

Abstract

Glycosyl cations are crucial intermediates formed during enzymatic and chemical glycosylation. The intrinsic high reactivity and short lifetime of these reaction intermediates make them very challenging to characterize using spectroscopic techniques. Herein, we report the use of collision induced dissociation tandem mass spectrometry to generate glycosyl cations in the gas phase followed by infrared ion spectroscopy using the FELIX infrared free electron laser. The experimentally observed IR spectra were compared to DFT calculated spectra enabling the detailed structural elucidation of elusive glycosyl oxocarbenium and dioxolenium ions.
The principal challenge in chemical oligosaccharide synthesis is the stereoselective synthesis of glycosidic bonds.1 Glycosidic bonds connecting monosaccharides can exist as α- or β-diastereomers. The most common approach to chemically prepare glycosidic bonds is via a nucleophilic substitution reaction between a glycosyl donor carrying an anomeric leaving group and a glycosyl acceptor containing a nucleophilic alcohol. Depending on the nature of the glycosyl donor, acceptor, and reaction parameters, the mechanism of a glycosylation is best described as a continuum between SN1-like and SN2-like reaction pathways (Scheme 1).2
Scheme 1. Reactive Intermediates in Glycosylation Reactions and Their Respective Reaction Pathways.
LG = Leaving group, E = Electrophile, A = Anion, P = Protecting group.
Stereoselective glycosylation can be achieved by the SN2-like displacement of a contact ion pair or covalent intermediate such as glycosyl triflates,3 sulfonium ions,4,5 and dioxolenium ions6 (Scheme 1). Reactions via this pathway are well studied as the reaction intermediates can be characterized by low temperature NMR spectroscopy.3,7,8 Loss of stereoselectivity during the displacement of the contact ion pair intermediates is often attributed to an alternative reaction pathway via the solvent separated ion pair, also known as the oxocarbenium ion. Hence, to fully understand the mechanistic pathways of glycosylation reactions, it is crucial to elucidate the structure of its intermediates such as glycosyl oxocarbenium ions. However, glycosyl oxocarbenium ions are very challenging to characterize due to their intrinsic high reactivity, short lifetimes, and equilibrium with the corresponding contact ion pair. Very recently, NMR spectroscopy of glycosyl oxocarbenium ions was reported in super acid solution.9 However, the harsh conditions used to generate the oxocarbenium ions are not compatible with all types of protecting groups commonly used in oligosaccharide synthesis. Tandem mass spectrometry (MS/MS) can be employed to generate glycosyl cations in complete isolation, and their gas-phase fragmentation has been extensively studied.10−14 Recently, the use of MS in combination with infrared (IR) ion spectroscopy has emerged as a powerful method to determine the gas-phase structures of molecular ions in MS/MS15−20 and to probe glycan structure.21−26
Herein we report the use of collisional dissociation to generate glycosyl cations followed by characterization using infrared (IR) ion spectroscopy. Electrospray ionization (ESI) of four glycosyl donors was employed to form a parent ion which was fragmented using collision induced dissociation (CID) affording the desired glycosyl cations. Crucially, the absence of a counterion and solvent led to “naked” cations that could be characterized using IR ion spectroscopy. The IR spectra showed specific diagnostic vibrational bands enabling the assignment of oxocarbenium and dioxolenium ions. In addition, a mixture of two dioxolenium ion isomers could be resolved thereby providing insight into the dynamics of these elusive intermediates.
We synthesized mannosyl donors 1–4 as substrates, and the commonly used benzyl ethers were replaced by methyl ethers in order to eliminate IR signals resulting from the aromatic systems (Scheme 2). Thioglycosides were selected, as they are frequently used glycosyl donors and have been used to study the formation and fragmentation of glycosyl cations using CID.27−29
Scheme 2. Structures of the Mannosyl Donors Used in This Study.

Ionization of mannosyl donor 1 was achieved using ESI resulting in the parent ion [M+NH4]+ with an m/z ratio of 458. Subsequent isolation and CID led to the formation of various fragment ions, most notably the mannosyl cation with an m/z ratio of 331 (Figure S1). This fragment ion was isolated in a quadrupole ion trap30 and subsequently characterized by IR ion spectroscopy using the FELIX laser operating in the 700–1850 cm–1 range (for details see Supporting Information). The mannosyl cation derived from 1, produced an IR spectrum containing a wealth of well-resolved and diagnostic peaks (Figure 1, black line). To explore the generality of this approach, gluco- and galactoside donors were also prepared, fragmented, and characterized by IR ion spectroscopy producing similar, yet distinct IR spectra (Figure S2).
Figure 1.
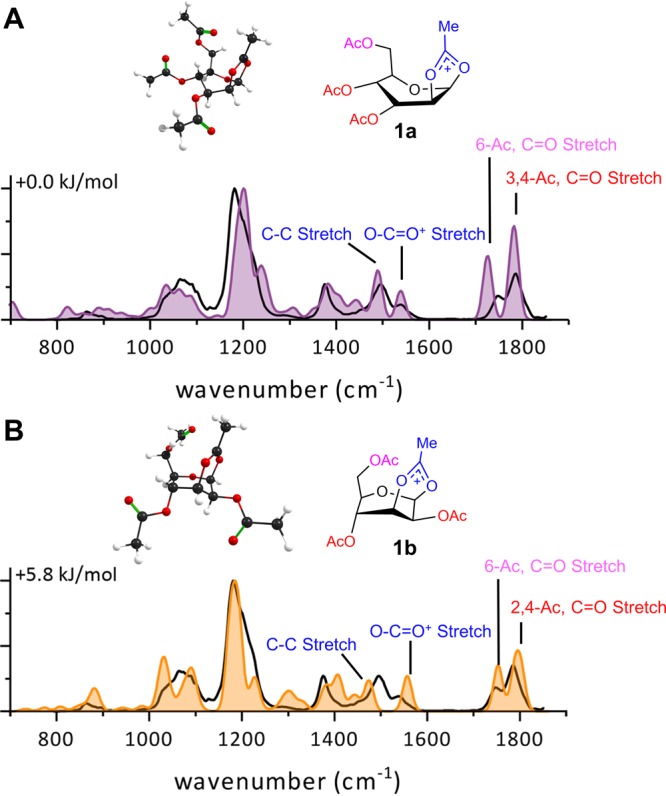
Comparison of the calculated spectra (filled) of 2-O-acetyl- (1a) or 3-O-acetyl- (1b) participation with the measured IR ion spectrum of mannosyl cation derived from 1 (black line in both panels).
To obtain structural information on the formed ions, IR ion spectroscopy is combined with high level ab initio calculations.31 Calculated IR spectra from selected low energy structures were compared with the experimental IR spectra for structural assignments.
In our spectral assessment, peak positions (cm–1) are most important since peak intensities in the calculated spectrum are based on linear IR absorption, whereas the measured spectra rely on multiple-photon absorption. Initial optimization indicated that DFT calculations at the B3LYP/6-31++G(d, p) level, with relative energies determined using MP2/6-311++G(2d, 2p), proved to provide the most representative spectra (Figure S3). It is well established that acetyl protected donors produce dioxolenium ions via neighboring group participation.32 The experimental IR spectrum was compared with the calculated IR spectra of the lowest energy isomers of four different structural families with m/z = 331. Participation of the C-2, C-3, C-4, or C-6 acetyl ester was considered together with the unstabilized oxocarbenium ion (Figure S4). The lowest energy structure resulted from C-2 acetyl participation followed by C-3 participation (+5.8 kJ/mol), while C-4/C-6 participation or no participation displays significant higher energies (+34.2–40.3 kJ/mol). Based on the energies themselves, the latter three structures are unlikely to be present. This is further supported by comparing the calculated and experimental IR spectra. The calculated C=O+ stretch of the oxocarbenium ion would appear at 1616 cm–1 (Figure S4); however, this signal is absent in the measured spectrum. The participation of acetyl esters can be assigned by the calculated O–C=O+ stretch (∼1540 cm–1) and the C–C stretch (∼1495 cm–1) modes of the dioxolenium ion, whereas the C=O stretch vibration of the nonparticipating acetyl esters are found at 1725–1800 cm–1. The C-6 acetyl C=O stretch (1725 cm–1) is distinct from the other esters and clearly visible in the measured IR spectrum. Calculations show that this signature is lost upon C-6 acetyl participation thereby excluding it (Figure S4). Dioxolenium ions resulting from either C-2 (1a) or C-3 (1b) ester participation both produce low energy structures (Figure 1). However, when comparing the calculated peaks originating from the O–C=O+ stretch and C–C stretch vibrations of the dioxolenium ion (1538 and 1496 cm–1), it is clear that C-2 participation is in much better agreement with the experimentally observed spectrum (1a, Figure 1A) than C-3 participation (1b, Figure 1B). Finally, the assignment that the activation of 1 leads to a C-2-dioxolenium ion is consistent with previous NMR studies.32 Hence, isobaric glycosyl cation fragments can be distinguished on the basis of their IR spectrum using DFT calculations.
Next, we investigated the characterization of the more challenging oxocarbenium ion using permethylated mannosyl donor 2 to limit the possibilities for neighboring group participation. ESI of 2 followed by CID resulted in the desired fragment ion with m/z = 219 (Figure S5). Subsequent IR ion spectroscopy yielded an IR spectrum rich in diagnostic peaks for structural characterization (Figure 2, black line). DFT calculations for oxocarbenium ions resulting from 2 yielded two main structural families which differ in energy by ∼13 kJ/mol (2a and 2b, Figure 2, color fill). Both structures are oxocarbenium ions, but differ in the conformation of the pyranose ring, 3E (2a) and 4H3 (2b). Most notably, the calculated C=O+ stretch, in 2a and 2b, corresponding to the C-1 oxocarbenium ion (1609 cm–1) was indeed observed in the measured spectrum at 1585 cm–1 (Figure 2A,B). The lowest energy structure, the 3E envelope, displayed the best fit with the observed spectrum. Previous calculations by Whitfield suggested a two conformer hypothesis with 4H3 and 3E conformations as the lowest energy states.33 The calculated spectra of the 3E (2a) and 4H3 (2b) conformers are very similar, but the additional signal at 1058 cm–1 (C-3–C-4 and C-2–O-2 stretch) predicted for the 4H3 conformer is absent in the measured spectrum (Figure 2B). In the calculated 3E conformation, the same C-3–C-4 and C-2–O-2 interactions are calculated at lower wavenumbers (991 cm–1) and are in better agreement with the measured spectrum. Together with the fact that the 4H3 has a higher energy, this suggests the 3E is the preferred conformer. These data indicate that elusive oxocarbenium ions can be generated and characterized at a highly detailed level using diagnostic peaks indicative of pyranose conformation.
Figure 2.

Comparison of the calculated spectra (filled) of the 3E (2a) or 4H3 (2b) oxocarbenium ion conformer with the measured IR ion spectrum of the m/z = 219 CID fragment of 2 (black line in both panels). Energies are relative to the 0.0 kJ/mol structure as reported in Figure S6.
The conformational flexibility of glycosyl oxocarbenium ions is an important factor determining the stereochemical outcome of a glycosylation reaction. Much effort has been directed to the creation of oxocarbenium ions that display a well-defined conformation to induce stereoselectivity via the preferential attack on one diastereotopic face.35−38 As a prime example, oxocarbenium ions derived from mannuronic acid esters 3 and 4 are expected to favor the 3H4 conformation due to stabilization of the oxocarbenium ion by the pseudo axial C-6 carboxylate ester.34,35 Subsequent pseudo axial attack is favorable, as it directly leads to the β-product in the chair conformation. In addition, it is also known that the α-triflate intermediate contact ion pair is formed in reactions with 3, leading to the β-product via SN2-like displacement.39 Hence, to elucidate the solvent separated ion pair side of this continuum, we characterized cations resulting from the fragmentation of 3 and 4.
Ionization and fragmentation resulted in the corresponding mannuronyl cations (Figure S7). Well-resolved IR spectra were obtained displaying distinct diagnostic peaks for permethylated mannuronic acid 3 (Figure 3A, black line). DFT-calculations indicated that participation of the C-6 ester provided the lowest energy structure 3a (Figure 3A, color fill).
Figure 3.
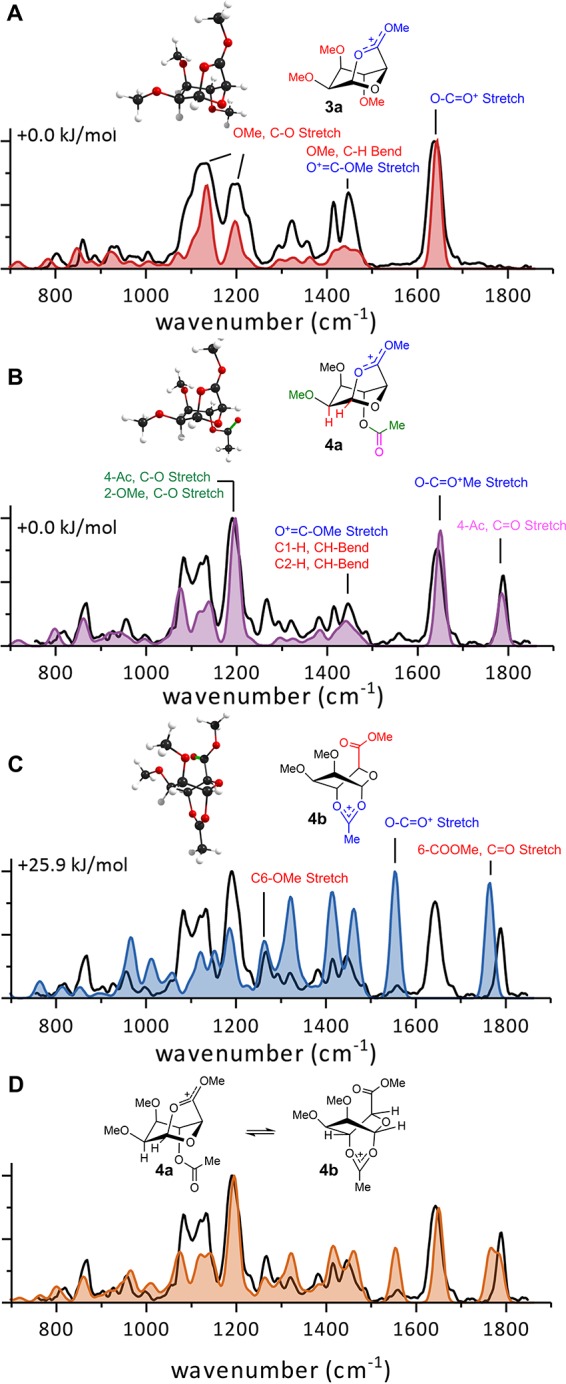
Comparison of calculated spectra (filled) with the measured IR ion spectra (black line) of mannuronic acids 3a, 4a, 4b (A–C) and a mixture of isomers 4a and 4b (D).
The measured spectrum is in good agreement with the calculated spectrum of this structure. The peak observed at 1637 cm–1 corresponds well with the calculated peak at 1644 cm–1 describing the diagnostic O–C=O+ stretch vibration. The C−H bend modes of the methyl ester located at ∼1409–1487 cm–1 in the stabilized oxocarbenium ion also have clear overlap (3a, Figure 3A). Furthermore, the various C−H bending modes belonging to the methoxy and ring hydrogens were identified in the calculated spectrum and showed clear overlap with peaks in the measured spectrum. Isomers in which the C-6 ester does not participate were also considered; however, as the characteristic calculated C=O stretch vibration at ∼1800 cm–1 is not observed in the experimental IR spectrum, we can discard these structures (Figure S8). Hence, these data strongly suggest C-6 ester participation upon activation of 3.
Finally, mannuronic acid ester donor 4 containing a C-4 ester was investigated. In this case, ester participation from both diastereotopic faces of the oxocarbenium ion should be possible. The IR spectra of the isolated ions of 4 were obtained and yield a well-resolved spectrum (Figure 3B–D, black line). Initial DFT calculations pointed toward structure 4a resulting from C-6 ester participation as observed with 3 (Figure 3B). All major calculated peaks such as C−H bending, C-6 O–C=O+ stretch, and 4-Ac C=O stretch could be assigned and showed excellent agreement between the experimental and theoretical IR spectrum (Figure 3B). However, distinct peaks in the measured spectrum observed at 1557 and 1267 cm–1 were not accounted for by the calculation. The computed spectrum of isomer 4b with C-4 acetyl participation shows clear activity at these vibrational frequencies (Figure 3C). The most characteristic peak calculated at 1554 cm–1 (4-Ac, O–C=O+ stretch) is highly diagnostic for C-4 acetyl participation whereas the peak at 1261 cm–1 (C-6–OMe stretch) belongs to the nonparticipating methyl ester. Mixing of the two calculated spectra belonging to both participation modes (4a and 4b) led to an excellent fit of the observed spectrum (Figure 3D). This result highlights the possibility to characterize a mixture of isomers using IR ion spectroscopy thereby providing crucial insight in the dynamics of glycosyl cations.
In conclusion, the use of mass spectrometry coupled with IR ion spectroscopy and quantum chemical calculations is a mild and versatile method to generate and structurally characterize elusive glycosyl cations in the gas phase. While we acknowledge that gas phase experiments are unlikely to be fully representative of glycosyl cation structure in solution, the absence of counterions and solvent allows for the detailed study of the intrinsic structure and dynamics of these intermediates. Furthermore, the IR spectra displayed a wealth of diagnostic peaks that could be used to elucidate conformational preferences as well as mixtures of isomeric dioxolenium ions. We therefore believe that this MS-IR based methodology will be valuable to advance our fundamental understanding SN1 pathways in glycosylation reactions and likely also other reactions proceeding through ionic intermediates.
Acknowledgments
This work was supported by an NWO VENI and ERC-Stg grant awarded to T.J.B. and an NWO VICI grant awarded to J.O. Calculations were carried out at the SurfSARA Cartesius cluster under NWO Rekentijd Contract 16327. We gratefully acknowledge the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO) for the support of the FELIX Laboratory.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b01236.
Experimental procedure and spectra for the synthesis of 1–4 as well as additional ESI and CID spectra and DFT calculations (PDF)
Author Contributions
§ H.E. and M.S. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Boltje T. J.; Buskas T.; Boons G.-J. Nat. Chem. 2009, 1, 611. 10.1038/nchem.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horenstein N. A. In Advances in Physical Organic Chemistry; Richard J. P., Ed.; Elsevier: New York, 2006; Vol. 41, p 275. [Google Scholar]
- Crich D. Acc. Chem. Res. 2010, 43, 1144. 10.1021/ar100035r. [DOI] [PubMed] [Google Scholar]
- Fang T.; Gu Y.; Huang W.; Boons G.-J. J. Am. Chem. Soc. 2016, 138, 3002. 10.1021/jacs.5b08436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mensink R. A.; Boltje T. J. Chem. - Eur. J. 2017, 23, 17637. 10.1002/chem.201700908. [DOI] [PubMed] [Google Scholar]
- Paulsen H.; Herold C.-P. Chem. Ber. 1970, 103, 2450. 10.1002/cber.19701030817. [DOI] [PubMed] [Google Scholar]
- Frihed T. G.; Bols M.; Pedersen C. M. Chem. Rev. 2015, 115, 4963. 10.1021/cr500434x. [DOI] [PubMed] [Google Scholar]
- Crich D.; Dai Z.; Gastaldi S. J. Org. Chem. 1999, 64, 5224. 10.1021/jo990424f. [DOI] [PubMed] [Google Scholar]
- Martin A.; Arda A.; Désiré J.; Martin-Mingot A.; Probst N.; Sinaÿ P.; Jiménez-Barbero J.; Thibaudeau S.; Blériot Y. Nat. Chem. 2016, 8, 186. 10.1038/nchem.2399. [DOI] [PubMed] [Google Scholar]
- Khanal N.; Masellis C.; Kamrath M. Z.; Clemmer D. E.; Rizzo T. R. Anal. Chem. 2017, 89, 7601. 10.1021/acs.analchem.7b01467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina E. R.; Eizaguirre A.; Haldys V.; Urban D.; Doisneau G.; Bourdreux Y.; Beau J.-M.; Salpin J.-Y.; Spezia R. ChemPhysChem 2017, 18, 2812. 10.1002/cphc.201700202. [DOI] [PubMed] [Google Scholar]
- Rabus J. M.; Abutokaikah M. T.; Ross R. T.; Bythell B. J. Phys. Chem. Chem. Phys. 2017, 19, 25643. 10.1039/C7CP04738J. [DOI] [PubMed] [Google Scholar]
- Chen J.-L.; Nguan H. S.; Hsu P.-J.; Tsai S.-T.; Liew C. Y.; Kuo J.-L.; Hu W.-P.; Ni C.-K. Phys. Chem. Chem. Phys. 2017, 19, 15454. 10.1039/C7CP02393F. [DOI] [PubMed] [Google Scholar]
- Bythell B. J.; Abutokaikah M. T.; Wagoner A. R.; Guan S.; Rabus J. M. J. Am. Soc. Mass Spectrom. 2017, 28, 688. 10.1007/s13361-016-1530-x. [DOI] [PubMed] [Google Scholar]
- Martens J.; Grzetic J.; Berden G.; Oomens J. Nat. Commun. 2016, 7, 11754. 10.1038/ncomms11754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick A. L.; Stedwell C. N.; Polfer N. C. Anal. Chem. 2014, 86, 5547. 10.1021/ac500992f. [DOI] [PubMed] [Google Scholar]
- Seo J.; Warnke S.; Pagel K.; Bowers M. T.; von Helden G. Nat. Chem. 2017, 9, 1263. 10.1038/nchem.2821. [DOI] [PubMed] [Google Scholar]
- Zhu Y.; Roy H. A.; Cunningham N. A.; Strobehn S. F.; Gao J.; Munshi M. U.; Berden G.; Oomens J.; Rodgers M. T. Phys. Chem. Chem. Phys. 2017, 19, 17637. 10.1039/C7CP02377D. [DOI] [PubMed] [Google Scholar]
- Zhu Y.; Hamlow L. A.; He C. C.; Lee J. K.; Gao J.; Berden G.; Oomens J.; Rodgers M. T. J. Phys. Chem. B 2017, 121, 4048. 10.1021/acs.jpcb.7b02906. [DOI] [PubMed] [Google Scholar]
- Zhu Y.; Yang Z.; Rodgers M. T. J. Am. Soc. Mass Spectrom. 2017, 28, 2602. 10.1007/s13361-017-1780-2. [DOI] [PubMed] [Google Scholar]
- Gray C. J.; Schindler B.; Migas L. G.; Pičmanová M.; Allouche A. R.; Green A. P.; Mandal S.; Motawia M. S.; Sánchez-Pérez R.; Bjarnholt N.; Møller B. L.; Rijs A. M.; Barran P. E.; Compagnon I.; Eyers C. E.; Flitsch S. L. Anal. Chem. 2017, 89, 4540. 10.1021/acs.analchem.6b04998. [DOI] [PubMed] [Google Scholar]
- Schindler B.; Barnes L.; Renois G.; Gray C.; Chambert S.; Fort S.; Flitsch S.; Loison C.; Allouche A.-R.; Compagnon I. Nat. Commun. 2017, 8, 973. 10.1038/s41467-017-01179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens J.; Berden G.; van Outersterp R. E.; Kluijtmans L. A. J.; Engelke U. F.; van Karnebeek C. D. M.; Wevers R. A.; Oomens J. Sci. Rep. 2017, 7, 3363. 10.1038/s41598-017-03387-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucha E.; Florez A. I. G.; Marianski M.; Thomas D. A.; Hoffmann W.; Struwe W. B.; Hahm H. S.; Gewinner S.; Schollkopf W.; Seeberger P. H.; von Helden G.; Pagel K. Angew. Chem., Int. Ed. 2017, 56, 11248. 10.1002/anie.201702896. [DOI] [PubMed] [Google Scholar]
- Contreras C. S.; Polfer N. C.; Oomens J.; Steill J. D.; Bendiak B.; Eyler J. R. Int. J. Mass Spectrom. 2012, 330-332, 285. 10.1016/j.ijms.2012.09.010. [DOI] [Google Scholar]
- Cocinero E. J.; Carcabal P.; Vaden T. D.; Simons J. P.; Davis B. G. Nature 2011, 469, 76. 10.1038/nature09693. [DOI] [PubMed] [Google Scholar]
- Kancharla P. K.; Navuluri C.; Crich D. Angew. Chem., Int. Ed. 2012, 51, 11105. 10.1002/anie.201204400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denekamp C.; Sandlers Y. J. Mass Spectrom. 2005, 40, 1055. 10.1002/jms.880. [DOI] [PubMed] [Google Scholar]
- Denekamp C.; Sandlers Y. J. Mass Spectrom. 2005, 40, 765. 10.1002/jms.848. [DOI] [PubMed] [Google Scholar]
- Martens J.; Berden G.; Gebhardt C. R.; Oomens J. Rev. Sci. Instrum. 2016, 87, 103108. 10.1063/1.4964703. [DOI] [PubMed] [Google Scholar]
- Rijs A. M.; Oomens J. In Gas-Phase IR Spectroscopy and Structure of Biological Molecules; Rijs A. M., Oomens J., Eds.; Springer International Publishing: Cham, 2015; p 1. [Google Scholar]
- Perlin A. S. Can. J. Chem. 1963, 41, 399. 10.1139/v63-058. [DOI] [Google Scholar]
- Nukada T.; Bérces A.; Wang L.; Zgierski M. Z.; Whitfield D. M. Carbohydr. Res. 2005, 340, 841. 10.1016/j.carres.2004.12.021. [DOI] [PubMed] [Google Scholar]
- Walvoort M. T. C.; Dinkelaar J.; van den Bos L. J.; Lodder G.; Overkleeft H. S.; Codée J. D. C.; van der Marel G. A. Carbohydr. Res. 2010, 345, 1252. 10.1016/j.carres.2010.02.027. [DOI] [PubMed] [Google Scholar]
- Codée J. D. C.; Walvoort M. T. C.; de Jong A.-R.; Lodder G.; Overkleeft H. S.; van der Marel G. A. J. Carbohydr. Chem. 2011, 30, 438. 10.1080/07328303.2011.624284. [DOI] [Google Scholar]
- Zhu X.; Kawatkar S.; Rao Y.; Boons G.-J. J. Am. Chem. Soc. 2006, 128, 11948. 10.1021/ja0629817. [DOI] [PubMed] [Google Scholar]
- Abe H.; Shuto S.; Matsuda A. J. Am. Chem. Soc. 2001, 123, 11870. 10.1021/ja011321t. [DOI] [PubMed] [Google Scholar]
- Ayala L.; Lucero C. G.; Romero J. A. C.; Tabacco S. A.; Woerpel K. A. J. Am. Chem. Soc. 2003, 125, 15521. 10.1021/ja037935a. [DOI] [PubMed] [Google Scholar]
- Walvoort M. T. C.; Lodder G.; Mazurek J.; Overkleeft H. S.; Codée J. D. C.; van der Marel G. A. J. Am. Chem. Soc. 2009, 131, 12080. 10.1021/ja905008p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.