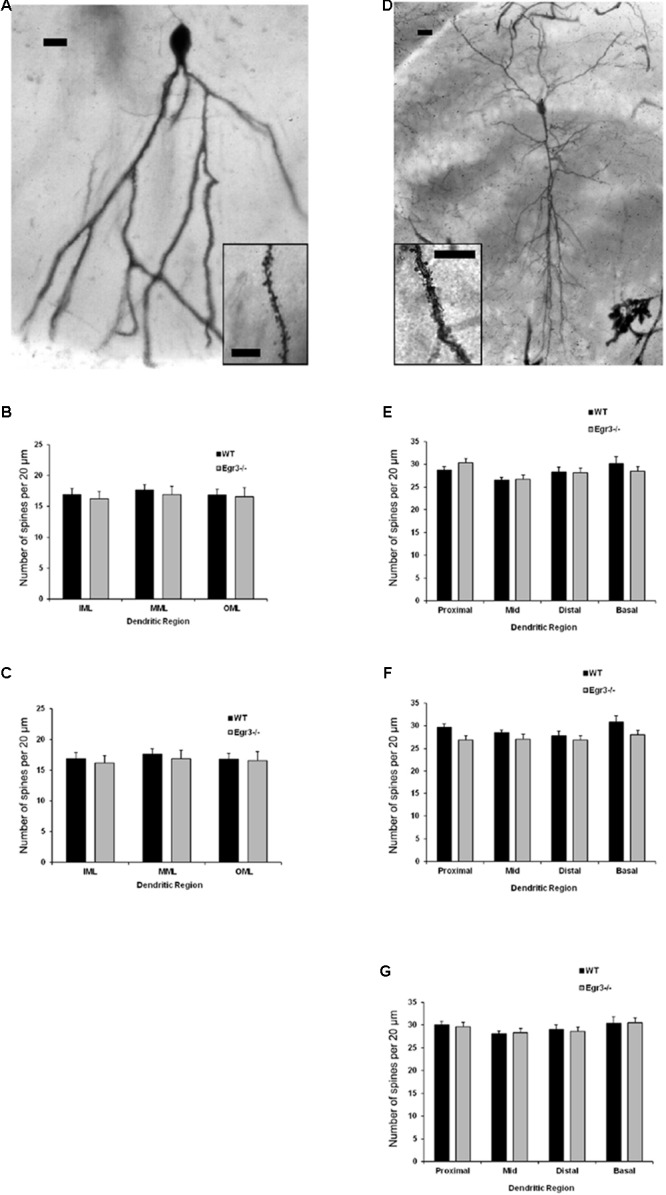
Egr3 is not required for normal development of neuronal morphology. Samples of Golgi-Cox staining in (A) granule cells and (D) pyramidal cells shows impregnation of both gross dendritic structure (scale bar = 20 μm) as well as spines (inset, scale bar = 10 μm). Quantitative data show no statistically significant differences in either dendritic morphology or spine number between WT (black) and Egr3–/– (gray) mice in the (B) DGsp, (C) DGip, (E) CA1, (F) CA3a/b, or (G) CA3c regions.
Table 2.
Dendritic arborization in hippocampal cells of WT and Egr3-/- mice.
| WT |
Egr3-/- |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CA1 |
CA3a/b |
CA3c |
CA1 |
CA3a/b |
CA3c |
||||||||
| Apical | Basal | Apical | Basal | Apical | Basal | Apical | Basal | Apical | Basal | Apical | Basal | ||
| Branching | 1 | 1.30 ± 0.211 | 2.50 ± 0.47 | 2.53 ± 0.42 | 3.35 ± 1.07 | 1.77 ± 0.38 | 4.73 ± 0.85 | 1.13 ± 0.07 | 4.00 ± 0.10 | 2.80 ± 0.23 | 2.13 ± 0.22 | 2.01 ± 0.92 | 3.04 ± 0.17 |
| Order | 2 | 2.19 ± 0.55 | 4.75 ± 0.47 | 3.28 ± 0.27 | 3.50 ± 0.87 | 2.50 ± 0.84 | 4.38 ± 0.94 | 3.66 ± 0.63 | 6.10 ± 0.64 | 3.10 ± 0.17 | 2.50 ± 0.14 | 3.80 ± 1.04 | 3.46 ± 0.41 |
| 3 | 5.89 ± 0.32 | 7.00 ± 0.74 | 3.05 ± 0.84 | 5.90 ± 0.87 | 2.53 ± 0.85 | 5.12 ± 0.78 | 6.08 ± 0.46 | 7.28 ± 0.88 | 4.00 ± 0.32 | 3.96 ± 0.17 | 3.20 ± 0.42 | 4.79 ± 0.22 | |
| 4 | 4.57 ± 0.39 | 7.00 ± 0.65 | 2.23 ± 0.31 | 5.50 ± 1.03 | 1.30 ± 0.78 | 5.30 ± 0.12 | 5.71 ± 0.34 | 5.23 ± 0.01 | 2.50 ± 0.27 | 3.38 ± 0.65 | 1.30 ± 0.16 | 2.83 ± 0.20 | |
| 5 | 6.02 ± 0.90 | 5.75 ± 1.08 | 3.03 ± 0.45 | 4.40 ± 1.03 | 2.37 ± 1.18 | 4.90 ± 0.10 | 4.30 ± 0.32 | 2.90 ± 0.06 | 1.80 ± 0.10 | 1.13 ± 0.07 | 2.00 ± 0.32 | 0.67 ± 0.19 | |
| 6 | 2.00 ± 0.49 | 2.50 ± 0.95 | 1.70 ± 0.37 | 2.10 ± 0.07 | 0.83 ± 0.59 | 1.80 ± 0.09 | 2.17 ± 0.31 | 1.15 ± 0.38 | 1.40 ± 0.23 | 0.50 ± 0.14 | 0.60 ± 0.12 | 0.29 ± 0.02 | |
| 7 | 1.57 ± 0.31 | 1.00 ± 0.47 | 1.60 ± 0.92 | 0.27 ± 0.19 | 0.89 ± 0.23 | 0.01 ± 0.01 | 4.00 ± 0.32 | 0.51 ± 0.43 | |||||
| 8+ | 0.84 ± 0.27 | 0.25 ± 0.12 | 0.50 ± 0.29 | – | 0.57 ± 0.33 | – | – | – | |||||
| Total | 24.51 ± 2.48 | 30.75 ± 4.90 | 19.90 ± 3.68 | 16.48 ± 2.73 | 16.17 ± 3.65 | 16.80 ± 2.77 | 22.94 ± 1.54 | 25.19 ± 4.98 | 21.60 ± 3.12 | 15.93 ± 3.34 | 14.95 ± 1.99 | 13.10 ± 0.64 | |
| Scholl (μm) | 20 | 2.03 ± 0.14 | 1.50 ± 0.70 | 1.70 ± 0.17 | 6.78 ± 0.56 | 1.73 ± 0.30 | 6.93 ± 0.19 | 1.91 ± 0.20 | 7.48 ± 1.00 | 1.70 ± 0.17 | 5.80 ± 0.46 | 1.38 ± 0.33 | 5.78 ± 0.01 |
| 40 | 3.50 ± 0.12 | 3.75 ± 0.60 | 2.58 ± 0.68 | 8.78 ± 1.14 | 3.68 ± 0.62 | 9.73 ± 0.30 | 3.58 ± 0.17 | 10.88 ± 0.65 | 2.58 ± 0.68 | 6.80 ± 0.46 | 2.75 ± 0.72 | 6.95 ± 0.84 | |
| 60 | 5.19 ± 0.22 | 5.00 ± 2.14 | 3.63 ± 0.36 | 7.15 ± 1.36 | 5.28 ± 0.27 | 10.43 ± 0.48 | 5.04 ± 0.31 | 10.03 ± 0.45 | 3.63 ± 0.36 | 5.58 ± 0.48 | 3.25 ± 0.82 | 5.25 ± 1.01 | |
| 80 | 4.51 ± 0.26 | 4.75 ± 1.72 | 4.85 ± 0.38 | 4.63 ± 2.09 | 6.60 ± 0.35 | 6.68 ± 1.20 | 5.61 ± 0.06 | 8.53 ± 0.74 | 4.85 ± 0.38 | 3.78 ± 0.59 | 5.25 ± 1.43 | 3.83 ± 0.91 | |
| 100 | 4.22 ± 0.57 | 6.00 ± 3.51 | 4.98 ± 0.45 | 1.85 ± 0.95 | 6.45 ± 1.13 | 5.45 ± 1.18 | 5.79 ± 0.53 | 4.00 ± 1.15 | 4.98 ± 0.45 | 1.73 ± 0.27 | 5.34 ± 1.24 | 2.75 ± 0.72 | |
| 120 | 5.18 ± 0.32 | 7.00 ± 3.79 | 4.65 ± 0.66 | 0.85 ± 0.38 | 4.35 ± 0.49 | 3.15 ± 0.78 | 5.78 ± 0.38 | 2.00 ± 0.58 | 4.65 ± 0.66 | 0.95 ± 0.26 | 6.13 ± 1.47 | 0.65 ± 0.09 | |
| 140 | 4.55 ± 0.45 | 4.25 ± 2.04 | 5.08 ± 0.76 | 0.50 ± 0.29 | 3.48 ± 0.16 | 1.35 ± 0.66 | 5.27 ± 0.59 | 0.85 ± 0.20 | 5.08 ± 0.76 | 0.20 ± 0.12 | 6.75 ± 1.67 | 0.19 ± 0.10 | |
| 160 | 5.63 ± 0.60 | 4.75 ± 1.98 | 4.48 ± 1.00 | – | 3.38 ± 0.36 | – | 5.08 ± 0.70 | 0.50 ± 0.15 | 4.48 ± 1.00 | – | 6.50 ± 1.90 | – | |
| 180 | 6.00 ± 0.53 | 4.50 ± 0.68 | 3.88 ± 1.23 | – | 2.50 ± 0.29 | – | 4.88 ± 0.65 | 0.19 ± 0.06 | 3.88 ± 1.23 | – | 4.13 ± 1.30 | – | |
| 200 | 5.09 ± 0.41 | 3.50 ± 0.24 | 3.00 ± 1.15 | – | 1.68 ± 0.53 | – | 4.73 ± 0.57 | 0.10 ± 0.06 | 3.00 ± 1.15 | – | 3.00 ± 0.94 | – | |
| 220 | 3.50 ± 0.41 | 2.75 ± 0.24 | 2.08 ± 0.76 | – | 1.35 ± 0.49 | – | 4.46 ± 0.48 | 0.03 ± 0.09 | 2.08 ± 0.76 | – | 1.50 ± 0.47 | – | |
| 240 | 1.99 ± 0.21 | 1.75 ± 0.98 | 1.90 ± 0.52 | – | 1.15 ± 0.38 | – | 2.32 ± 0.10 | – | 1.90 ± 0.52 | – | 1.00 ± 0.26 | – | |
| 260 | 1.11 ± 0.10 | 0.50 ± 0.28 | 1.28 ± 0.30 | – | 0.40 ± 0.23 | – | 1.40 ± 0.02 | – | 1.28 ± 0.30 | – | 0.50 ± 0.16 | – | |
| 280 | 0.69 ± 0.18 | –1 | – | – | 0.20 ± 0.12 | – | 0.58 ± 0.17 | – | – | – | 0.13 ± 0.06 | – | |
| 300 | 0.30 ± 0.06 | – | – | – | – | – | 0.13 ± 0.07 | – | – | – | – | – | |
| 320 | 0.11 ± 0.09 | – | – | – | – | – | – | – | – | – | – | – | |
| Total | 52.00 ± 9.93 | 39.10 ± 19.02 | 44.95 ± 4.88 | 26.30 ± 4.33 | 42.60 ± 4.23 | 33.08 ± 4.93 | 52.63 ± 8.30 | 35.25 ± 6.33 | 44.95 ± 4.88 | 24.83 ± 3.64 | 42.15 ± 3.94 | 35.20 ± 4.58 | |
1All data are reported as mean ±SE. 2Denotes that no observations could be made in this category.
Table 3.
Dendritic arborization in granule cells of WT and Egr3-/- mice.
| WT |
Egr3-/- |
||||
|---|---|---|---|---|---|
| DGsp | DGip | DGsp | DGip | ||
| Branching | 1 | 2.05 ± 0.171 | 2.04 ± 0.10 | 2.23 ± 0.12 | 2.08 ± 0.50 |
| Order | 2 | 4.13 ± 0.36 | 3.89 ± 0.28 | 4.65 ± 0.22 | 3.93 ± 0.47 |
| 3 | 5.08 ± 0.39 | 4.91 ± 0.21 | 5.55 ± 0.27 | 5.03 ± 0.68 | |
| 4 | 3.63 ± 0.16 | 3.58 ± 0.77 | 4.38 ± 0.35 | 4.28 ± 1.02 | |
| 5 | 1.225 ± 0.25 | 1.70 ± 0.39 | 1.50 ± 0.36 | 1.55 ± 1.15 | |
| 6 | 0.45 ± 0.17 | 0.40 ± 0.21 | 0.63 ± 0.31 | 0.35 ± 0.37 | |
| 7 | 0.45 ± 0.16 | 0.40 ± 0.21 | 0.53 ± 0.26 | 0.30 ± 0.37 | |
| 8+ | 0.09 ± 0.04 | 0.01 ± 0.01 | 0.10 ± 0.06 | 0.05 ± 0.01 | |
| Total | 17.00 ± 0.61 | 16.91 ± 1.20 | 19.55 ± 1.36 | 17.55 ± 2.74 | |
| Scholl (μm) | 20 | 2.85 ± 0.28 | 3.13 ± 0.85 | 3.08 ± 0.26 | 3.27 ± 0.28 |
| 40 | 4.425 ± 0.20 | 4.48 ± 1.20 | 4.55 ± 0.38 | 4.39 ± 0.43 | |
| 60 | 4.7 ± 0.16 | 5.83 ± 0.73 | 5.58 ± 0.49 | 5.53 ± 0.45 | |
| 80 | 5.73 ± 0.20 | 6.05 ± 0.71 | 6.32 ± 0.38 | 5.94 ± 0.35 | |
| 100 | 6.10 ± 0.16 | 6.33 ± 0.84 | 6.80 ± 0.39 | 6.02 ± 0.58 | |
| 120 | 6.85 ± 0.19 | 6.63 ± 0.62 | 7.20 ± 0.31 | 6.52 ± 0.48 | |
| 140 | 6.975 ± 0.21 | 6.50 ± 0.76 | 7.75 ± 0.49 | 6.55 ± 0.38 | |
| 160 | 7.05 ± 0.26 | 6.30 ± 0.85 | 7.90 ± 0.40 | 6.50 ± 0.41 | |
| 180 | 6.98 ± 0.28 | 5.65 ± 1.20 | 7.85 ± 0.46 | 5.95 ± 0.38 | |
| 200 | 6.58 ± 0.27 | 5.08 ± 0.93 | 7.48 ± 0.50 | 5.45 ± 0.49 | |
| 220 | 6.53 ± 0.23 | 4.48 ± 0.96 | 6.83 ± 0.50 | 5.40 ± 0.49 | |
| 240 | 6.10 ± 0.18 | 3.48 ± 1.34 | 6.43 ± 0.44 | 4.36 ± 0.45 | |
| 260 | 5.28 ± 0.25 | 2.50 ± 1.45 | 5.95 ± 0.31 | 3.55 ± 0.50 | |
| 280 | 4.65 ± 0.33 | 1.70 ± 1.80 | 5.15 ± 0.28 | 2.29 ± 0.37 | |
| 300 | 3.5 ± 0.27 | 0.95 ± 1.83 | 3.80 ± 0.23 | 1.37 ± 0.38 | |
| 320 | 2.3 ± 0.20 | 0.40 ± 0.97 | 2.48 ± 0.29 | 0.64 ± 0.29 | |
| 340 | 1.5 ± 0.44 | 0.25 ± 0.96 | 1.88 ± 0.55 | 0.48 ± 0.19 | |
| Total | 88.08 ± 2.15 | 69.68 ± 10.45 | 97.01 ± 4.26 | 74.17 ± 5.41 | |
1All data are reported as mean ±SE.
Spine Density Is Not Affected by Loss of Egr3
Consistent with the dendritic analysis, ANOVAs showed no significant differences in spine density in any brain region (Figure 6). In pyramidal cells, no significant difference on the basis of genotype in the density of basal spines in stratum oriens [F(1,24) = 0.35, p = 0.5588], in apical spines in stratum radiatum proximal [F(1,24) = 0.64, p = 0.43] or distal [F(1,24) = 0.35, p = 0.56] to the soma, nor in stratum lacunosum/moleculare [F(1,24) = 1.31, p = 0.26]. Similarly, no significant differences were observed on the basis of hippocampal region (i.e., CA1, CA3a/b, and CA3c) basal spines in stratum oriens [F(2,24) = 0.92, p = 0.41], in stratum radiatum proximal [F(2,24) = 0.42, p = 0.66] or distal [F(2,24) = 0.27, p = 0.77] to the soma, nor in stratum lacunosum/moleculare [F(2,24) = 0.5624, p = 0.5772]. Similarly, no significant interactions were observed between region and genotype (p > 0.05 in all cases).
In granule cells, there was no significant effect of genotype on the density of spines in the IML [F(1,16) = 0.31, p = 0.58], MML [F(1,16) = 0.14, p = 0.71], or OML [F(1,16) = 0.33, p = 0.58]. Similarly, no significant differences in spine density were observed on the basis of blade of the DG (i.e., DGsp vs. DGip) in IML [F(1,16) = 0.28, p = 0.64], MML [F(1,16) = 0.33, p = 0.57], or OML [F(1,16) = 0.76, p = 0.30]. Similarly, no significant interactions were observed between region and genotype (p > 0.05 in all cases).
Discussion
Egr3 is an IEG transcription factor that is induced in response to environmental stimuli (Thompson et al., 2010; Maple et al., 2015). We have hypothesized that dysfunction of Egr3 could disrupt the brain’s normal neurobiological response to stress, resulting in increased risk to develop mental illnesses such as schizophrenia and bipolar disorder (Gallitano-Mendel et al., 2007, 2008; Williams et al., 2012; Huentelman et al., 2015; Pfaffenseller et al., 2016; Marballi and Gallitano, 2018). In support of this hypothesis, our prior work has shown that Egr3-/- mice display behavioral abnormalities consistent with animal models of psychotic disorders that can be reversed by administration of medications used to treat these illnesses in humans (Gallitano-Mendel et al., 2007, 2008). In addition, Egr3 has been associated with risk for schizophrenia in Japanese, Korean, Han Chinese populations, and populations of European Descent (Yamada et al., 2007; Kim et al., 2010; Zhang et al., 2012; Huentelman et al., 2015), and Egr3 expression is decreased in the brains of patients with schizophrenia, compared with controls (Mexal et al., 2005; Yamada et al., 2007). Bioinformatics approaches have identified Egr3 as a central gene in a network of transcription factors and microRNAs implicated in schizophrenia risk (Guo et al., 2010), as well as a master regulator of genes differentially expressed in the brains of bipolar disorder patients in two independent cohorts (Pfaffenseller et al., 2016). Together, these findings suggest that disruption of Egr3 activity, or function of other proteins that act either upstream or downstream in the EGR3 pathway, may mediate both the genetic and environmental factors that contribute to risk for severe mental illnesses (Marballi and Gallitano, 2018).
Since EGR3 functions as a transcription factor, we hypothesized that the downstream target genes regulated by EGR3 may also influence risk for psychotic disorders. IEGs, including Egr3, are expressed at low levels at baseline. We therefore conducted ECS to induce rapid, high-level, expression of Egr3 (O’Donovan et al., 1998). We used a microarray approach to compare genes expressed 1 h following ECS in the hippocampus of WT, compared to Egr3-/-, mice to identify EGR3-dependent target genes. This study revealed increased expression of Bdnf, in the WT mice, but not in the Egr3-/- mice, indicating that ECS-induced Bdnf expression requires Egr3.
BDNF is a neurotrophic factor that promotes growth and differentiation of neurons and synapses (Barde et al., 1987; Binder and Scharfman, 2004; Bennett and Lagopoulos, 2014). In addition, BDNF has been found to play a role in the therapeutic effectiveness of treatments for psychiatric illnesses. Animals treated with antidepressant medications demonstrate increased expression of Bdnf in the hippocampus (Bjorkholm and Monteggia, 2016). Disruption of BDNF function is implicated in the pathology underlying numerous psychiatric disorders, ranging from schizophrenia and bipolar disorder to depression (Grande et al., 2010; Lee and Kim, 2010; Autry and Monteggia, 2012). Reduction of hippocampal BDNF attenuates the effect of antidepressants (Monteggia et al., 2004; Adachi et al., 2008), and infusion of BDNF into the hippocampus reverses depression-like behavior in rodents (Shirayama et al., 2002).
The mouse Bdnf gene consists of nine exons; eight of which are in the 5′ untranslated region of the gene (exons I–VIII) and only one of which encodes protein (exon IX; Aid et al., 2007). Different Bdnf splice variants are formed from a combination of one of the 5′ untranslated exons (I–VIII) with the common protein-coding exon (exon IX; Liu et al., 2005; McDowell et al., 2010). Two alternative polyadenylation signals result in transcription termination and result in either the short (0.3 kB) or long (2.9 kB) 3′ UTR of Bdnf (Timmusk et al., 1993).
Prior studies have shown that activity-dependent transcription of Bdnf can be mediated by calcium influx (Shieh and Ghosh, 1999; West et al., 2001; Aid et al., 2007; Hong et al., 2008; West, 2008). Calcium responsive elements, including cAMP-responsive element-binding (CREB) protein (West et al., 2001), Carf (Tao et al., 2002), calcium-dependent phosphorylation of the methyl CpG binding protein 2 (MeCP2), and the basic helix-loop-helix upstream signaling factors (USFs; Chen et al., 2003), positively regulate expression of Bdnf. In contrast, stressful environmental stimuli, ischemia (Lindvall et al., 1992), and prenatal stress (Boersma et al., 2014) lead to decreased expression levels of Bdnf in the hippocampus.
One of the stimuli known to activate Bdnf is ECS. ECS is the experimental equivalent of ECT, a treatment that has been in use for more than 80 years, and that remains the most effective treatment for severe mood disorders and psychotic disorders with a mood component (Fink, 2001, 2014; Pagnin et al., 2004; Rosenquist et al., 2014). ECT results in increased levels of BDNF in peripheral blood in humans (Bocchio-Chiavetto et al., 2006). Studies in rodents demonstrate that ECS induces high levels of BDNF expression in the hippocampus (Altar et al., 2004). ECS also induces expression of IEGs, including Egr3, as well as other growth factors in addition to BDNF, and stimulates neurogenesis and dendritic sprouting in the hippocampus (Duman and Vaidya, 1998; Malberg et al., 2000; Altar et al., 2004; Perera et al., 2007; Kato, 2009; Youssef and McCall, 2014; Thomann et al., 2017).
Our findings show that hippocampal induction of Bdnf in response to ECS requires Egr3. The presence of EGR3 binding sites in the Bdnf promoter suggests the possibility that EGR3, a transcription factor, may directly regulate expression of Bdnf. However, since the current study did not confirm binding of EGR3 to these sites in vivo, we are not able to determine this at this time.
Exon IV in particular seems to be an important component of activity-dependent Bdnf expression, as deletion of exon IV impedes both ECS-induced, and sleep deprivation-mediated, activation of several Bdnf transcripts in vivo (Martinowich et al., 2011). Notably, both ECS and sleep deprivation activate expression of Egr3 as well (Thompson et al., 2010; Maple et al., 2015). Deletion of exon IV also causes impairments in spatial memory reversal and fear memory extinction (Sakata et al., 2013), deficits in GABAergic transmission in the cortex (Sakata et al., 2009), and impairment of hippocampal synaptic plasticity (Sakata et al., 2013) in vivo.
Another important activity-induced exon in the hippocampus is exon VI (Chiaruttini et al., 2008). Both Bdnf exon IV and VI contribute significantly to BDNF protein levels in the hippocampus (Maynard et al., 2016). We found that both exon IV and VI expression failed to show induction in Egr3-/- mice following ECS compared to WT mice. Follow-up bioinformatic analysis of the 4 kb proximal upstream region of Bdnf using TFBind revealed eight high-probability EGR3 binding sites (Table 1). Further studies will be important to confirm the binding of EGR3 to these sites and confirm the specific conditions and cell types in which they occur.
Psychotic disorders are characterized by cognitive impairments (Goff et al., 2011). In animals, disrupting the expression of BDNF results in deficits in learning, memory, attention, executive function, and cognition (Sharma and Antonova, 2003). Both Egr3 and BDNF play critical roles in hippocampal synaptic plasticity, a process associated with memory formation. Specifically, long-term depression, a form of hippocampal plasticity that is facilitated by novelty and stress exposure, requires Egr3, and is stimulated by the immature form of BDNF, proBDNF (Kim et al., 1996; Woo et al., 2005; Gallitano-Mendel et al., 2007; Yang et al., 2014; Bukalo et al., 2016). The results of these, and many other studies, have led to the hypothesis that BDNF may be a critical molecule in mediating the therapeutic effects of psychiatric therapies, including ECT (Bocchio-Chiavetto et al., 2006; Li et al., 2016).
One limitation of our study is that it employed mice that lack Egr3 function throughout development and life. It is therefore possible that the failure of ECS to induce Bdnf expression in Egr3-/- mice could be explained by a developmental deficit of the cells that normally express Bdnf. However, our neuroanatomical studies demonstrated no regional differences in neuronal counts in the DG or CA regions of the hippocampus in Egr3-/- mice. Furthermore, no differences were found in the spine density or branch order of pyramidal cells, or in the density DG granule cells.
The absence of neuroanatomical deficits in Egr3-/- mice is supported by our prior findings of normal activation of Arc expression in DG granule cells following exposure to a novel environment (Maple et al., 2017) and prior studies demonstrating seizure-induced Arc expression in the hippocampus of Egr3-/- mice (Li et al., 2005). However, this Arc expression dissipates over several hours in Egr3-/- DG cells, compared to WT mice in which Arc expression perdures for up to 8 h (Maple et al., 2017). Thus, the hippocampal DG granule cells of Egr3-/- mice are capable of expressing Arc in response to activity, but maintenance of this expression requires Egr3. Our results indicate that the numbers of DG cells remain consistent in the WT and Egr3-/- mice, which suggests that the loss of Bdnf in these cells is due to failed activation in the absence of Egr3.
Another limitation is that, although we show that Bdnf fails to be induced following ECS in the Egr3-/- mice, and the Bdnf promoter contains putative EGR3 binding sites, we cannot rule out the possibility that an intermediary factor regulates expression of Bdnf. Future studies will employ chromatin immunoprecipitation (ChIP) to assess binding of EGR3 to the Bdnf promoter in the hippocampus, as well as luciferase reporter assays to demonstrate functionality of this binding in vitro.
The expression of IEGs is, by definition, independent of protein synthesis. Since exons III and IV of the mouse BDNF have been reported to have IEG activity (Lauterborn et al., 1996; Binder and Scharfman, 2004; Sakata et al., 2009), this indicates that Bdnf transcription should be dependent upon transcription factor proteins that are already present and poised for activation. However, in our study, we demonstrate that Egr3, an IEG transcription factor, mediates Bdnf mRNA expression in response to ECS. Specifically, we found that Egr3 is required for ECS-induced expression of Bdnf exons IV and VI, with a larger effect on exon IV, in the hippocampus. Since our study examined gene expression 1 h following ECS, it is possible that Bdnf is initially activated in the brains of Egr3-/- mice, but fails to be maintained in the absence of Egr3, as seen with Arc expression in prior studies by our group and others (Li et al., 2005; Maple et al., 2017). If this is the case, then the induced IEG Bdnf transcripts would have to be degraded within 1 h following ECS, a timeframe more rapid than that of Arc.
Our studies identify the novel requirement of EGR3 for hippocampal Bdnf expression in response to ECS. We further show that EGR3 is required for seizure-induced expression of Bdnf transcripts containing exons IV and VI. Notably, these two isoforms are the greatest contributors to total BDNF protein in the hippocampus (Maynard et al., 2016). These data suggest the possibility that EGR3 may play a role in other forms of activity-dependent Bdnf induction in the hippocampus, such as that induced by high-frequency stimulation or exposure to novelty. However, this will have to be tested in future studies. This finding is of interest as both Egr3 and Bdnf share several common features. These include essential roles in hippocampal long-term depression (Gallitano-Mendel et al., 2007; Novkovic et al., 2015; Bukalo et al., 2016) calcium-signaling-dependent activation (Mittelstadt and Ashwell, 1998; Shieh et al., 1998), and genetic association with schizophrenia (Gratacos et al., 2007; Yamada et al., 2007; Kim et al., 2010; Ning et al., 2012; Zhang et al., 2012; Huentelman et al., 2015). We have hypothesized that both Bdnf and Egr3 are crucial components of a biological pathway implicated in mental illness risk (Marballi and Gallitano, 2018). It will be interesting to determine whether activity-dependent, EGR3-mediated BDNF expression contributes to the therapeutic effects of ECT on the mood and psychotic symptoms of patients with severe psychiatric illnesses.
Author Contributions
MC accumulated images and data for the manuscript and assisted in analyzing NeuN counts. BR contributed to data analysis and image collection for the hippocampal neuroanatomical studies. SB analyzed the original microarray study, identified downstream targets of the immediate early gene, Egr3, and developed figures and tables for the binding regions of EGR3 within the Bdnf promoter. KKM validated the work of SB, performed validation studies in the female cohorts of mice, analyzed the qRT-PCR results, formulated graphs, and assisted in the conceptualization of the experiments and the development of the manuscript. AG funded the research with her NIMH funds, conceptualized the project, and developed the hypothesis of immediate early genes synergistically acting with genetic factors to contribute toward the pathogenesis of schizophrenia. DM utilized his funds obtained from the Natural Sciences and Engineering Research Council of Canada, collaborated with AG to establish the theory of regulation of Bdnf by Egr3, and contributed directly to the manuscript with his writing, editing, and neuroanatomical studies. KTM performed ECS, collected tissue, performed in situ hybridization, analyzed the results, and assisted in writing the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Andrew McBride, for technical assistance, Janet Campbell, M.S., for assistance with ECS and animal colony management, Stanley J. Watson, M.D., Ph.D., and members of his laboratory Jennifer Fitzpatrick and Ali Gheidi, Ph.D., at the Molecular and Behavioral Neuroscience Institute, University of Michigan, Ann Arbor, for generous donation of the mouse BDNF plasmid and extensive technical instruction.
Funding. This work was supported by the US National Institute of Mental Health award R01MH097803 and R21MH113154 (AG) and the Natural Sciences and Engineering Research Council of Canada (DM).
References
- Abercrombie M. (1946). Estimation of nuclear population from microtome sections. 94 239–247. 10.1002/ar.1090940210 [DOI] [PubMed] [Google Scholar]
- Adachi M., Barrot M., Autry A. E., Theobald D., Monteggia L. M. (2008). Selective loss of brain-derived neurotrophic factor in the dentate gyrus attenuates antidepressant efficacy. 63 642–649. 10.1016/j.biopsych.2007.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aid T., Kazantseva A., Piirsoo M., Palm K., Timmusk T. (2007). Mouse and rat BDNF gene structure and expression revisited. 85 525–535. 10.1002/jnr.21139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altar C. A., Laeng P., Jurata L. W., Brockman J. A., Lemire A., Bullard J., et al. (2004). Electroconvulsive seizures regulate gene expression of distinct neurotrophic signaling pathways. 24 2667–2677. 10.1523/JNEUROSCI.5377-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altar C. A., Whitehead R. E., Chen R., Wortwein G., Madsen T. M. (2003). Effects of electroconvulsive seizures and antidepressant drugs on brain-derived neurotrophic factor protein in rat brain. 54 703–709. 10.1016/S0006-3223(03)00073-8 [DOI] [PubMed] [Google Scholar]
- Autry A. E., Monteggia L. M. (2012). Brain-derived neurotrophic factor and neuropsychiatric disorders. 64 238–258. 10.1124/pr.111.005108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barde Y. A., Davies A. M., Johnson J. E., Lindsay R. M., Thoenen H. (1987). Brain derived neurotrophic factor. 71 185–189. 10.1016/S0079-6123(08)61823-3 [DOI] [PubMed] [Google Scholar]
- Bennett M. R., Lagopoulos J. (2014). Stress and trauma: BDNF control of dendritic-spine formation and regression. 112 80–99. 10.1016/j.pneurobio.2013.10.005 [DOI] [PubMed] [Google Scholar]
- Binder D. K., Scharfman H. E. (2004). Brain-derived neurotrophic factor. 22 123–131. 10.1080/08977190410001723308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkholm C., Monteggia L. M. (2016). BDNF - a key transducer of antidepressant effects. 102 72–79. 10.1016/j.neuropharm.2015.10.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocchio-Chiavetto L., Zanardini R., Bortolomasi M., Abate M., Segala M., Giacopuzzi M., et al. (2006). Electroconvulsive Therapy (ECT) increases serum Brain Derived Neurotrophic Factor (BDNF) in drug resistant depressed patients. 16 620–624. 10.1016/j.euroneuro.2006.04.010 [DOI] [PubMed] [Google Scholar]
- Boersma G. J., Lee R. S., Cordner Z. A., Ewald E. R., Purcell R. H., Moghadam A. A., et al. (2014). Prenatal stress decreases Bdnf expression and increases methylation of Bdnf exon IV in rats. 9 437–447. 10.4161/epi.27558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukalo O., Lee P. R., Fields R. D. (2016). BDNF mRNA abundance regulated by antidromic action potentials and AP-LTD in hippocampus. 635 97–102. 10.1016/j.neulet.2016.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W. G., Chang Q., Lin Y., Meissner A., West A. E., Griffith E. C., et al. (2003). Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. 302 885–889. 10.1126/science.1086446 [DOI] [PubMed] [Google Scholar]
- Chiaruttini C., Sonego M., Baj G., Simonato M., Tongiorgi E. (2008). BDNF mRNA splice variants display activity-dependent targeting to distinct hippocampal laminae. 37 11–19. 10.1016/j.mcn.2007.08.011 [DOI] [PubMed] [Google Scholar]
- Davis K. L., Stewart D. G., Friedman J. I., Buchsbaum M., Harvey P. D., Hof P. R., et al. (2003). White matter changes in schizophrenia: evidence for myelin-related dysfunction. 60 443–456. 10.1001/archpsyc.60.5.443 [DOI] [PubMed] [Google Scholar]
- Duman R. S., Vaidya V. A. (1998). Molecular and cellular actions of chronic electroconvulsive seizures. 14 181–193. 10.1097/00124509-199809000-00004 [DOI] [PubMed] [Google Scholar]
- Durany N., Michel T., Zochling R., Boissl K. W., Cruz-Sanchez F. F., Riederer P., et al. (2001). Brain-derived neurotrophic factor and neurotrophin 3 in schizophrenic psychoses. 52 79–86. 10.1016/S0920-9964(00)00084-0 [DOI] [PubMed] [Google Scholar]
- Dwork A. J., Mancevski B., Rosoklija G. (2007). White matter and cognitive function in schizophrenia. 10 513–536. 10.1017/S1461145707007638 [DOI] [PubMed] [Google Scholar]
- Fahmy R. G., Dass C. R., Sun L. Q., Chesterman C. N., Khachigian L. M. (2003). Transcription factor Egr-1 supports FGF-dependent angiogenesis during neovascularization and tumor growth. 9 1026–1032. 10.1038/nm905 [DOI] [PubMed] [Google Scholar]
- Fahmy R. G., Khachigian L. M. (2007). Suppression of growth factor expression and human vascular smooth muscle cell growth by small interfering RNA targeting EGR-1. 100 1526–1535. 10.1002/jcb.21145 [DOI] [PubMed] [Google Scholar]
- Fink M. (2001). Convulsive therapy: a review of the first 55 years. 63 1–15. 10.1016/S0165-0327(00)00367-0 [DOI] [PubMed] [Google Scholar]
- Fink M. (2014). The seizure, not electricity, is essential in convulsive therapy: the flurothyl experience. 30 91–93. 10.1097/YCT.0000000000000110 [DOI] [PubMed] [Google Scholar]
- Forbes N. F., Carrick L. A., McIntosh A. M., Lawrie S. M. (2009). Working memory in schizophrenia: a meta-analysis. 39 889–905. 10.1017/S0033291708004558 [DOI] [PubMed] [Google Scholar]
- Gallitano A. L., Satvat E., Gil M., Marrone D. F. (2016). Distinct dendritic morphology across the blades of the rodent dentate gyrus. 70 277–282. 10.1002/syn.21900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallitano-Mendel A., Izumi Y., Tokuda K., Zorumski C. F., Howell M. P., Muglia L. J., et al. (2007). The immediate early gene early growth response gene 3 mediates adaptation to stress and novelty. 148 633–643. 10.1016/j.neuroscience.2007.05.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallitano-Mendel A., Wozniak D. F., Pehek E. A., Milbrandt J. (2008). Mice lacking the immediate early gene Egr3 respond to the anti-aggressive effects of clozapine yet are relatively resistant to its sedating effects. 33 1266–1275. 10.1038/sj.npp.1301505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb R., Kolb B. (1998). A method for vibratome sectioning of Golgi-Cox stained whole rat brain. 79 1–4. 10.1016/S0165-0270(97)00163-5 [DOI] [PubMed] [Google Scholar]
- Goff D. C., Hill M., Barch D. (2011). The treatment of cognitive impairment in schizophrenia. 99 245–253. 10.1016/j.pbb.2010.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grande I., Fries G. R., Kunz M., Kapczinski F. (2010). The role of BDNF as a mediator of neuroplasticity in bipolar disorder. 7 243–250. 10.4306/pi.2010.7.4.243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratacos M., Gonzalez J. R., Mercader J. M., de Cid R., Urretavizcaya M., Estivill X. (2007). Brain-derived neurotrophic factor Val66Met and psychiatric disorders: meta-analysis of case-control studies confirm association to substance-related disorders, eating disorders, and schizophrenia. 61 911–922. 10.1016/j.biopsych.2006.08.025 [DOI] [PubMed] [Google Scholar]
- Guo A. Y., Sun J., Jia P., Zhao Z. (2010). A novel microRNA and transcription factor mediated regulatory network in schizophrenia. 4:10. 10.1186/1752-0509-4-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson D. R., Gottesman I. I. (2005). Theories of schizophrenia: a genetic-inflammatory-vascular synthesis. 6:7. 10.1186/1471-2350-6-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong E. J., McCord A. E., Greenberg M. E. (2008). A biological function for the neuronal activity-dependent component of Bdnf transcription in the development of cortical inhibition. 60 610–624. 10.1016/j.neuron.2008.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huentelman M. J., Muppana L., Corneveaux J. J., Dinu V., Pruzin J. J., Reiman R., et al. (2015). Association of SNPs in EGR3 and ARC with schizophrenia supports a biological pathway for schizophrenia risk. 10:e0135076. 10.1371/journal.pone.0135076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inta D., Lima-Ojeda J. M., Lau T., Tang W., Dormann C., Sprengel R., et al. (2013). Electroconvulsive therapy induces neurogenesis in frontal rat brain areas. 8:e69869. 10.1371/journal.pone.0069869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitt D. C., Zukin S. R. (1991). Recent advances in the phencyclidine model of schizophrenia. 148 1301–1308. 10.1176/ajp.148.10.1301 [DOI] [PubMed] [Google Scholar]
- Jessen K. R., Mirsky R. (2002). Signals that determine Schwann cell identity. 200 367–376. 10.1046/j.1469-7580.2002.00046.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato N. (2009). Neurophysiological mechanisms of electroconvulsive therapy for depression. 64 3–11. 10.1016/j.neures.2009.01.014 [DOI] [PubMed] [Google Scholar]
- Kim J. J., Foy M. R., Thompson R. F. (1996). Behavioral stress modifies hippocampal plasticity through N-methyl-D-aspartate receptor activation. 93 4750–4753. 10.1073/pnas.93.10.4750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. H., Song J. Y., Joo E. J., Lee K. Y., Ahn Y. M., Kim Y. S. (2010). EGR3 as a potential susceptibility gene for schizophrenia in Korea. 153B 1355–1360. 10.1002/ajmg.b.31115 [DOI] [PubMed] [Google Scholar]
- Lauterborn J. C., Rivera S., Stinis C. T., Hayes V. Y., Isackson P. J., Gall C. M. (1996). Differential effects of protein synthesis inhibition on the activity-dependent expression of BDNF transcripts: evidence for immediate-early gene responses from specific promoters. 16 7428–7436. 10.1523/JNEUROSCI.16-23-07428.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le H., Zeng F., Xu L., Liu X., Huang Y. (2013). The role of CD133 expression in the carcinogenesis and prognosis of patients with lung cancer. 8 1511–1518. 10.3892/mmr.2013.1667 [DOI] [PubMed] [Google Scholar]
- Lee B. H., Kim Y. K. (2010). The roles of BDNF in the pathophysiology of major depression and in antidepressant treatment. 7 231–235. 10.4306/pi.2010.7.4.231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Ye F., Xiao W., Tang X., Sha W., Zhang X., et al. (2016). Increased serum brain-derived neurotrophic factor levels following electroconvulsive therapy or antipsychotic treatment in patients with schizophrenia. 36 23–28. 10.1016/j.eurpsy.2016.03.005 [DOI] [PubMed] [Google Scholar]
- Li L., Carter J., Gao X., Whitehead J., Tourtellotte W. G. (2005). The neuroplasticity-associated arc gene is a direct transcriptional target of early growth response (EGR) transcription factors. 25 10286–10300. 10.1128/MCB.25.23.10286-10300.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindvall O., Ernfors P., Bengzon J., Kokaia Z., Smith M. L., Siesjo B. K., et al. (1992). Differential regulation of mRNAs for nerve growth factor, brain-derived neurotrophic factor, and neurotrophin 3 in the adult rat brain following cerebral ischemia and hypoglycemic coma. 89 648–652. 10.1073/pnas.89.2.648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q. R., Walther D., Drgon T., Polesskaya O., Lesnick T. G., Strain K. J., et al. (2005). Human brain derived neurotrophic factor (BDNF) genes, splicing patterns, and assessments of associations with substance abuse and Parkinson’s Disease. 134B 93–103. 10.1002/ajmg.b.30109 [DOI] [PubMed] [Google Scholar]
- Malberg J. E., Eisch A. J., Nestler E. J., Duman R. S. (2000). Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. 20 9104–9110. 10.1523/JNEUROSCI.20-24-09104.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maple A., Lackie R. E., Elizalde D. I., Grella S. L., Damphousse C. C., Xa C., et al. (2017). Attenuated late-phase ARC transcription in the dentate gyrus of mice lacking Egr3. 2017:6063048. 10.1155/2017/6063048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maple A. M., Zhao X., Elizalde D. I., McBride A. K., Gallitano A. L. (2015). Htr2a expression responds rapidly to environmental stimuli in an Egr3-dependent manner. 6 1137–1142. 10.1021/acschemneuro.5b00031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marballi K. K., Gallitano A. L. (2018). Immediate early genes anchor a biological pathway of proteins required for memory formation, long-term depression and risk for schizophrenia. 12:23. 10.3389/fnbeh.2018.00023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinowich K., Schloesser R. J., Jimenez D. V., Weinberger D. R., Lu B. (2011). Activity-dependent brain-derived neurotrophic factor expression regulates cortistatin-interneurons and sleep behavior. 4:11. 10.1186/1756-6606-4-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard K. R., Hill J. L., Calcaterra N. E., Palko M. E., Kardian A., Paredes D., et al. (2016). Functional role of BDNF production from unique promoters in aggression and serotonin signaling. 41 1943–1955. 10.1038/npp.2015.349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowell K. A., Hutchinson A. N., Wong-Goodrich S. J., Presby M. M., Su D., Rodriguiz R. M., et al. (2010). Reduced cortical BDNF expression and aberrant memory in Carf knock-out mice. 30 7453–7465. 10.1523/JNEUROSCI.3997-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechtcheriakova D., Wlachos A., Holzmuller H., Binder B. R., Hofer E. (1999). Vascular endothelial cell growth factor-induced tissue factor expression in endothelial cells is mediated by EGR-1. 93 3811–3823. [PubMed] [Google Scholar]
- Mexal S., Frank M., Berger R., Adams C. E., Ross R. G., Freedman R., et al. (2005). Differential modulation of gene expression in the NMDA postsynaptic density of schizophrenic and control smokers. 139 317–332. 10.1016/j.molbrainres.2005.06.006 [DOI] [PubMed] [Google Scholar]
- Mittelstadt P. R., Ashwell J. D. (1998). Cyclosporin A-sensitive transcription factor Egr-3 regulates Fas ligand expression. 18 3744–3751. 10.1128/MCB.18.7.3744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moises H. W., Zoega T., Gottesman I. I. (2002). The glial growth factors deficiency and synaptic destabilization hypothesis of schizophrenia. 2:8 10.1186/1471-244X-2-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteggia L. M., Barrot M., Powell C. M., Berton O., Galanis V., Gemelli T., et al. (2004). Essential role of brain-derived neurotrophic factor in adult hippocampal function. 101 10827–10832. 10.1073/pnas.0402141101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning Q. L., Ma X. D., Jiao L. Z., Niu X. R., Li J. P., Wang B., et al. (2012). [A family-based association study of the EGR3 gene polymorphisms and schizophrenia]. 34 307–314. 10.3724/SP.J.1005.2012.00307 [DOI] [PubMed] [Google Scholar]
- Novkovic T., Mittmann T., Manahan-Vaughan D. (2015). BDNF contributes to the facilitation of hippocampal synaptic plasticity and learning enabled by environmental enrichment. 25 1–15. 10.1002/hipo.22342 [DOI] [PubMed] [Google Scholar]
- O’Donovan K. J., Wilkens E. P., Baraban J. M. (1998). Sequential expression of Egr-1 and Egr-3 in hippocampal granule cells following electroconvulsive stimulation. 70 1241–1248. 10.1046/j.1471-4159.1998.70031241.x [DOI] [PubMed] [Google Scholar]
- Olney J. W., Newcomer J. W., Farber N. B. (1999). NMDA receptor hypofunction model of schizophrenia. 33 523–533. 10.1016/S0022-3956(99)00029-1 [DOI] [PubMed] [Google Scholar]
- Pagnin D., de Queiroz V., Pini S., Cassano G. B. (2004). Efficacy of ECT in depression: a meta-analytic review. 20 13–20. 10.1097/00124509-200403000-00004 [DOI] [PubMed] [Google Scholar]
- Paxinos G., Franklin K. B. J. (2001). 2nd Edn. Cambridge, MA: Academic Press. [Google Scholar]
- Perera T. D., Coplan J. D., Lisanby S. H., Lipira C. M., Arif M., Carpio C., et al. (2007). Antidepressant-induced neurogenesis in the hippocampus of adult nonhuman primates. 27 4894–4901. 10.1523/JNEUROSCI.0237-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffenseller B., da Silva Magalhaes P. V., De Bastiani M. A., Castro M. A., Gallitano A. L., Kapczinski F., et al. (2016). Differential expression of transcriptional regulatory units in the prefrontal cortex of patients with bipolar disorder: potential role of early growth response gene 3. 6:e805. 10.1038/tp.2016.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich M., Liefeld T., Gould J., Lerner J., Tamayo P., Mesirov J. P. (2006). GenePattern 2.0. 38 500–501. 10.1038/ng0506-500 [DOI] [PubMed] [Google Scholar]
- Roberts D. S., Hu Y., Lund I. V., Brooks-Kayal A. R., Russek S. J. (2006). Brain-derived neurotrophic factor (BDNF)-induced synthesis of early growth response factor 3 (Egr3) controls the levels of type A GABA receptor alpha 4 subunits in hippocampal neurons. 281 29431–29435. 10.1074/jbc.C600167200 [DOI] [PubMed] [Google Scholar]
- Roberts D. S., Raol Y. H., Bandyopadhyay S., Lund I. V., Budreck E. C., Passini M. A., et al. (2005). Egr3 stimulation of GABRA4 promoter activity as a mechanism for seizure-induced up-regulation of GABA(A) receptor alpha4 subunit expression. 102 11894–11899. 10.1073/pnas.0501434102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenquist P. B., Miller B., Pillai A. (2014). The antipsychotic effects of ECT: a review of possible mechanisms. 30 125–131. 10.1097/YCT.0000000000000131 [DOI] [PubMed] [Google Scholar]
- Sakata K., Martinowich K., Woo N. H., Schloesser R. J., Jimenez D. V., Ji Y., et al. (2013). Role of activity-dependent BDNF expression in hippocampal-prefrontal cortical regulation of behavioral perseverance. 110 15103–15108. 10.1073/pnas.1222872110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata K., Woo N. H., Martinowich K., Greene J. S., Schloesser R. J., Shen L., et al. (2009). Critical role of promoter IV-driven BDNF transcription in GABAergic transmission and synaptic plasticity in the prefrontal cortex. 106 5942–5947. 10.1073/pnas.0811431106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014). Biological insights from 108 schizophrenia-associated genetic loci. 511 421–427. 10.1038/nature13595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma T., Antonova L. (2003). Cognitive function in schizophrenia. Deficits, functional consequences, and future treatment. 26 25–40. 10.1016/S0193-953X(02)00084-9 [DOI] [PubMed] [Google Scholar]
- Shieh P. B., Ghosh A. (1999). Molecular mechanisms underlying activity-dependent regulation of BDNF expression. 41 127–134. [DOI] [PubMed] [Google Scholar]
- Shieh P. B., Hu S. C., Bobb K., Timmusk T., Ghosh A. (1998). Identification of a signaling pathway involved in calcium regulation of BDNF expression. 20 727–740. 10.1016/S0896-6273(00)81011-9 [DOI] [PubMed] [Google Scholar]
- Shirayama Y., Chen A. C., Nakagawa S., Russell D. S., Duman R. S. (2002). Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. 22 3251–3261. 10.1523/JNEUROSCI.22-08-03251.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sholl D. A. (1953). Dendritic organization in the neurons of the visual and motor cortices of the cat. 87 387–406. [PMC free article] [PubMed] [Google Scholar]
- Tallaksen-Greene S. J., Sadagurski M., Zeng L., Mauch R., Perkins M., Banduseela V. C., et al. (2014). Differential effects of delayed aging on phenotype and striatal pathology in a murine model of Huntington disease. 34 15658–15668. 10.1523/JNEUROSCI.1830-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao X., West A. E., Chen W. G., Corfas G., Greenberg M. E. (2002). A calcium-responsive transcription factor, CaRF, that regulates neuronal activity-dependent expression of BDNF. 33 383–395. 10.1016/S0896-6273(01)00561-X [DOI] [PubMed] [Google Scholar]
- Thomann P. A., Wolf R. C., Nolte H. M., Hirjak D., Hofer S., Seidl U., et al. (2017). Neuromodulation in response to electroconvulsive therapy in schizophrenia and major depression. 10 637–644. 10.1016/j.brs.2017.01.578 [DOI] [PubMed] [Google Scholar]
- Thompson C. L., Wisor J. P., Lee C. K., Pathak S. D., Gerashchenko D., Smith K. A., et al. (2010). Molecular and anatomical signatures of sleep deprivation in the mouse brain. 4:165. 10.3389/fnins.2010.00165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmusk T., Palm K., Metsis M., Reintam T., Paalme V., Saarma M., et al. (1993). Multiple promoters direct tissue-specific expression of the rat BDNF gene. 10 475–489. 10.1016/0896-6273(93)90335-O [DOI] [PubMed] [Google Scholar]
- Tourtellotte W. G., Milbrandt J. (1998). Sensory ataxia and muscle spindle agenesis in mice lacking the transcription factor Egr3. 20 87–91. 10.1038/1757 [DOI] [PubMed] [Google Scholar]
- Tsunoda T., Takagi T. (1999). Estimating transcription factor bindability on DNA. 15 622–630. 10.1093/bioinformatics/15.7.622 [DOI] [PubMed] [Google Scholar]
- van Pelt J., Verwer R. W., Uylings H. B. (1986). Application of growth models to the topology of neuronal branching patterns. 18 153–165. 10.1016/0165-0270(86)90117-2 [DOI] [PubMed] [Google Scholar]
- van Strien N. M., Cappaert N., Witter M. P. (2009). The anatomy of memory: an interactive overview of the parahippocampal-hippocampal network. 10 272–282. 10.1038/nrn2614 [DOI] [PubMed] [Google Scholar]
- West A. E. (2008). Biological functions of activity-dependent transcription revealed. 60 523–525. 10.1016/j.neuron.2008.11.008 [DOI] [PubMed] [Google Scholar]
- West A. E., Chen W. G., Dalva M. B., Dolmetsch R. E., Kornhauser J. M., Shaywitz A. J., et al. (2001). Calcium regulation of neuronal gene expression. 98 11024–11031. 10.1073/pnas.191352298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams A. A., Ingram W. M., Levine S., Resnik J., Kamel C. M., Lish J. R., et al. (2012). Reduced levels of serotonin 2A receptors underlie resistance of Egr3-deficient mice to locomotor suppression by clozapine. 37 2285–2298. 10.1038/npp.2012.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo N. H., Teng H. K., Siao C. J., Chiaruttini C., Pang P. T., Milner T. A., et al. (2005). Activation of p75NTR by proBDNF facilitates hippocampal long-term depression. 8 1069–1077. 10.1038/nn1510 [DOI] [PubMed] [Google Scholar]
- Yamada K., Gerber D. J., Iwayama Y., Ohnishi T., Ohba H., Toyota T., et al. (2007). Genetic analysis of the calcineurin pathway identifies members of the EGR gene family, specifically EGR3, as potential susceptibility candidates in schizophrenia. 104 2815–2820. 10.1073/pnas.0610765104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Harte-Hargrove L. C., Siao C. J., Marinic T., Clarke R., Ma Q., et al. (2014). proBDNF negatively regulates neuronal remodeling, synaptic transmission, and synaptic plasticity in hippocampus. 7 796–806. 10.1016/j.celrep.2014.03.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youssef N. A., McCall W. V. (2014). Relapse prevention after index electroconvulsive therapy in treatment-resistant depression. 26 288–296. [PMC free article] [PubMed] [Google Scholar]
- Zakharyan R., Atshemyan S., Gevorgyan A., Boyajyan A. (2014). Nerve growth factor and its receptor in schizophrenia. 1 24–29. 10.1016/j.bbacli.2014.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R., Lu S., Meng L., Min Z., Tian J., Valenzuela R. K., et al. (2012). Genetic evidence for the association between the early growth response 3 (EGR3) gene and schizophrenia. 7:e30237. 10.1371/journal.pone.0030237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng F., Wang H. (2009). NMDA-mediated and self-induced bdnf exon IV transcriptions are differentially regulated in cultured cortical neurons. 54 385–392. 10.1016/j.neuint.2009.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]