

Abstract
Fibroblast growth factor (FGF) signaling has been implicated in the regulation of osteogenesis in both intramembranous and endochondral ossifications. In the developing palate, the anterior bony palate forms by direct differentiation of cranial neural crest (CNC)–derived mesenchymal cells, but the signals that regulate the osteogenic cell fate in the developing palate remain unclear. In the present study, we investigated the potential role of FGF signaling in osteogenic fate determination of the palatal mesenchymal cells. We showed that locally activated FGF8 signaling in the anterior palate using a Shox2Cre knock-in allele and an R26RFgf8 allele leads to a unique palatal defect: a complete loss of the palatine process of the maxilla as well as formation of ectopic cartilaginous tissues in the anterior palate. This aberrant developmental process was accompanied by a significantly elevated level of cell proliferation, which contributes to an abnormally thickened palatal tissue, where the palatine process of the maxilla would normally form, and by a complete inhibition of Osterix expression, which accounts for the lack of bone formation. The coexpression of Runx2 initially with Sox9 and subsequently with Col II in the ectopic cartilaginous tissues indicates a conversion of osteogenic fate to a chondrogenic one. Consistent with the unique palatal phenotype, RNA-Sequencing analysis revealed that the augmented FGF8 signaling downregulated genes involved in ossification, biomineral tissue development, and bone mineralization but upregulated genes involved in cell proliferation, cartilage development, and cell fate commitment, which was further supported by quantitative real-time reverse transcription polymerase chain reaction validation of selected genes. Our results demonstrate that FGF8 signaling functions as a negative regulator of osteogenic fate and is sufficient to convert a subset of CNC cell-derived mesenchymal cells into cartilage in the anterior hard palate, which will have implications in future directed differentiation of CNC-derived precursor cells for clinical application.
Keywords: neural crest, fibroblast growth factors, osteogenesis, chondrogenesis, cell transdifferentiation, morphogenesis
Introduction
Intramembranous and endochondral ossifications are 2 mechanisms that mediate bone formation in vertebrate skeletal development (Zelzer and Olsen 2003). Mesenchymal cells either differentiate directly into osteoblasts (intramembranous ossification) or differentiate into chondrocytes and lay down a cartilaginous model that is later replaced by bone (endochondral ossification). In the craniofacial region, skeletons develop from cranial neural crest (CNC) cell–derived chondrocytes and osteoblasts, but most of the craniofacial bones, including the skull, maxillomandibular bone, and palate, form through intramembranous ossification (Santagati and Rijli 2003). It is generally accepted that cell fate determination is controlled by multiple signaling pathways and specific transcriptional factors. Runx2 controls osteogenic fate of osteoprogenitor cells (Pratap et al. 2003), while Sox9 is required for chondrogenic fate (Mori-Akiyama et al. 2003). Osterix, the downstream transcription factor of Runx2, marks osteoblasts and is required to ensure full differentiation of osteoblasts (Nakashima et al. 2002). Inactivation of key transcription factors for osteogenesis could also lead to cell fate change, as exemplified by the presence of ectopic chondrocytes in the regions where intramembranous bone would form in mice lacking Osterix and abnormal chondrogenic differentiation at the expense of osteoblast differentiation in mesenchymal progenitor cells lacking β-catenin (Nakashima et al. 2002; Day et al. 2005).
Fibroblast growth factor (FGF) signaling has been implicated in the regulation of craniofacial skeletal development. Mutations in FGF receptors, including FGFR1, FGFR2, and FGFR3, result in craniosynostosis and maxillary hypoplasia (Malcolm and Reardon 1996; Wilkie 1997; Burke et al. 1998). Fgf18 is required for both osteogenesis and chondrogenesis, as disruption of Fgf18 has led to defective calvarial and long bones (Ohbayashi et al. 2002). Although Fgfr3 inactivation caused skeletal abnormalities and reduced bone density (Yu et al. 2003), Fgfr3 overactivation in both humans and mice also resulted in mandibular bone hypoplasia and dysmorphogenesis (Biosse Duplan et al. 2016), indicating the importance of finely tuned FGF signaling in osteogenesis. In the developing craniofacial region, Fgf8 was shown to be essential for the first pharyngeal arch development in humans and mice, evidenced by bilateral cleft palate in humans carrying D73H missense mutation in FGF8 and defective maxillomandibular bone formation in Fgf8 hypomorphic Fgf8neo/– mice (Abu-Issa et al. 2002; Riley et al. 2007). We showed previously that ectopic activation of Fgf8 in CNC cells in mice inhibits differentiation of CNC-derived mesenchymal cells in the orofacial region and sustains their progenitor status (Shao et al. 2015). Interestingly, in vitro cell differentiation assays demonstrated that FGF8 signaling promotes differentiation of adipogenic, chondrogenic, or neurogenic differentiation but inhibits osteogenic differentiation of CNC-derived mesenchymal cells, suggesting a role for FGF signaling in cell fate determination of CNC-derived cells (Shao et al. 2015).
In mice, the secondary palate develops from the maxillary prominences at embryonic day 11.5 (E11.5). After bilateral palatal shelves contact and fusion around E14.5, osteoblastic differentiation occurs subsequently in the anterior two-thirds of the secondary palate and forms the bony hard palate (Bush and Jiang 2012). The palatine process of the maxilla makes the anterior three-quarters of the hard palate, with the horizontal plate of the palatine bone forming the rest. In the developing palate, short stature homeobox 2 gene (Shox2) is specifically expressed in the anterior palatal mesenchyme from E11.5 on and is required for normal palate development and palatal bone formation (Yu et al. 2005; Gu et al. 2008). In the present study, we took advantage of a Shox2Cre allele (Sun et al. 2013) to explore the role of augmented FGF8 signaling in cell fate determination of CNC-derived mesenchymal cells specifically in the anterior hard palate.
Materials and Methods
Animals
The generation and genotyping protocols of R26RFgf8, Shox2Cre, and R26RmTmG mice have been described previously (Muzumdar et al. 2007; Lin et al. 2013; Sun et al. 2013). The animal experiments in this study were approved by the Institutional Animal Care and Use Committee, Tulane University.
Embryo Collection, Histology, Immunofluorescence, X-gal Staining, and Skeletal Staining
Embryonic heads harvested from timed pregnant female mice or newborn pups were fixed in 4% paraformaldehyde (PFA) at 4°C, paraffin embedded, and sectioned at 8 µm for Azon red/Aniline blue, Alcian blue, or immunofluorescent staining as described previously (Gu et al. 2008; Li et al. 2013; Ye et al. 2015). Information for antibodies used in this study is provided in the Appendix. Alcian blue/Alizarin red skeletal staining was performed as described previously (Bobick and Cobb 2012). Unless specifically indicated, all experiments were repeated at least 3 times.
RNA-Sequencing and Quantitative Reverse Transcription Polymerase Chain Reaction Analyses
For RNA-Seq, the future palatine process of the maxilla was dissected out from E14.5 and E16.5 Shox2Cre;R26RFgf8 and control mice (Shox2Cre/+) (n = 3 for each group), respectively, and subjected to RNA extraction (RNeasy Micro Kit, cat. 74004; Qiagen). RNAs were quantified using a Qubit 2.0 Fluorometric Quantitation system (Life Technologies). The libraries for RNA-Seq were prepared with TruSeq RNA Sample Preparation Kit v2 (cat. RS-122-2001; Illumina) following the manufacturer’s instruction. Libraries were pooled and sequenced on the Illumina HiSeq 4000 platform using the 100-bp pairend-read configuration. Reads were aligned to NCBI37/mm9 genome with HISAT2 (Pertea et al. 2016). For each library, raw counts for each annotated gene were obtained using the featureCounts software from the Subread package (Liao et al. 2013). Differentially expressed genes were identified using DESeq2 (Love et al. 2014). The clusterProfiler was used to perform gene ontology (GO) analysis (Yu et al. 2012). The RNA-Seq data were deposited in the Gene Expression Omnibus (GEO) database with accession number GSE101909.
For quantitative reverse transcription polymerase chain reaction (RT-PCR), the future palatine process of the maxilla was isolated from Shox2Cre;R26RFgf8 and wild-type mice (n = 6 for each genotype) at E14.5 and E16.5, respectively, and subjected to RNA extraction (RNeasy Micro Kit; Qiagen). The RNAs were subsequently reversely transcribed into complementary DNAs (cDNAs). SYBR green and gene-specific primers (Appendix Table) were used and transcript levels were examined by a 7500 Fast Real-Time PCR System (Applied Biosystems). Statistical difference of the quantitative RT-PCR (qPCR) was analyzed by analysis of variance (ANOVA), and results were presented as mean ± standard deviation. P < 0.05 was considered significant.
Results
A Unique Shox2+ Mesenchymal Cell Lineage Contributes to the Anterior Hard Palate
We showed previously that Shox2 is specifically expressed in the anterior palate from E11.5 on and is required for correct bone formation in the hard palate (Yu et al. 2005; Gu et al. 2008). To establish a fate map of Shox2+ cells during palate development, we compounded the R26RmTmG allele with a Shox2-Cre knock-in allele (Shox2Cre) that has been shown to delete floxed DNA fragments with high efficiency in our previous studies (Sun et al. 2013; Ye et al. 2015; Ye et al. 2016). Closer examination and comparison of the Shox2Cre-labeled domain with stained palatal bone at postnatal day 0 (P0) revealed restricted Shox2+ cells in the palatine process of the maxilla that we further divided into the anterior part (ma-a) and posterior part (ma-p) (Fig. 1A, A′). To our surprise, the Shox2+ domain did not overlap with the palatine bone. To confirm these observations, we conducted coimmunostaining of green fluorescent protein (GFP) that was activated by Shox2Cre and Runx2 on the developing Shox2Cre; R26RmTmG palate from E13.5 to E15.5. The results revealed a restricted Shox2 expression only in the palatal mesenchyme throughout the stages examined (Fig. 1), contradicting our previous report that Shox2 is also expressed in the palatal epithelium, a result that was likely caused by overstained in situ hybridization (Yu et al. 2005). At E13.5, most mesenchymal cells in the ma-a domain were Shox2 positive, while in the ma-p domain, Shox2+ cells were present only in the regions adjacent to the medial edge epithelium and the future oral side, in a complementary manner to Runx2 expression domains in the maxilla (Fig. 1B, B′). Shox2+ cells were not found in the future palatine region at this stage (Fig. 1B′′). At E14.5 and E15.5, an osteogenic lineage of Shox2+ cells was found in the ma-a domain only, as revealed by coexpression of Runx2 and Shox2Cre-labeled mGFP (Fig. 1C, D). However, in the ma-p domain, Shox2+ cells were present in the midline and ventral regions in a complementary manner to the future ossification sites as marked by Runx2 expression (Fig. 1C′, D′). Similarly, Shox2+ cells were not found in the future palatine domain (Fig. 1C′′, D′′).
Figure 1.

Lineage tracing of Shox2+ cells in the anterior hard palate. (A–A′) Comparison of whole-mount skeletal staining and fluorescent images of P0 Shox2Cre;R26RmTmG palate (oral view) reveals localization of Shox2+ cells in the palatine process of the maxilla, which is further divided into the anterior part (ma-a) and the posterior part (ma-p) but not in the palatine. (B–D′′) Double immunofluorescent staining on sections of the developing Shox2Cre;R26RmTmG palate shows expression patterns of Runx2 and mGFP at E13.5 (B–B′′), E14.5 (C–C′′), and E15.5 (D–D′′). Note that the expression of Runx2 and mGFP overlaps in the ma-a domain (C, D) but exhibits a complementary localization in the ma-p part (C′, D′). Shox2+ cells are not present in the future palatine domain (C′′, D′′). Scale bars = 1 mm (A–A′); 50 µm (B–B′′, C–D′); 100 µm (C′′, D′′). ma-a, the anterior part of the palatine process of the maxilla; ma-p, the posterior part of the palatine process of the maxilla.
Enhanced FGF Signaling Causes Complete Absence of the Palatine Process of the Maxilla
Multiple FGF ligands and receptors are expressed in the early developing palatal shelves, implicating FGF signaling in palate patterning and cell fate determination. While it was reported that enhanced FGF8 signaling in the CNC lineage inhibited osteogenic differentiation, ectopic activation of FGF8 signaling by Wnt-Cre or Osr2-Cre led to lethality at mid-gestation or complete cleft palate (Shao et al. 2015; Wu et al. 2015), preventing analysis of osteogenic fate and structure of the hard palate. We therefore took advantage of the Shox2Cre allele to locally activate FGF8 signaling in the anterior palate by compounding the Shox2Cre allele with the R26RFgf8 allele (Lin et al. 2013). Although Shox2Cre;R26RFgf8 mice died soon after birth, likely due to severe cardiac defects (Appendix Fig. 1), the mutants did not exhibit a cleft palate defect (Fig. 2B). However, whole-mount skeletal staining unraveled a unique palatal defect: a complete loss of the palatine process of the maxilla (Fig. 2B′) at 100% penetrance (n = 12). The mutant mice also exhibited virtual loss of the stylopodial bone and severely truncated zeugopods (Appendix Fig. 1). Histological analysis of Shox2Cre;R26RFgf8 mice at E16.5 and P0 showed an absent osteogenesis in the palatine process of the maxilla (Fig. 2C–F′). However, in the palatine domain, osteogenic differentiation occurred normally in mutants compared to controls (Fig. 2C′′–F′′). To investigate which downstream signaling pathway(s) and FGF receptor(s) may be involved in mediating the effect of elevated FGF8 signaling, we conducted immunostaining and qRT-PCR assays. We found that elevated FGF8 signaling did not alter the levels of pERK1/2 and PLCγ in the palate at E14.5 and E15 (data not shown). The expression levels of Fgfr1-4 were also comparable in the control and mutant palate at E13.5 to E16.5 (data not shown). However, PI3K levels were largely reduced in the mutant palatine process of the maxilla domain at E15 (Appendix Fig. 2), suggesting a potential engagement of decreased PI3K in the inhibition of osteogenesis. In addition, the mutant anterior palate where the palatine process of the maxilla would form became much thickened compared to controls. Interestingly, ectopic cartilage-like tissues were identified in the mutant ma-p domain, assessed by histology and Alcian blue staining (Fig. 2D′, F′ and insets). These observations demonstrate that elevated FGF8 signaling inhibits osteogenic differentiation of the anterior palatal mesenchymal cells and induces formation of ectopic cartilage-like tissues.
Figure 2.

Augmented fibroblast growth factor 8 (FGF8) signaling leads to loss of the palatine process of the maxilla. (A–B′) Oral views and whole-mount skeletal preparations of palatal bone of controls and Shox2Cre;R26RFgf8 mice demonstrate the lack of cleft palate defect but a complete loss of the palatine process of the maxilla (marked by asterisk) in the mutant. (C–F′′) Histological analyses of control and Shox2Cre;R26RFgf8 palate at E16.5 and P0 show aberrant thickening and complete lack of osteogenesis in the ma-a (D, F) and the ma-p (D′, F′) in the mutants compared to the controls (C, C′, E, E′). However, osteogenesis in the palatine appears identical in both control and mutant (C′′–F′′). Cartilage-like structures (yellow arrowheads) were found in the ma-p region in the mutants (D′, F′). Cartilaginous tissues were assessed by histology (inset in D′) and by Alcian blue staining (inset in F′, which came from a separate experiment). Green arrowheads point to forming bones in the ma-a and ma-p. ma-a, the anterior part of the palatine process of the maxilla; ma-p, the posterior part of the palatine process of the maxilla. Scale bars = 1 mm (A–B′) and 400 µm (C–F′′).
Augmented Fgf8 Alters Cellular Behavior and Cell Fate in the Palatine Process of the Maxilla
To determine the cellular mechanisms contributing to the aberrant thickening of the palatine process of the maxilla region in Shox2Cre;R26RFgf8 mice, we conducted a cell proliferation assay using Ki67 antibody at E14.5 before the thickened phenotype became recognizable. The results demonstrated a statistically significantly increased number of Ki67+ cells in the mutants compared to controls (Fig. 3; P < 0.05). However, the levels of cell apoptosis, as assayed by anti–cleaved caspase-3 antibody staining, were found comparable between mutants and controls (data not shown).
Figure 3.
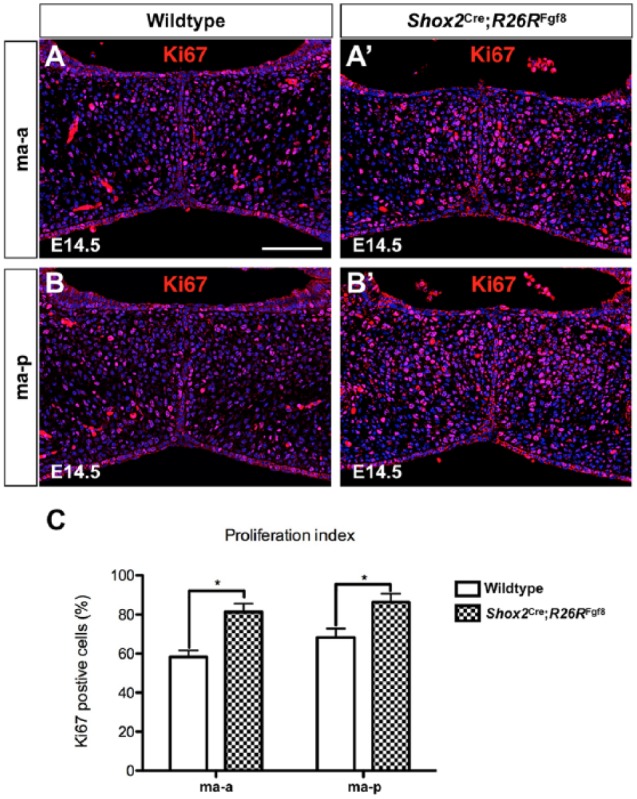
Abnormal cell proliferation in the palatine process of the maxilla of Shox2Cre;R26RFgf8 mice. (A–B′) Ki67 staining on sections of E14.5 control (A, B) and Shox2Cre;R26RFgf8 palate (A′, B′) at the levels of the ma-a (A, A′) and ma-p (B, B′). (C) Quantification of Ki67-positive cells in control (n = 3) and Shox2Cre;R26RFgf8 (n = 3) palates at E14.5. ma-a, the anterior part of the palatine process of the maxilla; ma-p, the posterior part of the palatine process of the maxilla. Error bars represent standard deviation. *P < 0.05.
To ensure that enhanced FGF8 signaling inhibited the osteogenic fate of the anterior palatal mesenchyme, we analyzed the expression of Runx2, a marker for osteoblast precursors (Fujita et al. 2004), and Osterix, an established osteogenic fate marker (Nakashima et al. 2002). In controls at E14.5, the expression of Runx2 and Osterix was initially detected in an overlapped manner in the future ossification centers within the ma-a and ma-p parts (Fig. 4A, B). However, in the mutants, Runx2 expression was inhibited significantly in the ma-a domain but reduced at a moderate level in the ma-p region, while Osterix expression was inhibited completely (Fig. 4A′, B′). The same results were also observed at E15.5 (data not shown), indicating the lack of osteogenic fate in the mutant anterior palatal mesenchyme. FGF signaling is known to act upstream of Runx2 (Komori 2011), but in the context of the anterior palatal mesenchyme, elevated FGF signaling appears to contribute to Osterix repression via an additional pathway as well, because Runx2 expression was only moderately inhibited in the mutant ma-p domain.
Figure 4.

Inhibition of osteogenesis and formation of ectopic cartilage by locally enhanced fibroblast growth factor 8 (FGF8) signaling in the hard palate. (A–B′) Immunostaining shows coexpression of Runx2 and Osterix in the future ossification centers of the ma-a and ma-p regions of the E14.5 control palate (A, B) but a complete absence of Osterix expression in both regions in Shox2Cre;R26RFgf8 mice (A′, B′). Runx2 expression is also reduced significantly in the ma-a domain but decreased at a moderate level in the ma-p region of the mutant. Red arrowheads point to Runx2+/Osterix+ cells; red asterisks mark the sites where Osterix+ cells would have presented in the mutants. (C–D′) Coimmunostaining shows ectopic activation of Sox9 that is coexpressed with Runx2 in the mutant ma-p domain but not the ma-a part at E15 compared to controls. (E–F′) Coimmunostaining of Runx2 and Col II on the palatine process of the maxilla of the E16.5 control (E, F) and Shox2Cre;R26RFgf8 mice (E′, F′). The results show ectopic expression of Col II in a subset of Runx2+ palatal mesenchymal cells in the ma-p portion (F′). (G–H′′) Coimmunostaining on the palatine process of the maxilla of control and Shox2Cre;R26RFgf8;R26RmTmG mice at P0 shows strong Runx2 expression in ossifying the palatine process of the maxilla in control mice but an almost complete absent Runx2 expression in the mutant mice (G, G′, H, H′). However, strong Col II expression is seen in the mutant ma-p domain only, which does not overlap with Shox2+ cells (G′ G′′, H′, H′′). Green arrowheads point to ossification sites. Yellow arrowheads point to Col II+ cells. Green asterisks mark the regions where ossification would have begun in the mutants. ma-a, the anterior part of the palatine process of the maxilla; ma-p, the posterior part of the palatine process of the maxilla. Scale bars = 50 µm.
To confirm that the ectopic cartilaginous tissues in the mutant ma-p domain were indeed chondrogenic fate, we performed coimmunostaining of Runx2 and Sox9 at E15 before the cartilage-like structures became discernible. We found that Sox9 expression was ectopically activated in the mutant ma-p domain, overlapping with Runx2 (Fig. 4D′). Immunostaining of type II Collagen (Col II) further verified the chondrogenic fate of the cartilage-like tissues (Fig. 4F′, H′). Runx2 expression was found in Col II+ cells in the mutant ma-p domain at E16.5 (Fig. 4F′) but was absent in Col II+ cells at P0 (Fig. 4H). The coexpression of Runx2, initially with Sox9 and subsequently with Col II, indicates an osteogenic to chondrogenic cell fate change. Double immunostaining of mGFP and Col II on the Shox2Cre;R26RFgf8;R26RmTmG palate showed that Shox2+ cells did not contribute to the Col II+ cells, indicating a paracrine effect of overexpressed FGF8 (Fig. 4H′′). The different response to FGF8 signaling in terms of ectopic cartilage formation in the ma-p part but not the ma-a domain suggests a distinct property or origin of these 2 groups of cells. This is also supported by the results of the Shox2Cre lineage tracing study in which Shox2+ cells were found within the future ossification centers in the ma-a part but were present in a complementary manner to the future ossification sites in the ma-p region.
FGF8 Signaling Inhibits Osteogenic Genes but Upregulates Chondrogenic Genes
To establish a genome-wide gene expression profile regulated by elevated FGF8 signaling in the palatal mesenchymal cells, we conducted RNA-Seq on the future palatine process of the maxilla from control (Shox2Cre) and Shox2Cre;R26RFgf8 embryos at E14.5 and E16.5. Comparison of RNA-Seq data between controls and mutants revealed highly activated transgenic Fgf8 in the anterior palate, which was verified by qRT-PCR (Fig. 5A). GO analysis showed that in the mutant palate, downregulated genes are primarily involved in ossification and upregulated genes are primarily implicated in cell proliferation and cartilage development (Fig. 5B). Among those genes with altered expression, we identified a number of genes that are crucial for bone and cartilage development, as well as genes that are implicated in cell proliferation and cell fate commitment, as exemplified in Figure 5C. We further validated RNA-Seq results by qPCR on some selected genes that are implicated in cell cycle, osteogenic differentiation, mineralization, and chondrogenic differentiation (Fig. 5D). The RNA-Seq results are therefore consistent with the phenotype observed in the mutants and provide a genome-wide molecular basis for the effects of FGF8 signaling on cell fate determination, cell proliferation, and osteogenic or cartilaginous differentiation of palatal mesenchymal cells.
Figure 5.

RNA-Seq analysis identifies gene expression profile regulated by fibroblast growth factor (FGF) signaling. (A) RNA-Seq and quantitative reverse transcription polymerase chain reaction (PCR) results demonstrate significantly elevated level of Fgf8 expression in the anterior palate of E14.5 and E16.5 Shox2Cre;R26RFgf8 embryos compared with controls. (B) Gene ontology analysis shows that genes that were downregulated in the Shox2Cre;R26RFgf8 palate are primarily those involved in ossification. Upregulated genes in the mutant palate include those that positively regulate cell proliferation and cartilage development. (C) Heatmaps show Z scores (interpreted as a measure of SD away from the mean) for some selected genes from E14.5 and E16.5 RNA-Seq data. Downregulated genes in the Shox2Cre;R26RFgf8 palate include osteogenic differentiation factors (Runx2, Sp7, Col1a1, Bglap, Mn1, Phospho1, Asxl2, Dlx5, Smad5), and the upregulated genes include cell cycle and proliferation (Myc, Cdk4, Cdk6,Wnt5a, Prrx1, Sox9), chondrogenic (Sox9, Col2a1, Col9a1, Dlx2, Acan), and cell fate commitment factors (Prrx1, Trp53, Tbx3, Wnt5a, Sox5, Sox9). The color scheme is based on the Z scores, with upregulation in red, downregulation in blue, and undetermined directionality in gray. (D) Validation of RNA-Seq data by quantitative PCR shows decreased expression levels of selected genes implicated in osteoblast differentiation and elevated expression levels of genes involved in the regulation of cell cycle and chondrogenesis. *P < 0.05.
Discussion
In this study, we present evidence that augmented FGF8 signaling in the developing anterior palate inhibits osteogenesis and alters cell fate of a population of CNC-derived palatal mesenchyme. CNC-derived cells play a central role in craniofacial development and regeneration, being able to differentiate into a broad range of tissues and participating in the formation of multiple craniofacial organs (Santagati and Rijli 2003; Le Douarin et al. 2004). Proper fate determination and differentiation of CNC-derived cells require precise regulations by signaling molecules from several families of growth factors (Minoux and Rijli 2010). FGF8 has been implicated in the induction, migration, and differentiation of neural crest cells. It is also essential for craniofacial development, evidenced by structure loss in mice carrying Fgf8 inactivation in the first branchial arch (Trumpp et al. 1999; Abu-Issa et al. 2002). Fgf8 ectopic expression by the Wnt1Cre or Osr2CreKI allele resulted in severe craniofacial defects or complete cleft palate, respectively (Shao et al. 2015; Wu et al. 2015), indicating the importance of finely tuned FGF8 signaling in craniofacial development. Interestingly, FGF8 can exert dual effects on osteogenic differentiation. For example, osteogenic differentiation assays on mouse bone marrow mesenchymal stem cells, the mouse myoblast cell line, and the rat osteogenic cell line support a positive role for FGF8 (Valta et al. 2006; Omoteyama and Takagi 2009), but FGF8 could inhibit osteogenic differentiation in primary rat osteogenic cells and mouse CNC-derived cells (Lin et al. 2009; Shao et al. 2015). In our current study, the complete lack of Osterix+ cells in the future domain of the palatine process of the maxilla where the transgenic Fgf8 was activated demonstrates that FGF8 signaling exerts its inhibitory effect on osteogenic fate determination of the palatal mesenchymal cells. In addition to its inhibitory effect on osteogenic fate determination, FGF8 could also restrain the full differentiation of osteoblasts. This conclusion is based on the observations that Fgf8 overexpression in differentiated osteoblasts, activated by the Dmp1Cre allele that is expressed in late osteoblasts (Kalajzic et al. 2004), caused osteopenia in both the maxilla and mandible (Appendix Fig. 3). The negative effect of FGF8 signaling on Osterix expression and osteogenic fate could be mediated partially by the downregulation of Runx2, because the expression of Runx2 in the mutant ma-p domain was reduced only moderately.
Consistent with our previous report that FGF8 signaling favors chondrogenic differentiation of CNC-derived cells in vitro in cell culture (Shao et al. 2015), the augmented FGF8 signaling in the anterior palate not only inhibited osteogenesis but also induced ectopic cartilage formation in the place of palatal bone. It has been demonstrated that cell fate plasticity remains sustained in Runx2+ osteoblast precursors during long bone formation (Fujita et al. 2004). In line with this notion, our observations that ectopic Sox9 and Col II expression is restricted to the Runx2+ cells in the anterior palate of Shox2Cre;R26RFgf8 mice further indicate a cell fate change from Runx2+ osteoblast precursors to Col II–positive chondrocytes. It was reported previously that FGF9 signaling can induce endochondral ossification in the cranial mesenchyme by converting the osteogenic differentiation program to the cartilaginous differentiation program of mesoderm-derived cranial mesenchymal cells (Govindarajan and Overbeek 2006). Although the functional mechanisms warrant future investigation, FGF signaling appears to play similar role in promoting chondrogenic fate in mesenchymal cells with both CNC and mesodermal origins.
In line with the palatal phenotype seen in Shox2Cre;R26RFgf8 mice, our RNA-Seq results reveal a dramatic downregulation of genes involved in osteogenesis, such as Runx2, Sp7, and Bglap, in the Fgf8 overexpressing palate, compared to controls. On the other hand, genes that are implicated in cartilage development, including Sox9 and Col2a1, were significantly upregulated in the Shox2Cre;R26RFgf8 palate. Together with the upregulation of genes involved in the regulation of cell proliferation that account for the increased cell proliferation rate in the Shox2Cre;R26RFgf8 palate, our RNA-Seq data, validated by qRT-PCR on some selected genes, provide a molecular basis for the production of the phenotype.
In sum, our results present evidence that augmented FGF8 signaling alters cell proliferation, cell fate determination, and differentiation of CNC-derived osteoblast precursors in the anterior palate. Although the gene overexpression approach has its limitation for studying normal gene function in development, given the fact that multiple FGF ligands and receptors are expressed in the early developing palate, our results implicate the importance of FGF signaling in the regulation of cell fate determination and differentiation of osteoblast precursors during palatogenesis.
Author Contributions
J. Xu, contributed to conception and design, drafted the manuscript; Z. Huang, contributed to data acquisition, analysis, and interpretation, critically revised the manuscript; X. Tan, W. Wang, H. Li, contributed to data acquisition and analysis, critically revised the manuscript; Y. Zhang, W. Tian, contributed to design, critically revised the manuscript; T. Hu, Y.P. Chen, contributed to conception, critically revised the manuscript. All authors gave final approval and agreed to be accountable for all aspects of the work.
Supplementary Material
Footnotes
A supplemental appendix to this article is available online.
This work was supported by the National Institutes of Health (NIH) grants (R01 DE026482 and R01 DE024152) to Y.P. Chen, a grant (81700933) from the National Natural Science Foundation of China and a grant (2015J05063) from the Natural Science Foundation of Fujian Province of China to Z. Huang, and a grant (2017YFA0104800) from The National Key Research and Development Program of China to W. Tian. J. Xu was supported by a fellowship from the China Scholarship Council.
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Abu-Issa R, Smyth G, Smoak I, Yamamura K, Meyers EN. 2002. Fgf8 is required for pharyngeal arch and cardiovascular development in the mouse. Development. 129(19):4613–4625. [DOI] [PubMed] [Google Scholar]
- Biosse Duplan M, Komla-Ebri D, Heuzé Y, Estibals V, Gaudas E, Kaci N, Benoist-Lasselin C, Zerah M, Kramer I, Kneissel M, et al. 2016. Meckel’s and condylar cartilages anomalies in achondroplasia result in defective development and growth of the mandible. Hum Mol Genet. 25(14):2997–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobick BE, Cobb J. 2012. Shox2 regulates progression through chondrogenesis in the mouse proximal limb. J Cell Sci. 125(Pt 24):6071–6083. [DOI] [PubMed] [Google Scholar]
- Burke D, Wilkes D, Blundell TL, Malcolm S. 1998. Fibroblast growth factor receptors: lessons from the genes. Trends Biochem Sci. 23(2):59–62. [DOI] [PubMed] [Google Scholar]
- Bush JO, Jiang RL. 2012. Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development. 139(2):231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day TF, Guo X, Garrett-Beal L, Yang Y. 2005. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell. 8(5):739–750. [DOI] [PubMed] [Google Scholar]
- Fujita T, Azuma Y, Fukuyama R, Hattori Y, Yoshida C, Koida M, Ogita K, Komori T. 2004. Runx2 induces osteoblast and chondrocyte differentiation and enhances their migration by coupling with PI3K-Akt signaling. J Cell Biol. 166(1):85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindarajan V, Overbeek PA. 2006. FGF9 can induce endochondral ossification in cranial mesenchyme. BMC Dev Biol. 6:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu S, Wei N, Yu X, Jiang Y, Fei J, Chen Y. 2008. Mice with an anterior cleft of the palate survive neonatal lethality. Dev Dyn. 237(5):1509–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalajzic I, Braut A, Guo D, Jiang X, Kronenberg MS, Mina M, Harris MA, Harris SE, Rowe DW. 2004. Dentin matrix protein 1 expression during osteoblastic differentiation, generation of an osteocyte GFP-transgene. Bone. 35(1):74–82. [DOI] [PubMed] [Google Scholar]
- Komori T. 2011. Signaling networks in RUNX2-dependent bone development. J Cell Biochem. 112(3):750–755. [DOI] [PubMed] [Google Scholar]
- Le Douarin NM, Creuzet S, Couly G, Dupin E. 2004. Neural crest cell plasticity and its limits. Development. 131(19):4637–4650. [DOI] [PubMed] [Google Scholar]
- Li L, Wang Y, Lin M, Yuan G, Yang G, Zheng Y, Chen YP. 2013. Augmented BMPRIA-mediated BMP signaling in cranial neural crest lineage leads to cleft palate formation and delayed tooth differentiation. PLoS One. 8(6):e66107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. 2013. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 30(7):923–930. [DOI] [PubMed] [Google Scholar]
- Lin C, Yin Y, Bell SM, Veith GM, Chen H, Huh SH, Ornitz DM, Ma L. 2013. Delineating a conserved genetic cassette promoting outgrowth of body appendages. PLoS Genet. 9(1):e1003231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JM, Callon KE, Lin JS, Watson M, Empson V, Tong PC, Grey A, Naot D, Green CR, Reid IR, et al. 2009. Actions of fibroblast growth factor-8 in bone cells in vitro. Am J Physiol Endocrinol Metab. 297(1):E142–E150. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malcolm S, Reardon W. 1996. Fibroblast growth factor receptor-2 mutations in craniosynostosis. Ann N Y Acad Sci. 785:164–170. [DOI] [PubMed] [Google Scholar]
- Minoux M, Rijli FM. 2010. Molecular mechanisms of cranial neural crest cell migration and patterning in craniofacial development. Development. 137(16):2605–2621. [DOI] [PubMed] [Google Scholar]
- Mori-Akiyama Y, Akiyama H, Rowitch DH, de Crombrugghe B. 2003. Sox9 is required for determination of the chondrogenic cell lineage in the cranial neural crest. Proc Natl Acad Sci U S A. 100(16):9360–9365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. 2007. A global double-fluorescent Cre reporter mouse. Genesis. 45(9):593–605. [DOI] [PubMed] [Google Scholar]
- Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, de Crombrugghe B. 2002. The novel zinc finger containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 108(1):17–29. [DOI] [PubMed] [Google Scholar]
- Ohbayashi N, Shibayama M, Kurotaki Y, Imanishi M, Fujimori T, Itoh N, Takada S. 2002. FGF18 is required for normal cell proliferation and differentiation during osteogenesis and chondrogenesis. Genes Dev. 16(7):870–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omoteyama K, Takagi M. 2009. FGF8 regulates myogenesis and induces Runx2 expression and osteoblast differentiation in cultured cells. J Cell Biochem. 106(4):546–552. [DOI] [PubMed] [Google Scholar]
- Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. 2016. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 11(9):1650–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratap J, Galindo M, Zaidi SK, Vradii D, Bhat BM, Robinson JA, Choi JY, Komori T, Stein JL, Lian JB, et al. 2003. Cell growth regulatory role of Runx2 during proliferative expansion of preosteoblasts. Cancer Res. 63(17):5357–5362. [PubMed] [Google Scholar]
- Riley BM, Mansilla MA, Ma J, Daack-Hirsch S, Maher BS, Raffensperger LM, Russo ET, Vieira AR, Dode C, Mohammadi M, et al. 2007. Impaired FGF signaling contributes to cleft lip and palate. Proc Natl Acad Sci U S A. 104(11):4512–4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santagati F, Rijli FM. 2003. Cranial neural crest and the building of the vertebrate head. Nat Rev Neurosci. 4(10):806–818. [DOI] [PubMed] [Google Scholar]
- Shao M, Liu C, Song Y, Ye W, He W, Yuan G, Gu S, Lin C, Ma L, Zhang Y, et al. 2015. FGF8 signaling sustains progenitor status and multipotency of cranial neural crest-derived mesenchymal cells in vivo and in vitro. J Mol Cell Biol. 7(5):441–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Zhang T, Liu C, Gu S, Chen Y. 2013. Generation of Shox2-Cre allele for tissue specific manipulation of genes in the developing heart, palate, and limb. Genesis. 51(7):515–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trumpp A, Depew MJ, Rubenstein JL, Bishop JM, Martin GR. 1999. Cre-mediated gene inactivation demonstrates that FGF8 is required for cell survival and patterning of the first branchial arch. Genes Dev. 13(23):3136–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valta MP, Hentunen T, Qu Q, Valve EM, Harjula A, Seppänen JA, Väänänen HK, Härkönen PL. 2006. Regulation of osteoblast differentiation: a novel function for fibroblast growth factor 8. Endocrinology. 147(5):2171–2182. [DOI] [PubMed] [Google Scholar]
- Wilkie AO. 1997. Craniosynostosis: genes and mechanisms. Hum Mol Genet. 6(10):1647–1656. [DOI] [PubMed] [Google Scholar]
- Wu W, Gu S, Sun C, He W, Xie X, Li X, Ye W, Qin C, Chen Y, Xiao J, et al. 2015. Altered FGF signaling pathways impair cell proliferation and elevation of palate shelves. PLoS One. 10(9):e0136951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye W, Song Y, Huang Z, Osterwalder M, Ljubojevic A, Xu J, Bobick B, Abassah-Oppong S, Ruan N, Shamby R, et al. 2016. A unique stylopod patterning mechanism by Shox2-controlled osteogenesis. Development. 143(14):2548–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye W, Wang J, Song Y, Yu D, Sun C, Liu C, Chen F, Zhang Y, Wang F, Harvey RP, et al. 2015. A common Shox2-Nkx2-5 antagonistic mechanism primes the pacemaker cell fate in the pulmonary vein myocardium and sinoatrial node. Development. 142(14):2521–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang L, Han Y, He Q. 2012. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 16(5):284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, Towler DA, Ornitz DM. 2003. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development. 130(13):3063–3074. [DOI] [PubMed] [Google Scholar]
- Yu L, Gu S, Alappat S, Song Y, Yan M, Zhang X, Zhang G, Jiang Y, Zhang Z, Zhang Y, et al. 2005. Shox2-deficient mice exhibit a rare type of incomplete clefting of the secondary palate. Development. 132(19):4397–4406. [DOI] [PubMed] [Google Scholar]
- Zelzer E, Olsen BR. 2003. The genetic basis for skeletal diseases. Nature. 423(6937):343–348. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.