

Abstract
Cancer stem cells (CSCs) are a sub-population of cancer cells capable of self-renewal, proliferation, differentiation, plastic adaptation and immune regulation, thereby mediating tumorigenesis, metastasis and therapy resistance. CSCs are associated with cancer progression and clinical outcomes of cancer patients. Therefore successfully targeting CSCs is required to eradicate and cure cancer. Functional regulators of stem cell (stemness) signaling pathways in human cancers have brought new opportunities to target CSCs and reframe cancer-targeting strategies in clinical settings. However, challenges remain due to a lack of complete understanding of CSC plasticity/heterogeneity and limited efficacy of individual stemness inhibitors in cancer treatment. In this article, we review the CSC signaling pathways and the current state of CSC-targeting therapeutics in combinatory treatment in clinical trials.
Keywords: cancer stem cells, immune evasion, targeted therapy
I Cancer stem cells as the underlying root of cancer
Cancer remains one of the most fatal diseases worldwide with approximately 14 million new cases and 8.2 million cancer related deaths every year [1]. Despite advances in treatment, cancer relapse, distant metastases and drug resistance continue to kill patients. Work over the past twenty years has identified a subset of cancer cells with tumorigenic and stem cell properties called cancer stem cells (CSCs) as the underlying root for tumor initiation, metastasis, relapse and drug resistance in both liquid and solid tumors [2-13].
The defining property of these CSCs is self-renewal contributed by both genetic mutations and abnormal epigenetic alternations [14]. The cell of origin of CSCs vary across different cancer types [15]. It has been hypothesized that CSCs originate from somatic stem cells, partially-differentiated progenitors, or differentiated cells that acquire stemness through various mechanisms [16-18]. CSCs may derive from transformation of non-cancerous stem cell populations within a tissue that are already capable of self-renewal and differentiation, such as hematopoietic stem cells, but may also arise from progenitor populations that have begun differentiation and are not considered capable of permanent self-renewal, such as common myeloid progenitors [19].
In this review we list major methods and markers used to identify CSCs, discuss key pathways that support CSC functions, and summarize current approaches utilizing these pathway targets to combat CSCs and cancers in the clinic. We also address challenges of CSC research and clinical applications. Finally, we highlight future directions and explore important novel avenues of research and how they apply to CSCs, including biomarker evaluation, immunotherapies and combination therapies, and big data-driven bioinformatics.
II CSCs are identified as essential therapeutic targets
The first evidence of mouse CSCs tracks back to 1937 when single transplanted mouse leukemic cells regenerated leukemia in recipient mice [20]. Human CSCs were first identified in the 1990s when Dick’s group transplanted human acute myeloid leukemia (AML) into severe combined immunodeficiency disease (SCID) mice or NOD/SCID mice [21, 22], and found that only a fraction of the total leukemic cell population (CD34++CD38−) could regenerate AML in recipient mice due to their capacity for self-renewal, proliferation and differentiation into other cancer cells. Since this discovery, the gold standard assay to identify the tumorigenic properties of human CSCs has been their ability to generate human tumors in immune-deficient mice. Such use of in vivo CSC functional assays has helped create the alternative interchangeably-used terms in the literature, such as “tumor-initiating cell” and “tumorigenic cell” to describe putative CSCs. In Box I, we list common methods to identify CSCs, including cell sorting-based transplantation [4, 21-29], lineage tracing [15, 30-34], barcode tracing [35], and other functional assays [36] for representative CSC markers in solid tumors, hematologic malignancies, and metastases. It is anticipated that pooled CRISPR/Cas-mediated gene editing and modulation would also be used in identifying tumorigenic drivers and regulator markers of CSCs in various cancers.
Box I. Identification methods and markers of CSCs.
CSC Identification Methods
Fluorescence-activated cell sorting (FACS) and serial transplantations in vivo [4, 21-29]. This is the first method used to identify human CSCs xenografting sorted cells into immune-deficient mice.
Lineage tracing of CSCs with putative promoter-driven reporters in vivo [15, 30-34]. It examines the CSCs in intact tumor microenvironment without dissociation and sorting-mediated disruption. It is suitable for syngeneic mouse tumor models and can study immune –CSC interactions.
Barcode-tagging and tracing via RNA-sequencing to identify metastatic CSCs [35]. It enables simultaneous tracing of all tagged cancer cells for systematic, large scale, high throughput analyses. Chuang et al [35] used this approach and identified CD109 and the Jak-Stat pathway as critical drivers of lung cancer metastasis.
Other complementary assays in vitro, such as spheres and reporter systems [36] which normally require validation analyses in vivo.
Representative CSC Markers
| Tumor Type | CSC markers |
|---|---|
| Solid tumors | |
| Brain [149, 150] | CD133, CD49f, CD90, CD44 |
| Breast [4] | CD44, CD24 (negative/low), EpCAM, ALDH |
| Colon [25, 27, 151] | CD133, CD44, CD44v6, CD166, EpCAM, CD24, Lgr5 |
| Gastric [152-159] | ALDH1, CD24/CD44, CD54/CD44, EpCAM/CD44, CD71-negative, CD90, CD133 |
| Lung [26, 160] | Sca 1, CD34, CCA, CD133, ABCG2, |
| Melanoma [38, 161] | CD271, CD20 |
| Pancreatic [134] | CD133, CD44, EpCAM, CD24 |
| Prostate [162] | CD133, CD44, CD24 (negative) |
| Skin [163] | SOX2 |
| Hematologic maligencies | |
| Acute myeloid Leukemia [22] | CD34, CD38 |
| Leukemia [164] | CD34, CD38-negative, CD71-negative, CD90-negative, CD117-negative, CD123 |
| Metastases | |
| Pancreas [165] | CD133, CXCR4 |
| Breast [10, 39] | CD44, CD36 |
| Melanoma [39] | CD44, CD36 |
| Lung [35] | CD109 |
| Colon [49, 148] | CD110, LGR5 |
ABCG2, ATP-binding cassette subfamily G member 2; ALDH, aldehyde dehydrogenase; CCA, Clara cell-specific marker; CSC, cancer stem cell; CXCR4, C-X-C chemokine receptor type 4; EpCAM, epithelial cell adhesion molecule; LGR5, Leucine-rich repeat-containing G-protein coupled receptor 5.
Notably, there has been a debate whether CSC models exist in melanoma. Studies by the Morrison group examined a list of markers, but failed to identify any that enrich CSCs from human melanoma or differentiate the tumorigenic capacity of individual melanoma cells [37]. In contrast, the Weissman group was able to successfully identify a specific CSC surface marker, CD271 in human melanoma [38]. The debating results illustrate some of the limitations of using biased markers to identify CSCs. Recent findings from Pascual et al. identified the fatty acid receptor CD36 as a target and marker of metastasis-initiating cells (CD44bright) in melanoma and breast cancer since they rely on dietary lipids to promote metastasis [39]. In addition, heterogeneous CSC markers might be associated with distinct breast cancer subtypes, such as CD44+/CD24− phenotype with basal-like or mesenchymal-like cancers [40, 41].
As the defining feature of CSCs is self-renewal, CSCs are often compared with the counterpart of non-malignant stem cells. Much like traditional stem cells, CSCs have the capacity to: 1) self-renew, 2) proliferate by asymmetric or symmetric divisions, and 3) differentiate into several cell types of the specific tissue or tumor. CSCs are often linked to altered proliferation, pluripotency (giving rise to phenotypically diverse differentiated tumor cell populations), persistence (with the ability to maintain a reservoir within the tumor), and resistance to traditional chemotherapy agents as well as radiation [42]. Distinct features have been observed in various CSCs versus other cancer cells, such as epithelial-mesenchymal transition [43], increased resistance to oxidative stress [44], induced reprogramming [45-48], enhanced metabolic alterations [49, 50] and improved DNA damage repair [51, 52]. More recently, the capacity and mechanisms of CSC regulators such as β-catenin and Myc in immune evasion and suppression have been investigated [53, 54], highlighting the necessity and potential of CSC-targeting effects in increasing the efficacy of immunotherapies.
The extent of similarity between CSCs and normal stem cells could cause toxicity as therapeutics are developed to target CSC stemness. The number of normal stem cells is usually regulated through strict balance of proliferation, differentiation, and apoptosis [42] while CSCs are abnormally regulated due to their genetic and epigenetic alterations [55]. One potential solution could be to target stemness linked to the genetic and abnormal epigenetic alterations (such as fusion genes PTPRK-RSPO3) specific to CSCs or cancer cells, allowing for a much more targeted therapy while sparing normal cells [56].
III Signaling pathways of CSC therapeutic targets
Over the last few decades the signaling pathways that regulate CSCs have been identified [57], facilitating the development of novel CSC targeted strategies. The best characterized CSC pathways include, but are not limited to, Sonic Hedgehog (Hh)/Patched (Ptch)/Smoothened (Smo), Notch/Delta-like ligand (DLL), CXC chemokine receptor1-2/CXCL8/FAK, and Wnt, which may also affect downstream effectors including the transcription factor activators and transcription factors such as β-catenin, STAT3, and NANOG. Figure 1 gives examples of some of the CSC signaling pathways that are currently being pursued as potential clinical targets to treat a number of cancer types. While advances in drug discovery have led to the development of several molecules targeting CSC regulatory players, several concerns have been raised in terms of their safety, efficacy and clinical impact. First, CSCs share expression of many genes and signaling pathways involved in regulating differentiation and self-renewal with normal stem cells. Therefore, the use of these drugs raise concerns about toxicity and off target effects. Secondly, the redundancy in the regulatory pathways creates challenging hurdles that can lead to limited efficacy. To tackle such obstacles clinical trials are currently utilizing high-throughput screening of combinatory approaches to target CSCs (such as NCT02654964). Thirdly, it is not clear whether targeting CSCs alone is sufficient to treat cancers. As cellular plasticity may exist to switch the lineage fate of CSCs [58, 59] or to de-differentiate cancer cells, it would be beneficial to target both CSCs and differentiated cancer cells using complementary, combinational therapies. Therapeutic interventions that target these cancer stemness pathways and show clear evidence of activity will help define the clinical impact of each putative signaling pathway in CSC maintenance and pool expansion.
Figure 1. Targeting CSC pathways with newly developed drugs.
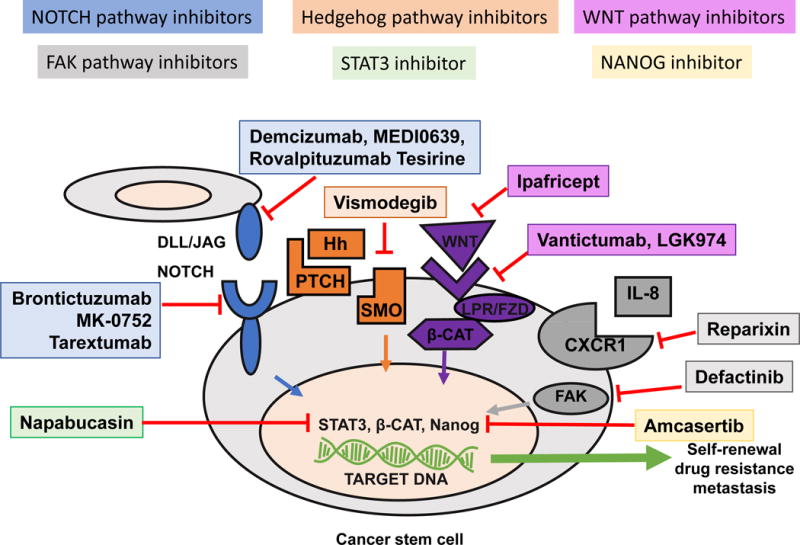
The schematic displays targeting strategies to combat CSCs by blocking stemness signaling pathways. NOTCH receptors can be inhibited by brontictuzumab, MK-0752, and tarextumab while NOTCH ligands can be inhibited by demcizumab, MEDI0639, and rovalpituzumab tersirine. These agents all block NOTCH signaling, ultimately preventing intracellular Notch cleavage and translocation into the nucleus. The hedgehog pathway can be blocked using the smoothened receptor antagonist Vismodegib and Sonidegib, which prevent smoothened activation and downstream signaling. WNT ligands can be inhibited by ipafricept and WNT receptors can be inhibited by vantictumab and LGK974. Reparixin can block FAK signaling by inhibiting CXCR1 thereby blocking IL-8 binding which normally triggers FAK phosphorylation and downstream signaling. FAK can also be targeted directly by defactinib. Finally, napabucasin can preventSTAT3 transcriptional activation while amcasertib inhibits Nanog transcriptional activation. While these agents are designed to limit CSCs by inhibiting mechanisms of self-renewal, drug resistance, and metastasis, their efficacy in the randomized trials has yet to be demonstrated. Additional innovative strategies are sought to target CSC plasticity and heterogeneity, including immunotherapies that overcome CSC-mediated immune suppression and evasion with diverse CSC recognition and targeting, as well as big data and super computation-based bioinformatic intelligence.
Examples of CSC-targeted therapies that are in various stages of clinical development are presented in Table 1. Early phase clinical trials have shown promise for agents that target CSC pathways and preliminary results of single-arm studies have been reported as shown in Table 2. In general, CSC-targeted therapies were associated with manageable toxicities and the most common adverse reactions included Grade 1-3 fatigue, hematological events, and gastrointestinal events (Table 2). In addition to these early-phase studies, a number of CSC-targeted therapies are undergoing evaluation in randomized clinical trials (Table 3). Many of these studies are ongoing with no results published to date. Notably, these identified signaling pathways with developed therapeutics might not represent the complete picture of CSC regulation. Among the targeted CSC signaling pathways, Sonic Hedgehog pathway inhibitors have shown promising cancer-treating efficacy [60-62]. While many of the other approaches are combining with standard-of-care chemotherapy to treat cancer patients, the clinical efficacy of these individual pathway inhibitors is yet to be demonstrated in randomized clinical trials. Additional innovative strategies are sought to target CSCs in which plasticity and heterogeneity might be the major hurdles to defeat. Immunotherapies that overcome CSC-mediated immune suppression and evasion with diverse CSC recognition and targeting strategies could be one of the promising directions.
Table 1.
Clinical drugs targeting CSC pathways.
| Targeted CSC Therapy | Targeted CSC Pathway(s) | Mechanism of Action Notes |
|---|---|---|
| Brontictuzumab (OMP-52M51) http://www.oncomed.com/ | Notch | An anti-Notch1 antibody |
| Demcizumab (OMP-21M18) http://www.oncomed.com/ | Notch/DLL-4 | A humanized IgG2 antibody that targets DLL-4 |
| MEDI0639 https://www.medimmune.com/ | Notch/DLL-4 | An immunoglobulin G1 lambda (IgG1λ) monoclonal antibody that selectively binds to DLL-4 |
| MK-0752 http://www.merck.com/ | Notch/γ-secretase | A γ-secretase inhibitor |
| Rovalpituzumab tesirine (SC16LD6.5) https://www.abbvie.com/landing/stemcentrx.html | Notch/DLL-3 | An anti-DLL3 antibody drug conjugate |
| Tarextumab (OMP-59R5) http://www.oncomed.com/ | Notch | An anti-Notch-2/-3 antibody |
| Vismodegib https://www.gene.com/ | Hedgehog/Smoothened | An antagonist of the Smoothened receptor, part of the Hh pathway; FDA-approved for treatment of basal cell carcinoma |
| Defactinib (VS-6063) http://www.verastem.com/ | FAK | A focal adhesion kinase (FAK) inhibitor |
| Reparixin http://www.dompe.com/en/ | CXCR1 | An inhibitor of human CXCR1/R2 receptor activation |
| Ipafricept (OMP-54F28) http://www.oncomed.com/ | Wnt | A fusion protein formed with a truncated Frizzled-8 receptor and the Fc fragment of human immunoglobulin IgG1 that acts as a decoy receptor for Wnt ligands |
| LGK974 [166] | Wnt/Porcupine | Porcupine inhibitor |
| Vantictumab (OMP-18R5) http://www.oncomed.com/ | Wnt/Frizzled | A fully human IgG2 monoclonal antibody targeting the Frizzled receptors 1, 2, 5, 7 and 8 |
| Napabucasin (BBI608) http://www.bostonbiomedical.com | STAT3/β-catenin/NANOG | A CSC inhibitor that targets STAT3 |
| Amcasertib (BBI503) http://www.bostonbiomedical.com/ | NANOG | A multi-kinase inhibitor of NANOG pathway |
Table 2.
Early phase (1/1b/2), single-arm trials of CSC-targeted therapies.
| Date initiated | Estimated Completion date | Treatment | Tumor Type | NCT# | Outcomes |
|---|---|---|---|---|---|
| 03-01-10 | 10-2015 | Vismodegib + nab-p + gem [167] | Pancreatic | NCT01088815 | DCR: 42/49; grade ≥3 AE: neutropenia (37.5%), anemia (21.4%), neuropathy (16.1%), fatigue (9.4%) |
| 04-01-10 | 10-2014 | MK-0752 + gem [83] | Pancreatic | NCT01098344 | PR: 11/18 AE: diarrhea, nausea, vomiting, fatigue, thrombocytopena, transaminitis |
| 08-25-10 | 09-2016 | Demcizumab + carbo/pem [84] | NSCLC | NCT01189968 | Well tolerated from initial report; regimen in ongoing randomized phase 2 study (final report to be published) AE: fatigue, nausea, hypertension |
| 02-09-11 | 08-2015 | MK-0752 + ridaforolimus [82] | HNSCC | NCT01295632 | CR: 1/15; PR: 1/15; SD: 1/15; AE: stomatitis, diarrhea, decreased appetite, hyperglycaemia, thrombocytopenia, asthenia and rash |
| 12-01-11 | 01-2020 | LGK974 + EGFR inhibitor + B-raf inhibitor PDR001 | Solid tumors | NCT01351103 | To be determined |
| 04-05-12 | 03-2017 | MEDI0639 [87] | Solid tumors | NCT01577745 | PR: 1/19; SD: 7/19; AE: increased aspartate aminotransferase, increased brain natriuretic peptid, and fatigue |
| 05-29-12 | 08-2016 | Ipafricept [168] | Solid tumors | NCT01608867 | |
| 07-11-12 | 04-2016 | Tarextumab + nab-p + gem [92] | Pancreatic | NCT01647828 | No grade ≥ 3 AEs; SD: 6/25 |
| 01-17-13 | 12-2015 | Napabucasin[169] | Solid tumors | NCT01775423 | Well tolerated; 15 mg/kg dose selected for phase 2; AE: diarrhea, fatigue, anemia |
| 01-24-13 | 12-2017 | Amcasertib (BBI503) [126] | CRC | NCT01781455 | DCR: 56% (in pts with high NANOG expression) AE: diarrhea, abdominal pain, fatigue, nausea |
| 01-24-13 | 07-2016 | Brontictuzumab (OMP-52M51) [89, 90] | Solid and hematologic tumors | NCT01778439 | DCR (CR+PR+SD) in biomarker-positive for NANOG (BP+) pts:56%; DCR in biomarker-negative (BP-) pts:13% (P=.040); OS in BP+ pts: 38 weeks; OS in BP- pts: 16 weeks |
| 05-16-13 | 04-2018 | Tarextumab + etoposide/platinum [89, 90, 93] | SCLC | NCT01859741 | Well tolerated; AE: diarrhea; final report is not yet published |
| 07-11-13 | 01-2017 | Rovalpituzumab tesirine (SC16LD6.5) [88] | SCLC | NCT01901653 | Well tolerated; 15 mg/kg dose selected for phase 2; AE: diarrhea, fatigue, nausea, anemia;ORR: 84% |
| 08-30-14 | 06-2018 | Napabucasin + paclitaxel [170] | Pancreatic | NCT02231723 | OR: 11/60; DCR: 46/65; AE: thrombocytopenia, pleural effusion, increased lipase |
carbo=carboplatin; CRC=colorectal cancer; CR=complete response; OR=objective response; ORR= overall response rate; pts= patients; DCR=disease control rate; DLT=dose-limiting toxicity; AE= most common treatment-related adverse effects; MTD=maximum tolerated dose; GEJ=gastroesophageal junction; gem=gemcitabine; nab-p=nab-paclitaxel; pem=pemetrexed; NSCLC=non-small cell lung cancer; SCLC=small cell lung cancer, HNSCC=head and neck squamous cell carcinoma
Table 3.
Recently completed or ongoing randomized trials (phase 2/3) of CSC-targeted therapies.
| Trial Name Treatment | Tumor Type | NCT# | Phase | Outcomes |
|---|---|---|---|---|
| ALPINE [171] Tarextumab + gem/nab-p vs gem/nab-p + placebo | Pancreatic | NCT01647828 | 2 | Study completed; Addition of TRXT to Nab-P+Gem did not improve OS in 1st line mPC. A potential detrimental effect on PFS and ORR was seen in subjects with N3 < 25%ile (ASCO 2017 Abstract 279) |
| BRIGHTER [117] Napabucasin + paclitaxel vs paclitaxel + placebo | Gastric/GEJ | NCT02178956 | 3 | Study is ongoing (Active, not recruiting) |
| CO.23 [172][116] Napabucasin + BSC vs placebo + BSC | CRC | NCT01830621 | 3 | Study closed; No significant difference between treatment groups AE: diarrhea |
| COMMAND [98] Defactinib vs placebo | Pleural mesothelioma | NCT01870609 | 2 | The study was terminated; interim analysis-DSMB stated good safety profile but lack of efficacy; results not yet published |
| DENALI Demcizumab + carbo/pem vs carbo/pem + placebo | NSCLC | NCT02259582 | 2 | Study is ongoing, (Active, not recruiting) |
| fRida [101] Reparixin + paclitaxel vs paclitaxel + placebo | Breast | NCT02370238 | 2 | Study is currently recruiting |
| PINNACLE Tarextumab + etop/platinum vs etop/platinum + placebo | SCLC | NCT01859741 | 2 | Study is ongoing (Active, not recruiting) |
| YOSEMITE Demcizumab + gem/nab-p vs gem/nab-p + placebo | Pancreatic | NCT02289898 | 2 | Study is ongoing (Active, not recruiting) |
BSC=best supportive care; carbo=carboplatin; CRC=colorectal cancer; CR=complete response; DCR=disease control rate; AE= most severe adverse effect; etop=etoposide; GEJ=gastroesophageal junction; gem=gemcitabine; nab-p=nab-paclitaxel; NSCLC=non-small cell lung cancer; SCLC=small cell lung cancer
IIIa. The Sonic Hedgehog signaling pathway
The Sonic Hedgehog (Hh) pathway is crucial to development and patterning during embryogenesis and essential for the maintenance of CSCs [63]. Activation of the Hh pathway occurs when Hh protein ligand binds to and inhibits a transmembrane protein called Patched (Ptch), leading to activation of the protein receptor Smoothened (Smo) which regulates target genes involved in metastasis, proliferation, survival, and pathway auto-regulation. Numerous human malignancies are associated with Hh deregulation and have a profound effect on the outcome of treatment [64]. The Hh pathway may be important for the maintenance of CSCs in various tumor types, including glioma, small cell lung cancer, non-small cell lung cancer, colorectal, pancreatic, and gastric cancers [65-68]. As such, inhibiting any step of the Hh signaling pathway may reduce CSCs and overcome treatment resistance.
The most extensive studies on the pathway regarding CSCs have been done in basal cell carcinoma (BCC) and medulloblastoma [69]. Constitutive activation of Hh pathway via Ptch1 deletion in various hair follicle stem cell populations have demonstrated BCC-like neoplasms, suggesting the emergence of CSCs in BCC results from constitutive Hh signaling [70]. In medulloblastomas, 30% of all sporadic cases are a result of Hh pathway mutations, where dormant, therapy resistant SOX2+ CSCs have been shown to be the key drivers of this population of medulloblastomas [71]. Two SMO inhibitors (LDE225/Sonidegib and GDC-0449/Vismodegib) have received FDA approval for treating basal cell carcinoma [72].
While there are many small drug inhibitors targeting Smo, vismodegib is currently being used in the clinic to effectively treat BCC [61]. Early results from vismodegib clinical trials suggest that it is effective at treating BCC in patients whose tumors display an activated Hh pathway, as assayed by the presence of GLI1 or PTCH2 [73]. Vismodegib is also being used in Phase 2 clinical trials to treat patients with Hh pathway mutations resulting in medulloblastoma [62].
IIIb. Notch signaling pathway
The Notch signaling pathway is one of the most extensively studied signaling pathways in CSC biology. [74]. The Notch pathway is activated via ligand-receptor interactions of 4 receptors (Notch-1-4) and 5 Notch ligands (Delta-like-ligand [DLL]-1, -3, -4 and Jagged-1, -2), leading to gamma-secretase-mediated cleavage of Notch, nuclear translocation of Notch Intracellular Domain (NICD), and NICD-transactivated target gene expression. In different cancers, the CSC-regulated Notch signaling can elicit either tumorigenic phenotypes (cervical, lung, colon, head and neck, prostate, brain/nerve, breast, and pancreatic cancer) or tumor-suppressive phenotypes (hepatocellular carcinoma, skin, and small cell lung cancer) [75-77]. Still, it is unclear how this dual role of Notch impacts clinical targeting of CSCs.
Elevated levels of Notch are often expressed in medulloblastoma and breast CSCs, making them ideal targets for Notch inhibition [78-80]. Studies show gamma-secretase inhibitor, GSI-18, prevents Notch receptor from becoming cleaved and translocated into the nucleus for transcriptional activation. GSI-18 can deplete CSC populations from medulloblastoma cell lines [79]. A different gamma-secretase inhibitor MK-0752 can also reduce CSCs and improve the activity of docetaxel in breast cancer cell lines [81]. Early staged clinical trials assessing the efficacy of MK-0752 in combination with other drugs, such as MTOR inhibitors or chemotherapies, demonstrate that while gamma secretase inhibitors can limit CSC populations in preclinical studies, it is still challenging to completely eradicate CSC subgroups in clinical settings of head and neck squamous cell carcinoma (HNSCC) and pancreatic ductal adenocarcinoma (PDAC) [82, 83]. Complete response in these studies was only observed in 1 out of 15 patients with HNSCC (Table 3) [82]. Partial response was observed in 1 out of the 15 patients with HNSCC and 11 out of 18 patients with PDAC [82, 83]. While partial response was promising for the PDAC patients, time to subsequent disease progression was only 38 weeks, suggesting the need for more effective therapies.
In effort to limit Notch signaling abnormalities, an array of antibodies have been designed to target distinct components of the pathway. The first therapeutic drug to selectively target the Notch pathway and enter clinical trials was the anti-DLL4 antibody, demcizumab. Demcizumab has been expected to block tumor angiogenesis, diminish CSCs, and alleviate immune suppression. A phase 1b clinical trial, combining demcizumab with chemotherapies, carboplatin and permetrexed, was well tolerated by patients [84, 85]. The combination is now being evaluated in phase 2 clinical trials (NCT02289898) which however has not appeared to meet its expected endpoint based on the OncoMed updates in 2017 [86]. MEDI0639, also an anti-DLL4 antibody, was evaluated in phase 1 clinical trials for solid tumors, but achieved minimal disease control [87]. DLL3 has also been shown to be a promising target to limit CSCs. A recent phase 1 clinical trial using the anti-DLL3 drug rovalpituzumab tesirine (SC16LD6.5) was designed to treat small-cell lung cancer patients who frequently express high levels of DLL3 [88]. This trial confirmed 18% of patients achieved the objective response, including 38% of patients with high DLL3 expression levels [88]. This study demonstrates that despite patients expressing high levels of DLL3, combinatorial approaches may be more effective to combat pathway redundancies.
The anti-Notch1 antibody, brontictuzumab has been evaluated in a phase 1 clinical trial as a single agent in patients with advanced solid tumors or blood cancers [89, 90]. This early study shows brontictuzumab is generally well tolerated and demonstrates moderate anti-tumor activity in patients. Finally, tarextumab is an anti-Notch2/3 antibody that can trigger tumor cell differentiation, limit angiogenesis, and delay tumor recurrence after cytotoxic therapies [91]. After being well tolerated by patients in phase 1b clinical trials, tarextumab has recently entered phase 2 clinical trials in combination with either etoposide and platinum or nab-paclitaxel and gemcitabine to combat untreated extensive small-cell lung cancer and metastatic pancreatic cancer, respectively (Tables 2 and 3) [92, 93]. Encouraging anti-tumor activity of tarextumab was observed for both phase 1b studies, however, final safety, immunogenicity, and correlation to clinical response are not yet determined.
IIIc. FAK signaling pathway
Focal adhesion kinase (FAK) is a cytoplasmic non-receptor tyrosine kinase that plays a significant role in tumor cell survival by orchestrating cellular signaling through integrins and growth factor receptors and functions in multiple steps of tumorigenesis [94]. This includes regulation of cell motility, invasion, cell survival, and transcription that promotes epithelial-mesenchymal transition. FAK also contributes to breast CSC proliferation and cell survival in a kinase-independent manner [95-97].
In vitro, FAK inhibitors exhibit enhanced activity in combination with cytotoxic drugs or agents targeting angiogenesis, such as sunitinib [94]. Preclinical data supports using FAK inhibition to treat tumors with upregulated CSC pathways [94]. A recently completed phase 2 randomized clinical trial assessed the FAK inhibitor defactinib as a maintenance therapy in patients with malignant pleural mesothelioma (NCT01870609) [98]. Patients who had 4 or more cycles of chemotherapy were recruited with the purpose of defactinib preventing tumors from reappearing by CSC initiation [98]. However, defactinib lacks efficacy as a single agent to control malignant mesothelioma [98]. A future promising direction might be utilizing FAK inhibitors with immune checkpoint inhibitors to target immunosuppressive pancreatic cancer microenvironment [99].
The binding of IL-8 to CXCR1 can also promote CSCs by triggering FAK phosphorylation. This activated cascade can then phosphorylate AKT and activate the WNT pathway for self-renewal. FAK signaling has been proposed to protect CSCs from a FASL/FAS-mediated bystander effect by limiting downstream signaling of FAS during chemotherapy in models of breast cancer in vitro and in vivo, suggesting a combined chemotherapy and CXCR1 inhibitor regimen could provide a strategy to effectively target breast CSC compartments [100]. Currently, an ongoing phase 2 clinical trial is testing this concept using paclitaxel in combination with the CXCR1 inhibitor reparixin to treat breast cancer patients after a phase 1b clinical study showed safety and tolerability, as well as promising responses in triple negative breast cancer patients [101].
IIId. Wnt signaling pathway
The canonical Wnt signaling pathway is another developmental cascade involved in multiple biological processes including regulation of gene transcription for embryogenesis, development, cell proliferation, survival, renewal, and differentiation [102-104]. Lineage tracing assays in intestinal adenomas demonstrated that a small fraction of tumor cells positive for Lgr5, a Wnt target gene associated with somatic stem cells in the colon, and these tumor cells were largely responsible for the abnormal growths [31]. Other lineage tracing studies of Wnt target Axin2 and Lgr5 have also been utilized to identify cells with self-renewing capacity in several tissue types [105-107]. Still, the reach of Wnt signaling seems limitless, playing a role in numerous cancers. Deletion of β-catenin has limited tumorigenesis in mixed lineage leukemia and facilitated tumor regression by depleting CSCs in epidermal tumors [33, 108]. Thus, targeting CSC Wnt/β-catenin signaling or further downstream transcription factors in this pathway are attractive strategies for CSC-targeted cancer therapy.
Due to the cascading nature of the pathway, Wnt inhibitors have emerged to target various components of the pathway. For example, vantictumab (OMP-18R5) blocks Wnt signaling by binding multiple FZD receptors (FZD1,2,5,7, and 8) and has been found to decrease CSC numbers and proliferation in patient-derived xenograft (PDX) models of breast, pancreatic, and lung cancers [109]. Currently, vantictumab is being administered in phase 1b clinical trials to breast, pancreatic, and lung cancer patients in combination with standard chemotherapy regimens (clinicaltrials.gov).
Another Wnt inhibitor also in Phase 1b clinical trials is Ipafricept which binds and sequesters Wnt directly to block the cascade [110] (clinicaltrials.gov). Lipid modification is necessary for all Wnt proteins, therefore targeting the enzyme, Porcupine (Porcn), which facilitates lipid modifications of Wnt as it binds Fzd or as it is secreted from a ligand expressing cell, works as an alternative form to sequester Wnt from activating the pathway. Porcn inhibitor LGK974 is currently being used in combination with EGFR and B-raf inhibitors to treat colorectal cancer patients with aberrant Wnt signaling in Phase 1b clinical trials [111]. Additionally, dual targeting of the Wnt pathway and microtubule function shows considerable promise as an effective therapy against cancer pathologies [112].
IIIe. Signal transducer and activator of transcription 3 (STAT3) signaling
STAT3 is one of the seven STAT family members that have been identified and primarily activated by Janus kinases (JAK)-mediated phosphorylation of a specific tyrosine residue [113]. Once activated, dimerized STATs translocate to the nucleus and initiate transcription of the target genes. STAT3 is excessively active in many cancers and plays a major role in tumor growth and metastasis [114], notably converting non-CSCs to CSCs by upregulating the expression of the reprograming factor OCT 3/4, and subsequent expression of several stem cell genes and suppressed cell commitment genes [114, 115]. Targeting STAT3 activation inhibits tumor growth and metastasis both in-vitro and in-vivo without affecting normal cells, therefore suggesting that STAT3 could be a valid molecular target for cancer therapy [114]. A phase 3 study recently completed the evaluation of the STAT3 inhibitor napabucasin (BBI608) plus best supportive care treatment in advanced colorectal carcinoma (CO.23 study) [116]. Although this monotherapy study was stopped early due to minimal differences between the napabucasin and placebo treatment groups, patients who showed expression of pSTAT3 in their tissue samples had improved survival, suggesting napabucasin may be useful for selective patients with targeted biomarker expression [116]. Currently, another phase 3 study is assessing combinational napabucasin and paclitaxel treatments in patients with advanced gastric or gastroesophageal junction cancer who progressed after initial chemotherapy treatment (BRIGHTER study) [117].
IIIf. NANOG signaling
NANOG is a homeodomain-containing transcription factor that is part of the key set of transcription factors involved in the maintenance of pluripotency and self-renewal in undifferentiated embryonic stem cells [118]. In CSCs of various cancers, NANOG expression levels are higher including breast, cervix, oral, kidney, prostate, lung, gastric, brain, pancreas, and ovarian cancer [47, 119-125]. Increased expression of NANOG is shown as an indicator of a poor prognosis for ovarian, colorectal, and breast cancer patients [118]. In oral squamous cell and lung adenocarcinoma, higher expression of NANOG was associated with advanced cancer stage and shorter patient survival rate. NANOG expression has also been shown to be higher in CSCs than in other cancer cells in several types of cancers [118]. CD133+ cancer cells express significantly higher levels of NANOG in comparison to CD133− cancer cells. Whereas, in prostate cancer cell lines, NANOG induction results in upregulation of CD133 and ALDH1. Functional studies have shown that NANOG is not only a marker for CSCs, but also promotes CSC characteristics across multiple cancers. Overexpression of NANOG in cancer cells has been linked to increased proliferation rate in vitro and tumor growth in vivo [118].
Amacasertib (BBI503) which targets multiple kinases in the NANOG pathway is currently being evaluated in clinical trials (NCT02232633, NCT02232620, NCT02232646, and NCT02432690). Since colorectal cancer patients often express high levels of NANOG which is associated with poor prognosis, a phase 1 clinical trial used amacasertib to treat patients (NCT01781455). This study demonstrated a disease control rate of 56% in patients with high NANOG expression compared to 13% in patients that were NANOG negative [126]. These results highlight the potential benefits of CSC biomarkers and how they can help personalize patient care.
IV. CSCs and resistance to therapy
The percentage of CSCs in a tumor is determined mainly by: 1) the characteristics of the CSC population that initiated tumorigenesis, 2) the “niche” or microenvironment, and 3) the frequency with which new or additional CSCs are created and non-CSCs are removed. Tumor type or stage of progression may determine the percentage of CSCs, where larger numbers of CSCs may indicate: 1) worse clinical outcomes, 2) higher rate of proliferation, 3) greater genetic instability, and 4) lack of differentiation. In addition, CSCs gain selective advantage in distinct microenvironment conditions, such as hypoxic environments or in the presence of chemotherapy [127]. CSCs have numerous properties that allow them to persist through chemotherapy, radiation and other treatments [2, 24, 28, 53, 58, 59, 128]. First, CSCs have plastic cell cycle kinetics, allowing a sub-population of CSCs to remain quiescent even as others are proliferating. Thus, certain CSCs are able to evade the numerous therapies that target rapidly proliferating cells [129]. Additionally, CSCs have more efficient DNA repair mechanisms than other cancer cells [24, 130, 131], and exposure to DNA damage (through ionizing radiation, for example) triggers recruitment of cell cycle inhibitors, allowing more time for the DNA damage to be repaired [51, 52]. Furthermore, CSCs express transporter proteins that can pump chemotherapy agents out of the cells [132]. CSCs express high levels of metabolic regulators, such as ALDH and oxidant scavengers, which are able to metabolize chemotherapeutic agents or reactive oxygen species (ROS) [2] to reduce damage from ROS [2]. Finally, stemness regulators such as SOX2 and EZH2 work in the absence of p53 and RB1 to confer lineage plasticity, allowing CSCs to survive anti-tumor therapies [58, 59]. Thus, the intrinsic heterogeneity, plasticity and adaptive resistance of CSCs to stress contribute to the protection of CSCs, allow them to foster the tumor mass and establish secondary tumors, and provide resistance to anti-tumor therapies [127, 133]. Whether the current CSC identification and targeting strategies are sufficient and effective enough to overcome a wide array of mechanisms CSCs possess that enable them to resist anti-tumor therapies is an open question. More importantly, it is uncertain whether these properties would also enable CSCs to resist the newly-developed and to-be-developed anti-CSC therapeutics.
Studies using CSC-targeted therapies as monotherapy have shown promise in early phase studies, but so far, have not shown adequate activity in randomized trials. One solution would be combining anti-CSC approaches with other therapies to not only eliminate the bulk of dividing and differentiated components of the tumor responsible for tumor-associated symptoms, but also to remove the minority CSC population that is resistant to therapies, fuels tumor growth, and contributes to disease relapse and metastases [134]. Indeed many of the randomized trials have combined the listed anti-CSC therapeutics with standard-of-care chemotherapies (Table 3). Another solution might be that the CSC hypothesis requires adjustments about how CSCs are diagnosed and treated in tumor. Possible adjustments could include evaluations of CSC biomarkers based on their levels of pluripotency (dedifferentiation), plasticity and heterogeneity instead of certain pathway markers, development of efficacious approaches to selectively target CSCs without affecting normal SCs, and blocking the mechanisms of CSC resistance to anti-tumor treatment using innovative targeting techniques, such as immunotherapy.
V. Immunotherapy and CSCs
Recent studies demonstrated the immune suppressing roles of cancer stemness regulators such as β-catenin in the Wnt signaling pathway [53]. cMyc as one of the pluripotency transcription factors upregulates both innate immune inhibitor CD47 and adaptive immune checkpoint molecule PD-L1 in human tumor cells [54]. This suggests that CSCs might not only passively evade immune attacks, but actively suppress immune responses. Durable cancer-controlling effects of immune checkpoint blockade antibodies have brought a new era of cancer immunotherapy, leading to more successful eradication of CSCs and other cancer cells in the clinic. Targeting CSCs using immunotherapy can take the form of either generating an immune response directed at CSC-specific antigens, blocking the function of those CSC-specific proteins or by inhibiting components of the immune checkpoint system. Inhibition of immune checkpoints could thereby raise the immune system’s sensitivity to tumor/CSC antigens and trigger a response by the immune system against cells that would normally be unaffected. We will summarize below a few CSC-specific antigen therapies tested against CD133, and CD47 as well as immune checkpoint blockade therapy using anti-PD-1 and anti-CTLA-4 antibodies.
Recently, Huang et al.[135] produced a bi-specific antibody against CD3 and CD133, binding cytokine-induced killer (CIK) CD3+ cells to CD133+ CSCs from pancreas and hepatic cancers. This antibody should bring CSCs into contact with CIK cells, targeting the CD133+ cells for death. In nude mice, CIK cells bound to this antibody effected the destruction of CD133+ tumor cells more than unbound CIK cells or cells bound to only a CD3-specific antibody, suggesting that the antibody was successfully recruiting the immune system to attack CSCs.
CD47 has been identified as a surface marker for all cancer cells, but has been of particular clinical interest as a marker of leukemic stem cells [136] and other CSCs [137-142]. CD47 functions as a potent anti-phagocytic agent, allowing cells to evade the innate immune system. When leukemia CSCs express lower levels of CD47, they are preferentially phagocytosed by macrophages, meaning that CD47+ leukemia cells predominate [143]. Blockade of CD47 could therefore be a potent treatment targeting leukemia CSCs, and has been shown to work as such by Liu et al. [144] in PDX models of leukemia.
Targeting the immune checkpoint molecules of PD-1 and CTLA-4 with blocking antibodies has proven to be a productive treatment for some patients, particularly among those with lung cancer and melanoma of high mutation rates. Immune checkpoint blockade inhibits the interaction of PD-1 and CTLA-4 with their ligands, allowing cytotoxic T cells to attack cancer cells despite the presence of inhibitory ligands on cancer cells. Although CSCs may express many immunogenic antigens, the expression of PD-1 ligand PD-L1 and CTLA-4 ligands CD80 and CD86 prevent the cytotoxic activity of the immune system from attacking them [145]. The durable response that some patients have to immune checkpoint blockade therapy may be attributable to its effect on CSC populations, although this hypothesis has yet to be proven. However, current immune checkpoint blockade to inhibit PD-1 and CTLA4 pathways is only suitable for a small set of cancer types and subtypes with relatively high mutation loads. To effectively target CSCs in the majority of cancers with low mutation frequencies, new immune-regulating approaches are to be discovered and explored.
VI Future trends and strategies of targeting CSCs –bioinformatics and big data
Identifying targets for novel drugs in CSCs is complicated by both their relative rarity in a tumor sample and the difficult task of accurately characterizing them. New developments in high-throughput sequencing, including single-cell sequencing, will help provide a better definition of CSCs and may identify new targets for treatment. By sequencing individual tumor cells and looking for populations with expression patterns associated with stemness, relatively complete genomic and epigenomic profiles of CSCs can be identified without labor-intensive and condition-compromised tumor initiation assays.
Tirosh et al.[146] performed single-cell RNA-seq on whole primary lesions from six oligodendroglioma patients, hoping to identify intratumoral heterogeneity and potentially characterize CSCs from within those lesions. Among all cancer cells, they identified three common expression patterns – one with genes associated with oligodendrocytic markers, one with genes associated with astrocytic markers, and a third that had more intermediate expression patterns for oligodendocytic genes and astrocytic genes, but also with genes important for neuronal development and proliferation, suggesting a stem-cell like population. Expression patterns from within this sub-population may be useful for the identification of novel drug targets for oligodendroglioma CSCs. Another method is barcode-tagging and tracing in vivo used to identify CD109+ metastatic CSCs in lung cancer [35].
We anticipate that the understanding and targeting of CSCs will be inevitably tailored and accelerated by big interactive data-driven artificial intelligence and deep learning. Esteva et al. recently utilized deep learning algorithms to develop convolutional neural networks to identify skin cancer with dermatologist-level capacity [147]. With the recent boom of multi–omic studies, machine-learning and bioinformatic approaches, big data mining-assisted decision making has emerged as a new tool to study cancer and would benefit ongoing and future CSC-targeting trials. However, there continues to be a translational disconnect between the research bench and the clinic. Despite the numerous ongoing clinical trials, only 4-5% of cancer patients are enrolled. Major hurdles include a lack of data sharing, overwhelming disorganization, insufficient data access, and unbridged collaboration between specialized data-mining experts and cancer biologists. Much of the data gathered is not being used at a maximum potential. For this reason, moving towards a more uniform system of data collection and dissemination may mobilize and integrate the entire cancer community’s effort to identify and target CSCs, find cure, and stop cancer.
VII. Concluding Remarks
Cancer stem cells (CSCs) are a sub-population of cancer cells capable of self-renewal, proliferation, differentiation, plastic adaptation and immune regulation, thereby mediating tumorigenesis, therapy resistance and metastasis. Successful cancer treatment is complex and involves direct anticancer activity of eliminating both CSCs and other tumor cells, changing immune responses, and altering the tumor microenvironment. Therapies directed only toward differentiated cancer cells risk relapse and proliferation of more aggressive tumor cells due to not eliminating the pool of CSCs. In the recent trials, anti-CSC therapies have been benefited in combination with other approaches targeting bulk tumor cells, such as radiation and chemotherapies. However, lack of efficacy has been the hurdle for many individual pathway-specific anti-CSC therapeutics, thus demanding better identification and targeting strategies to specifically, effectively, and rapidly eliminate CSCs and tumor burden, as well as limit toxic side effects on normal SCs and organs.
Moving forward, the CSC model has profound implications for the development of new treatments for cancer. Many current therapies are based on the ability to shrink or de-bulk tumors as assessed by Response Evaluation Criteria in Solid Tumors (RECIST). Unfortunately, tumor shrinkage does not correlate well with patient survival for many tumor types. Therefore, using tumor shrinkage as an indicator of activity for an agent that targets CSCs in either preclinical models or phase 1/2 clinical trials may have to be re-examined. In addition, development of adjuvant therapies may have to be completely reanalyzed based on the CSC model (see outstanding questions). A recent study demonstrated that targeting Lgr5+ CSCs are more critical for metastasis than the primary colon cancer [148]. It is likely that targeting CSCs is more crucial to remove secondary tumors than for shrinking primary tumors.
Many of the listed CSC-targeting therapeutics have not yet shown promising efficacy as single agents to treat cancer in randomized clinical trials. Not only is there a need to develop the innovative CSC-targeted therapies, much work still needs to be done to evaluate combinations of CSC-targeted therapies with other therapies, such as immunotherapy that provide the most benefit in a particular tumor type. As many identifiers of CSCs are surface markers, use of antibodies to invoke an immune response may be represented as another tactic but requires targeting heterogeneous CSC populations in combination with the blockade of immune checkpoints, including PD-1 and CTLA4. Finally, the particular choice of CSC-targeted therapy may depend on better assays and strategies to detect and target CSCs. Thus, validated biomarker assays for CSC defining and specific features for self-renewal, plasticity and motility will need to be developed and these assays will help to define patient populations who will benefit most from a particular CSC-targeted therapy.
While the heterogeneity, evolution, plasticity and motility of CSCs need to be better recognized in both cancer initiation and progression, we need to keep in mind that understanding and targeting CSCs must be the entire cancer community’s team effort. Open and shared resources with integrated and interactive big data on CSCs will empower us to take advantage of super computation-based artificial intelligence, thereby hopefully outsmarting intelligent CSCs and other cancer cells to defeat cancer.
Acknowledgments
This manuscript has been partially supported by NIH/NCI R00CA160638, DOD BCRP W81XWH-16-1-0021, ACS127951-RSG-15-025-01-CSM, and Komen CCR15332826 (H.L.). We are thankful to Link Health Group and Boston Biomedical Pharma, Inc for their help in editorial and scientific discussions.
Footnotes
No conflict of interest.
References
- 1.McGuire S. World Cancer Report 2014. Geneva, Switzerland: World Health Organization, International Agency for Research on Cancer, WHO Press, 2015. Adv Nutr. 2016;7(2):418–9. doi: 10.3945/an.116.012211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Diehn M, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458(7239):780–3. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li X, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008;100(9):672–9. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- 4.Al-Hajj M, et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Charafe-Jauffret E, et al. Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clin Cancer Res. 2010;16(1):45–55. doi: 10.1158/1078-0432.CCR-09-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Giordano A, et al. Clinical relevance of cancer stem cells in bone marrow of early breast cancer patients. Ann Oncol. 2013;24(10):2515–21. doi: 10.1093/annonc/mdt223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Idowu MO, et al. CD44(+)/CD24(-/low) cancer stem/progenitor cells are more abundant in triple-negative invasive breast carcinoma phenotype and are associated with poor outcome. Hum Pathol. 2012;43(3):364–73. doi: 10.1016/j.humpath.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 8.Lin Y, et al. CD44+/CD24- phenotype contributes to malignant relapse following surgical resection and chemotherapy in patients with invasive ductal carcinoma. J Exp Clin Cancer Res. 2012;31:59. doi: 10.1186/1756-9966-31-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Owens TW, Naylor MJ. Breast cancer stem cells. Front Physiol. 2013;4:225. doi: 10.3389/fphys.2013.00225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu H, et al. Cancer stem cells from human breast tumors are involved in spontaneous metastases in orthotopic mouse models. Proc Natl Acad Sci U S A. 2010;107(42):18115–20. doi: 10.1073/pnas.1006732107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu S, Wicha MS. Targeting breast cancer stem cells. J Clin Oncol. 2010;28(25):4006–12. doi: 10.1200/JCO.2009.27.5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Korkaya H, Wicha MS. HER-2, notch, and breast cancer stem cells: targeting an axis of evil. Clin Cancer Res. 2009;15(6):1845–7. doi: 10.1158/1078-0432.CCR-08-3087. [DOI] [PubMed] [Google Scholar]
- 13.Adorno-Cruz V, et al. Cancer stem cells: targeting the roots of cancer, seeds of metastasis, and sources of therapy resistance. Cancer Res. 2015;75(6):924–9. doi: 10.1158/0008-5472.CAN-14-3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Brien CA, et al. Cancer stem cells and self-renewal. Clin Cancer Res. 2010;16(12):3113–20. doi: 10.1158/1078-0432.CCR-09-2824. [DOI] [PubMed] [Google Scholar]
- 15.Driessens G, et al. Defining the mode of tumour growth by clonal analysis. Nature. 2012;488(7412):527–30. doi: 10.1038/nature11344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jamieson CH, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351(7):657–67. doi: 10.1056/NEJMoa040258. [DOI] [PubMed] [Google Scholar]
- 17.Lim E, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med. 2009;15(8):907–13. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- 18.Sugiarto S, et al. Asymmetry-defective oligodendrocyte progenitors are glioma precursors. Cancer Cell. 2011;20(3):328–40. doi: 10.1016/j.ccr.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reya T, et al. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 20.Furth J, Kahn MC. The transmission of leukemia of mice with a single cell. Cancer Res (original Am J Cancer) 1937;(31):276–282. [Google Scholar]
- 21.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730–7. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 22.Lapidot T, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367(6464):645–8. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 23.Zabala M, et al. Chapter 2 - Overview: Cancer Stem Cell Self-Renewal A2 - Liu, Huiping. In: Lathia JD, editor. Cancer Stem Cells. Academic Press; 2016. pp. 25–58. [Google Scholar]
- 24.Bao S, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–60. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 25.O’Brien CA, et al. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445(7123):106–10. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 26.Kim CF, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121(6):823–35. doi: 10.1016/j.cell.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 27.Prince ME, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci U S A. 2007;104(3):973–8. doi: 10.1073/pnas.0610117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kurtova AV, et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature. 2015;517(7533):209–13. doi: 10.1038/nature14034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chan KS, et al. Identification, molecular characterization, clinical prognosis, and therapeutic targeting of human bladder tumor-initiating cells. Proc Natl Acad Sci U S A. 2009;106(33):14016–21. doi: 10.1073/pnas.0906549106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kretzschmar K, Watt FM. Lineage tracing. Cell. 2012;148(1-2):33–45. doi: 10.1016/j.cell.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 31.Schepers AG, et al. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science. 2012;337(6095):730–5. doi: 10.1126/science.1224676. [DOI] [PubMed] [Google Scholar]
- 32.Zong H, et al. Mosaic analysis with double markers in mice. Cell. 2005;121(3):479–92. doi: 10.1016/j.cell.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 33.Malanchi I, et al. Cutaneous cancer stem cell maintenance is dependent on beta-catenin signalling. Nature. 2008;452(7187):650–3. doi: 10.1038/nature06835. [DOI] [PubMed] [Google Scholar]
- 34.Barker N, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457(7229):608–11. doi: 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- 35.Chuang CH, et al. Molecular definition of a metastatic lung cancer state reveals a targetable CD109-Janus kinase-Stat axis. Nat Med. 2017;23(3):291–300. doi: 10.1038/nm.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jarrar A, et al. Cancer Stem Cells. Academic Press; 2016. Chapter 3 - Enrichment and Interrogation of Cancer Stem Cells; pp. 59–98. [Google Scholar]
- 37.Quintana E, et al. Efficient tumour formation by single human melanoma cells. Nature. 2008;456(7222):593–8. doi: 10.1038/nature07567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boiko AD, et al. Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature. 2010;466(7302):133–7. doi: 10.1038/nature09161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pascual G, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature. 2017;541(7635):41–45. doi: 10.1038/nature20791. [DOI] [PubMed] [Google Scholar]
- 40.Horimoto Y, et al. Combination of Cancer Stem Cell Markers CD44 and CD24 Is Superior to ALDH1 as a Prognostic Indicator in Breast Cancer Patients with Distant Metastases. PLoS One. 2016;11(10):e0165253. doi: 10.1371/journal.pone.0165253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ricardo S, et al. Breast cancer stem cell markers CD44, CD24 and ALDH1: expression distribution within intrinsic molecular subtype. J Clin Pathol. 2011;64(11):937–46. doi: 10.1136/jcp.2011.090456. [DOI] [PubMed] [Google Scholar]
- 42.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14(3):275–91. doi: 10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 43.Mani SA, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–15. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ishimoto T, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell. 2011;19(3):387–400. doi: 10.1016/j.ccr.2011.01.038. [DOI] [PubMed] [Google Scholar]
- 45.Gidekel S, et al. Oct-3/4 is a dose-dependent oncogenic fate determinant. Cancer Cell. 2003;4(5):361–70. doi: 10.1016/s1535-6108(03)00270-8. [DOI] [PubMed] [Google Scholar]
- 46.Bass AJ, et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet. 2009;41(11):1238–42. doi: 10.1038/ng.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chiou SH, et al. Coexpression of Oct4 and Nanog enhances malignancy in lung adenocarcinoma by inducing cancer stem cell-like properties and epithelial-mesenchymal transdifferentiation. Cancer Res. 2010;70(24):10433–44. doi: 10.1158/0008-5472.CAN-10-2638. [DOI] [PubMed] [Google Scholar]
- 48.Friedmann-Morvinski D, Verma IM. Dedifferentiation and reprogramming: origins of cancer stem cells. EMBO Rep. 2014;15(3):244–53. doi: 10.1002/embr.201338254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu Z, et al. TPO-Induced Metabolic Reprogramming Drives Liver Metastasis of Colorectal Cancer CD110+ Tumor-Initiating Cells. Cell Stem Cell. 2015;17(1):47–59. doi: 10.1016/j.stem.2015.05.016. [DOI] [PubMed] [Google Scholar]
- 50.Shen YA, et al. Metabolic reprogramming orchestrates cancer stem cell properties in nasopharyngeal carcinoma. Cell Cycle. 2015;14(1):86–98. doi: 10.4161/15384101.2014.974419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Skvortsov S, et al. Crosstalk between DNA repair and cancer stem cell (CSC) associated intracellular pathways. Semin Cancer Biol. 2015;31:36–42. doi: 10.1016/j.semcancer.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 52.Maugeri-Sacca M, et al. DNA damage repair pathways in cancer stem cells. Mol Cancer Ther. 2012;11(8):1627–36. doi: 10.1158/1535-7163.MCT-11-1040. [DOI] [PubMed] [Google Scholar]
- 53.Spranger S, et al. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015;523(7559):231–5. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 54.Casey SC, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. 2016;352(6282):227–31. doi: 10.1126/science.aac9935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bao B, et al. Overview of cancer stem cells (CSCs) and mechanisms of their regulation: implications for cancer therapy. Curr Protoc Pharmacol Chapter. 2013;14 doi: 10.1002/0471141755.ph1425s61. Unit 14 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Storm EE, et al. Targeting PTPRK-RSPO3 colon tumours promotes differentiation and loss of stem-cell function. Nature. 2016;529(7584):97–100. doi: 10.1038/nature16466. [DOI] [PubMed] [Google Scholar]
- 57.Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells - what challenges do they pose? Nat Rev Drug Discov. 2014;13(7):497–512. doi: 10.1038/nrd4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mu P, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science. 2017;355(6320):84–88. doi: 10.1126/science.aah4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ku SY, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science. 2017;355(6320):78–83. doi: 10.1126/science.aah4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Beachy PA, et al. Tissue repair and stem cell renewal in carcinogenesis. Nature. 2004;432(7015):324–31. doi: 10.1038/nature03100. [DOI] [PubMed] [Google Scholar]
- 61.Von Hoff DD, et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med. 2009;361(12):1164–72. doi: 10.1056/NEJMoa0905360. [DOI] [PubMed] [Google Scholar]
- 62.Robinson GW, et al. Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog-Subgroup Medulloblastoma: Results From Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J Clin Oncol. 2015;33(24):2646–54. doi: 10.1200/JCO.2014.60.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev. 2008;22(18):2454–72. doi: 10.1101/gad.1693608. [DOI] [PubMed] [Google Scholar]
- 64.Merchant AA, Matsui W. Targeting Hedgehog–a cancer stem cell pathway. Clin Cancer Res. 2010;16(12):3130–40. doi: 10.1158/1078-0432.CCR-09-2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Song Z, et al. Sonic hedgehog pathway is essential for maintenance of cancer stem-like cells in human gastric cancer. PLoS One. 2011;6(3):e17687. doi: 10.1371/journal.pone.0017687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tang SN, et al. Inhibition of sonic hedgehog pathway and pluripotency maintaining factors regulate human pancreatic cancer stem cell characteristics. Int J Cancer. 2012;131(1):30–40. doi: 10.1002/ijc.26323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tang JY, et al. Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome. N Engl J Med. 2012;366(23):2180–8. doi: 10.1056/NEJMoa1113538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zabala M, L N, Qian D, van Weele LJ, Heiser D, Clarke MF. Overview: Cancer Stem cell Self Renewal (Chapter 2) In: Liu H, editor. Cancer Stem Cells. 2016. [Google Scholar]
- 69.Li Y, et al. Targeting the Hedgehog signaling pathway for cancer therapy. Expert Opin Ther Targets. 2012;16(1):49–66. doi: 10.1517/14728222.2011.617367. [DOI] [PubMed] [Google Scholar]
- 70.Peterson SC, et al. Basal cell carcinoma preferentially arises from stem cells within hair follicle and mechanosensory niches. Cell Stem Cell. 2015;16(4):400–12. doi: 10.1016/j.stem.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vanner RJ, et al. Quiescent sox2(+) cells drive hierarchical growth and relapse in sonic hedgehog subgroup medulloblastoma. Cancer Cell. 2014;26(1):33–47. doi: 10.1016/j.ccr.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rimkus TK, et al. Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors. Cancers (Basel) 2016;8(2) doi: 10.3390/cancers8020022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sekulic A, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366(23):2171–9. doi: 10.1056/NEJMoa1113713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Soltanian S, Matin MM. Cancer stem cells and cancer therapy. Tumour Biol. 2011;32(3):425–40. doi: 10.1007/s13277-011-0155-8. [DOI] [PubMed] [Google Scholar]
- 75.Lobry C, et al. Oncogenic and tumor suppressor functions of Notch in cancer: it’s NOTCH what you think. J Exp Med. 2011;208(10):1931–5. doi: 10.1084/jem.20111855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Radtke F, Raj K. The role of Notch in tumorigenesis: oncogene or tumour suppressor? Nat Rev Cancer. 2003;3(10):756–67. doi: 10.1038/nrc1186. [DOI] [PubMed] [Google Scholar]
- 77.Harrison H, et al. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 2010;70(2):709–18. doi: 10.1158/0008-5472.CAN-09-1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fan X, et al. Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res. 2006;66(15):7445–52. doi: 10.1158/0008-5472.CAN-06-0858. [DOI] [PubMed] [Google Scholar]
- 79.Fan X, et al. Notch pathway blockade targets medulloblastoma-initiating cancer stem cells. Cancer Research. 2006;66(8 Supplement):1053–1053. [Google Scholar]
- 80.Acar A, et al. A Role for Notch Signalling in Breast Cancer and Endocrine Resistance. Stem Cells Int. 2016;2016:2498764. doi: 10.1155/2016/2498764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schott AF, et al. Preclinical and clinical studies of gamma secretase inhibitors with docetaxel on human breast tumors. Clin Cancer Res. 2013;19(6):1512–24. doi: 10.1158/1078-0432.CCR-11-3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Piha-Paul SA, et al. Results of a phase 1 trial combining ridaforolimus and MK-0752 in patients with advanced solid tumours. Eur J Cancer. 2015;51(14):1865–73. doi: 10.1016/j.ejca.2015.06.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cook N, et al. A phase I trial of the ɣ-secretase inhibitor (GSI) MK-0752 in combination with gemcitabine in patients with pancreatic ductal adenocarcinoma (PDAC). Annual Meeting of the American Society of Clinical Oncology; Chicago, IL. 2015. p. abstr 4116. [Google Scholar]
- 84.Kotasek D, et al. A phase 1b study of the anti-cancer stem cell agent demcizumab (DEM), pemetrexed (PEM) & carboplatin (CARBO) in pts with 1st line non-squamous NSCLC. Annual Meeting of the American Society of Clinical Oncology; Chicago, IL. 2015. p. abstr 8045. [Google Scholar]
- 85.Smith DC, et al. A phase I dose escalation and expansion study of the anticancer stem cell agent demcizumab (anti-DLL4) in patients with previously treated solid tumors. Clin Cancer Res. 2014;20(24):6295–303. doi: 10.1158/1078-0432.CCR-14-1373. [DOI] [PubMed] [Google Scholar]
- 86.Inc O. In: OncoMed’s Phase 2 Demcizumab Pancreatic Cancer Trial Misses Primary Endpoint. Corral M, editor. OncoMed Inc; 2017. [Google Scholar]
- 87.Falchook GS, et al. Phase I study of MEDI0639 in patients with advanced solid tumors. Annual Meeting of the American Society of Clinical Oncology; Chicago, IL. 2015. p. abstr 3024. [Google Scholar]
- 88.Rudin CM, et al. Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: a first-in-human, first-in-class, open-label, phase 1 study. Lancet Oncol. 2017;18(1):42–51. doi: 10.1016/S1470-2045(16)30565-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Casulo C, et al. Safety and Preliminary Efficacy Results of a Phase I First-in-Human Study of the Novel Notch-1 Targeting Antibody Brontictuzumab (OMP-52M51) Administered Intravenously to Patients with Hematologic Malignancies. Blood. 2016;128(22):5108–5108. [Google Scholar]
- 90.Davis SL, et al. Abstract B48: A first-in-human Phase I study of the novel cancer stem cell (CSC) targeting antibody OMP-52M51 (anti-Notch1) administered intravenously to patients with certain advanced solid tumors. Molecular Cancer Therapeutics. 2013;12(11 Supplement):B48–B48. [Google Scholar]
- 91.Yen WC, et al. Targeting Notch signaling with a Notch2/Notch3 antagonist (tarextumab) inhibits tumor growth and decreases tumor-initiating cell frequency. Clin Cancer Res. 2015;21(9):2084–95. doi: 10.1158/1078-0432.CCR-14-2808. [DOI] [PubMed] [Google Scholar]
- 92.O’Reilly EM, et al. Final results of phase Ib of anticancer stem cell antibody tarextumab (OMP-59R5, TRXT, anti-Notch 2/3) in combination with nab-paclitaxel and gemcitabine (Nab-P+Gem) in patients (pts) with untreated metastatic pancreatic cancer (mPC). Annual Meeting of the American Society of Clinical Oncology; Chicago, IL. 2015. p. abstr 278. [Google Scholar]
- 93.Pietanza MC, et al. Final results of phase Ib of tarextumab (TRXT, OMP-59R5, anti-Notch2/3) in combination with etoposide and platinum (EP) in patients (pts) with untreated extensive-stage small-cell lung cancer (ED-SCLC) Annual Meeting of the American Society of Clinical Oncology. 2015:abstr 7508. [Google Scholar]
- 94.Sulzmaier FJ, et al. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer. 2014;14(9):598–610. doi: 10.1038/nrc3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cance WG, Golubovskaya VM. Focal adhesion kinase versus p53: apoptosis or survival? Sci Signal. 2008;1(20):pe22. doi: 10.1126/stke.120pe22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lim ST, et al. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol Cell. 2008;29(1):9–22. doi: 10.1016/j.molcel.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Luo M, et al. Distinct FAK activities determine progenitor and mammary stem cell characteristics. Cancer Res. 2013;73(17):5591–602. doi: 10.1158/0008-5472.CAN-13-1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fennell DA, et al. Annual Meeting of the American Society of Clinical Oncology. Chicago, IL: 2014. p. abstr TPS7611. [Google Scholar]
- 99.Jiang H, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. 2016;22(8):851–60. doi: 10.1038/nm.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ginestier C, et al. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J Clin Invest. 2010;120(2):485–97. doi: 10.1172/JCI39397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Joshi NS, et al. Regulatory T Cells in Tumor-Associated Tertiary Lymphoid Structures Suppress Anti-tumor T Cell Responses. Immunity. 2015;43(3):579–90. doi: 10.1016/j.immuni.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kelly KF, et al. beta-catenin enhances Oct-4 activity and reinforces pluripotency through a TCF-independent mechanism. Cell Stem Cell. 2011;8(2):214–27. doi: 10.1016/j.stem.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.ten Berge D, et al. Embryonic stem cells require Wnt proteins to prevent differentiation to epiblast stem cells. Nat Cell Biol. 2011;13(9):1070–5. doi: 10.1038/ncb2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Willert K, et al. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 2003;423(6938):448–52. doi: 10.1038/nature01611. [DOI] [PubMed] [Google Scholar]
- 105.Barker N, et al. Lgr5(+ve) stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell. 2010;6(1):25–36. doi: 10.1016/j.stem.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 106.Barker N, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449(7165):1003–7. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 107.van Amerongen R, et al. Developmental stage and time dictate the fate of Wnt/beta-catenin-responsive stem cells in the mammary gland. Cell Stem Cell. 2012;11(3):387–400. doi: 10.1016/j.stem.2012.05.023. [DOI] [PubMed] [Google Scholar]
- 108.Yeung J, et al. beta-Catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell. 2010;18(6):606–18. doi: 10.1016/j.ccr.2010.10.032. [DOI] [PubMed] [Google Scholar]
- 109.Gurney A, et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc Natl Acad Sci U S A. 2012;109(29):11717–22. doi: 10.1073/pnas.1120068109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yeung P, et al. Wnt pathway antagonist OMP-54F28 (FZD8-Fc) inhibits tumor growth and reduces tumor-initiating cell frequency in patient-derived hepatocellular carcinoma and ovarian cancer xenograft models. Abstract Presented at American Association for Cancer Research Annual Meeting. 2014 [Google Scholar]
- 111.Liu J, et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc Natl Acad Sci U S A. 2013;110(50):20224–9. doi: 10.1073/pnas.1314239110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yen WC, et al. Cancer Research. AMER ASSOC CANCER RESEARCH 615 CHESTNUT ST, 17TH FLOOR, PHILADELPHIA, PA 19106-4404; USA: 2015. Enhanced antitumor efficacy by sequential application of Wnt pathway antagonists in combination with taxanes. [Google Scholar]
- 113.Kamran MZ, et al. Role of STAT3 in cancer metastasis and translational advances. Biomed Res Int. 2013;2013:421821. doi: 10.1155/2013/421821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhao D, et al. VEGF drives cancer-initiating stem cells through VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene. 2015;34(24):3107–19. doi: 10.1038/onc.2014.257. [DOI] [PubMed] [Google Scholar]
- 115.Cedar H, Bergman Y. Epigenetic silencing during early lineage commitment. 2009 [PubMed] [Google Scholar]
- 116.Jonker DJ, et al. A randomized phase III study of napabucasin [BBI608] (NAPA) vs placebo (PBO) in patients (pts) with pretreated advanced colorectal cancer (ACRC): the CCTG/AGITG CO.23 trial. Annals of Oncology. 2016;27 [Google Scholar]
- 117.Shah MA, et al. The BRIGHTER trial: A phase III randomized double-blind study of BBI608 + weekly paclitaxel versus placebo (PBO) + weekly paclitaxel in patients (pts) with pretreated advanced gastric and gastro-esophageal junction (GEJ) adenocarcinoma. Annual Meeting of the American Society of Clinical Oncology. 2015:abstr TPS4139. [Google Scholar]
- 118.Wang ML, et al. Targeting cancer stem cells: emerging role of Nanog transcription factor. Onco Targets Ther. 2013;6:1207–20. doi: 10.2147/OTT.S38114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bussolati B, et al. Identification of a tumor-initiating stem cell population in human renal carcinomas. Faseb j. 2008;22(10):3696–705. doi: 10.1096/fj.08-102590. [DOI] [PubMed] [Google Scholar]
- 120.Amsterdam A, et al. Localization of the stem cell markers LGR5 and Nanog in the normal and the cancerous human ovary and their inter-relationship. Acta Histochem. 2013;115(4):330–8. doi: 10.1016/j.acthis.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 121.Ezeh UI, et al. Human embryonic stem cell genes OCT4, NANOG, STELLAR, and GDF3 are expressed in both seminoma and breast carcinoma. Cancer. 2005;104(10):2255–65. doi: 10.1002/cncr.21432. [DOI] [PubMed] [Google Scholar]
- 122.Lin T, et al. Overexpression of Nanog protein is associated with poor prognosis in gastric adenocarcinoma. Med Oncol. 2012;29(2):878–85. doi: 10.1007/s12032-011-9860-9. [DOI] [PubMed] [Google Scholar]
- 123.Guo Y, et al. Expression profile of embryonic stem cell-associated genes Oct4, Sox2 and Nanog in human gliomas. Histopathology. 2011;59(4):763–75. doi: 10.1111/j.1365-2559.2011.03993.x. [DOI] [PubMed] [Google Scholar]
- 124.Wen J, et al. Oct4 and Nanog expression is associated with early stages of pancreatic carcinogenesis. Pancreas. 2010;39(5):622–6. doi: 10.1097/MPA.0b013e3181c75f5e. [DOI] [PubMed] [Google Scholar]
- 125.Chiou SH, et al. Positive correlations of Oct-4 and Nanog in oral cancer stem-like cells and high-grade oral squamous cell carcinoma. Clin Cancer Res. 2008;14(13):4085–95. doi: 10.1158/1078-0432.CCR-07-4404. [DOI] [PubMed] [Google Scholar]
- 126.Jonker DJ, et al. Phase 1 extension study of BBI503, a first-in-class cancer stemness kinase inhibitor, in patients with advanced colorectal cancer. Annual Meeting of the American Society of Clinical Oncology; Chicago, IL. 2015. p. abstr 3615. [Google Scholar]
- 127.Chen K, et al. Understanding and targeting cancer stem cells: therapeutic implications and challenges. Acta Pharmacol Sin. 2013;34(6):732–40. doi: 10.1038/aps.2013.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Colak S, Medema JP. Cancer stem cells–important players in tumor therapy resistance. Febs j. 2014;281(21):4779–91. doi: 10.1111/febs.13023. [DOI] [PubMed] [Google Scholar]
- 129.Moitra K. Overcoming Multidrug Resistance in Cancer Stem Cells. Biomed Res Int. 2015;2015:635745. doi: 10.1155/2015/635745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhang M, et al. Identification of tumor-initiating cells in a p53-null mouse model of breast cancer. Cancer Res. 2008;68(12):4674–82. doi: 10.1158/0008-5472.CAN-07-6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhang M, et al. Selective targeting of radiation-resistant tumor-initiating cells. Proc Natl Acad Sci U S A. 2010;107(8):3522–7. doi: 10.1073/pnas.0910179107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Dean M. ABC transporters, drug resistance, and cancer stem cells. J Mammary Gland Biol Neoplasia. 2009;14(1):3–9. doi: 10.1007/s10911-009-9109-9. [DOI] [PubMed] [Google Scholar]
- 133.Wahl GM, Spike BT. Cell state plasticity, stem cells, EMT, and the generation of intra-tumoral heterogeneity. NPJ Breast Cancer. 2017;3:14. doi: 10.1038/s41523-017-0012-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Li F, et al. Beyond tumorigenesis: cancer stem cells in metastasis. Cell Res. 2007;17(1):3–14. doi: 10.1038/sj.cr.7310118. [DOI] [PubMed] [Google Scholar]
- 135.Huang J, et al. Cytokine-induced killer (CIK) cells bound with anti-CD3/anti-CD133 bispecific antibodies target CD133(high) cancer stem cells in vitro and in vivo. Clin Immunol. 2013;149(1):156–68. doi: 10.1016/j.clim.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 136.Jaiswal S, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138(2):271–85. doi: 10.1016/j.cell.2009.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Betancur PA, et al. A CD47-associated super-enhancer links pro-inflammatory signalling to CD47 upregulation in breast cancer. Nat Commun. 2017;8:14802. doi: 10.1038/ncomms14802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ngo M, et al. Antibody Therapy Targeting CD47 and CD271 Effectively Suppresses Melanoma Metastasis in Patient-Derived Xenografts. Cell Rep. 2016;16(6):1701–16. doi: 10.1016/j.celrep.2016.07.004. [DOI] [PubMed] [Google Scholar]
- 139.Weiskopf K, et al. CD47-blocking immunotherapies stimulate macrophage-mediated destruction of small-cell lung cancer. J Clin Invest. 2016;126(7):2610–20. doi: 10.1172/JCI81603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Edris B, et al. Antibody therapy targeting the CD47 protein is effective in a model of aggressive metastatic leiomyosarcoma. Proc Natl Acad Sci U S A. 2012;109(17):6656–61. doi: 10.1073/pnas.1121629109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Willingham SB, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U S A. 2012;109(17):6662–7. doi: 10.1073/pnas.1121623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chao MP, et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell. 2010;142(5):699–713. doi: 10.1016/j.cell.2010.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Jaiswal S, et al. Macrophages as mediators of tumor immunosurveillance. Trends Immunol. 2010;31(6):212–9. doi: 10.1016/j.it.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Liu J, et al. Pre-Clinical Development of a Humanized Anti-CD47 Antibody with Anti-Cancer Therapeutic Potential. PLoS One. 2015;10(9):e0137345. doi: 10.1371/journal.pone.0137345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–64. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tirosh I, et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature. 2016;539(7628):309–313. doi: 10.1038/nature20123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Esteva A, et al. Dermatologist-level classification of skin cancer with deep neural networks. Nature. 2017;542(7639):115–118. doi: 10.1038/nature21056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.de Sousa e Melo F, et al. A distinct role for Lgr5+ stem cells in primary and metastatic colon cancer. Nature. 2017;543(7647):676–680. doi: 10.1038/nature21713. [DOI] [PubMed] [Google Scholar]
- 149.Singh SK, et al. Identification of human brain tumour initiating cells. Nature. 2004;432(7015):396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 150.Pietras A, et al. Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell. 2014;14(3):357–69. doi: 10.1016/j.stem.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Todaro M, et al. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell. 2014;14(3):342–56. doi: 10.1016/j.stem.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 152.Katsuno Y, et al. Coordinated expression of REG4 and aldehyde dehydrogenase 1 regulating tumourigenic capacity of diffuse-type gastric carcinoma-initiating cells is inhibited by TGF-beta. J Pathol. 2012;228(3):391–404. doi: 10.1002/path.4020. [DOI] [PubMed] [Google Scholar]
- 153.Takaishi S, et al. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells. 2009;27(5):1006–20. doi: 10.1002/stem.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Chen T, et al. Identification and expansion of cancer stem cells in tumor tissues and peripheral blood derived from gastric adenocarcinoma patients. Cell Res. 2012;22(1):248–58. doi: 10.1038/cr.2011.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Han ME, et al. Cancer spheres from gastric cancer patients provide an ideal model system for cancer stem cell research. Cell Mol Life Sci. 2011;68(21):3589–605. doi: 10.1007/s00018-011-0672-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Jiang J, et al. Trastuzumab (herceptin) targets gastric cancer stem cells characterized by CD90 phenotype. Oncogene. 2012;31(6):671–82. doi: 10.1038/onc.2011.282. [DOI] [PubMed] [Google Scholar]
- 157.Ohkuma M, et al. Absence of CD71 transferrin receptor characterizes human gastric adenosquamous carcinoma stem cells. Ann Surg Oncol. 2012;19(4):1357–64. doi: 10.1245/s10434-011-1739-7. [DOI] [PubMed] [Google Scholar]
- 158.Jiang Y, et al. Expressions of putative cancer stem cell markers ABCB1, ABCG2, and CD133 are correlated with the degree of differentiation of gastric cancer. Gastric Cancer. 2012;15(4):440–50. doi: 10.1007/s10120-012-0140-y. [DOI] [PubMed] [Google Scholar]
- 159.Hashimoto K, et al. Expression of CD133 in the cytoplasm is associated with cancer progression and poor prognosis in gastric cancer. Gastric Cancer. 2014;17(1):97–106. doi: 10.1007/s10120-013-0255-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Eramo A, et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008;15(3):504–14. doi: 10.1038/sj.cdd.4402283. [DOI] [PubMed] [Google Scholar]
- 161.Fang D, et al. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005;65(20):9328–37. doi: 10.1158/0008-5472.CAN-05-1343. [DOI] [PubMed] [Google Scholar]
- 162.Du L, et al. CD44 is of functional importance for colorectal cancer stem cells. Clin Cancer Res. 2008;14(21):6751–60. doi: 10.1158/1078-0432.CCR-08-1034. [DOI] [PubMed] [Google Scholar]
- 163.Boumahdi S, et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature. 2014;511(7508):246–50. doi: 10.1038/nature13305. [DOI] [PubMed] [Google Scholar]
- 164.Guzman ML, Jordan CT. Considerations for targeting malignant stem cells in leukemia. Cancer Control. 2004;11(2):97–104. doi: 10.1177/107327480401100216. [DOI] [PubMed] [Google Scholar]
- 165.Hermann PC, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1(3):313–23. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 166.Liu J, et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(50):20224–20229. doi: 10.1073/pnas.1314239110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Jesus-Acosta AD, et al. A phase II study of vismodegib, a hedgehog (Hh) pathway inhibitor, combined with gemcitabine and nab-paclitaxel (nab-P) in patients (pts) with untreated metastatic pancreatic ductal adenocarcinoma (PDA) Gastrointestinal Cancers Symposium (ASCO) 2014:abstr 257. [Google Scholar]
- 168.Jimeno A, et al. A first-in-human phase 1 study of anticancer stem cell agent OMP-54F28 (FZD8-Fc), decoy receptor for WNT ligands, in patients with advanced solid tumors. Annual Meeting of the American Society of Clinical Oncology. 2014:abstr 2505. doi: 10.1158/1078-0432.CCR-17-2157. [DOI] [PubMed] [Google Scholar]
- 169.Jonker DJ, et al. A phase I extension study of BBI608, a first-in-class cancer stem cell (CSC) inhibitor, in patients with advanced solid tumors. Annual Meeting of the American Society of Clinical Oncology; Chicago, IL. 2014. p. abstr 2546. [Google Scholar]
- 170.Bekaii-Saab TS, et al. A phase Ib/II study of BBI608 combined with weekly paclitaxel in advanced pancreatic cancer. Gastrointestinal Cancer Symposium (ASCO); San Francisco, CA. 2016. p. abstr 196. [Google Scholar]
- 171.A Phase 1b/2 Study of OMP-59R5 in Combination With Nab-Paclitaxel and Gemcitabine in Subjects With Previously Untreated Stage IV Pancreatic Cancer (ALPINE) 2016 https://clinicaltrials.gov/ct2/show/NCT01647828, (accessed 2016)
- 172.Jonker DJ, et al. The NCIC CTG and AGITG CO.23 trial: A phase III randomized study of BBI608 plus best supportive care (BSC) versus placebo (PBO) plus BSC in patients (Pts) with pretreated advanced colorectal carcinoma (CRC) Annual Meeting of the American Society of Clinical Oncology. 2014:abstr TPS3660. [Google Scholar]