

Abstract
Diabetes mellitus increases periodontitis and pathogenicity of the oral microbiome. To further understand mechanisms through which diabetes affects periodontitis, we examined its impact on periodontal ligament fibroblasts in vivo and in vitro. Periodontitis was induced by inoculation of Porphyromonas gingivalis and Fusobacterium nucleatum in normoglycemic and diabetic mice. Diabetes, induced by multiple low-dose injections of streptozotocin increased osteoclast numbers and recruitment of neutrophils to the periodontal ligament, which could be accounted for by increased CXC motif chemokine 2 (CXCL2) and receptor activator of nuclear factor kappa B ligand (RANKL) expression by these cells. Diabetes also stimulated a significant increase in nuclear factor kappa B (NF-κB) expression and activation in periodontal ligament (PDL) fibroblasts. Surprisingly, we found that PDL fibroblasts express a 2.3-kb regulatory unit of Col1α1 (collagen type 1, alpha 1) promoter typical of osteoblasts. Diabetes-enhanced CXCL2 and RANKL expression in PDL fibroblasts was rescued in transgenic mice with lineage-specific NF-κB inhibition controlled by this regulatory element. In vitro, high glucose increased NF-κB transcriptional activity, NF-κB nuclear localization, and RANKL expression in PDL fibroblasts, which was reduced by NF-κB inhibition. Thus, diabetes induces changes in PDL fibroblast gene expression that can enhance neutrophil recruitment and bone resorption, which may be explained by high glucose–induced NF-κB activation. Furthermore, PDL fibroblasts express a regulatory element in vivo that is typical of committed osteoblasts.
Keywords: bone resorption, chemokines, cytokines, inflammation, periodontal diseases, osteoclasts
Introduction
Periodontitis is a chronic inflammatory disease and the most common cause of tooth loss in adults (Silva et al. 2015). It is associated with a dental plaque biofilm on the tooth surface, which activates the host immune response and causes alveolar bone destruction and collagen degradation (Hasturk and Kantarci 2015; Xiao et al. 2016). In periodontitis, bacteria and their products stimulate a host response resulting in the generation of proinflammatory molecules and receptor activator of nuclear factor kappa B ligand (RANKL) that stimulate periodontal bone resorption. Several studies demonstrated that inhibition of RANKL reduces alveolar bone loss (Teng et al. 2000; Taubman and Kawai 2001; Jin et al. 2007), indicating its essential role in periodontal bone remodeling. Chemokines are also proinflammatory and stimulate recruitment of leukocytes to sites of inflammation (Graves et al. 2011; Scott and Krauss 2012; Sahingur and Yeudall 2015).
Diabetes mellitus enhances periodontal inflammation to increase the risk and severity of periodontal diseases (Lalla and Papapanou 2011; Chapple et al. 2013; Wu et al. 2015). Diabetes may affect periodontal inflammation by inducing a greater inflammatory response in host cells to given microbial challenge (Salvi et al. 1997; Naguib et al. 2004) or by increasing the pathogenicity of the oral microbiome (Zhou et al. 2013; Xiao et al. 2017). Diabetes induces greater bone loss through a more persistent inflammatory infiltrate, increased osteoclast mediated resorption, and reduced bone coupling (Liu et al. 2006; Duarte et al. 2007; Wu et al. 2015).
Dysregulation of nuclear factor kappa B (NF-κB) may be linked to several diseases that cause osteolysis including periodontitis (Xu et al. 2009; Abu-Amer 2013). NF-κB activation is inhibited by inhibitor of nuclear factor kappa B (IκB). Activation of IκB kinase (IKK) leads to IκB degradation, freeing NF-κB to enter the nucleus and induce gene transcription to stimulate an inflammatory response. Inhibition of NF-κB in osteoblast lineage cells reduces bone resorption and enhances coupled bone formation to reduce periodontal bone loss (Pacios et al. 2015).
Periodontal ligament (PDL) fibroblasts may participate in periodontal inflammation by releasing cytokines and chemokines (Garlet et al. 2008; Jönsson et al. 2009; Zhang et al. 2016). PDL cells are fibroblast-like cells but also exhibit some osteoblastic features. A small portion of PDL cells show high expressions of alkaline phosphatase and bone-associated proteins (Somerman et al. 1990; Giannopoulou and Cimasoni 1996). Fibroblasts can be distinguished from osteoblasts by expression of a 2.3-kb regulatory unit in the collagen type 1, alpha 1 (Col1α1) promoter that is typically restricted to osteoblasts and osteocytes (Dacquin et al. 2002; Braut et al. 2003). Moreover, this regulatory element has been used to generate transgenic (Col1α1.IKK-DN) mice that express a dominant negative mutant of IKK in osteoblast-lineage cells (Liu et al. 2004; Chang et al. 2009; Pacios et al. 2015). We investigated the potential contribution of PDL fibroblasts to diabetes-enhanced periodontitis by examining NF-κB activation and expression of genes that are downstream of NF-κB. Surprisingly, we found that PDL fibroblasts express the 2.3-kb Col1α1 regulatory element and that inflammatory and osteoclastogenic effects of diabetes on PDL fibroblasts can be rescued by lineage-specific NF-κB inhibition in transgenic Col1α1.IKK-DN mice. These studies further establish the phenotype of PDL fibroblasts in vivo and provide evidence regarding how they may contribute to diabetes-enhanced periodontitis.
Material and Methods
Animal Models
Heterozygous 2.3Col1α1–green fluorescent protein (GFP) mice (B6.Cg-Tg[Col1α1*2.3-GFP]1Rowe/J, Stock No. 013134) were purchased from Jackson Laboratory and examined at 3 to 12 wk of age. Mandibles were fixed in 4% paraformaldehyde and decalcified at 4 °C, and CryoJane frozen sections were prepared (Jing et al. 2016). Fluorescent images were captured with an SP5 Leica confocal microscope. Protocols were approved by the Institutional Animal Care and Use Committee at Texas A&M College of Dentistry.
Periodontal disease studies were approved by the Institutional Animal Care and Use Committee at University of Pennsylvania. Lineage-specific inhibition of IKK was achieved in transgenic mice (Col1α1.IKKDN) as described (Chang et al. 2009). Diabetes was induced by intraperitoneal streptozotocin injection (Sigma-Aldrich) for 5 consecutive days (Zhang et al. 2015). The glucose levels of mice with streptozotocin injection exceeded 220 mg/dL for at least 6 wk (see Appendix Table). Mice received antibiotics (sulfamethoxazole and trimethoprim) before oral inoculation of Porphyromonas gingivalis and Fusobacterium nucleatum (2 × 109 colony-forming units) for 2 wk as previously described (Wu et al. 2016) or vehicle (2% methylcellulose) alone. Mice were euthanized 6 wk after oral inoculation. Mandibles were examined by micro–computed tomography (micro-CT) and then decalcified in 10% EDTA for 4 wk. After paraffin embedding, 4-μm sagittal sections that included the first and second molars were prepared as described (Wu et al. 2016).
Micro-CT and Histomorphometric Analysis
The bone between the first and second molars was quantified by micro-CT (μCT35; SCANCO Medical) and in hematoxylin and eosin–stained sections as described (Pacios et al. 2015). Polymorphonuclear leukocytes (PMNs) were counted in PDL and gingival connective tissue in hematoxylin and eosin–stained sections (Kang et al. 2012). Osteoclasts were identified as large multinucleated cells located on the surface of alveolar bone in tartrate-resistant acid phosphatase–stained sections (Liu et al. 2006). Images were captured with a Nikon Eclipse 90i microscope, and NIS Elements-AR software (Nikon) was used for analysis.
Detection of NF-κB p65, RANKL, and CXCL2 in PDL and Gingival Connective Tissue by Immunofluorescence
NF-κB, RANKL, and CXC motif chemokine 2 (CXCL2) expressions were assessed by immunofluorescence with antibodies specific for NF-κB p65 (Cell Signaling Technology), RANKL (Santa Cruz), or CXCL2 (Thermo Fisher Scientific). Sections went through antigen retrieval and were then incubated with primary antibody or matched control IgG at 4 °C overnight, followed by incubation with biotinylated secondary antibody (Vector Laboratories), avidin-biotin-peroxidase complex (ABC Reagent; Vector Laboratories), and tyramide signal amplification (PerkinElmer). Immune complexes were localized by incubation with Alexa Fluor 546–conjugated streptavidin (Invitrogen) and mounting medium containing DAPI (Abcam). Fluorescent images were captured and analyzed at 40× magnification with NIS Elements software (Nikon). Nuclear localization of NF-κB p65 was identified by colocalization of NF-κB p65 antibody and DAPI nuclear stain. Fibroblastic cells in the gingiva and PDL were identified by their location and typical fusiform-shaped nucleus in DAPI images, which distinguishes them from bone-lining cells, leukocytes, or endothelial cells. Quantitation was carried out, and the percentage of immunopositive fibroblastic cells in the PDL or gingiva was measured.
NF-κB p65, Phospho-IκBs, and RANKL Expressions in hPDLCs In Vitro
Human PDL cells were generously provided by Dr. Songtao Shi, University of Pennsylvania, and cultured in α-MEM (Mrozik et al. 2017). Cells were incubated with high glucose (25 mM) and/or tumor necrosis factor alpha (TNF-α) (10 ng/mL) for 24 h. Some cells were preincubated with BAY-117082 (Santa Cruz) for 1 h, which was continued after stimulation with high glucose or TNF. mRNA levels of NF-κB p65 and RANKL were determined by real-time quantitative reverse transcription polymerase chain reaction normalized to ribosomal protein L32 with Fast SYBR Green Master Mix (Applied Biosystem). The amplification was monitored by melting curve analysis. Primer sequences were as follows:
NF-κB p65: forward, 5′-GAGACATCCTTCCGCAAACT-3′; reverse, 5′-TCCTTCCTGCCCATAATCA-3′
RANKL: forward, 5′-TGATTCATGTAGGAGAATTAAA CAGG-3′; reverse, 5′-GATGTGCTGTGATCCAACGA-3′
L32: forward, 5′-TGACAACAGGGTTCGTAGAAGAT-3′; reverse, 5′-GTTCTTGGAGGAAACATTGTGAG-3′
Protein levels of phospho-IκBs (RayBiotech) and RANKL (R&D Systems) were assessed by ELISA. NF-κB transcriptional activity was measured with a dual-luciferase reporter assay System (Promega) normalized with a control Renilla luciferase reporter in each assay.
Statistical Analysis
Statistical analysis was performed with SPSS 20.0 software (IBM). In vivo experiments included 7 to 10 mice for each group with a similar distribution of males and females. One specimen was examined from each mouse per study. Each in vitro experiment was repeated at least 3 times with similar results. Analysis of histologic sections was performed by an examiner who evaluated specimens randomly and was blinded to the specimen group. Significance between wild-type and transgenic mice was determined by 2-tailed Student’s t test, and differences among multiple groups was established by analysis of variance with Tukey’s post hoc test. The significance level was set at P < 0.05.
Results
Localization of GFP in Periodontal Tissue of 2.3Col1α1-GFP Transgenic Mice
Expression of 2.3Col1α1-GFP in the PDL was observed by green fluorescence. Approximately 70% of PDL fibroblasts were GFP positive. No detectable GFP signal was found in epithelium or gingival connective tissue (Fig. 1A). The expression of this promoter element in the majority of PDL fibroblasts was surprising given previous reports of its restricted expression to osteoblast lineage cells (Dacquin et al. 2002; Braut et al. 2003).
Figure 1.

Diabetes and oral infection induce NF-κB activation in PDL fibroblasts, which could be inhibited by the 2.3-kb Col1α1 promoter gene typical of osteoblast lineage cells. (A) Localization of GFP in periodontal tissue of 2.3Col1α1-GFP transgenic mice. 2.3Col1α1-GFP signal was detected in PDL and alveolar bone with expression by PDL fibroblasts, bone-lining cells, and osteocytes. (B) NF-κB p65 expression by PDL fibroblasts by immunofluorescence. (C) NF-κB p65 expression was measured in vivo by immunofluorescence as percentage of immunopositive fibroblastic cells in the PDL. (D) NF-κB p65 nuclear localization was measured in vivo by colocalization of NF-κB p65 (red) and DAPI (blue) nuclear staining and presented as the percentage of fibroblastic cells in the PDL with NF-κB p65 detected in the nucleus. (E) Total NF-κB p65 expression was measured by immunofluorescence in vivo and presented as the percentage of immunopositive fibroblastic cells in gingiva. (F) NF-κB p65 nuclear localization was measured in vivo by colocalization of NF-κB p65 (red) and DAPI (blue) nuclear staining and presented as the percentage of fibroblastic cells in the gingiva with NF-κB p65 detected in the nucleus. *P < 0.05 (vs. uninfected normoglycemic group). **P < 0.05 (vs. matched wild-type group). †P < 0.05 (vs. matched normoglycemic group). Col1α1, collagen type 1, alpha 1; GFP, green fluorescent protein; NF-κB, nuclear factor kappa B; PDL, periodontal ligament. Error bars indicate SEM.
Diabetes and Oral Infection Induce NF-κB Activation in PDL Fibroblasts
Mice were challenged with oral bacterial inoculation. The expression and nuclear localization of the NF-κB subunit p65 in PDL fibroblasts were examined by immunofluorescence (Fig. 1B–F). Diabetes alone induced a 59% increase in NF-κB expression and a 90% increase in NF-κB nuclear localization in PDL fibroblasts when compared with normoglycemic animals (P < 0.05; Fig. 1C, D), indicating increased basal inflammation in PDL fibroblasts caused by diabetes. Infection in wild-type mice (vs. uninfected wild-type mice) induced a 103% increase in PDL fibroblasts that expressed NF-κB and a 229% increase in NF-κB nuclear localization (P < 0.05). When compared with baseline, infected diabetic mice had a 186% increase in NF-κB expression and a 346% increase in NF-κB nuclear localization (P < 0.05). In contrast, the levels of NF-κB were not increased by diabetes or infection in transgenic mice (P > 0.05). Similar increases were noted in gingival fibroblasts in wild-type mice, and there was no difference between transgenic and wild-type mice (P > 0.05; Fig. 1E, F).
To investigate mechanisms by which diabetes may affect the PDL, human PDL cells (hPDLCs) were incubated in high glucose media (25 mM), media supplemented with TNF-α (10 ng/mL), or both, since each is elevated in high glucose conditions (Pacios et al. 2015). NF-κB mRNA levels in hPDLCs were enhanced 58% by high glucose, 108% by TNF-α, and 213% by a combination of high glucose and TNF-α (P < 0.05; Fig. 2A). High glucose and TNF stimulated a 51% increase in NF-κB mRNA levels in hPDLCs when compared with TNF alone (P < 0.05). Increases in NF-κB mRNA levels stimulated by high glucose, TNF-α, or both were completely blocked by the NF-κB inhibitor BAY-117082 (P < 0.05). NF-κB is activated by phosphorylation of IκB. High glucose and TNF-α stimulated an 80% and 130% increase in IκB phosphorylation, respectively, as determined by ELISA (P < 0.05; Fig. 2B). A luciferase reporter assay demonstrated that high glucose induced a 251% increase in NF-κB transcriptional activity in hPDLCs, while TNF-α stimulated a 439% increase (P < 0.05; Fig. 2C). High glucose further increased the NF-κB transcriptional activity in hPDLCs by 93% when compared with TNF alone (P < 0.05). The increase in NF-κB transcriptional activity and IκB phosphorylation was largely suppressed when the NF-κB inhibitor BAY-117082 was applied (P < 0.05).
Figure 2.
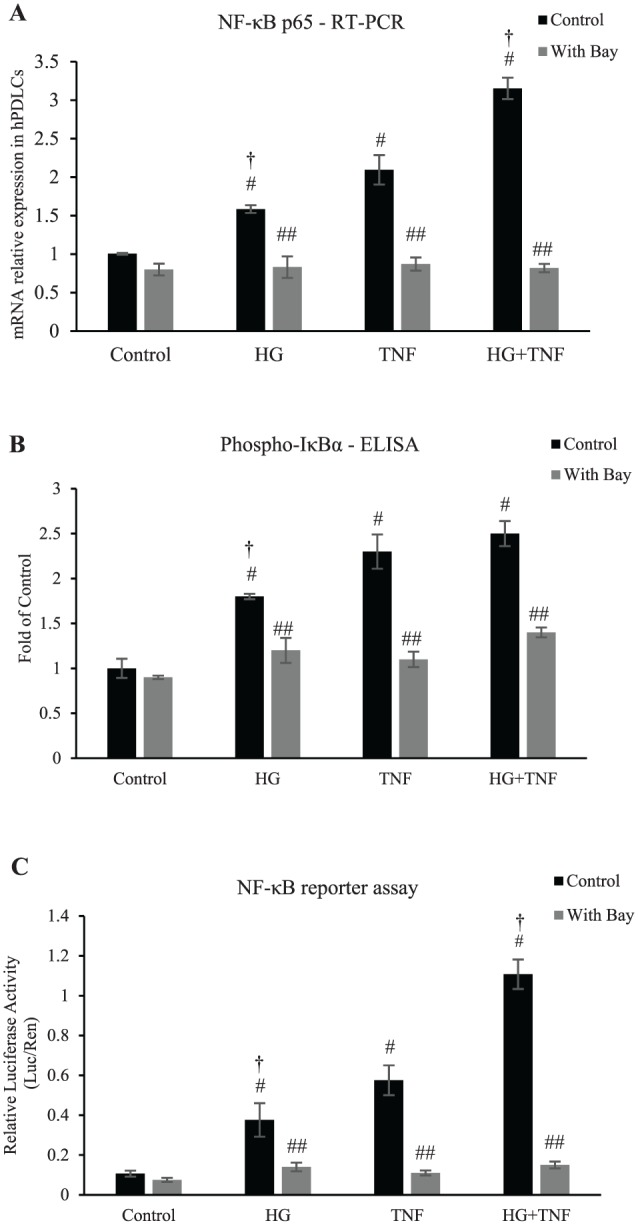
High glucose and TNF-α induce NF-κB activation in human PDL cells in vitro. (A–C) High glucose and TNF-α stimulate NF-κB activity in hPDLCs, which can be blocked by BAY-117082. Cells were cultured with stimulation of high glucose and/or TNF-α with or without BAY-117082. (A) Total NF-κB mRNA were assessed by qRT-PCR when stimulated with high glucose and/or TNF-α with or without BAY-117082 for 3 d. (B) Phospho-IκBs in hPDLCs was measured by ELISA assay when stimulated with high glucose and/or TNF-α with or without BAY-117082 for 24 h. (C) NF-κB transcriptional activity was measured with a luciferase reporter assay. Luciferase activity was normalized by Renilla control. #P < 0.05 (vs. untreated control group). ##P < 0.05 (vs. matched control group). †P < 0.05 (vs. matched normoglycemic group). hPDLC, human periodontal ligament cell; IκB, inhibitor of nuclear factor kappa B; NF-κB, nuclear factor kappa B; PDL, periodontal ligament; qRT-PCR, real-time quantitative reverse transcription polymerase chain reaction. Error bars indicate SEM.
Transgenic Mice Have Reduced RANKL Expression in PDL Fibroblasts, Not Gingival Fibroblasts
Expression of RANKL was measured in PDL and gingival fibroblasts (Fig. 3A, B). Diabetes increased by 145% the number of PDL fibroblasts expressing RANKL in uninfected wild-type animals versus normoglycemic controls (P < 0.05; Fig. 3A). Oral inoculation of bacteria stimulated a 177% increase in RANKL in the PDL fibroblasts as compared with baseline (P < 0.05), which was increased 44% by diabetes (P < 0.05). This number was significantly reduced in transgenic mice (P < 0.05). Diabetes and oral infection also increased RANKL expression in gingival fibroblasts of both wild-type and transgenic mice (Fig. 3B).
Figure 3.

Transgenic mice with lineage-specific inhibition of NF-κB have reduced RANKL expression in PDL fibroblasts not gingival fibroblasts. (A) RANKL expression by fibroblastic cells in the PDL was measured by immunofluorescence. (B) RANKL expression by gingival fibroblasts was measured by immunofluorescence. (C) RANKL mRNA levels were assessed by qRT-PCR when stimulated with high glucose and/or TNF-α with or without the NF-κB inhibitor BAY-117082. (D) RANKL protein levels in hPDLCs were measured by ELISA assay when stimulated with high glucose and/or TNF-α with or without BAY-117082 for 3 d. *P < 0.05 (vs. uninfected normoglycemic group). **P < 0.05 (vs. matched wild-type group). #P < 0.05 (vs. untreated control group). ##P < 0.05 (vs. matched control group). †P < 0.05 (vs. matched normoglycemic group). hPDLC, human periodontal ligament cell; NF-κB, nuclear factor kappa B; PDL, periodontal ligament; RANKL, receptor activator of NF-κB ligand; qRT-PCR, real-time quantitative reverse transcription polymerase chain reaction. Error bars indicate SEM.
RANKL induction by high glucose in hPDLCs was measured in vitro (Fig. 3C, D). High glucose stimulated a 74% increase in RANKL mRNA levels; TNF, a 110% increase; and the combination of high glucose and TNF-α, a 215% increase (P < 0.05; Fig. 3C). ELISA analysis revealed a similar pattern at the protein level (P < 0.05; Fig. 3D). RANKL expression in hPDLCs induced by high glucose, TNF-α, and high glucose plus TNF-α was blocked by the NF-κB inhibitor BAY-117082 at the mRNA and protein levels (P < 0.05; Fig. 3C, D).
Inhibition of NF-κB Activation Decreases PMN Numbers in the PDL and Expression of CXCL2
To determine whether diabetes enhances the recruitment of PMNs to the PDL, we examined CXCL2 and PMN numbers in wild-type and transgenic diabetic mice (Fig. 4A–D). Diabetes stimulated a 211% increase in PMN numbers in wild-type mice, and oral infection induced a 215% increase as compared with normoglycemic, uninfected controls (P < 0.05; Fig. 4A). In infected wild-type mice, diabetes further stimulated a 74% increase in PMNs in the PDL (P < 0.05). PMN numbers in PDL were significantly lower in matched transgenic mice (P < 0.05). In contrast, diabetes and oral inoculation significantly increased PMN numbers in gingival connective tissue of both wild-type and transgenic groups (Fig. 4B).
Figure 4.

Inhibition of NF-κB activation decreases PMN numbers and CXCL2 expression in PDL. Inflammatory status was examined in PDL and gingival connective tissue by quantifying the number of PMNs, which were identified by their spherical shape and multilobed nuclei. (A) PMN numbers/mm2 in the PDL space were measured between molars in hematoxylin and eosin–stained sections. (B) PMN numbers/mm2 were measured in gingival connective tissue between molars in hematoxylin and eosin–stained sections. (C) CXCL2 expression by PDL fibroblasts was measured by immunofluorescence via an antibody to CXCL2. (D) CXCL2 expression in gingival fibroblasts was measured by immunofluorescence via an antibody to CXCL2. (E) Polymorphonuclear neutrophil leukocytes (PMNs; arrows) in periodontal ligament of wild-type and transgenic mice. *P < 0.05 (vs. uninfected normoglycemic group). **P < 0.05 (vs. matched wild-type group). †P < 0.05 (vs. matched normoglycemic group). CXCL2, CXC motif chemokine 2; NF-κB, nuclear factor kappa B; PDL, periodontal ligament; PMN, polymorphonuclear leukocyte. Error bars indicate SEM.
We next examined expression of the neutrophil chemoattractant CXCL2 in PDL fibroblasts by quantitative immunofluorescence (Fig. 4C, D). CXCL2 in PDL fibroblasts of diabetic mice was 236% higher than corresponding normoglycemic mice (P < 0.05; Fig. 4C). Oral inoculation increased the expression of CXCL2 by 254% in normoglycemic wild-type mice versus uninfected controls (P < 0.05). Infected diabetic mice had the largest increase, 446% (P < 0.05). The increased CXCL2 expression by PDL fibroblasts was largely blocked in mice lacking NF-κB activation. In gingival fibroblasts, diabetes similarly induced an increase in both uninfected and infected wild-type mice (P < 0.05; Fig. 4D). Similar increases were observed in gingival fibroblasts of transgenic mice.
Normoglycemic and Diabetic Transgenic Mice with Dominant Negative Inhibition of NF-κB Have Reduced Osteoclast Numbers and Alveolar Bone Loss
The loss of periodontal bone is mediated by osteoclasts. Tartrate-resistant acid phosphatase staining showed that diabetes alone and oral infection alone induced a 225% increase and 307% increase in osteoclasts, respectively (P < 0.05; Fig. 5A, D), with the highest increase, 548%, in infected diabetic mice (P < 0.05). However, in transgenic mice with diabetes or infection, the osteoclast numbers did not increase in comparison with matched controls (P > 0.05). The effect of oral infection and diabetes on alveolar bone loss was examined by micro-CT and histologic analysis. Diabetes in the absence of infection induced a 20% loss of bone (P < 0.05; Fig. 5B, E) and oral inoculation, a 24% loss (P < 0.05). In diabetic mice, infection caused a 44% loss of bone, which was almost double that of the normoglycemic infected group (P < 0.05). In contrast, no bone loss was observed in transgenic mice (P > 0.05). Similar results were obtained in histologic sections (Fig. 5C, F).
Figure 5.

Normoglycemic and diabetic transgenic mice with dominant negative inhibition of NF-κB have reduced osteoclast numbers and alveolar bone loss. Diabetes and periodontitis were induced in wild-type and transgenic (Col1α1.IKKDN) mice by streptozotocin injection and oral inoculation. All mice were euthanized 6 wk after completion of oral bacterial inoculation. (A, D) TRAP analysis were carried out to measure the osteoclasts (arrows) identified as multinucleated cells harboring on the bone surface. (B, E) Micro-CT analysis of remaining bone (the different lengths of arrows indicate the difference in alveolar bone height) between the molars in the mandible. (C, F) Bone area was measured in H&E-stained sections between the molars in the mandible. *P < 0.05 (vs. uninfected normoglycemic group). **P < 0.05 (vs. matched wild-type group). †P < 0.05 (vs. matched normoglycemic group). H&E, hematoxylin and eosin; micro-CT, micro–computed tomography; NF-κB, nuclear factor kappa B; TRAP, tartrate-resistant acid phosphatase. Error bars indicate SEM.
Discussion
We previously demonstrated that osteoblast lineage cells play an important role in bacteria-induced periodontitis mediated by NF-κB signaling (Pacios et al. 2015). In the current study, we surprisingly noted that the PDL fibroblasts strongly expressed the 2.3-kb Col1α1 promotor element. This finding extends our previous work and indicates that PDL fibroblasts, with osteoblast lineage cells, contribute to bone loss as an important source of RANKL in bacteria-induced periodontitis. Most unexpected was GFP staining in the Col 2.3 reporter mice observed in the majority of PDL fibroblasts. In agreement with GFP reporter results was the inhibition of NF-κB, CXCL2, and RANKL expression in a different mouse line that expressed a dominant negative IKK under the control of the same 2.3-kb Col1α1 promoter element. Previous reports indicated that a small percentage of PDL fibroblasts exhibit an osteoblast-like phenotype characterized by the expression of alkaline phosphatase and bone-related proteins (Giannopoulou and Cimasoni 1996). In addition, a low expression of the 2.3-kb Col1α1 promoter was detected in tendon fibroblasts in Col 2.3 reporter mice (Liu et al. 2004). Thus, the data presented here suggest that PDL fibroblasts may be more committed to the osteoblast lineage than previously recognized.
We demonstrated here that diabetes stimulates expression and nuclear localization of the NF-κB p65 subunit in PDL fibroblasts, which was suppressed in transgenic mice. When hPDLCs were stimulated with high glucose and TNF in vitro, both of which are elevated by diabetes, there was increased phospho-IκB, NF-κB p65, and NF-κB transcriptional activity. The increase of each was reduced by the NF-κB inhibitor BAY-117082, consistent with in vivo data for NF-κB in transgenic versus wild-type PDL fibroblasts. The results suggest a mechanism whereby high glucose and TNF stimulate NF-κB activation in these cells, which may account for the increased NF-κB activation in PDL fibroblasts observed in diabetic mice. A previous report showing that hyperglycemia is associated with activation of the NF-κB pathway supports this interpretation (Kato et al. 2016).
Diabetes contributes to alveolar bone loss. PDL cells are an important source of osteoprogenitors (Lim et al. 2014). Thus, inflammation could affect both anabolic and catabolic events in the PDL. For example, it could reduce bone coupling by limiting the differentiation of PDL cells to osteoblasts to inhibit bone formation following an episode of resorption. PDL cells could also contribute to osteoclast formation through generation of RANKL, thus promoting a catabolic event. Multiple low-dose streptozotocin injections were used to induce diabetes since it is a well-established model that mimics human type 1 diabetes by inducing β-cell dysfunction and insulin deficiency. This model was shown in many diabetic complications to reflect the impact of streptozotocin on β cells rather than direct toxic effects in other tissues (Shehata et al. 2011). Our results indicate that the effect of diabetes on PDL fibroblasts may significantly contribute to the enhanced susceptibility of individuals with diabetes to periodontitis. This is based on findings that diabetes increased the expression of CXCL2 and RANKL in PDL fibroblasts of mice with or without inoculation of periodontal pathogens. Gingival fibroblasts may play a critical role in the inflammatory events that precede RANKL expression by PDL and osteoblast lineage cells. However, the results point to PDL and osteoblast lineage cells as being particularly important in RANKL-induced bone loss. Furthermore, high glucose alone stimulated RANKL expression at the mRNA and protein levels in these cells and further enhanced the levels induced by TNF. This increase was NF-κB dependent, agreeing well with in vivo results. Diabetes significantly enhanced PMN recruitment to the PDL, which was largely blocked in transgenic mice. Taken together, these results reinforce the concept that diabetes mellitus activates PDL fibroblasts to contribute to inflammation and RANKL expression in close proximity to bone (Duarte et al. 2007; Belibasakis and Bostanci 2012; Feng et al. 2012).
Author Contributions
J. Zheng, S. Chen, contributed to data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; M.L. Albiero, G.H.A. Vieira, J. Wang, J.Q. Feng, contributed to data acquisition and analysis, critically revised the manuscript; D.T. Graves, contributed to conception, design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplementary Material
Footnotes
A supplemental appendix to this article is available online.
This study was supported by grants from the National Institute of Dental and Craniofacial Research, R01DE021921 and R01DE 017732.
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Abu-Amer Y. 2013. NF-κB signaling and bone resorption. Osteoporos Int. 24(9):2377–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belibasakis GN, Bostanci N. 2012. The RANKL-OPG system in clinical periodontology. J Clin Periodontol. 39(3):239–248. [DOI] [PubMed] [Google Scholar]
- Braut A, Kollar EJ, Mina M. 2003. Analysis of the odontogenic and osteogenic potentials of dental pulp in vivo using a Col1a1-2.3-GFP transgene. Int J Dev Biol. 47(4):281–292. [PubMed] [Google Scholar]
- Chang J, Wang Z, Tang E, Fan Z, McCauley L, Franceschi R, Guan K, Krebsbach PH, Wang CY. 2009. Inhibition of osteoblastic bone formation by nuclear factor-kappaB. Nat Med. 15(6):682–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapple IL, Genco R; Working Group 2 of the Joint EFP/AAP Workshop. 2013. Diabetes and periodontal diseases: consensus report of the Joint EFP/AAP Workshop on Periodontitis and Systemic Diseases. J Periodontol. 84 Suppl 4:S106–S112. [DOI] [PubMed] [Google Scholar]
- Dacquin R, Starbuck M, Schinke T, Karsenty G. 2002. Mouse alpha1(i)-collagen promoter is the best known promoter to drive efficient Cre recombinase expression in osteoblast. Dev Dyn. 224(2):245–251. [DOI] [PubMed] [Google Scholar]
- Duarte PM, Neto JB, Casati MZ, Sallum EA, Nociti FH., Jr. 2007. Diabetes modulates gene expression in the gingival tissues of patients with chronic periodontitis. Oral Dis. 13(6):594–599. [DOI] [PubMed] [Google Scholar]
- Feng Y, Liu JQ, Liu HC. 2012. AMP-activated protein kinase acts as a negative regulator of high glucose-induced RANKL expression in human periodontal ligament cells. Chin Med J (Engl). 125(18):3298–3304. [PubMed] [Google Scholar]
- Garlet TP, Coelho U, Repeke CE, Silva JS, Cunha Fde Q, Garlet GP. 2008. Differential expression of osteoblast and osteoclast chemmoatractants in compression and tension sides during orthodontic movement. Cytokine. 42(3):330–335. [DOI] [PubMed] [Google Scholar]
- Giannopoulou C, Cimasoni G. 1996. Functional characteristics of gingival and periodontal ligament fibroblasts. J Dent Res. 75(3):895–902. [DOI] [PubMed] [Google Scholar]
- Graves DT, Oates T, Garlet GP. 2011. Review of osteoimmunology and the host response in endodontic and periodontal lesions. J Oral Microbiol. 3. doi: 10.3402/jom.v3i0.5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasturk H, Kantarci A. 2015. Activation and resolution of periodontal inflammation and its systemic impact. Periodontol 2000. 69(1):255–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Q, Cirelli JA, Park CH, Sugai JV, Taba M, Jr, Kostenuik PJ, Giannobile WV. 2007. RANKL inhibition through osteoprotegerin blocks bone loss in experimental periodontitis. J Periodontol. 78(7):1300–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing Y, Hinton RJ, Chan KS, Feng JQ. 2016. Co-localization of cell lineage markers and the tomato signal. J Vis Exp. 118. doi: 10.3791/54982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jönsson D, Amisten S, Bratthall G, Holm A, Nilsson BO. 2009. LPS induces GROalpha chemokine production via NF-kappaB in oral fibroblasts. Inflamm Res. 58(11):791–796. [DOI] [PubMed] [Google Scholar]
- Kang J, de Brito Bezerra B, Pacios S, Andriankaja O, Li Y, Tsiagbe V, Schreiner H, Fine DH, Graves DT. 2012. Aggregatibacter actinomycetemcomitans infection enhances apoptosis in vivo through a caspase-3-dependent mechanism in experimental periodontitis. Infect Immun. 80(6):2247–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Taguchi Y, Tominaga K, Kimura D, Yamawaki I, Noguchi M, Yamauchi N, Tamura I, Tanaka A, Umeda M. 2016. High glucose concentrations suppress the proliferation of human periodontal ligament stem cells and their differentiation into osteoblasts. J Periodontol. 87(4):e44–e51. [DOI] [PubMed] [Google Scholar]
- Lalla E, Papapanou PN. 2011. Diabetes mellitus and periodontitis: a tale of two common interrelated diseases. Nat Rev Endocrinol. 7(12):738–748. [DOI] [PubMed] [Google Scholar]
- Lim WH, Liu B, Mah SJ, Chen S, Helms JA. 2014. The molecular and cellular effects of ageing on the periodontal ligament. J Clin Periodontol. 41(10):935–942. [DOI] [PubMed] [Google Scholar]
- Liu F, Woitge HW, Braut A, Kronenberg MS, Lichtler AC, Mina M, Kream BE. 2004. Expression and activity of osteoblast-targeted Cre recombinase transgenes in murine skeletal tissues. Int J Dev Biol. 48(7):645–653. [DOI] [PubMed] [Google Scholar]
- Liu R, Bal HS, Desta T, Krothapalli N, Alyassi M, Luan Q, Graves DT. 2006. Diabetes enhances periodontal bone loss through enhanced resorption and diminished bone formation. J Dent Res. 85(6):510–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrozik K, Gronthos S, Shi S, Bartold PM. 2017. A method to isolate, purify, and characterize human periodontal ligament stem cells. Methods Mol Biol. 1537:413–427. [DOI] [PubMed] [Google Scholar]
- Naguib G, Al-Mashat H, Desta T, Graves DT. 2004. Diabetes prolongs the inflammatory response to a bacterial stimulus through cytokine dysregulation. J Invest Dermatol. 123(1):87–92. [DOI] [PubMed] [Google Scholar]
- Pacios S, Xiao W, Mattos M, Lim J, Tarapore RS, Alsadun S, Yu B, Wang CY, Graves DT. 2015. Osteoblast lineage cells play an essential role in periodontal bone loss through activation of nuclear factor-kappa B. Sci Rep. 5:16694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahingur SE, Yeudall WA. 2015. Chemokine function in periodontal disease and oral cavity cancer. Front Immunol. 6:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvi GE, Yalda B, Collins JG, Jones BH, Smith FW, Arnold RR, Offenbacher S. 1997. Inflammatory mediator response as a potential risk marker for periodontal diseases in insulin-dependent diabetes mellitus patients. J Periodontol. 68(2):127–135. [DOI] [PubMed] [Google Scholar]
- Scott DA, Krauss J. 2012. Neutrophils in periodontal inflammation. Front Oral Biol. 15:56–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shehata AM, Quintanilla-Fend L, Bettio S, Singh CB, Ammon HP. 2011. Prevention of multiple low-dose streptozotocin (MLD-STZ) diabetes in mice by an extract from gum resin of Boswellia serrata (BE). Phytomedicine. 18(12):1037–1044. [DOI] [PubMed] [Google Scholar]
- Silva N, Abusleme L, Bravo D, Dutzan N, Garcia-Sesnich J, Vernal R, Hernandez M, Gamonal J. 2015. Host response mechanisms in periodontal diseases. J Appl Oral Sci. 23(3):329–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somerman MJ, Young MF, Foster RA, Moehring JM, Imm G, Sauk JJ. 1990. Characteristics of human periodontal ligament cells in vitro. Arch Oral Biol. 35(3):241–247. [DOI] [PubMed] [Google Scholar]
- Taubman MA, Kawai T. 2001. Involvement of T-lymphocytes in periodontal disease and in direct and indirect induction of bone resorption. Crit Rev Oral Biol Med. 12(2):125–135. [DOI] [PubMed] [Google Scholar]
- Teng YT, Nguyen H, Gao X, Kong YY, Gorczynski RM, Singh B, Ellen RP, Penninger JM. 2000. Functional human T-cell immunity and osteoprotegerin ligand control alveolar bone destruction in periodontal infection. J Clin Invest. 106(6):R59–R67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Dong G, Xiao W, Xiao E, Miao F, Syverson A, Missaghian N, Vafa R, Cabrera-Ortega AA, Rossa C, Jr, et al. 2016. Effect of aging on periodontal inflammation, microbial colonization, and disease susceptibility. J Dent Res. 95(4):460–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YY, Xiao E, Graves DT. 2015. Diabetes mellitus related bone metabolism and periodontal disease. Int J Oral Sci. 7(2):63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao E, Mattos M, Vieira GHA, Chen S, Correa JD, Wu Y, Albiero ML, Bittinger K, Graves DT. 2017. Diabetes enhances IL-17 expression and alters the oral microbiome to increase its pathogenicity. Cell Host Microbe. 22(1):120–128.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W, Li S, Pacios S, Wang Y, Graves DT. 2016. Bone remodeling under pathological conditions. Front Oral Biol. 18:17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Wu HF, Ang ES, Yip K, Woloszyn M, Zheng MH, Tan RX. 2009. NF-kappaB modulators in osteolytic bone diseases. Cytokine Growth Factor Rev. 20(1):7–17. [DOI] [PubMed] [Google Scholar]
- Zhang C, Ponugoti B, Tian C, Xu F, Tarapore R, Batres A, Alsadun S, Lim J, Dong G, Graves DT. 2015. Foxo1 differentially regulates both normal and diabetic wound healing. J Cell Biol. 209(2):289–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Ding Y, Rao GZ, Miao D. 2016. Effects of IL-10 and glucose on expression of OPG and RANKL in human periodontal ligament fibroblasts. Braz J Med Biol Res. 49(4):e4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Rong R, Munro D, Zhu C, Gao X, Zhang Q, Dong Q. 2013. Investigation of the effect of type 2 diabetes mellitus on subgingival plaque microbiota by high-throughput 16S rDNA pyrosequencing. PLoS One. 8(4):e61516. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.