

Abstract
Key events that occur during inflammation include the recruitment, adhesion, and transmigration of leukocytes from the circulation to the site of inflammation. These events are modulated by chemokines, integrins, and selectins and the interaction of these molecules with glycosaminoglycans, predominantly heparan sulfate (HS). The development of HS/heparin mimetics that interfere or inhibit the interactions that occur between glycosaminoglycans and modulators of inflammation holds great potential for use as anti-inflammatory therapeutics. This review will detail the role of HS in the events that occur during inflammation, their interaction and modulation of inflammatory mediators, and the current advances in the development of HS/heparin mimetics as anti-inflammatory biotherapeutics.
Keywords: extracellular matrix, glycosaminoglycans, heparan sulfate, heparin, inflammation, innate immunity
Introduction
The inflammatory response is the body’s reaction to invading foreign material or injury. Cells of the innate immune system include tissue-resident macrophages, mast cells, dendritic cells, and circulating leukocytes. Pathogen-associated molecular patterns (PAMPs) as well as damage-associated molecular patterns (DAMPs) released from injured cells are recognized by receptors on the surface of immune cells that initiate signaling cascades that make up the inflammatory response. One of these most noted events is the recruitment, adhesion, and transmigration of leukocytes at the site of inflammation. Leukocytes, or white blood cells, are derived from hematopoietic stem cells and include neutrophils, eosinophils, basophils, lymphocytes, and monocytes. There are numerous mediators involved in the inflammatory response, including chemokines, selectins, and cell surface receptors. One family of molecules that is involved with the control of numerous events that occur during inflammation are glycosaminoglycans (GAGs), most notably heparan sulfate (HS). GAGs are long polysaccharide structures that are made up of repeating disaccharide units. GAGs are made up of four main subgroups: (1) HS/heparin, (2) chondroitin/dermatan sulfate (CS/DS), (3) keratan sulfate, and (4) hyaluronic acid or hyaluronan (HA). With the exception of HA, GAGs are found covalently attached to the core protein of proteoglycans (PGs); their attached GAG chains are sulfated to variable degrees and at various ring positions providing a heterogeneous population of structures with the ability to bind growth factors which modulate various biological processes,1 including inflammation.2 While HS is produced by virtually all cells, heparin is produced exclusively by mast cells and decorates the PG serglycin that is stored in α-granules and released upon activation (Fig. 1). While HS and heparin are similar in structure, heparin tends to contain a higher degree of sulfation when compared with HS. Heparin is widely used as an anticoagulant. Interestingly, 3-O-sulfotransferase 1 (Hs3st1) knockout mice, removing the enzyme responsible for producing the anticoagulant structures in heparin, do not display a procoagulant phenotype,3 suggesting that this enzyme and the structures it produces may have a different role in biology. HS is involved in mediating the activities of leukocytes in inflammation, due to its wider distribution in tissues compared with heparin. Due to the fundamental similarities in structure between HS and heparin, heparin is capable of interfering with the role of HS in inflammation, and this has enabled the use of heparin as an HS mimetic. Thus, given the central role of HS in inflammatory processes, the development of HS/heparin mimetics as anti-inflammatory therapeutics has tremendous potential.
Figure 1.

The role of HS in inflammation. Key events that occur during inflammation include the recruitment, adhesion, crawling, and transmigration of leukocytes from the circulation to the site of inflammation. The recruitment of leukocytes to the site of inflammation occurs between P-selectin on endothelial cells and PSGL-1 on the surface of leukocytes, and L-selectin and HS that decorates HSPGs on the endothelial cell surface. Interaction of G-protein-coupled receptors on the surface of leukocytes with chemokines presented by cell surface HSPGs results in the activation of integrins that bind to cell surface receptors on endothelial cells, including ICAM. Migration or crawling of leukocytes is driven by chemokine gradients, presented by HSPGs, along the surface of the endothelial cells. This is followed by the transmigration of leukocytes, either transcellular or pericellular, to the site of inflammation. Cells at the site of inflammation include macrophages and mast cells. Macrophages following inflammatory stimulation release chemokines that stimulate upregulation of P-selectin on the surface of endothelial cells. On activation, mast cells release the contents of their α-granules that contain chemokines and proteases. Heparin decorates the PG serglycin stored in the α-granules of mast cells that is also released on activation. Abbreviations: HS, heparan sulfate; PSGL-1, P-selectin glycoprotein ligand-1; ICAM, intracellular adhesion molecule; PG, proteoglycan; HSPG, heparan sulfate proteoglycan.
HS Biosynthesis
The biosynthesis of HS is a complex process involving many sequential steps to modify regions of the emerging HS chains which are highly heterogeneous.4–6 The co-ordination of these HS biosynthetic steps occurs in a specific spatiotemporal manner by biosynthetic enzymes under developmental regulation to control HS fine structure in a tissue-specific manner. HS is attached to the core proteins of a number of modular extracellular, cell membrane–associated, and intracellular secretory vesicle PGs.7 These include perlecan, agrin, collagen type XVIII, syndecan 1–4, glypican 1–6, and serglycin. HS also occurs on several part-time PGs including betaglycan, a 110-kDa fibroblast PG; neuropilin, a 130-kDa endothelial cell heparan sulfate proteoglycan (HSPG); CD-44 V3, a variant HA receptor produced by lymphocytes; and epican, a CD-44 variant synthesized by keratinocytes.
HS biosynthesis is initiated by the attachment of xylose to specific serine residues in HSPG core proteins leading to the formation of a linkage tetrasaccharide, glucuronic acid-galactose-galactose-xylose (GlcA-Gal-Gal-Xyl), where xylose can be phosphorylated at C2. Formation of the linkage region occurs through sequential xylosyl transferase-1 and 2 (XYLT1 and 2), galactosyl transferase-1 and 2 (GALT1 and 2), and glucuronyl transferase-1 (GLCAT1) activities. Assembly of the HS chain is initiated upon the attachment of N-acetylglucosamine (GlcNAc) to the linkage module by N-acetylglucosaminyl transferase-1 (EXTL3), an enzyme complex of HS copolymerase (N-acetyl glucosaminyl glucuronyl transferase-1 and 2, EXT 1, 2); then, GlcA and GlcNAc are added sequentially to the nascent HS chain.4,6 The HS chain then undergoes deacetylation of clusters of GlcNAc residues and sulfation of the generated free amino groups by up to four members of the N-deacetylase-N-sulfotransferases (NDST) enzymes. The C5 epimerase (HSGLCE) epimerizes D-GlcA adjacent to N-GlcNS residues at C5 to form l-IdoA, a series of O-sulfotransferases then add sulfate, uronyl 2-O-sulfotransferase (HS2ST) attaches sulfate at C2 of the IdoA, and 6-O-sulfotransferases (Hs6st1, 2, 3) add sulfate at C6 of N-GlcNS (and less frequently to GlcNAc residues). Seven 3-O-sulfotransferases (Hs3st 1, 2, 3a, 3b, 4, 5, 6) add sulfate at C3 of N-sulfated or non-substituted GlcNH2 residues although this is a relatively rare sulfation motif.5,8 Sulfation along the HS chain is therefore not uniform but contains highly modified areas reminiscent of heparin of high sulfation (NS domains) and areas of unmodified low sulfation (N-acetylated or NA domains) domains, which are not present in heparin. After sulfation, the primary structure of the HS chains of HSPGs can be further modified by SULF1 and 2, endosulfatase enzymes that remove sulfate groups from C6 of GlcNH2 sulfate in HS or by the action of heparanase or extracellular proteases; this releases any bound growth factors or cytokines.9 It is the heterogeneous nature of HS and its diverse structural forms that facilitates its binding to various mediators of inflammation, and the ability to modulate and control events that occur throughout this biological process.
The Role of HS in Inflammation
Key events that occur during inflammation include the recruitment, adhesion, rolling, and transmigration of leukocytes from the circulation (Fig. 1). During these processes, there are many molecules involved that modulate these events including chemokines, integrins, selectins, and enzymes. The initial stages of inflammation may be stimulated by PAMPs or DAMPs, and vary between tissues. PAMPs released from pathogens include lipopolysaccharides or endotoxins. These stimulate the endothelium resulting in the upregulation of selectins expressed on the surface of endothelial cells, including P- and E-selectin. DAMPs are released from necrotic cells; these include DNA or RNA, heat shock proteins, and HA oligosaccharides, which stimulate the innate immune system via the vasculature resulting in the extravasation of leukocytes into tissues.10 Interactions that occur during this recruitment process involve P-selectin, P-selectin glycoprotein ligand (PSGL)-1,11 as well as L-selectin, expressed on leukocytes, and GAGs. HS is involved in the binding and release of various mediators that modulate all stages of leukocyte recruitment, adhesion, rolling, and transmigration.2,12
Infiltration of leukocytes and immune cells to the site of inflammation is due to the presentation of chemokines along the endothelium. HS is present on the surface of the cells covalently coupled to syndecan and glypican PGs that interact with integrins, as well as in intracellular α-granules decorating serglycin, which bind the chemokines as inactive precursors within neutrophils and monocytes. HS also decorates the basal laminae of endothelial cells covalently attached to the PGs, perlecan, agrin, and type XV and XVIII collagens, where they bind to endothelial cell growth factors and act as a reservoir. It is thought that a major role for HS is in the promotion of extravasation and migration of inflammatory cells from the vasculature into tissues, where it establishes and provides cytokine gradients facilitating the communication between bone marrow and progenitor inflammatory cells in the tissues. Adhesion of leukocytes to activated endothelial cells involves the activation of cell surface receptors and molecules including intracellular adhesion molecule (ICAM)-1 on endothelial cells and binding of chemokines to the G-protein-coupled receptors on the leukocyte surface,13 facilitated by the interaction of chemokines attached to cell surface HSPGs. There are a number of integrins expressed on the surface of leukocytes which promote their adherence to the endothelium, including Mac-1,14,15 which binds to both HS and heparin but not to other GAGs.16 The immobilization of macrophage inflammatory protein (MIP)-1β, also known as CCL4, on endothelial PGs results in adhesion of T cells.17 It is not only the GAGs attached to cell surface PGs that are capable of chemokine signaling, soluble GAGs also modulate the binding of chemokines to their receptors, modeled in vitro with heparin, HS, and CS/DS.18 In addition, platelets bind to activated endothelial cells but do so via interactions with the C-type lectin, P-selectin. Platelets also interact with leukocytes through P-selectin,19 facilitating their adhesion to the endothelium.20 This binding supports the rolling action of platelets to activated endothelium and may involve HS. Heparin inhibits the movement of these cells via a P-selectin-dependent manner21; however, the precise mechanism of action has not yet been elucidated. Chemokines not only modulate the adhesion of immune cells to GAG chains on the endothelial cell surface, but they also modify and increase the expression of HSPGs on the surface of endothelial cells.22
Once immune cells have adhered to the surface of the endothelium, they migrate, also known as crawling, across the endothelial surface before transmigration occurs. This crawling process is modulated by GAG chains of various PGs on the endothelial cell surface directing immune cell migration23–25 and is driven by chemokine gradients.26,27 The extravasation of leukocytes from the circulation culminates with transmigration in either a transcellular or pericellular manner.28–30 HSPGs are also involved in the transendothelial migration of immune cells. Silencing of a key enzyme involved in HS biosynthesis, EXT1, reduces neutrophil transendothelial migration but does not affect T-cell migration.31 Following transendothelial migration, immune cells are required to move through the endothelial basement membrane which is rich in type IV collagen, laminin, and perlecan. Upon leukocyte activation, endothelial cell gap junctions become more permeable and the leukocytes move between the endothelial cells via interactions with selectins, integrins, and other cell surface molecules such as the cluster of differentiation (CD) molecule, CD99, to cross the basement membrane at regions where laminin and type IV collagen levels are reduced. These same regions do not show decreased perlecan,32 suggesting that the degradation of HS on perlecan by heparanase may stimulate the movement of leukocytes from the circulation into tissues.
Mediators of Inflammation and Their Interaction With HS
The activities of various inflammatory mediators, including chemokines and selectins, are mediated by interactions with GAGs including HS. The interactions between HS and these mediators will be reviewed in the following sections.
Chemokines
Cytokines are signaling molecules produced by immune cells, endothelial cells, and fibroblasts. Chemokines are a type of chemoattractant cytokine. Chemokines are small, predominantly basic molecules that regulate the migration of leukocytes during inflammation, through interactions with G-protein-coupled receptors. Chemokines are grouped into two main subfamilies, referred to as CC and CXC, which are named on the basis of whether the first two cysteine residues are adjacent (CC) or contain an amino acid between them (CXC).33 The structural diversity of GAGs and chemokines gives rise to their specificity and ability to fine-tune biological processes in vivo. Such interactions have a crucial role in the activity of chemokines and the recruitment and infiltration of inflammatory cells including lymphocytes34 and neutrophils.27 Chemokines, in a general sense, interact with GAGs via clusters of basic amino acids, where four different modes of binding between chemokines and HS have been shown.35 The oligomerization of chemokines plays a role in the interaction with HS, and GAGs in general. Chemokine oligomerization can modulate the binding affinity to HS by altering the confirmation of GAG binding sites36 and increase the binding surface as compared with monomeric forms.37
CCL5, also known as RANTES, is predominantly produced by T cells and plays a role in the migration of leukocytes into inflammatory sites. GAG–chemokine interactions have important roles to play in this process; CCL5–GAG interactions are essential for the creation of haptotactic gradients that are responsible for the migration of leukocytes. The BBXB motif (basic/basic/x/basic amino acid, x = any amino acid) present in CC chemokines is essential for the interaction between CCL5 and heparin disaccharide structures.38 Specifically, the BBXB motif 44Arg-Lys-Asn-Arg47 (44RKNR47) present in CCL5 is responsible for interaction with heparin.39 Mutation of the 44RKNR47 motif results in decreased binding of CCL5 to heparin in vitro and a reduction in cell infiltration into the peritoneal cavity following intraperitoneal injection.40 The structure and availability of CCL5, and other chemokines, and whether they are present as oligomers modulate interactions with GAGs.18,41,42 While the 44RKNR47 motif in CCL5 binds to heparin, when CCL5 is oligomerized, this motif is buried, and in this case, the primary heparin binding site of CCL5 is 55Lys-Lys-Trp-Val-Arg59 (55KKWVR59).36 The interaction between CCL5 and its receptor present on monocytes, CCR1, can be inhibited by HS/heparin.43 The interaction between CCL5 and GAGs is strongly modulated by pH and by GAG sulfation with CCL5 binding to heparin and HS, as well as CS but with variable affinity.44
The chemokine CXCL8 is also the human neutrophil chemoattractant interleukin (IL)-8 and binds to GAGs on the surface of endothelial cells where it plays a crucial role in the infiltration of neutrophils to the site of inflammation. The binding affinity of monomeric and dimeric CXCL8 to a range of HS and heparin oligosaccharide structures reveals differing affinities depending on the oligosaccharide length.45 Both monomeric and dimeric forms of CXCL8 bind to HS, though the dimeric form has been shown to have higher binding affinity.46 HS/heparin interactions with CXCL8 stabilize its tertiary structure preventing it from unfolding.45 This involves the amino acid residues Arg60, Lys64, Lys67, and Arg68 which are present in the C-terminal α-helix in CXCL8 and Lys20 within the proximal loop.47 These residues, along with His18, are essential in the binding of CXCL8 to GAG oligosaccharides, in addition to Lys15, Arg47, Lys23, and Lys54.46 Like CCL5, CXCL8 displays variable binding affinities to GAGs, depending on whether they are free in solution or associated with the cell surface.18 CXCL8 has chemoattractant properties in zebrafish48 where it formed a gradient stabilized by interactions with HSPGs in the vasculature, to modulate neutrophil infiltration by a haptotactic gradient. Monomeric and dimeric CXCL8–GAG interactions are tissue specific in a mouse model, and demonstrate subtle differences in how neutrophils are recruited in inflammation in cell-specific tissue contexts.49
CXCL1 is also a member of the neutrophil attracting chemokine family. Similar to CXCL8 and other neutrophil-activating chemokines, dimeric CXCL1 is a highly efficient neutrophil chemoattractant, and its association with GAGs has an important role in this activity.49,50 GAGs also stabilize dimeric CXCL151 and protect it from protease cleavage sites in the CXCL1 dimer overlap.51 These sites include residues within the N-loop and C-terminal helix of CXCL152 and CXCL8.46 Secondary GAG binding sites are the N-terminus, 40s turn, and β-domain in CXCL1.52 GAG binding to these residues disrupts the interaction of CXCL1 with its receptor52 and may also involve further N-loop and N-terminal residues.53,54 Generation of CXCL1 mutants with disrupted GAG binding sites have a reduced ability to recruit neutrophils in vivo.55
Monocyte and macrophage chemoattractant chemokines that interact with the chemokine receptor CCR2 include CCL2 (monocyte chemoattractant protein [MCP]-1), and CCL7 (MCP-3).56,57 GAG binding sites in CCL2 include Arg18, Lys19, Arg24, and Lys49 with a significant but small contribution from Lys58 and His66.42 Preparation of CCL2 mutants has identified residues critical for interactions with heparin.40 Interaction of CCL2, 3, and 7 alters with HS sulfation where O-sulfation rather than N-sulfation is more important in such chemokine–GAG interactions.58 CCL2, CCL7, CCL8, and CCL13 are all members of the MCP family (56%–71% sequence identity) and bind to the CCR2 receptor. Monomeric forms of CCL7 have been shown to be incapable of oligomerization in the presence of heparin59 or HS,60 whereas CCL2 and CCL8 oligomerize when free in solution, and this is increased in the presence of GAGs.42,59 CCL7 as a monomer binds more strongly to GAGs compared with monomeric CCL2,60 possibly due to an extended GAG binding site in CCL7 (BXBXXB), where it is within the 40s loop,61 together with the other within the N-loop and the C terminus.60 The infiltration of neutrophils and macrophages into inflamed tissues can result in the generation of reactive oxygen62 and nitrogen63 species which can modify CCL264 and CCL5.65 Nitrated CCL2 displays reduced monocyte chemoattractant properties probably due to a reduction in binding to heparin compared with wild-type (WT) CCL2.66
Selectin
L-, P-, and E- selectin are cell surface adhesion molecules expressed by leukocytes, platelets, and endothelial cells, with P-selectin being expressed together with E-selectin on endothelial cells. Interactions between selectins and GAGs expressed on endothelial cells control the recruitment and rolling of inflammatory cells, particularly leukocytes during inflammation. Stimulated endothelial cells, via tumor necrosis factor (TNF)-α, promote inflammation; however, when treated with heparinase I, which removes HS/heparin chains from cell surface PGs, the attachment of monocytes under flow conditions was inhibited.67 The binding of L-selectin to HS/heparin in a rodent model of kidney transplantation together with prolonged ischemia and reperfusion (I/R) replicates early inflammatory events in tissues. Localization of L-selectin (Fig. 2) in the kidney microvasculature, using CD31 as a marker (Fig. 2B, D, and F), was absent following treatment with heparitinase (Fig. 2E and F).68 Nitrous acid treatment, which cleaves HS/heparin at unsubstituted amine groups within some of the glucosamine residues, also reduced the binding of L-selectin indicating the involvement of these residues in these interactions.69
Figure 2.

Localization of L-selectin in renal tissue. L-selectin ligands (A–F, green) on tissue sections of either contralateral rat kidney (A, B) or kidney 24 hr after I/R (C– D). In (B), (D), and (F), double staining is shown to identify endothelium using anti-CD31 antibody (red). Arrows indicate the presence of L-selectin ligands associated with interstitial capillaries, and high-power magnification indicates that these ligands are localized in the microvascular basement membrane (D, inset). L-selectin ligands were shown to be HSPGs using heparitinase I pretreatment of sequential I/R tissue sections (E, F). Scale bars = 50 µm. Reprinted with permission from Celie et al.68 Abbreviation: I/R, ischemia and reperfusion.
Endothelial cell–derived PGs are laid down in basement membranes in specific locations. Co-localization of L-selectin with three HSPGs in renal basement membrane (Fig. 3) have been identified as perlecan (Fig. 3A), agrin (Fig. 3B), and collagen type XVIII (Fig. 3C).70 While co-localization was shown, Celie et al.70 demonstrated that collagen XVIII was the only HSPG able to bind to L-selectin and that it was bound to its HS chains as binding was removed following heparitinase treatment.71
Figure 3.
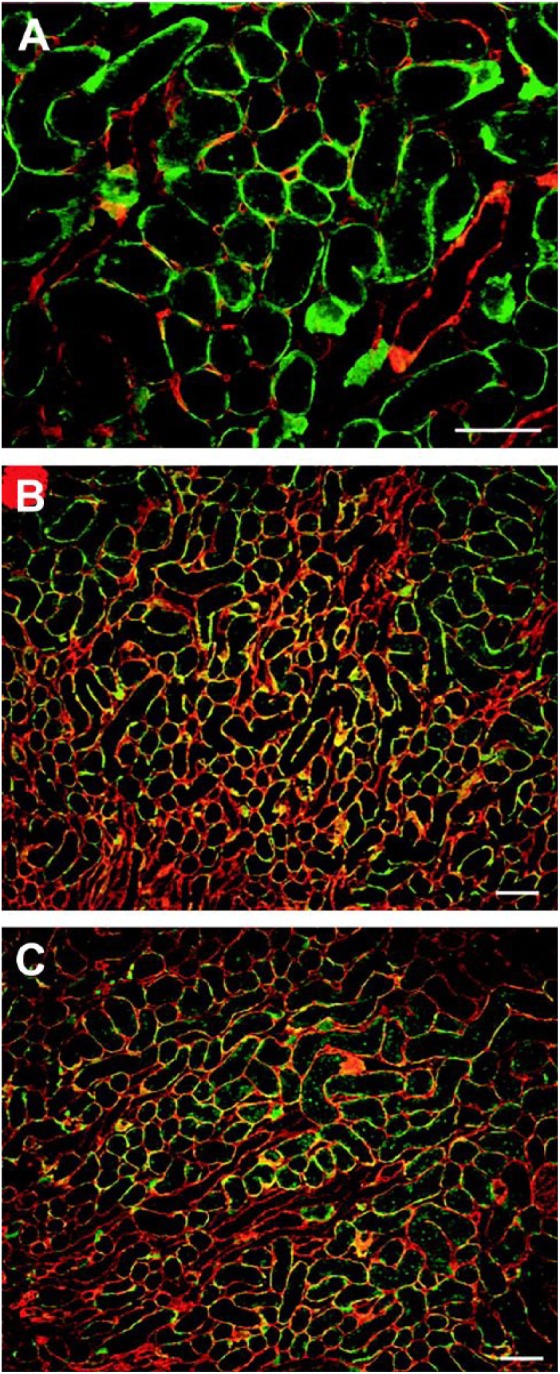
Renal L-selectin ligands partially colocalize with perlecan, agrin, and collagen type XVIII. Sections of normal rat kidney were probed for L-selectin (A–C, green) and anti-perlecan (A, red), anti-agrin (B, red), or anti-collagen type XVIII (C, red). Bar = 100 μm. Reprinted with permission Celie et al.70
As already discussed, differences in GAG structure and how this modulates selectin binding have been investigated in rodent models in which different HS/heparin biosynthesis enzymes have been inactivated. The enzymes NDST1 and NDST2 are the most widely distributed of the NDST enzymes, and are responsible for the deacetylation and sulfation of GlcNAc residues within HS.72 The inactivation of Ndst1 and Ndst2 in endothelial cells and leukocytes results in a decreased HS sulfation and a decrease in neutrophil infiltration into the peritoneal cavity of mutant mice compared with WT mice.73,74 Endothelial cells from Ndst1 mutant mice had reduced L-selectin binding, whereas cells derived from Ndst2 mutant mice displayed no reduction in L-selectin binding. Inactivation of Hs2st, an enzyme acting downstream from Ndst1 and which couples sulfate to IdoA at the C2 position, resulted in an increase in neutrophil recruitment into the peritoneal cavity.75 Endothelial cells from Hs2st–/– mice also showed increased L-selectin binding concomitant with a reduction in the rolling velocity of neutrophils on endothelial cells.75 L-, E-, and P-selectins play a role in leukocyte rolling and adhesion to the endothelium during inflammation, and their activities can be modulated by GAGs. The presence of heparin was shown to inhibit E-selectin and reduce P-selectin binding to endothelial cells.76 Modulation of L- and P-selectin binding was dependent on the structure, including the amount and position of the sulfate groups on heparin. The adhesion of monocytes to endothelial cells in vitro was inhibited by the presence of different heparin disaccharides that varied in the degree and position of sulfation.77 Furthermore, P- or L-selectin-deficient mice have an impaired inflammatory response that was further reduced following a heparin injection.77
Heparanase
Heparanase is an endo-β-glucuronidase and the only known mammalian glycosidase capable of cleaving HS chains. Mammalian heparanase differs from the bacterial heparitinases; the latter depolymerize HS/heparin by eliminative cleavage resulting in a ring opening reaction, which leaves an unsaturated bond, whereas heparanase undertakes a glycosidic cleavage of HS via a hydrolase reaction. HS cleavage by heparanase occurs at a limited number of sites along the HS chain, resulting in the production of HS-oligosaccharide fragments that are 10–20 saccharide units in length.78 As described above, HS interacts with chemokines, creating gradients, and selectins that recruit and guide leukocytes to sites of inflammation. Heparanase plays a significant role in inflammation cleaving cell surface HS disrupting interactions with chemokines and selectins that recruit leukocytes.79 Elevated heparanase expression has been shown in several inflammatory pathologies and is described in the sections below. The role that heparanase plays in a number of disease states has been investigated using heparanase-deficient (Hpse-KO)80 and heparanase-overexpressing (Hpa-tg)81 transgenic mice.
Heparanase activity is elevated in synovial fluid and synovium of rheumatoid arthritic patients compared with osteoarthritic patients.82 Heparanase in the urine of patients with type 1 or type 2 diabetes,83 and elevated levels of insulin and glucose in patients with type 2 diabetes, induce the secretion of active heparanase by kidney cells.84 Heparanase is also associated with the onset of type 1 diabetes where autoimmune cells attach and damage the islet β cells.85
The expression of heparanase is elevated and localized to epithelial cells in psoriatic lesions compared with normal skin (Fig. 4A).86 Furthermore, after induction of psoriasis in Hga-tg mice, an increase in macrophage infiltration occurs within psoriatic lesions, but not in WT mice86; similar observations have also been reported in psoriatic patients.87 Differing levels of heparanase activity in acute and chronic inflammatory bowel disease correlate with high expression levels of heparanase in Crohn’s disease (Fig. 4B(iii)) and ulcerative colitis (Fig. 4B(iv)), but not in normal tissue (Fig. 4B(i)) or in patients with infectious colitis (Fig. 4B(ii)) where heparanase expression was not detected.88
Figure 4.

Localization of heparanase. (A) Presence of heparanase in psoriatic lesions: (i) normal skin tissues and (ii) psoriatic tissue. Original magnification ×200. Reprinted with permission Lerner et al.86 (B) Presence of heparanase in inflammatory bowel disease: (i) normal, (ii) infectious colitis, (iii) Crohn’s disease, and (iv) ulcerative colitis. Original magnification ×200. Reprinted with permission Waterman et al.88 Tissue sections in both (A) and (B) were probed for the presence of heparanase with an anti-heparanase antibody and the presence of heparanase indicated by brown (A) and red (B) staining.
Heparanase is produced and secreted by platelets,89 neutrophils,90 and mast cells.91 Upon degranulation, heparanase release is associated with diapedesis and extravasation of inflammatory cells.92,93 During extravasation of leukocytes, a major obstacle to the successful migration of leukocytes into the tissues is the subendothelial basement membrane. In a model of delayed-type hypersensitivity, increased heparanase activity by activated endothelial cells resulted in damage to the subendothelial basement membrane enabling vessel leakage and the migration of inflammatory cells into the site of inflammation.94 Expression of heparanase by dendritic cells demonstrated that immature cells expressed heparanase in the nuclei and cytoplasm. Upon maturation, heparanase expression was localized to the plasma membranes concentrated in membrane extensions, where its localization was thought to facilitate extracellular matrix degradation and cellular transmigration.95 In chronic inflammation, stimulation of macrophages by heparanase initiates a vicious cycle where infiltrated macrophages further produce heparanase and recruit more macrophages to the site of inflammation.96 A characteristic feature of diabetic kidney disease is the activation and recruitment of macrophages, by TNF-α secretion,97 where TNF-α staining correlates with macrophage infiltration.98
The role of heparanase in the autoimmune disease, Alzheimer’s, is thought to be due to the degradation of HS chains that are responsible for the infiltration of inflammatory cells, which clear amyloid deposits. A delay in amyloid clearance in the brain of heparanase-overexpressing mice, however, coincided with an impaired recruitment of inflammatory cells.99 Heparanase is reported to increase inflammatory cell infiltration in the case of the recruitment of monocytes into the peritoneum after the induction of inflammation; however leukocyte or endothelial heparanase is not required for either T-cell or neutrophil extravasation.100
Degradation of HS by heparanase modifies/remodels the glycocalyx. In a model of inflammatory lung disease, activation of endothelial cells and elevated expression of endothelial heparanase result in a modification to the endothelial cell surface layer, and a loss of HS, which was prevented by the use of heparanase inhibitors.101 Degradation of the endothelial surface layer and removal of HS chains increased neutrophil adhesion by increasing the availability of endothelial cell surface adhesion molecules.101 The heparanase-mediated loss of the glycocalyx was suggested to contribute to the development of septic acute lung injury.101 The heparanase-mediated degradation of the glycocalyx has also been shown to be associated with the onset of inflammatory conditions including acute kidney102 and intestinal injury103 both of which were associated with sepsis.
The role of heparanase in the inflammatory response also involves indirect effects related to the regulation of inflammatory chemokines. The addition of heparanase to primary macrophages in vitro increased levels of TNF-α, CCL2, matrix metalloproteinase (MMP)-9, and IL-1. An increase in TNF-α and IL-1 also occurs after addition of heparanase to monocytes. This effect is thought to be mediated through the Toll-like receptor (TLR) pathways. The addition of heparanase to macrophages isolated from either TLR-2, TLR-4, or TLR-2/TLR-4-deficient mice failed to elicit an increased release of TNF-α.104 Ex vivo treatment of blood-derived mononuclear cells with heparanase also resulted in the release of inflammatory chemokines including TNF-α, CXCL8, IL-6, and IL-10.105 Furthermore, soluble HS signals through the TLR pathways, suggesting that the action of heparanase and generation of HS oligosaccharides may also upregulate cytokine production via the TLR pathways.105
Therapeutic Use of HS/Heparin Mimetics and Heparanase Inhibitors
There is ample evidence indicating that chemokines require HS to carry out their full range of functional activities during innate immunity. Supporting the hypothesis that molecules that interfere with the inflammatory promoting activities of HS are of therapeutic interest. The approaches explored to date include the use of heparin, HS mimetics, heparanase, and other small molecule inhibitors as detailed below.
Heparin
Many studies have explored the therapeutic use of heparin to inhibit the activity of HS in multiple stages of innate immunity. The anti-inflammatory activities of HS involve interactions with chemokines, cytokines, and selectins. Soluble heparin can compete with the binding of these molecules to HS on the surface of the endothelium.106,107 In addition, heparin can interfere with leukocyte recruitment to and transmigration through the endothelium, which uses L-selectin binding to endothelial cell surface HS73,77,108 and inhibits the expression of T lymphocyte–derived heparanase.109 These activities have also been demonstrated by the use of low-molecular-weight heparin, which can block leukocyte rolling, adhesion, and extravasation through activated endothelium.110 The efficacy of low-molecular-weight heparin to reduce inflammatory markers in vivo has been reported.111
Although these studies indicate the potential of heparin as an anti-inflammatory agent, it has not progressed to the clinic due to its structural diversity and its ability to perturb many biological processes, most notably coagulation. These data suggest that more defined molecules that can interfere with the activities of HS may be useful in the development of anti-inflammatory agents. Progress in this space has included the demonstration that heparin oligosaccharides, containing four or more monosaccharide residues without anticoagulant activity, are effective at inhibiting the function of L-selectin mediated neutrophil adhesion.112 In addition, 2,3-desulfated heparin inhibited neutrophil recruitment and leukocyte adhesion to the endothelium in a rat peritoneal model of inflammation113,114 while N-desulfated heparin inhibited leukocyte adhesion.115
HS/Heparin Mimetics
Due to the ability of heparin to interfere with the activities of HS in innate immunity, a range of heparin/HS mimetics have been explored to provide these functions without anticoagulant activity. Mouse knockout studies have aided in the design of these mimetics and shown that Ndst1-deficient endothelial cells produce HS with low levels of sulfation and do not take part in inflammatory processes the same way as WT endothelial cells.73 Most HS/heparin mimetics are carbohydrates or derivatives thereof. For example, trestatin A sulfate, a sulfated derivative of trestatin A, a non-uronic pseudo-nonasaccharide extracted from Streptomyces dimorphogenes, is effective in inhibiting L-selectin-mediated neutrophil interactions with the activated endothelium including almost completely blocking leukocyte rolling along rat mesenteric postcapillary venules and neutrophil migration into the thioglycollate-inflamed peritoneum of BALB/c mice.116 Low-molecular-weight fucoidin, a sulfated polysaccharide from brown algae, reduced the expression of proinflammatory cytokines including IL-6, TGF-β, TNF-α, ICAM-1, JNK, and mitogen-activated protein kinase (MAPK) in vivo117 and can modulate the expression of L-selectin.118 Fucosylated CS extracted from the sea cucumber Acaudina molpadioides can bind to L-selectin and inhibit the migration of neutrophils through an endothelial cell layer in vitro.119 Cyclitol-based pseudo-sugars coupled with an alkyl chain spacer of 8–10 carbon atoms were effective in inhibiting the activity of CXCL8, and interestingly, the sulfated derivatives did not enhance this inhibition.120 These findings support previous studies indicating that HS fragments containing two highly sulfated hexasaccharides joined by a non- or undersulfated dodecasaccharide linker that can bind to CXCL8.121 In a more synthetic approach, heparin-like synthetic polymers, called regenerating agents (RGTAs), preserved heparin-binding growth factor availability by inhibiting heparanase activity and thus protecting HS.122
Heparanase Inhibitors
Multiple approaches have been explored to inhibit heparanase activity including the use of modified heparins including periodate-oxidized, carboxyl-reduced, and N-desulfated, acetylated heparins.123 Phosphomannopentaose sulfate (PI-88) contains a mixture of phosphomannopentaose and phosphomannotetraose sulfates and maltohexaose sulfate which inhibit heparanase activity124; PI-88 decreases heparanase activity in pancreatic islets, protecting non-obese diabetic mice from developing type 1 diabetes.85 While the ability of PI-88 to bind a range of chemokines has not been extensively explored, it is reported to bind with high affinity to fibroblast growth factor-2, hepatocyte growth factor and vascular endothelial growth factor125; thus, it is plausible that PI-88 may also be involved in binding chemokines involved in innate immunity. However, PI-88 does not inhibit the activity of CXCL8.120 A more synthetic approach has explored the components (R)-3-hexadecanoyl-5-hydroxymethyltetronic acid (RK-682) and a benzylated version, 4-benzyl-RK-682, and show that both inhibit the activity of heparanase.126
Small Molecule Inhibitors of HS–Chemokine Interactions
An alternative to HS/heparin mimetics are small peptides that resemble the HS binding sites of cytokines and chemokines. One example is a peptide that corresponds to the HS-binding region of interferon-γ that has been effective in delaying the rejection of skin allografts.127 Chemokine variants that do not bind to HS have also been explored and shown not to oligomerize and signal without the HS binding region.40,128
Conclusions
The role that HS plays in inflammation is dependent on its structure, and ability to modulate interactions with mediators of inflammation such as chemokines, selectins, and enzymes. The fine structure of HS and its interaction with inflammatory mediators are of great importance to understanding how HS acts in such processes. As such, the use of heparin as a model to investigate these properties is not always appropriate. This statement is evident in the development of HS/heparin mimetics for anti-inflammatory biotherapeutics, to ensure the desired patient outcome. As researchers within this space continue to gain a more in-depth understanding of the interactions that occur between HS/heparin and mediators of inflammation, this will allow the continuing development of anti-inflammatory biotherapeutics that target these interactions.
Footnotes
Competing Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: All authors (BLF, MSL, JM, and JMW) contributed to the literature review and manuscript preparation.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by funding from the Australian Research Council under the Linkage Project (LP140101056) scheme.
Contributor Information
Brooke L. Farrugia, Graduate School of Biomedical Engineering, University of New South Wales Sydney, Sydney, New South Wales, Australia.
Megan S. Lord, Graduate School of Biomedical Engineering, University of New South Wales Sydney, Sydney, New South Wales, Australia
James Melrose, Graduate School of Biomedical Engineering, University of New South Wales Sydney, Sydney, New South Wales, Australia; Raymond Purves Bone and Joint Research Laboratory, Kolling Institute, Northern Sydney Local Health District, St. Leonards, New South Wales, Australia; Sydney Medical School–Northern, Royal North Shore Hospital, The University of Sydney, St. Leonards, New South Wales, Australia.
John M. Whitelock, Graduate School of Biomedical Engineering, University of New South Wales Sydney, Sydney, New South Wales, Australia
Literature Cited
- 1. Brickman YG, Ford MD, Gallagher JT, Nurcombe V, Bartlett PF, Turnbull JE. Structural modification of fibroblast growth factor-binding heparan sulfate at a determinative stage of neural development. J Biol Chem. 1998;273(8):4350–9. [DOI] [PubMed] [Google Scholar]
- 2. Parish CR. The role of heparan sulphate in inflammation. Nat Rev Immunol. 2006;6(9):633–43. [DOI] [PubMed] [Google Scholar]
- 3. HajMohammadi S, Enjyoji K, Princivalle M, Christi P, Lech M, Beeler D, Rayburn H, Schwartz JJ, Barzegar S, de Agostini AI, Post MJ, Rosenberg RD, Shworak NW. Normal levels of anticoagulant heparan sulfate are not essential for normal hemostasis. J Clin Invest. 2003;111(7):989–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007;446(7139):1030–7. [DOI] [PubMed] [Google Scholar]
- 5. Lindahl U, Kusche-Gullberg M, Kjellen L. Regulated diversity of heparan sulfate. J Biol Chem. 1998;273(39):24979–82. [DOI] [PubMed] [Google Scholar]
- 6. Sarrazin S, Lamanna WC, Esko JD. Heparan sulfate proteoglycans. Cold Spring Harb Perspect Biol. 2011;3(7):a004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Iozzo RV, Schaefer L. Proteoglycan form and function: a comprehensive nomenclature of proteoglycans. Matrix Biol. 2015;42:11–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kreuger J, Kjellen L. Heparan sulfate biosynthesis: regulation and variability. J Biol Chem. 2012;60(12):898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Whitelock JM, Murdoch AD, Iozzo RV, Underwood PA. The degradation of human endothelial cell-derived perlecan and release of bound basic fibroblast growth factor by stromelysin, collagenase, plasmin, and heparanases. J Biol Chem. 1996;271(17):10079–86. [DOI] [PubMed] [Google Scholar]
- 10. Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8(4):279–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McEver RP, Cummings RD. Perspectives series: cell adhesion in vascular biology. Role of PSGL-1 binding to selectins in leukocyte recruitment. J Clin Invest. 1997;100(3):485–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Archana VK, Sampath KK, Ann-Kathrin U, Martin G. Heparan sulphate as a regulator of leukocyte recruitment in inflammation. Curr Protein Pept Sci. 2015;16(1):77–86. [DOI] [PubMed] [Google Scholar]
- 13. Wright RD, Cooper D. Glycobiology of leukocyte trafficking in inflammation. Glycobiology. 2014;24(12):1242–51. [DOI] [PubMed] [Google Scholar]
- 14. Carlos TM, Harlan JM. Membrane proteins involved in phagocyte adherence to endothelium. Immunol Rev. 1990;114(1):5–28. [DOI] [PubMed] [Google Scholar]
- 15. Springer TA. Adhesion receptors of the immune system. Nature. 1990;346(6283):425–34. [DOI] [PubMed] [Google Scholar]
- 16. Diamond MS, Alon R, Parkos CA, Quinn MT, Springer TA. Heparin is an adhesive ligand for the leukocyte integrin Mac-1 (CD11b/CD1). J Cell Biol. 1995;130(6):1473–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tanaka Y, Adams DH, Hubscher S, Hirano H, Siebenlist U, Shaw S. T-cell adhesion induced by proteoglycan-immobilized cytokine MIP-1β. Nature. 1993;361(6407):79–82. [DOI] [PubMed] [Google Scholar]
- 18. Kuschert GSV, Coulin F, Power CA, Proudfoot AEI, Hubbard RE, Hoogewerf AJ, Wells TNC. Glycosaminoglycans interact selectively with chemokines and modulate receptor binding and cellular responses. Biochemistry. 1999;38(39):12959–68. [DOI] [PubMed] [Google Scholar]
- 19. Evangelista V, Manarini S, Sideri R, Rotondo S, Martelli N, Piccoli A, Totani L, Piccardoni P, Vestweber D, de Gaetano G, Cerletti C. Platelet/polymorphonuclear leukocyte interaction: P-selectin triggers protein-tyrosine phosphorylation-dependent CD11b/CD18 adhesion: role of PSGL-1 as a signaling molecule. Blood. 1999;93(3):876–85. [PubMed] [Google Scholar]
- 20. Mine S, Fujisaki T, Suematsu M, Tanaka Y. Activated platelets and endothelial cell interaction with neutrophils under flow conditions. Intern Med. 2001;40(11):1085–92. [DOI] [PubMed] [Google Scholar]
- 21. Shao B, Wahrenbrock MG, Yao L, David T, Coughlin SR, Xia L, Varki A, McEver RP. Carcinoma mucins trigger reciprocal activation of platelets and neutrophils in a murine model of Trousseau syndrome. Blood. 2011;118(15):4015–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rops AL, van den Hoven MJ, Baselmans MM, Lensen JF, Wijnhoven TJ, van den Heuvel LP, van Kuppevelt TH, Berden JH, van der Vlag J. Heparan sulfate domains on cultured activated glomerular endothelial cells mediate leukocyte trafficking. Kidney Int. 2008;73(1):52–62. [DOI] [PubMed] [Google Scholar]
- 23. Weber M, Hauschild R, Schwarz J, Moussion C, de Vries I, Legler DF, Luther SA, Bollenbach T, Sixt M. Interstitial dendritic cell guidance by haptotactic chemokine gradients. Science. 2013;339(6117):328–32. [DOI] [PubMed] [Google Scholar]
- 24. Rot A. Chemokine patterning by glycosaminoglycans and interceptors. Front Biosci. 2010;15(2):645–60. [DOI] [PubMed] [Google Scholar]
- 25. Tanino Y, Coombe DR, Gill SE, Kett WC, Kajikawa O, Proudfoot AEI, Wells TNC, Parks WC, Wight TN, Martin TR. and others. Kinetics of chemokine-glycosaminoglycan interactions control neutrophil migration into the airspaces of the lungs. J Immunol. 2010;184(5):2677–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Patel DD, Koopmann W, Imai T, Whichard LP, Yoshie O, Krangel MS. Chemokines have diverse abilities to form solid phase gradients. J Clin Immunol. 2001;99(1):43–52. [DOI] [PubMed] [Google Scholar]
- 27. Massena S, Christoffersson G, Hjertström E, Zcharia E, Vlodavsky I, Ausmees N, Rolny C, Li J-P, Phillipson M. A chemotactic gradient sequestered on endothelial heparan sulfate induces directional intraluminal crawling of neutrophils. Blood. 2010;116(11):1924–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Carman CV, Springer TA. A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J Cell Biol. 2004;167(2):377–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Carman CV, Sage PT, Sciuto TE, de la Fuente MA, Geha RS, Ochs Hans D, Dvorak HF, Dvorak AM, Springer TA. Transcellular diapedesis is initiated by invasive podosomes. Immunity. 2007;26(6):784–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Küppers V, Vestweber D, Schulte D. Locking endothelial junctions blocks leukocyte extravasation, but not in all tissues. Tissue Barriers. 2013;1(1):e23805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stoler-Barak L, Barzilai S, Zauberman A, Alon R. Transendothelial migration of effector T cells across inflamed endothelial barriers does not require heparan sulfate proteoglycans. Int Immunol. 2014;26(6):315–24. [DOI] [PubMed] [Google Scholar]
- 32. Wang S, Voisin M-B, Larbi KY, Dangerfield J, Scheiermann C, Tran M, Maxwell PH, Sorokin L, Nourshargh S. Venular basement membranes contain specific matrix protein low expression regions that act as exit points for emigrating neutrophils. J Exp Med. 2006;203(6):1519–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zlotnik A, Yoshie O. Chemokines: A new classification system and their role in immunity. Immunity. 2000;12(2):121–27. [DOI] [PubMed] [Google Scholar]
- 34. Bao X, Moseman EA, Saito H, Petryanik B, Thiriot A, Hatakeyama S, Ito Y, Kawashima H, Yamaguchi Y, Lowe JB, von Andrian UH, Fukuda M. Endothelial heparan sulfate controls chemokine presentation in recruitment of lymphocytes and dendritic cells to lymph nodes. Immunity. 2010;33(5):817–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lortat-Jacob H, Grosdidier A, Imberty A. Structural diversity of heparan sulfate binding domains in chemokines. Proc Natl Acad Sci U S A. 2002;99(3):1229–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liang WG, Triandafillou CG, Huang TY, Zulueta MML, Banerjee S, Dinner AR, Hung SC, Tang WJ. Structural basis for oligomerization and glycosaminoglycan binding of CCL5 and CCL3. Proc Natl Acad Sci U S A. 2016;113(18):5000–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dyer DP, Salanga CL, Volkman BF, Kawamura T, Handel TM. The dependence of chemokine-glycosaminoglycan interactions on chemokine oligomerization. Glycobiology. 2016;26(3):312–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shaw JP, Johnson Z, Borlat F, Zwahlen C, Kungl A, Roulin K, Harrenga A, Wells TNC, Proudfoot AEI. The X-ray structure of RANTES. Structure. 2004;12(11):2081–93. [DOI] [PubMed] [Google Scholar]
- 39. Proudfoot AEI, Fritchley S, Borlat F, Shaw JP, Vilbois F, Zwahlen C, Trkola A, Marchant D, Clapham PR, Wells TNC. The BBXB motif of RANTES is the principal site for heparin binding and controls receptor selectivity. J Biol Chem. 2001;276(14):10620–6. [DOI] [PubMed] [Google Scholar]
- 40. Proudfoot AEI, Handel TM, Johnson Z, Lau EK, LiWang P, Clark-Lewis I, Borlat F, Wells TNC, Kosco-Vilbois MH. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc Natl Acad Sci U S A. 2003;100(4):1885–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hoogewerf AJ, Kuschert GSV, Proudfoot AEI, Borlat F, Clark-Lewis I, Power CA, Wells TNC. Glycosaminoglycans mediate cell surface oligomerization of chemokines. Biochemistry. 1997;36(44):13570–8. [DOI] [PubMed] [Google Scholar]
- 42. Lau EK, Paavola CD, Johnson Z, Gaudry JP, Geretti E, Borlat F, Kungl AJ, Proudfoot AE, Handel TM. Identification of the glycosaminoglycan binding site of the CC chemokine, MCP-1: implications for structure and function in vivo. J Biol Chem. 2004;279(21):22294–305. [DOI] [PubMed] [Google Scholar]
- 43. Singh A, Kett WC, Severin IC, Agyekum I, Duan J, Amster IJ, Proudfoot AEI, Coombe DR, Woods RJ. The interaction of heparin tetrasaccharides with chemokine CCL5 is modulated by sulfation pattern and pH. J Biol Chem. 2015;290(25):15421–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Martin L, Blanpain C, Garnier P, Wittamer V, Parmentier M, Vita C. Structural and functional analysis of the RANTES-glycosaminoglycans interactions. Biochemistry. 2001;40(21):6303–18. [DOI] [PubMed] [Google Scholar]
- 45. Goger B, Halden Y, Rek A, Mösl R, Pye D, Gallagher J, Kungl AJ. Different affinities of glycosaminoglycan oligosaccharides for monomeric and dimeric interleukin-8: a model for chemokine regulation at inflammatory sites. Biochemistry. 2002;41(5):1640–6. [DOI] [PubMed] [Google Scholar]
- 46. Joseph PR, Mosier PD, Desai UR, Rajarathnam K. Solution NMR characterization of chemokine CXCL8/IL-8 monomer and dimer binding to glycosaminoglycans: structural plasticity mediates differential binding interactions. Biochem J. 2015;472(1):121–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kuschert GSV, Hoogewerf AJ, Proudfoot AEI, Chung CW, Cooke RM, Hubbard RE, Wells TNC, Sanderson PN. Identification of a glycosaminoglycan binding surface on human interleukin-8. Biochemistry. 1998;37(32):11193–201. [DOI] [PubMed] [Google Scholar]
- 48. Sarris M, Masson JB, Maurin D, Van der Aa, Lieke M, Boudinot P, Lortat-Jacob H, Herbomel P. Inflammatory chemokines direct and restrict leukocyte migration within live tissues as glycan-bound gradients. Curr Biol. 2012;22(24):2375–82. [DOI] [PubMed] [Google Scholar]
- 49. Gangavarapu P, Rajagopalan L, Kolli D, Guerrero-Plata A, Garofalo RP, Rajarathnam K. The monomer-dimer equilibrium and glycosaminoglycan interactions of chemokine CXCL8 regulate tissue-specific neutrophil recruitment. J Leukoc Biol. 2012;91(2):259–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Das ST, Rajagopalan L, Guerrero-Plata A, Sai J, Richmond A, Garofalo RP, Rajarathnam K. Monomeric and dimeric CXCL8 are both essential for in vivo neutrophil recruitment. PLoS ONE. 2010;5(7):e11754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Poluri KM, Joseph PRB, Sawant KV, Rajarathnam K. Molecular basis of glycosaminoglycan heparin binding to the chemokine CXCL1 dimer. J Biol Chem. 2013;288(35):25143–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sepuru KM, Rajarathnam K. CXCL1/MGSA is a novel glycosaminoglycan (GAG)-binding chemokine: structural evidence for two distinct non-overlapping binding domains. J Biol Chem. 2016;291(8):4247–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Murphy PM. Neutrophil receptors for interleukin-8 and related CXC chemokines. Semin Hematol. 1997;34(4):311–18. [PubMed] [Google Scholar]
- 54. Frevert CW, Kinsella MG, Vathanaprida C, Goodman RB, Baskin DG, Proudfoot A, Wells TNC, Wight TN, Martin TR. Binding of interleukin-8 to heparan sulfate and chondroitin sulfate in lung tissue. Am J Respir Cell Mol Biol. 2003;28(4):464–72. [DOI] [PubMed] [Google Scholar]
- 55. Sawant KV, Poluri KM, Dutta AK, Sepuru KM, Troshkina A, Garofalo RP, Rajarathnam K. Chemokine CXCL1 mediated neutrophil recruitment: role of glycosaminoglycan interactions. Sci Rep. 2016;6:33123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res. 2009;29(6):313–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11(11):762–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schenauer MR, Yu Y, Sweeney MD, Leary JA. CCR2 chemokines bind selectively to acetylated heparan sulfate octasaccharides. J Biol Chem. 2007;282(35):25182–8. [DOI] [PubMed] [Google Scholar]
- 59. Yu Y, Sweeney MD, Saad OM, Crown SE, Handel TM, Leary JA. Chemokine-glycosaminoglycan binding: specificity for CCR2 ligand binding to highly sulfated oligosaccharides using FTICR mass spectrometry. J Biol Chem. 2005;280(37):32200–8. [DOI] [PubMed] [Google Scholar]
- 60. Salanga CL, Dyer DP, Kiselar JG, Gupta S, Chance MR, Handel TM. Multiple glycosaminoglycan-binding epitopes of monocyte chemoattractant protein-3/CCL7 enable it to function as a non-oligomerizing chemokine. J Biol Chem. 2014;289(21):14896–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ali S, Robertson H, Wain JH, Isaacs JD, Malik G, Kirby JA. A non-glycosaminoglycan-binding variant of CC chemokine ligand 7 (monocyte chemoattractant protein-3) antagonizes chemokine-mediated inflammation. J Immunol. 2005;175(2):1257–66. [DOI] [PubMed] [Google Scholar]
- 62. Dröge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82(1):47–95. [DOI] [PubMed] [Google Scholar]
- 63. Dedon PC, Tannenbaum SR. Reactive nitrogen species in the chemical biology of inflammation. Arch Biochem Biophys. 2004;423(1):12–22. [DOI] [PubMed] [Google Scholar]
- 64. Sato E, Simpson KL, Grisham MB, Koyama S, Robbins RA. Effects of reactive oxygen and nitrogen metabolites on MCP-1-induced monocyte chemotactic activity in vitro. Am J Physiol Lung Cell Mol Physiol. 1999;277(3):L543–9. [DOI] [PubMed] [Google Scholar]
- 65. Sato E, Simpson KL, Grisham MB, Koyama S, Robbins RA. Effects of reactive oxygen and nitrogen metabolites on RANTES- and IL-5-induced eosinophil chemotactic activity in vitro. Am J Pathol. 1999;155(2):591–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Barker CE, Thompson S, O’Boyle G, Lortat-Jacob H, Sheerin NS, Ali S, Kirby JA. CCL2 nitration is a negative regulator of chemokine-mediated inflammation. Sci Rep. 2017;7:44384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Giuffrè L, Cordey AS, Monai N, Tardy Y, Schapira M, Spertini O. Monocyte adhesion to activated aortic endothelium: role of L-selectin and heparan sulfate proteoglycans. J Cell Biol. 1997;136(4):945–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Celie JWAM, Rutjes NWP, Keuning ED, Soininen R, Heljasvaara R, Pihlajaniemi T, Dräger AM, Zweegman S, Kessler FL, Beelen RHJ, Florquin S, Aten J, van den Born J. Subendothelial heparan sulfate proteoglycans become major L-selectin and monocyte chemoattractant protein-1 ligands upon renal ischemia/reperfusion. Am J Pathol. 2007;170(6):1865–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Norgard-Sumnicht K, Varki A. Endothelial heparan sulfate proteoglycans that bind to L-selectin have glucosamine residues with unsubstituted amino groups. J Biol Chem. 1995;270(20):12012–24. [DOI] [PubMed] [Google Scholar]
- 70. Celie JWAM, Keuning ED, Beelen RHJ, Dräger AM, Zweegman S, Kessler FL, Soininen R, van den Born J. Identification of L-selectin binding heparan sulfates attached to collagen type XVIII. J Biol Chem. 2005;280(29):26965–73. [DOI] [PubMed] [Google Scholar]
- 71. Kawashima H, Watanabe N, Hirose M, Sun X, Atarashi K, Kimura T, Shikata K, Matsuda M, Ogawa D, Heljasvaara R, Rehn M, Pihlajaniemi T, Miyasaka M. Collagen XVIII, a basement membrane heparan sulfate proteoglycan, interacts with L-selectin and monocyte chemoattractant protein-1. J Biol Chem. 2003;278(15):13069–76. [DOI] [PubMed] [Google Scholar]
- 72. Grobe K, Ledin J, Ringvall M, Holmborn K, Forsberg E, Esko JD, Kjellén L. Heparan sulfate and development: differential roles of the N-acetylglucosamine N-deacetylase/N-sulfotransferase isozymes. Biochim Biophys Acta. 2002;1573(3):209–15. [DOI] [PubMed] [Google Scholar]
- 73. Wang L, Fuster M, Sriramarao P, Esko JD. Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat Immunol. 2005;6(9):902–10. [DOI] [PubMed] [Google Scholar]
- 74. Lwmm Rops A, Loeven MA, van Gemst JJ, Eversen I, Van Wijk XM, Dijkman HB, van Kuppevelt TH, Berden JHM, Rabelink TJ, Esko JD, van der Vlag J. Modulation of heparan sulfate in the glomerular endothelial glycocalyx decreases leukocyte influx during experimental glomerulonephritis. Kidney Int. 2014;86(5):932–42. [DOI] [PubMed] [Google Scholar]
- 75. Axelsson J, Xu D, Na Kang B, Nussbacher JK, Handel TM, Ley K, Sriramarao P, Esko JD. Inactivation of heparan sulfate 2-O-sulfotransferase accentuates neutrophil infiltration during acute inflammation in mice. Blood. 2012;120(8):1742–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Luo J, Kato M, Wang H, Bernfield M, Bischoff J. Heparan sulfate and chondroitin sulfate proteoglycans inhibit E-selectin binding to endothelial cells. J Cell Biochem. 2001;80(4):522–31. [PubMed] [Google Scholar]
- 77. Wang L, Brown JR, Varki A, Esko JD. Heparin’s anti-inflammatory effects require glucosamine 6-O-sulfation and are mediated by blockade of L- and P-selectins. J Clin Invest. 2002;110(1):127–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Pikas DS, Li JP, Vlodavsky I, Lindahl U. Substrate specificity of heparanases from human hepatoma and platelets. J Biol Chem. 1998;273(30):18770–7. [DOI] [PubMed] [Google Scholar]
- 79. Meirovitz A, Goldberg R, Binder A, Rubinstein AM, Hermano E, Elkin M. Heparanase in inflammation and inflammation-associated cancer. FEBS J. 2013;280(10):2307–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zcharia E, Jia J, Zhang X, Baraz L, Lindahl U, Peretz T, Vlodavsky I, Li JP. Newly generated heparanase knock-out mice unravel co-regulation of heparanase and matrix metalloproteinases. PLoS ONE. 2009;4(4):e5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zcharia E, Metzger S, Chajek-Shaul T, Aingorn H, Elkin M, Friedmann Y, Weinstein T, Li JP, Lindahl U, Vlodavsky I. Transgenic expression of mammalian heparanase uncovers physiological functions of heparan sulfate in tissue morphogenesis, vascularization, and feeding behavior. FASEB J. 2004;18(2):252–63. [DOI] [PubMed] [Google Scholar]
- 82. Li RW, Freeman C, Yu D, Hindmarsh EJ, Tymms KE, Parish CR, Smith PN. Dramatic regulation of heparanase activity and angiogenesis gene expression in synovium from patients with rheumatoid arthritis. Arthritis Rheumatol. 2008;58(6):1590–600. [DOI] [PubMed] [Google Scholar]
- 83. Rops ALWMM, van den Hoven MJ, Veldman BA, Salemink S, Vervoort G, Elving LD, Aten J, Wetzels JF, van der Vlag J, Berden JHM. Urinary heparanase activity in patients with Type 1 and Type 2 diabetes. Nephrol Dial Transplant. 2012;27(7):2853–61. [DOI] [PubMed] [Google Scholar]
- 84. Shafat I, Ilan N, Zoabi S, Vlodavsky I, Nakhoul F. Heparanase levels are elevated in the urine and plasma of type 2 diabetes patients and associate with blood glucose levels. PLoS ONE. 2011;6(2):e17312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ziolkowski AF, Popp SK, Freeman C, Parish CR, Simeonovic CJ. Heparan sulfate and heparanase play key roles in mouse β cell survival and autoimmune diabetes. J Clin Invest. 2012;122(1):132–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lerner I, Zcharia E, Neuman T, Hermano E, Rubinstein AM, Vlodavsky I, Elkin M. Heparanase is preferentially expressed in human psoriatic lesions and induces development of psoriasiform skin inflammation in mice. Cell Mol Life Sci. 2014;71(12):2347–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Fuentes-Duculan J, Suárez-Fariñas M, Zaba LC, Nograles KE, Pierson KC, Mitsui H, Pensabene CA, Kzhyshkowska J, Krueger JG, Lowes MA. A subpopulation of CD163-positive macrophages is classically activated in psoriasis. J Invest Dermatol. 2010;130(10):2412–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Waterman M, Ben-Izhak O, Eliakim R, Groisman G, Vlodavsky I, Ilan N. Heparanase upregulation by colonic epithelium in inflammatory bowel disease. Mod Pathol. 2006;20(1):8–14. [DOI] [PubMed] [Google Scholar]
- 89. Ihrcke NS, Parker W, Reissner KJ, Platt JL. Regulation of platelet heparanase during inflammation: role of pH and proteinases. J Cell Physio. 1998;175(3):255–67. [DOI] [PubMed] [Google Scholar]
- 90. Matzner Y, Bar-Ner M, Yahalom J, Ishai-Michaeli R, Fuks Z, Vlodavsky I. Degradation of heparan sulfate in the subendothelial extracellular matrix by a readily released heparanase from human neutrophils. Possible role in invasion through basement membranes. J Clin Invest. 1985;76(4):1306–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Bashkin P, Razin E, Eldor A, Vlodavsky I. Degranulating mast cells secrete an endoglycosidase that degrades heparan sulfate in subendothelial extracellular matrix. Blood. 1990;75(11):2204–12. [PubMed] [Google Scholar]
- 92. Vlodavsky I, Eldor A, Haimovitz-Friedman A, Matzner Y, Ishai-Michaeli R, Lider O, Naparstek Y, Cohen IR, Fuks Z. Expression of heparanase by platelets and circulating cells of the immune system: possible involvement in diapedesis and extravasation. Invasion Metastasis. 1992;12(2):112–27. [PubMed] [Google Scholar]
- 93. Bartlett MR, Underwood PA, Parish CR. Comparative analysis of the ability of leucocytes, endothelial cells and platelets to degrade the subendothelial basement membrane: evidence for cytokine dependence and detection of a novel sulfatase. Immunol Cell Biol. 1995;73(2):113–24. [DOI] [PubMed] [Google Scholar]
- 94. Edovitsky E, Lerner I, Zcharia E, Peretz T, Vlodavsky I, Elkin M. Role of endothelial heparanase in delayed-type hypersensitivity. Blood. 2006;107(9):3609–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Benhamron S, Nechushtan H, Verbovetski I, Krispin A, Abboud-Jarrous G, Zcharia E, Edovitsky E, Nahari E, Peretz T, Vlodavsky I, Mevorach D. Translocation of active heparanase to cell surface regulates degradation of extracellular matrix heparan sulfate upon transmigration of mature monocyte-derived dendritic cells. J Immunol. 2006;176(11):6417–24. [DOI] [PubMed] [Google Scholar]
- 96. Lerner I, Hermano E, Zcharia E, Rodkin D, Bulvik R, Doviner V, Rubinstein AM, Ishai-Michaeli R, Atzmon R, Sherman Y, Meirovitz A, Peretz T, Vlodavsky I, Elkin M. Heparanase powers a chronic inflammatory circuit that promotes colitis-associated tumorigenesis in mice. J Clin Invest. 2011;121(5):1709–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Goldberg R, Rubinstein AM, Gil N, Hermano E, Li JP, van der Vlag J, Atzmon R, Meirovitz A, Elkin M. Role of heparanase-driven inflammatory cascade in pathogenesis of diabetic nephropathy. Diabetes. 2014;63(12):4302–13. [DOI] [PubMed] [Google Scholar]
- 98. Chow FY, Nikolic-Paterson DJ, Atkins RC, Tesch GH. Macrophages in streptozotocin-induced diabetic nephropathy: potential role in renal fibrosis. Nephrol Dial Transplant. 2004;19(12):2987–96. [DOI] [PubMed] [Google Scholar]
- 99. Zhang X, Wang B, O’Callaghan P, Hjertström E, Jia J, Gong F, Zcharia E, Nilsson LNG, Lannfelt L, Vlodavsky I, Lindahl U, Li JP. Heparanase overexpression impairs inflammatory response and macrophage-mediated clearance of amyloid-β in murine brain. Acta Neuropathol. 2012;124(4):465–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Stoler-Barak L, Petrovich E, Aychek T, Gurevich I, Tal O, Hatzav M, Ilan N, Feigelson SW, Shakhar G, Vlodavsky I, Alon R. Heparanase of murine effector lymphocytes and neutrophils is not required for their diapedesis into sites of inflammation. FASEB J. 2015;29(5):2010–21. [DOI] [PubMed] [Google Scholar]
- 101. Schmidt EP, Yang Y, Janssen WJ, Gandjeva A, Perez MJ, Barthel L, Zemans RL, Bowman JC, Koyanagi DE, Yunt ZX, Smith LP, Cheng SS, Overdier KH, Thompson KR, Geraci MW, Douglas IS, Pearse DB, Tuder RM. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat Med. 2012;18(8):1217–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Lygizos MI, Yang Y, Altmann CJ, Okamura K, Hernando AA, Perez MJ, Smith LP, Koyanagi DE, Gandjeva A, Bhargava R, Tuder RM1, Faubel S2, Schmidt EP. Heparanase mediates renal dysfunction during early sepsis in mice. Physiol Rep. 2013;1(6):e00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Chen S, He Y, Hu Z, Lu S, Yin X, Ma X, Lv C, Jin G. Heparanase mediates intestinal inflammation and injury in a mouse model of sepsis. J Histochem Cytochem. 2017;65(4):241–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Blich M, Golan A, Arvatz G, Sebbag A, Shafat I, Sabo E, Cohen-Kaplan V, Petcherski S, Avniel-Polak S, Eitan A, Hammerman H, Aronson D, Axelman E, Ilan N, Nussbaum G, Vlodavsky I. Macrophage activation by heparanase is mediated by TLR-2 and TLR-4 and associates with plaque progression. Arterioscler Thromb Vasc Biol. 2013;33(2):e56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Goodall KJ, Poon IKH, Phipps S, Hulett MD. Soluble heparan sulfate fragments generated by heparanase trigger the release of pro-inflammatory cytokines through TLR-4. PLoS ONE. 2014;9(10):e109596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Lever R, Page CP. Novel drug development opportunities for heparin. Nat Rev Drug Discov. 2002;1(2):140–8. [DOI] [PubMed] [Google Scholar]
- 107. Tyrrell DJ, Horne AP, Holme KR, Preuss JMH, Page CP. Heparin in inflammation: potential therapeutic applications beyond anticoagulation. Adv Pharmacol. 1999;46:151–208. [DOI] [PubMed] [Google Scholar]
- 108. Johnson Z, Proudfoot AE, Handel TM. Interaction of chemokines and glycosaminoglycans: a new twist in the regulation of chemokine function with opportunities for therapeutic intervention. Cytokine Growth Factor Rev. 2005;16(6):625–36. [DOI] [PubMed] [Google Scholar]
- 109. Lider O, Mekori YA, Miller T, Bar-Tana R, Vlodavsky I, Baharav E, Cohen IR, Naparstek Y. Inhibition of T lymphocyte heparanase by heparin prevents T cell migration and T cell-mediated immunity. Eur J Immunol. 1990;20(3):493–9. [DOI] [PubMed] [Google Scholar]
- 110. Wan MX, Zhang XW, Törkvist L, Thorlacius H. Low molecular weight heparin inhibits tumor necrosis factor α-induced leukocyte rolling. Inflamm Res. 2001;50(12):581–4. [DOI] [PubMed] [Google Scholar]
- 111. Hoppensteadt D, Fareed J, Klein AL, Jasper SE, Apperson-Hansen C, Lieber EA, Katz WE, Malouf JF, Stoddard MF, Pape LA. Comparison of anticoagulant and anti-inflammatory responses using enoxaparin versus unfractionated heparin for transesophageal echocardiography-guided cardioversion of atrial fibrillation. Am J Cardiol. 2008;102(7):842–6. [DOI] [PubMed] [Google Scholar]
- 112. Nelson R, Cecconi O, Roberts W, Aruffo A, Linhardt R, Bevilacqua M. Heparin oligosaccharides bind L- and P-selectin and inhibit acute inflammation. Blood. 1993;82(11):3253–8. [PubMed] [Google Scholar]
- 113. Lever R, Smailbegovic A, Page CP. Locally available heparin modulates inflammatory cell recruitment in a manner independent of anticoagulant activity. Eur J Pharmacol. 2010;630(1):137–44. [DOI] [PubMed] [Google Scholar]
- 114. Lakshmi TSR, Shanmugasundaram N, Shanmuganathan S, Karthikeyan K, Meenakshi J, Babu M. Controlled release of 2, 3 desulfated heparin exerts its anti-inflammatory activity by effectively inhibiting E-selectin. J Biomed Mater Res A. 2010;95A(1):118–28. [DOI] [PubMed] [Google Scholar]
- 115. Wang JG, Mu JS, Zhu HS, Geng JG. N-desulfated non-anticoagulant heparin inhibits leukocyte adhesion and transmigration in vitro and attenuates acute peritonitis and ischemia and reperfusion injury in vivo. Inflamm Res. 2002;51(9):435–43. [DOI] [PubMed] [Google Scholar]
- 116. Xie X, Rivier AS, Zakrzewicz A, Bernimoulin M, Zeng XL, Wessel HP, Schapira M, Spertini O. Inhibition of selectin-mediated cell adhesion and prevention of acute inflammation by nonanticoagulant sulfated saccharides: studies with carboxyl-reduced and sulfated heparin and with trestatin a sulfate. J Biol Chem. 2000;275(44):34818–25. [DOI] [PubMed] [Google Scholar]
- 117. Xu Y, Zhang Q, Luo D, Wang J, Duan D. Low molecular weight fucoidan modulates P-selectin and alleviates diabetic nephropathy. Int J Biol Macromol. 2016;91:233–40. [DOI] [PubMed] [Google Scholar]
- 118. Carvalho ACS, Sousa RB, Franco ÁX, Costa JVG, Neves LM, Ribeiro RA, Sutton R, Criddle DN, Soares PMG, de Souza MHLP. Protective effects of fucoidan, a P- and L-selectin inhibitor, in murine acute pancreatitis. Pancreas. 2014;43(1):82–7. [DOI] [PubMed] [Google Scholar]
- 119. Panagos CG, Thomson DS, Moss C, Hughes AD, Kelly MS, Liu Y, Chai W, Venkatasamy R, Spina D, Page CP, Hogwood J, Woods RJ, Mulloy B, Bavington CD, Uhrín D. Fucosylated chondroitin sulfates from the body wall of the sea cucumber holothuria forskali: conformation, selectin binding, and biological activity. J Biol Chem. 2014;289(41):28284–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Freeman C, Liu L, Banwell MG, Brown KJ, Bezos A, Ferro V, Parish CR. Use of sulfated linked cyclitols as heparan sulfate mimetics to probe the heparin/heparan sulfate binding specificity of proteins. J Biol Chem. 2005;280(10):8842–9. [DOI] [PubMed] [Google Scholar]
- 121. Spillmann D, Witt D, Lindahl U. Defining the interleukin-8-binding domain of heparan sulfate. J Biol Chem. 1998;273(25):15487–93. [DOI] [PubMed] [Google Scholar]
- 122. Rouet V, Meddahi-Pellé A, Miao HQ, Vlodavsky I, Caruelle JP, Barritault D. Heparin-like synthetic polymers, named RGTAs, mimic biological effects of heparin in vitro. J Biomed Mater Res A. 2006;78A(4):792–7. [DOI] [PubMed] [Google Scholar]
- 123. Parish CR, Hindmarsh EJ, Bartlett MR, Staykova MA, Cowden WB, Willenborg DO. Treatment of central nervous system inflammation with inhibitors of basement membrane degradation. Immunol Cell Biol. 1998;76(1):104–13. [DOI] [PubMed] [Google Scholar]
- 124. Parish CR, Freeman C, Brown KJ, Francis DJ, Cowden WB. Identification of sulfated oligosaccharide-based inhibitors of tumor growth and metastasis using novel in vitro assays for angiogenesis and heparanase activity. Cancer Res. 1999;59(14):3433–41. [PubMed] [Google Scholar]
- 125. Khachigian LM, Parish CR. Phosphomannopentaose sulfate (PI-88): heparan sulfate mimetic with clinical potential in multiple vascular pathologies. Cardiovasc Drug Rev. 2004;22(1):1–6. [DOI] [PubMed] [Google Scholar]
- 126. Ishida K, Hirai G, Murakami K, Teruya T, Simizu S, Sodeoka M, Osada H. Structure-based design of a selective heparanase inhibitor as an antimetastatic agent. Mol Cancer Ther. 2004;3(9):1069–77. [PubMed] [Google Scholar]
- 127. Fernandez-Botran R, Gorantla V, Sun X, Ren X, Perez-Abadia G, Crespo FA, Oliver R, Orhun HI, Quan EE, Maldonado C, Ray M, Barker JH. Targeting of glycosaminoglycan-cytokine interactions as a novel therapeutic approach in allotransplantation. Transplantation. 2002;74(5):623–9. [DOI] [PubMed] [Google Scholar]
- 128. Johnson Z, Kosco-Vilbois MH, Herren S, Cirillo R, Muzio V, Zaratin P, Carbonatto M, Mack M, Smailbegovic A, Rose M. and others. Interference with heparin binding and oligomerization creates a novel anti-inflammatory strategy targeting the chemokine system. J Immunol. 2004;173(9):5776–85. [DOI] [PubMed] [Google Scholar]