

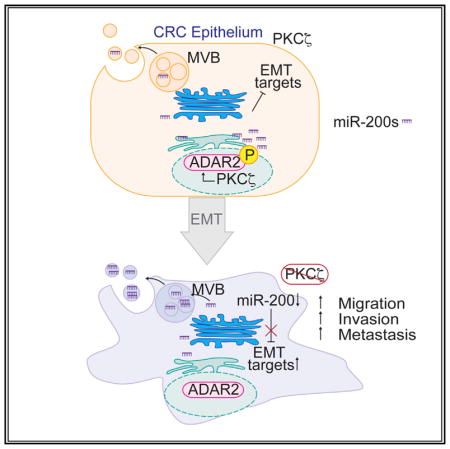
SUMMARY
Most colorectal cancer (CRC)-related deaths are due to liver metastases. PKCζ is a tumor suppressor in CRC with reduced expression in metastasis. Given the importance of microRNAs (miRNAs) in regulating cellular plasticity, we performed an unbiased screening and identified the miR-200 family as the most relevant miRNAs downregulated by PKCζ deficiency. The regulation of the intracellular levels of miR-200 by PKCζ is post-transcriptional and involves their secretion in extracellular vesicles. Here, we identified ADAR2 as a direct substrate of PKCζ in CRC cells. Phosphorylation of ADAR2 regulates its editing activity, which is required to maintain miR-200 steady-state levels, suggesting that the PKCζ/ADAR2 axis regulates miR-200 secretion through RNA editing. Loss of this axis results in epithelial-to-mesenchymal transition (EMT) and increased liver metastases, which can be inhibited in vivo by blocking miR-200 release. Therefore, the PKCζ/ADAR2 axis is a critical regulator of CRC metastases through modulation of miR-200 levels.
In Brief
Shelton et al. demonstrate that the loss of the tumor suppressor PKCζ in colorectal cancer cells results in the downregulation of miR-200, leading to increased epithelial-to-mesenchymal transition, cell invasion, and liver metastasis. This is mediated by an increase in miR-200 secretion through the inactivation of the RNA editing enzyme ADAR2, identifying a key vulnerability in liver metastasis.
INTRODUCTION
Colorectal cancer (CRC) is the third most common cancer diagnosed in the United States and ranks third in cancer-related deaths (Siegel et al., 2016). The main cause of death in patients with CRC is liver metastasis, with nearly 70% of CRC patients eventually developing a liver tumor (Mehlen and Puisieux, 2006). Moreover, metastasis accounts for more than 90% of all deaths in cancer patients (Mehlen and Puisieux, 2006). Recent evidence has demonstrated that plasticity is a key feature of metastatic cells, and that microRNAs (miRNAs) are important in regulating the tumor phenotype as it transitions from a benign adenoma, with epithelial characteristics, to an invasive tumor with mesenchymal and stem-cell-like properties (Pencheva and Tavazoie, 2013). The emergence of a consensus molecular subtyping for CRC has revealed that the stem-like/mesenchymal CRC subtype is associated with poor prognosis, aggressive cancer, and dysregulation of miRNAs (Guinney et al., 2015). Thus, understanding the mechanisms controlling the expression of miRNAs has emerged as a potential venue for the design of better strategies to treat metastasis.
Our laboratory recently identified protein kinase C ζ (PKCζ) as a tumor suppressor in CRC. PKCζ (encoded by the PRKCZ gene) is under-expressed in human CRC relative to normal colon tissue, and its levels are also considerably decreased in distant metastasis, which correlates with poor prognosis (Ma et al., 2013). In addition, cancer-associated loss-of-function mutations were found in the kinase domain of PKCζ, implicating the importance of the kinase activity for its role as a tumor suppressor (Galvez et al., 2009; Ma et al., 2013). One possibility when targeting tumor suppressors as therapeutic targets is to devise ways to restore their levels or activity. This can be challenging, and an alternative approach is to elucidate their mechanisms of action to identify cell vulnerabilities created by the loss of the tumor suppressor’s function. This is the rationale for investigating the signaling pathways unleashed during PKCζ inactivation in cancer. In this regard, our previous studies demonstrated that the loss of PKCζ allows the metabolic reprogramming of CRC cells under conditions of nutrient stress to promote cancer cell survival and tumorigenesis (Ma et al., 2013). Likewise, our data also demonstrated that PKCζ is a repressor of the β-catenin and Yap pathways in intestinal stem cells, which is important for intestinal regeneration and cancer (Llado et al., 2015). However, both types of signaling cascades are difficult to target, and key potential points of intervention need to be found. Whether PKCζ is not only involved in the repression of tumor initiation and survival but also restrains metastasis through the regulation of the expression of miRNAs has not yet been explored. Here, we show that PKCζ exerts its metastasis suppressor activity via activation of the RNA editing enzyme adenosine deaminases acting on RNA 2 (ADAR2), which regulates the intracellular levels of the miR-200 family of miRNAs.
RESULTS
Loss of PKCζ Is Associated with Decreased Expression of the miR-200 Family
Considering the importance of miRNAs in intestinal carcinogenesis, we explored whether the loss of PKCζ may lead to changes in the miRNA expression profile of CRC cells. To this effect, SW480 human CRC cells were infected with lentiviruses expressing short hairpin non-targeting (shNT) or shRNA selective for PKCζ (shPKCζ), and microarrays were used to create global gene-expression profiles of both mRNAs and miRNAs (GEO: GSE42186; Table S1). From this analysis, we found a list of 74 miRNAs that significantly changed in shPKCζ cells (Figure 1A). In parallel, we also analyzed the mRNA expression levels of shNT and shPKCζ cells and performed gene set enrichment analysis (GSEA) using the “C3 miRNA target motifs” gene set compilation to predict which miRNAs were active/inactive based on 3′ UTR miRNA-binding motifs present in mRNAs that were differentially expressed in shPKCζ cells (Figures 1A and S1A). To select the most biologically relevant miRNAs altered by PKCζ loss, we crossed these data with a profile of human miRNAs that were significantly changed in CRC patients with low PRKCZ mRNA expression as compared to those with high PRKCZ levels. To do that, we used The Cancer Genome Atlas network colon adenocarcinoma dataset (TCGA-COAD) and selected patients in the top and bottom 25% of PRKCZ mRNA expression levels. We then identified the miRNAs and mRNAs that were significantly altered in both groups and subjected them to GSEA using the “C3 miRNA target motifs” gene matrix (Figures 1A and S1A). Finally, we crossed the results from the analysis of the miRNA profile in shPKCζ cells with those of the human TCGA-COAD, which converged in the identification of miR-200b, miR-200c, and miR-429 as the most relevant miRNAs significantly downregulated under conditions of PKCζ deficiency (Figure S1B). The miRNAs identified in our unbiased approach are part of the miR-200 family, which consists of five evolutionarily conserved miRNAs (miR-200b, miR-200a, miR-429, miR-200c, and miR-141) that are localized in two polycistronic clusters: miR-200b/200a/429 on chromosome 1, and miR-200c/141 on chromosome 12 (Figure S1C). They share a high degree of sequence homology, with a difference of only 1 nt in their seed sequence: miR-141/200a (seed AACACUG) and miR-200b/200c/429 (seed AAUACUG). Importantly, the miR-200 family members have been described as key players in mediating stemness and epithelial-to-mesenchymal transition (EMT), processes that reportedly facilitate tumor metastasis (Hur et al., 2013; Wellner et al., 2009). Accordingly, we focused our efforts on this subset of miRNAs.
Figure 1. miR-200s Positively Correlate with PKCζ in Colorectal Cancer.

(A) Venn diagram of common miRNAs identified from an miRNA microarray using SW480 cells, TCGA colon adenocarcinoma miRNA expression profile, and GSEA-C3 analysis of the mRNAs from the datasets.
(B–F) qPCR of miRNAs and immunoblot of PKCζ in SW480 shNT and shPKCζ cells, n = 4 biological replicates (B); SW480 and SW620 cells, n = 4 biological replicates (C); WT and PKCζ−/− mouse intestinal organoids, n = 3 biological replicates (D); ileum tissue 3 days post irradiation from Lgr5 and Lgr5;PKCζf/f mice, n = 3 mice/group (E); and intestinal tumor organoids from Lgr5;APCf/f and Lgr5;PKCζf/fAPCf/f mice, n = 3 biological replicates (F).
(G) Kaplan-Meier overall survival analysis of TCGA colon adenocarcinoma patients.
(H) Correlation between PRKCZ gene expression and the miR-200b/a/429 cluster in TCGA colon adenocarcinoma patients; n = 233 patients.
Results are presented as mean ± SD. **p < 0.01, ***p < 0.001, ****p < 0.0001. See also Figure S1 and Table S1.
Because PKCζ is a tumor suppressor in intestinal cancer (Llado et al., 2015; Ma et al., 2013), we hypothesized that the downregulation of miR-200s could contribute to its tumor suppressor role. To start exploring this possibility, we first validated the findings of the miRNA profiling by measuring the expression levels of miR-200s by qPCR in shNT and shPKCζ cells. There was a significant downregulation of all the members of the miR-200 family in shPKCζ cells, with no changes in the levels of two unrelated miRNAs (miR-16-5p and miR-26a-5p) (Figure 1B). Similar results were obtained in the human CRC cell lines RKO and HCT116 (Figures S1D and S1E). Importantly, miR-200s, but not miR-16-5p, were also decreased in SW620 cells, a metastatic derivative from the same patient from which SW480 cells were derived and a CRC line expressing lower endogenous PKCζ levels (Figure 1C). We also noted a similar altered expression profile in intestinal organoids from PKCζ knockout (KO) mice (Figure 1D).
Lgr5+ intestinal stem cells (ISCs) are believed to be the cells of origin of intestinal cancer, and have recently been shown to be critical for CRC liver metastasis (Barker et al., 2007; de Sousa e Melo et al., 2017). This is noteworthy because PKCζ has been shown to regulate ISC function and tumorigenesis (Llado et al., 2015). Functional activity of ISCs can be measured by intestinal regeneration after acute damage, as modeled by irradiation (IR)-induced ablation of the intestinal epithelium (Llado et al., 2015). To this end, we monitored the levels of miR-200s in KO mice for PKCζ, looking specifically in the Lgr5+ ISC population (Lgr5;PKCζf/f) post IR. Interestingly, reduced levels of miR-200s upon PKCζ deficiency were concomitant with higher repopulation activity in Lgr5;PKCζf/f ISCs (Llado et al., 2015) (Figure 1E). Furthermore, our previous data demonstrated that the selective loss of PKCζ in Lgr5+ ISCs deficient for APC enhanced the tumor-forming potential and stem activity of these cells (Llado et al., 2015). Analysis of miR-200s in organoids derived from the intestinal tumors of these mice also revealed significant downregulation of these miRNAs in PKCζ-deficient organoids (Figure 1F). These data validated our findings from the miRNA profile and support the notion that downregulation of miR-200 family members might be a functionally important downstream event upon PKCζ loss.
Aberrant regulation of miR-200 family members has been reported to be associated with tumor progression, metastasis, and drug resistance in several types of cancers, including CRC (Diaz et al., 2014; Hur et al., 2013). In particular, high levels of miR-200a, miR-200c, and miR-429 have been shown to correlate with increased overall survival in CRC patients with stage I–III cancer (Diaz et al., 2014). Moreover, levels of miR-200s have been proposed as prognostic markers to predict beneficial response to adjuvant chemotherapy in CRC patients (Diaz et al., 2014). Notably, miR-200c and miR-200b were down-regulated at the invasive front of CRC tumors but upregulated in metastases. Our bioinformatics analysis of TCGA-COAD revealed that low expression of the miR-200b/a family members correlated with a worse patient prognosis in terms of overall survival and relapse (Figures 1G and S1F). More importantly, we found a significant positive correlation between PRKCZ mRNA levels and those of the miR-200b/a/429 cluster in the same human CRC dataset (Figure 1H). These results support our hypothesis that downregulation of miR-200s occurs upon PKCζ loss, and that this could be a critical event in intestinal carcinogenesis, not only in mice but also in human patients.
Loss of PKCζ Promotes EMT through the Regulation of miR-200 Levels
Because low expression of the miR-200 family members has been reported to promote the acquisition of stem cell properties and to induce EMT in tumor cells, we next determined the impact that PKCζ regulation of miR-200s has on the mRNA expression levels of markers of stemness and EMT in shNT and shPKCζ cells. Consistent with a critical role of PKCζ in miR-200 regulation, we found that shPKCζ cells displayed reduced epithelial markers with a concomitant increase in mesenchymal markers (Figure 2A). Similarly, loss of PKCζ resulted in the increased expression of genes associated with intestinal stemness (Figure 2A). Immunoblot analysis confirmed the reduced levels of E-cadherin concomitant with a gain in Zeb1, TGFβ, and CD44 in shPKCζ cells (Figure 2B). In keeping with these results, PKCζ knockdown induced a more mesenchymal morphology (Figure 2C). Importantly, these in vitro results were corroborated in patients by GSEA using TCGA-COAD, which established that PRKCZ expression negatively associates with EMT and stemness in human CRC patients (Figure 2D). These results strongly support a model whereby PKCζ deficiency promotes stemness and a more mesenchymal phenotype in CRC cells, through a mechanism that involves the control of the levels of miR-200s. In agreement with this, the mesenchymal characteristics of shPKCζ cells were severely abrogated when the levels of miR-200a/b were restored by the overexpression of miR-200a/b mimics. Indeed, immunofluorescence analysis revealed that the overexpression of miR-200a/b mimics rescued the loss of the epithelial marker E-cadherin (Figure 2E) and decreased the gain of the mesenchymal marker Zeb1 (Figure 2F). In association with these changes, we confirmed a decrease in mesenchymal transcripts (Figure 2G) and an increase in epithelial morphology (Figure 2H). These results establish that PKCζ is an important regulator of EMT through its ability to modulate the levels of miR-200s. These results are also relevant from the clinical point of view, because multiple studies have linked EMT and stemness with a poor-prognosis subtype in CRC patients (Guinney et al., 2015; Marisa et al., 2013; Sadanandam et al., 2013). Interestingly, when gene neighbors of PRKCZ were compared with the molecular signatures of three separate studies of CRC patients, we found that transcripts associated with PRKCZ expression negatively correlated with the EMT/stemness and poor-prognosis CRC patient groups (Figure 2I).
Figure 2. Loss of PKCζ Promotes EMT through miR-200s.

(A) qPCR of epithelial, mesenchymal, and stem-like genes in SW480 shNT and shPKCζ cells; n = 3 biological replicates.
(B) Immunoblot of EMT markers in SW480 shNT and shPKCζ cells; n = 3 biological replicates.
(C) Representative pictures of cell morphology of SW480 shNT and shPKCζ cells. Scale bar, 50 μm.
(D) GSEA plot of EMT, extracellular matrix remodeling, and stem-like signatures in low-versus high-PRKCZ-expressing tumors from TCGA-COAD. FDR, false discovery rate; NES, normalized enrichment score.
(E) Immunofluorescence for E-cadherin (left) and quantification of E-cadherin intensity (right) in SW480 shNT, shPKCζ, and shPKCζ cells rescued with miR-200a/b. Scale bar, 20 μm; n = 3 biological replicates.
(F) Immunofluorescence for Zeb1 (left) and quantification of Zeb1 intensity (right) in SW480 shNT, shPKCζ, and shPKCζ cells rescued with miR-200a/b. Scale bar, 20 μm; n = 3 biological replicates.
(G) qPCR of mesenchymal genes (left) and immunoblot of PKCζ (right) in SW480 shNT, shPKCζ, and shPKCζ cells rescued with miR-200a/b; n = 3 biological replicates.
(H) Representative pictures of cell morphology of SW480 shPKCζ cells rescued with miR-200a/b. Scale bar, 50 μm.
(I) Summary of the correlation between PRKCZ gene neighbors and CRC gene set signatures.
Results are presented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
PKCζ Regulates the Steady-State Level of miR-200s
We next investigated the mechanism whereby PKCζ deficiency leads to reduced intracellular levels of miR-200s. We first determined whether the expression of this subset of miRNAs was regulated transcriptionally or post-transcriptionally. miR-200b, miR-200a, and miR-429 are located in a single polycistronic transcript (Figure S2A). qPCR of the region encoding the primary miRNAs in this cluster revealed no significant alterations in the transcription of these miRNAs (Figure S2B). Furthermore, experiments using the full-length endogenous promoter of the miR-200b/a/429 cluster tagged with luciferase revealed no change in the transcription of this cluster when PKCζ was knocked down (Figure S2C), suggesting that the loss of PKCζ is most likely affecting the levels of miR-200s post-transcriptionally. No changes in the levels of the components of the RNA-induced silencing complex (RISC) were observed (Figures S2D–S2G).
Although we did not detect alterations in the pri-miRNA, northern blot analysis demonstrated a drastic reduction in the levels of mature miR-200b-3p and miR-200c-3p in shPKCζ cells despite the lack of changes in the precursor form (Figure 3A). No defect was observed in the nuclear export of pre-miR-200b (Figure 3B). Furthermore, results from an in vitro precursor-processing assay showed no defects in shPKCζ lysates for the generation of mature miR-200b (Figure 3C). All this, coupled with the fact that the decrease in mature miR-200b-3p in shPKCζ cells does not correlate with increased levels of pre-miR-200b (Figure 3A), strongly suggests that PKCζ is most likely affecting the steady-state levels of mature miR-200s rather than processing. To test this possibility, we treated shNT and shPKCζ cells with the transcriptional inhibitor actinomycin D and compared the levels of endogenous mature miR-200s (Figure 3D). After inhibiting de novo transcription of RNA, the steady-state levels of mature miR-200s decreased more rapidly in shPKCζ than in shNT cells (Figure 3D). No difference in the steady-state levels of miR-16-5p between the genotypes was observed (Figure 3D). These results demonstrate that PKCζ deficiency does not affect the synthesis or maturation of miR-200s but targets a step regulating the maintenance of their steady-state levels.
Figure 3. PKCζ Regulates the Steady-State Levels of the miR-200 Family.

(A) Northern blot of precursor and mature miR-200b in SW480 shNT and shPKCζ cells (left) and quantification of band intensities (right). U6 small nuclear RNA (snRNA), loading control; n = 3 biological replicates.
(B) Northern blot of precursor and mature miR-200b from nuclear and cytosolic fractions of SW480 shNT and shPKCζ cells (left) and quantification of band intensities, relative to U6 (nuclear) and tRNA (cytoplasmic) (right); n = 3 biological replicates.
(C) Northern blot from an in vitro precursor-processing reaction of miR-200b using SW480 shNT and shPKCζ cell lysates; n = 3 biological replicates.
(D) qPCR of miRNAs (left) and immunoblot of PKCζ levels (right) in SW480 shNT and shPKCζ cells after 10 μg/mL actinomycin D treatment for the indicated time points; n = 3 biological replicates.
Results are presented as mean ± SD. *p < 0.05, **p < 0.01, ****p < 0.0001. See also Figure S2.
PKCζ Regulates the Secretion of miR-200s
We next hypothesized that PKCζ deficiency might lead to decreased intracellular levels of miR-200s by promoting their secretion. In support of this possibility, we observed increased levels of miR-200s, but not miR-26a-5p, in the conditioned media from shPKCζ cells (Figure 4A), SW620 cells as compared to SW480 (Figure 4B), and PKCζ-deficient organoids (Figure 4C). Previous reports of miRNAs secreted via multivesicular bodies (MVBs) have demonstrated that they are present in extra-cellular vesicles (EVs) termed exosomes (Gibbings et al., 2009; McKenzie et al., 2016). When shNT and shPKCζ cells were treated with a chemical inhibitor of neutral sphingomyelinases, GW4869, which has been reported to be required for the release of EVs from MVBs (Kosaka et al., 2010; Trajkovic et al., 2008), we observed that the increased levels of miR-200s in the extra-cellular milieu of shPKCζ cells were reduced to control levels (Figure 4D). Importantly, the reduced intracellular levels of miR-200s in shPKCζ cells were rescued by the addition of GW4869 (Figure 4E). No decrease in cell viability or proliferation was observed in response to GW4869 treatment (Figures S3A and S3B). Likewise, knockdown of Rab27a, which is also required for the secretory machinery by promoting the docking of MVBs to the plasma membrane (Ostrowski et al., 2010), rescued the reduced intracellular levels of miR-200s in shPKCζ cells (Figure 4F). Consistent with miR-200s being secreted in EVs, we found significantly more miR-200s in EVs from shPKCζ cells than in shNT cells, which were inhibited by blocking secretion with GW4869 (Figure 4G). Nanoparticle tracking analysis (NTA) revealed that EVs prepared by two different methods had a size of approximately 100–150 nm, which is consistent with that of exosomes (Figures 4H and S3C). Of note, no changes were found in vesicle concentration in shPKCζ as compared to shNT cells (Figures 4H and S3C). In addition, immunoblot analysis of these vesicles confirmed the presence of exosome-enriched proteins, including the tetraspanin CD63 and members of the endosomal sorting complexes required for transport (TSG101, Flotillin-1, and Alix), as well as the absence of intracellular proteins, such as peroxiredoxin 3 and histone 3 (Figures 4I and S3D). Furthermore, increased levels of miR-200s were also observed in CD63+ EVs purified from shPKCζ cells as compared to those from shNT cells (Figures 4J and S3E). Efficient pull-down of CD63 was confirmed by western blot (Figure S3F). NTA and transmission electron microscopy (TEM) of purified CD63+ EVs showed sizes compatible with exosomes (Figures 4K and 4L). These results establish that the increased secretion of miR-200s by shPKCζ cells is through exosomes via MVBs. Because no differences were found in the amount of EVs secreted by shPKCζ as compared to that in shNT cells (Figures 4H and S4C), our results suggest that the higher secreted levels of miR-200s by PKCζ-deficient cells are most likely due to increased loading, rather than increased secretion of EVs.
Figure 4. PKCζ Regulates the Secretion of miR-200s in EVs.

(A–C) qPCR of extracellular miRNAs from SW480 shNT and shPKCζ cells, n = 4 biological replicates (A); SW480 and SW620 cells, n = 3 biological replicates (B); and intestinal tumor organoids from Lgr5;APCf/f and Lgr5;PKCζf/fAPCf/f mice, n = 3 biological replicates (C).
(D) qPCR of extracellular miRNAs from SW480 shNT and shPKCζ cells treated with vehicle control or 10 μM GW4869; n = 3 biological replicates.
(E) qPCR of intracellular miRNAs (left) and immunoblot of PKCζ (right) from SW480 shNT and shPKCζ cells treated with vehicle control or 10 μM GW4869; n = 4 biological replicates.
(F) qPCR of intracellular miRNAs (left) and immunoblot of Rab27a and PKCζ (right) from SW480 shNT and shPKCζ cells treated with 50 nM siRNA against Rab27a; n = 4 biological replicates.
(G) qPCR of miRNAs in total EVs from SW480 shNT, shPKCζ, and shPKCζ cells treated with vehicle control or 10 μM GW4869; n = 3 biological replicates.
(H) Size and concentration measurements of EVs from SW480 cells prepared by ultracentrifugation (UC) or ExoQuick; n = 3 biological replicates.
(I) Immunoblot of intracellular and EV markers in whole-cell lysates (WCL) and EVs from HCT116 shNT and shPKCζ cells; n = 2 biological replicates.
(J and K) qPCR of miRNAs in CD63+ EVs and immunoblot of CD63 after CD63 pull-down, n = 4 biological replicates (J), and size and concentration measurements of CD63+ EVs, n = 3 biological replicates (K), from SW480 shNT and shPKCζ cells.
(L) TEM of EVs from shNT and shPKCζ cells. Scale bars, 0.1 μm.
Results are presented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. See also Figure S3.
Loss of PKCζ Promotes Liver Metastasis through Secretion of miR-200s
In agreement with the connection of EMT/stemness with invasion and metastasis, we observed that shPKCζ cells displayed increased cell migration and invasion in vitro (Figure 5A). Therefore, we next investigated whether the loss of PKCζ could trigger a metastatic phenotype in CRC cells in vivo. Liver metastases were induced in immunodeficient mice by in vivo intrasplenic injection of SW480 shNT or shPKCζ cells followed by splenectomy, after which metastatic colonization was analyzed 28 days thereafter (Figure 5B). Mice injected with shPKCζ cells displayed higher liver metastasis burden and increased metastasis area than those injected with shNT cells (Figures 5C–5F). A quantitative analysis measuring human Alu sequences in the mouse liver confirmed these conclusions (Figure 5G). These results establish that PKCζ deficiency confers CRC cells with a metastatic phenotype. Because our model predicts that this is through the loading of miR-200s into exosomes, we next determined whether metastatic CRC patients had higher levels of miR-200s in EVs from their plasma. Notably, we found that plasma CD63+ EVs from CRC metastatic patients displayed higher levels of miR-200b-3p, but not of the unrelated miR-26a-5p, than those from non-metastatic CRC patients (Figure 5H). In order to determine the potential involvement of exosomes in the liver metastatic phenotype of PKCζ-deficient CRC cells, we repeated the intrasplenic model as above but included the treatment of mice for the first 5 days with GW4869 or DMSO, as a negative control (Figure 5I). Interestingly, GW4869-treated mice displayed reduced metastatic activity of shPKCζ cells that became comparable to that of shNT levels (Figures 5J–5N). As a readout of miR-200s, we analyzed the EMT phenotype in the liver metastases of this experiment. Importantly, the liver metastases induced by PKCζ-deficient cells displayed an EMT phenotype with loss of E-cadherin and gain of vimentin that was reverted by GW4869 treatment (Figures 5O–5Q). Altogether, these results demonstrate that PKCζ represses the metastatic potential of CRC cells by controlling the intracellular steady-state levels of miR-200s through the regulation of their secretion.
Figure 5. Loss of PKCζ Promotes Liver Metastasis.

(A) Cell migration and invasion assay (left) and immunoblot of PKCζ (right) in SW480 shNT and shPKCζ cells; n = 3 biological replicates.
(B–G) Experimental design for liver metastases and immunoblot of PKCζ (B) in SW480 shNT and shPKCζ cells used for splenic injection (n = 8 mice per group); bioluminescence imaging and liver photographs; scale bar, 1 cm (C); quantification of total photon counts (shNT, n = 7; shPKCζ, n = 8) (D); H&E staining of liver sections; scale bar, 200 μm (E); quantification of metastatic area relative to total liver area (F); and quantification of human metastases in mouse liver by qPCR (G).
(H) qPCR of miRNAs in exosomes from plasma of non-metastatic (n = 8) and metastatic (n = 6) patients.
(I–N) Experimental design for inhibiting CRC liver metastases with GW4869 and immunoblot of PKCζ (I) in SW480 shNT and shPKCζ cells used for splenic injection; bioluminescence imaging and liver photographs of mice injected with SW480 shNT (n = 4), shPKCζ (n = 4), or shPKCζ cells + GW4869 (n = 5); scale bar, 1 cm (J); quantification of total photon counts (K); H&E staining of liver sections; scale bar, 200 μm (L); quantification of human metastases in mouse liver by qPCR (M); and quantification of metastatic area relative to total liver area (N).
(O–Q) Immunofluorescence for E-cadherin and vimentin of liver metastases of the experiment described in (I) (n = 3); scale bars, 50 μm (O). Quantification of E-cadherin (P) and vimentin (Q) areas.
Results are presented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
PKCζ Phosphorylates and Activates ADAR2 to Promote the Accumulation of miR-200s
To explore how PKCζ specifically regulates the levels of mature miR-200s, we tested whether its kinase activity was required for that function. Thus, we reconstituted wild-type (WT) or a kinase-dead (KD) mutant of PKCζ into shPKCζ cells. Importantly, the expression of WT-PKCζ, but not the KD-PKCζ mutant, was able to rescue the levels of the miR-200 family (Figure 6A). Given that PKCζ’s kinase activity is required for that effect, we then carried out an unbiased screening for PKCζ substrates in SW480 cells stably expressing WT-PKCζ or its analog-sensitive (AS) PKCζ mutant, which utilizes a bulky ATP analog (benzyl-ATPγS) that cannot be used by endogenous kinases and transfers a thiophosphate onto its substrates (Figure S4A). Among the identified putative substrates, we selected ADAR2 for further analysis based on two criteria: (1) proteins involved in miRNA regulation; and (2) proteins known to regulate cell migration. ADAR2 is an enzyme that edits adenosine to inosine (A-to-I) nucleotides in RNA (Kawahara et al., 2007). An in vitro phosphorylation assay using recombinant PKCζ confirmed that ADAR2 is a bona fide PKCζ substrate (Figure 6B). In addition, we established that PKCζ interacts with ADAR2 in reciprocal co-transfection (Figures 6C and 6D) and in semi-endogenous (Figures 6E and 6F) experiments and, more importantly, in endogenous conditions (Figure 6G).
Figure 6. PKCζ Phosphorylates and Regulates ADAR2 Activity.

(A) qPCR of miR-200s (left) and immunoblot of PKCζ (right) in SW480 shNT and shPKCζ cells reconstituted with HA-PKCζWT or HA-PKCζKD; n = 4 biological replicates. HA, hemagglutinin.
(B) Immunoblot of in vitro phosphorylation reaction for ADAR2 and PKCζ; n = 3 biological replicates. GST, glutathione S-transferase.
(C and D) Reciprocal co-immunoprecipitation of GST-ADAR2 and Flag-PKCζ, IP anti-Flag (C) and anti-GST (D); n = 3 biological replicates.
(E and F) Reciprocal semi-endogenous co-immunoprecipitation of Flag-ADAR2 and PKCζ, IP anti-Flag (E) and anti-PKCζ (F); n = 2 biological replicates.
(G) Endogenous co-immunoprecipitation of PKCζ and ADAR2; n = 2 biological replicates.
(H) qPCR of miRNAs (left) and immunoblot of ADAR2 (right) in SW480 shNT and shADAR2 cells; n = 3 biological replicates.
(I) qPCR of miRNAs in CD63+ EVs from SW480 shNT and shADAR2 cell media; n = 4 biological replicates.
(J) qPCR of epithelial (left) and mesenchymal genes (right) in SW480 shNT and shADAR2 cells; n = 3 biological replicates.
(K) Immunoblot of EMT markers in SW480 shNT and shADAR2 cells; n = 3 biological replicates.
(L) Representative pictures of the cell morphology of SW480 shNT and shADAR2 cells. Scale bar, 50 μm.
(M) Immunofluorescence for E-cadherin (left) and quantification of E-cadherin intensity (right) in SW480 shNT and shADAR2 cells. Scale bar, 20 μm; n = 3 biological replicates.
(N) Cell migration and invasion assay in SW480 shNT and shADAR2 cells; n = 3 biological replicates.
(O) Diagram of the ADAR2 self-editing product.
(P) qPCR of ADAR2 self-editing in organoids of WT and PKCζ KO mice (left) and SW480 shNT and shPKCζ cells (right); n = 3 biological replicates.
(Q) Diagram of ADAR2 domains in relation to PKCζ phosphorylation sites.
(R) In vitro phosphorylation of GST-ADAR2WT and GST-ADAR2SS211/216AA; n = 2 biological replicates.
(S) qPCR of miR-200s (left) and immunoblot of Flag-ADAR2 (right) in SW480 cells stably expressing ADAR2 WT, ADAR2SS211/216AA, or ADAR2E396A; n = 3 biological replicates.
(T) Sanger sequencing results for COPAI164V editing in SW480 and SW620 cells (left), calibration curve of editing frequency (middle), and quantification of the editing frequency (right); n = 3 biological replicates.
Results are presented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. See also Figure S4.
Next, we investigated whether ADAR2 played a functional role in the secretion of the miR-200s and, consequently, the EMT phenotype. Upon knockdown of ADAR2, we observed a decrease in the intracellular levels of the miR-200s, concomitant with their increased secretion in CD63+ EVs (Figures 6H and 6I). No changes were observed in the size or concentration of the secreted vesicles (Figure S4B). Consistent with the loss of intra-cellular miR-200 levels, ADAR2-deficient cells had decreased expression of epithelial genes and increased expression of mesenchymal genes, both at mRNA and protein levels, as well as an enhanced mesenchymal morphology (Figures 6J–6L). The EMT phenotype upon ADAR2 deficiency was also observed by immunofluorescence of E-cadherin and Zeb1 (Figures 6M and S4C). Importantly, the overexpression of miR-200a/b mimics reversed the mesenchymal transition observed in ADAR2-deficient cells (Figure S4D). Similar to PKCζ-deficient cells, the loss of ADAR2 also induced increased migration and invasion (Figure 6N). These results demonstrate that ADAR2 is required for maintaining the intracellular levels of miR-200s and the epithelial morphology of CRC cells, and that ADAR2 loss recapitulates the phenotype of PKCζ-deficient cells.
ADAR2 is known to undergo self-editing of its own pre-mRNA to create an alternative splice site within intron 1, leading to the inclusion of 47 extra nucleotides that produces a frameshift in the coding region and results in a truncated, inactive protein (Rueter et al., 1999) (Figure 6O). In this way, ADAR2 controls its own activity by self-regulating its own mRNA and protein levels. Therefore, reduced levels of self-editing mRNA that results in increased ADAR2 protein levels are surrogate markers of inhibited ADAR2 activity. We hypothesized that PKCζ might be required for the activation of ADAR2. Consistent with this notion, we found lower levels of the self-editing product in intestinal organoids from PKCζ KO mice, and in SW480 shPKCζ cells, relative to their respective controls, demonstrating that PKCζ phosphorylation is critical for the maintenance of ADAR2 gene editing activity (Figure 6P). In agreement with this, we found that ADAR2 mRNA and protein levels were increased in shPKCζ cells (Figures S4E and S4F), as well as that ADAR2 protein levels were higher in tumors from Lgr5;PKCζf/fAPCf/f mice as compared to Lgr5;APCf/f (Figure S4G).
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis identified serines 211 and 216 as the targets of PKCζ phosphorylation on ADAR2 (Figure S4H), which are conserved across different species (Figure S4I). These residues lie between the two protein double-stranded RNA-binding motifs (DRBM1 and DRBM2; Figure 6Q), which are important domains for ADAR2 activity (Sansam et al., 2003), suggesting that phosphorylation of these residues by PKCζ may regulate ADAR2 activity. To determine the role of ADAR2 phosphorylation by PKCζ, we mutated these serine residues to alanine to create ADAR2SS211/216AA and confirmed that these were the amino acids phosphorylated by PKCζ using an in vitro kinase assay with purified proteins (Figure 6R). Importantly, expression of ADAR2SS211/216AA, but not ADAR2WT, in SW480 cells significantly decreased miR-200 levels, comparable to that produced by the expression of an enzymatically inactive form of ADAR2 (ADAR2E396A) that lacks RNA editing capacity (Figure 6S). Furthermore, incubation of SW480 cells expressing ADAR2SS211/216AA or ADAR2E396A with actinomycin D showed that the levels of endogenous mature miR-200s, but not those of miR-16-5p, were reduced more rapidly in mutant ADAR2-expressing cells than in ADAR2WT cells (Figure S4J). These results demonstrate that the activation of ADAR2 by PKCζ phosphorylation is a key mechanism for the regulation of the steady-state levels of miR-200s.
Consistent with PKCζ phosphorylation mediating ADAR2 activity, we observed hypo-editing of coatamer protein alpha (COPA), which has previously been reported to be a substrate of ADAR2 and to be hypo-edited in cancer (Chan et al., 2014). PKCζ-deficient cells displayed reduced COPA editing, which could be rescued by reconstituting SW480 shPKCζ cells with WT-PKCζ (Figure 6T). Similarly, COPA editing was significantly reduced in cells deficient in ADAR2, as well as in cells expressing the phospho-dead mutant ADAR2SS211/216AA or the catalytically inactive mutant ADAR2E396A (Figure 6T). Hypo-editing of COPA could also be confirmed in SW620 cells, which have reduced levels of endogenous PKCζ (Figure 6T). Moreover, COPA was found to be significantly hypo-edited in tumors from human CRC patients, relative to adjacent normal tissue from the same patients, and the editing levels were consistent with what was observed in vitro (Figure S4K). Consequently, this suggests that phosphorylation of ADAR2 by PKCζ is required for its activity, which is necessary for the regulation of the intracellular levels of miR-200s.
DISCUSSION
Here, we demonstrate that the loss of PKCζ in CRC cells is associated with lower ADAR2 activity, which promotes the loading of miR-200s into EVs, thereby decreasing their intracellular steady-state levels. This reduced PKCζ/ADAR2/miR-200 signaling pathway gives rise to an EMT/stemness phenotype, which explains the higher metastatic activity of PKCζ-deficient CRC cells. This is an unexpected mechanism for the regulation of miR-200s that has important repercussions in vivo because, as shown here, CRC patients with metastatic tumors have increased serum levels of exosomes containing miR-200s, indicating a potential non-invasive diagnostic marker for patients at risk of metastasis. We also demonstrate that the pharmacological inhibition of miR-200 secretion in vivo severely impairs CRC metastasis to the liver. This is of great potential therapeutic relevance, because it constitutes a proof of concept that targeting PKCζ downstream signals activated upon its loss of function can be useful anti-metastatic therapies.
The inhibition of ADAR2 activity upon loss of PKCζ as a mechanism for tumor invasiveness is a key finding for CRC, and is consistent with previous reports for ADAR2 acting as a tumor suppressor in other types of neoplasia. Most notably, ADAR2 editing activity has been shown to inhibit the growth and migration of astrocytomas, with decreased editing of the ionotropic glutamate receptors GluR-B and GluR-6, and the cell-cycle-regulating phosphatase CDC14B being implicated as a potential driver of the disease (Cenci et al., 2008; Galeano et al., 2013). However, the hypo-editing of these substrates is unlikely to be involved in the context of PKCζ-deficient CRC cells, considering that the ionotropic glutamate receptors are restricted to the central nervous system, and CRC cells deficient for PKCζ do not exhibit increased proliferation (Figure S3B). Importantly, ADAR2 has also been reported to be tumor suppressive in other solid tumors, such as gastric cancer, hepatocellular carcinoma, and esophageal squamous cell carcinoma (Chan et al., 2014, 2016; Chen et al., 2017), which underscores the importance of ADAR2 beyond CRC. Yet, despite being implicated as a tumor suppressor, little is known about the regulation of ADAR2 in cancers. In fact, although the phosphorylation of residues S26, S31, and T32 has been previously reported to regulate ADAR2 activity through a mechanism involving Pin1, a kinase was not identified in that study (Marcucci et al., 2011). Given the role that ADAR2 plays in inhibiting multiple types of cancer, our finding that ADAR2 is regulated by PKCζ is extremely important because it identifies a synthetic vulnerability of PKCζ-deficient tumors.
In this study, we also show that COPA, a previously identified substrate of ADAR2, is hypo-edited in CRC cells deficient for PKCζ, which further confirms the functional role of the PKCζ-ADAR2 connection. Interestingly, COPA has also been reported to be hypo-edited in multiple types of cancers (Chan et al., 2014, 2016; Han et al., 2015), which suggests that the editing of COPA may have a functional role in modulating tumor growth. COPA is part of the coat protein I (COPI) complex, which has been shown to mediate the retrieval of proteins from the Golgi back to the endoplasmic reticulum (ER) (Letourneur et al., 1994). This is noteworthy, because the ER has been reported to be the site of RISC formation and mRNA silencing, whereas the Golgi lies on the exit route from the ER to the secretory and endosomal pathways. These pathways have been implicated in RISC turnover as well as miRNA secretion (Gibbings et al., 2009; Lee et al., 2009; Stalder et al., 2013). Therefore, it is tempting to speculate that COPA editing may have a role in preventing the secretion of the miR-200s, although other substrates might also be important mediators of the functional consequence of losing the PKCζ/ADAR2 axis. In this regard, this study opens the important question of how the editing activity of ADAR2 regulates the secretion of miR-200s. One possibility could be through direct editing of miR-200 itself. However, we did not find changes in editing in miR-200s upon PKCζ deficiency in our system (not shown). Alternatively, ADAR2 editing could be impacting the levels of RNA-binding proteins or other components of the miRNA secretion machinery or competing endogenous RNAs. Therefore, the identification of the ADAR2 substrates mediating the selective secretion of miR-200 deserves further investigation, and will contribute to a better understanding of the mechanisms of miRNA secretion and the selective loading of miRNAs in extracellular vesicles.
Our finding that PKCζ deficiency promotes the circulation of miR-200s is of great significance, not only as a mechanism to downregulate their intracellular levels in the tumor cell but also because EVs and their miRNA content are being increasingly accepted as vehicles to promote metastasis (Becker et al., 2016). Similarly, the paracrine action of miR-200 secretion could allow the transition of the fibroblasts in the metastasis niche into cancer-associated fibroblasts (CAFs), which may increase the stromal response. This potential effect is highly pertinent, considering that an activated stroma is increasingly appreciated in CRC patients as a marker of poor prognosis and increased metastasis (Calon et al., 2015; Guinney et al., 2015; Isella et al., 2015). Altogether, the results reported here highlight the importance of identifying actionable signaling vulnerabilities unleashed upon the inactivation of tumor suppressors and establish PKCζ-derived signals controlling miR-200s as potentially important therapeutic targets in cancer metastasis.
EXPERIMENTAL PROCEDURES
A detailed description of the experimental procedures utilized in this work can be found in Supplemental Experimental Procedures. Primers used are described in Table S2.
Mice
PKCζ−/−, APCMin/+, Lgr5-EGFP-ires-creERT2 (Lgr5), Lgr5;PKCζf/f, Lgr5;APCf/f, and Lgr5;APCf/f;PKCζf/f mice were described previously (Llado et al., 2015). For the intestinal regeneration experiments, mice were handled as described previously (Llado et al., 2015). For specific ablation of PKCζ in Lgr5+ cells, 8-week-old mice were treated as described previously (Llado et al., 2015). Experimental mice were all males and aged 8 weeks (PKCζ−/−) or 5–8 weeks (NOD scid gamma [NSG]) at the time of experimentation. NSG mice were randomized between litters to prevent a bias toward age. Animal handling and experimental procedures were approved by the Sanford Burnham Prebys (SBP) Institutional Animal Care and Use Committee.
Patient Samples
Blood samples were collected in EDTA tubes (9 mL) immediately before surgery. Samples were centrifuged at 1,800 × g for 10 min. Plasma was collected, subjected to a second centrifugation at 3,000 × g for 10 min, aliquoted, and stored at −80°C until analysis. All donors were informed of the aim of the study and gave consent to donate their samples. Corresponding clinical data were obtained from medical records and de-identified. The study was approved by the Ethics and Health Research Committee of Fundación Jiménez Díaz University Hospital (CEIC-FJD).
Statistical Analysis
The significance level for statistical testing was set at p <0.05. For the animal studies, we used G*Power Data (Faul et al., 2007) analysis to ensure adequate power to detect a prespecified effect size. Specifically, a 2-sided t test with a sample size of 8 mice per group achieves approximately 80% power at a 2-tailed 0.05 significance level. No statistical method was used to predetermine any other sample sizes.
Supplementary Material
Highlights.
Loss of PKCζ results in increased secretion of miR-200s, EMT, and liver metastases
PKCζ phosphorylates and activates ADAR2 gene editing activity
PKCζ-phosphorylated ADAR2 promotes the accumulation of miR-200s
ADAR2 regulation is a key vulnerability of PKCζ-deficient cancer cells
Acknowledgments
Research was supported by grants from the NIH (R01DK108743, R01CA172025, and R01CA207177 to J.M.; R01CA192642 and R01CA218254 to M.T.D.-M.). We thank Dr. Tariq M. Rana and Dr. Rui Zhou (SBP Medical Discovery Institute) for helpful discussions on miRNAs. We thank Michael Leitges for providing PKCζ−/− mice. We thank Diantha LaVine for the artwork, Clayton Deighan from Malvern Instruments for the NTA readings, and Sarah Gilmour, the personnel of the Proteomics, Histology, Cell Imaging, Genomics, Animal Facility, and Viral Vectors Shared Resources at the SBP Medical Discovery Institute, and Malcolm R. Wood in the Core Microscopy Facility at the Scripps Research Institute for technical assistance.
Footnotes
DATA AND SOFTWARE AVAILABILITY
The accession number for the microarray data reported in this paper is GEO: GSE42186. All original, unprocessed data can be accessed at Mendeley Data (https://doi.org/10.17632/9wcwrfn7yh.2).
Supplemental Information includes Supplemental Experimental Procedures, four figures, and two tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.03.118.
AUTHOR CONTRIBUTIONS
P.M.S. and A.D. performed most of the experiments. L.M. performed the mi-croarray analysis. V.L. generated the organoid cultures and performed the in vivo irradiation experiments. Y.N. and H.K. performed the in vivo metastasis experiments. Y.N. and A.D. contributed to the immunofluorescence data. M.R.-C. contributed to the bioinformatics and phosphorylation analyses. A.C. performed the proteomic analysis. D.G.-O., M.G.-A., D.C.G.-O., and S.O.-L. collected and provided clinical samples. J.F.C. provided expertise in miRNA biogenesis. All authors discussed the results and commented on the manuscript. M.T.D.-M. and J.M. conceived and supervised the project. P.M.S., M.T.D.-M., and J.M. wrote the manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, Clevers H. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- Becker A, Thakur BK, Weiss JM, Kim HS, Peinado H, Lyden D. Extracellular vesicles in cancer: cell-to-cell mediators of metastasis. Cancer Cell. 2016;30:836–848. doi: 10.1016/j.ccell.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calon A, Lonardo E, Berenguer-Llergo A, Espinet E, Hernando-Mom-blona X, Iglesias M, Sevillano M, Palomo-Ponce S, Tauriello DV, Byrom D, et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat Genet. 2015;47:320–329. doi: 10.1038/ng.3225. [DOI] [PubMed] [Google Scholar]
- Cenci C, Barzotti R, Galeano F, Corbelli S, Rota R, Massimi L, Di Rocco C, O’Connell MA, Gallo A. Down-regulation of RNA editing in pediatric astrocytomas: ADAR2 editing activity inhibits cell migration and proliferation. J Biol Chem. 2008;283:7251–7260. doi: 10.1074/jbc.M708316200. [DOI] [PubMed] [Google Scholar]
- Chan THM, Lin CH, Qi L, Fei J, Li Y, Yong KJ, Liu M, Song Y, Chow RKK, Ng VHE, et al. A disrupted RNA editing balance mediated by ADARs (adenosine deaminases that act on RNA) in human hepatocellular carcinoma. Gut. 2014;63:832–843. doi: 10.1136/gutjnl-2012-304037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan TH, Qamra A, Tan KT, Guo J, Yang H, Qi L, Lin JS, Ng VH, Song Y, Hong H, et al. ADAR-mediated RNA editing predicts progression and prognosis of gastric cancer. Gastroenterology. 2016;151:637–650. e10. doi: 10.1053/j.gastro.2016.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YB, Liao XY, Zhang JB, Wang F, Qin HD, Zhang L, Shugart YY, Zeng YX, Jia WH. ADAR2 functions as a tumor suppressor via editing IGFBP7 in esophageal squamous cell carcinoma. Int J Oncol. 2017;50:622–630. doi: 10.3892/ijo.2016.3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Sousa e Melo F, Kurtova AV, Harnoss JM, Kljavin N, Hoeck JD, Hung J, Anderson JE, Storm EE, Modrusan Z, Koeppen H, et al. A distinct role for Lgr5+ stem cells in primary and metastatic colon cancer. Nature. 2017;543:676–680. doi: 10.1038/nature21713. [DOI] [PubMed] [Google Scholar]
- Diaz T, Tejero R, Moreno I, Ferrer G, Cordeiro A, Artells R, Navarro A, Hernandez R, Tapia G, Monzo M. Role of miR-200 family members in survival of colorectal cancer patients treated with fluoropyrimidines. J Surg Oncol. 2014;109:676–683. doi: 10.1002/jso.23572. [DOI] [PubMed] [Google Scholar]
- Faul F, Erdfelder E, Lang AG, Buchner A. G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods. 2007;39:175–191. doi: 10.3758/bf03193146. [DOI] [PubMed] [Google Scholar]
- Galeano F, Rossetti C, Tomaselli S, Cifaldi L, Lezzerini M, Pezzullo M, Boldrini R, Massimi L, Di Rocco CM, Locatelli F, Gallo A. ADAR2-editing activity inhibits glioblastoma growth through the modulation of the CDC14B/Skp2/p21/p27 axis. Oncogene. 2013;32:998–1009. doi: 10.1038/onc.2012.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvez AS, Duran A, Linares JF, Pathrose P, Castilla EA, Abu-Baker S, Leitges M, Diaz-Meco MT, Moscat J. Protein kinase Czeta represses the interleukin-6 promoter and impairs tumorigenesis in vivo. Mol Cell Biol. 2009;29:104–115. doi: 10.1128/MCB.01294-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbings DJ, Ciaudo C, Erhardt M, Voinnet O. Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat Cell Biol. 2009;11:1143–1149. doi: 10.1038/ncb1929. [DOI] [PubMed] [Google Scholar]
- Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda G, Angelino P, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21:1350–1356. doi: 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Diao L, Yu S, Xu X, Li J, Zhang R, Yang Y, Werner HMJ, Eterovic AK, Yuan Y, et al. The genomic landscape and clinical relevance of A-to-I RNA editing in human cancers. Cancer Cell. 2015;28:515–528. doi: 10.1016/j.ccell.2015.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur K, Toiyama Y, Takahashi M, Balaguer F, Nagasaka T, Koike J, Hemmi H, Koi M, Boland CR, Goel A. MicroRNA-200c modulates epithelial-to-mesenchymal transition (EMT) in human colorectal cancer metastasis. Gut. 2013;62:1315–1326. doi: 10.1136/gutjnl-2011-301846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isella C, Terrasi A, Bellomo SE, Petti C, Galatola G, Muratore A, Mellano A, Senetta R, Cassenti A, Sonetto C, et al. Stromal contribution to the colorectal cancer transcriptome. Nat Genet. 2015;47:312–319. doi: 10.1038/ng.3224. [DOI] [PubMed] [Google Scholar]
- Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, Nishikura K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science. 2007;315:1137–1140. doi: 10.1126/science.1138050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka N, Iguchi H, Yoshioka Y, Takeshita F, Matsuki Y, Ochiya T. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J Biol Chem. 2010;285:17442–17452. doi: 10.1074/jbc.M110.107821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Pressman S, Andress AP, Kim K, White JL, Cassidy JJ, Li X, Lubell K, Lim DH, Cho IS, et al. Silencing by small RNAs is linked to endosomal trafficking. Nat Cell Biol. 2009;11:1150–1156. doi: 10.1038/ncb1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letourneur F, Gaynor EC, Hennecke S, Démollière C, Duden R, Emr SD, Riezman H, Cosson P. Coatomer is essential for retrieval of dilysine-tagged proteins to the endoplasmic reticulum. Cell. 1994;79:1199–1207. doi: 10.1016/0092-8674(94)90011-6. [DOI] [PubMed] [Google Scholar]
- Llado V, Nakanishi Y, Duran A, Reina-Campos M, Shelton PM, Linares JF, Yajima T, Campos A, Aza-Blanc P, Leitges M, et al. Repression of intestinal stem cell function and tumorigenesis through direct phosphorylation of β-catenin and Yap by PKCζ. Cell Rep. 2015;10:740–754. doi: 10.1016/j.celrep.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Tao Y, Duran A, Llado V, Galvez A, Barger JF, Castilla EA, Chen J, Yajima T, Porollo A, et al. Control of nutrient stress-induced metabolic reprogramming by PKCζ in tumorigenesis. Cell. 2013;152:599–611. doi: 10.1016/j.cell.2012.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcucci R, Brindle J, Paro S, Casadio A, Hempel S, Morrice N, Bisso A, Keegan LP, Del Sal G, O’Connell MA. Pin1 and WWP2 regulate GluR2 Q/R site RNA editing by ADAR2 with opposing effects. EMBO J. 2011;30:4211–4222. doi: 10.1038/emboj.2011.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marisa L, de Reyniès A, Duval A, Selves J, Gaub MP, Vescovo L, Etienne-Grimaldi MC, Schiappa R, Guenot D, Ayadi M, et al. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med. 2013;10:e1001453. doi: 10.1371/journal.pmed.1001453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie AJ, Hoshino D, Hong NH, Cha DJ, Franklin JL, Coffey RJ, Patton JG, Weaver AM. KRAS-MEK signaling controls Ago2 sorting into exosomes. Cell Rep. 2016;15:978–987. doi: 10.1016/j.celrep.2016.03.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehlen P, Puisieux A. Metastasis: a question of life or death. Nat Rev Cancer. 2006;6:449–458. doi: 10.1038/nrc1886. [DOI] [PubMed] [Google Scholar]
- Ostrowski M, Carmo NB, Krumeich S, Fanget I, Raposo G, Savina A, Moita CF, Schauer K, Hume AN, Freitas RP, et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol. 2010;12:19–30. doi: 10.1038/ncb2000. [DOI] [PubMed] [Google Scholar]
- Pencheva N, Tavazoie SF. Control of metastatic progression by microRNA regulatory networks. Nat Cell Biol. 2013;15:546–554. doi: 10.1038/ncb2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueter SM, Dawson TR, Emeson RB. Regulation of alternative splicing by RNA editing. Nature. 1999;399:75–80. doi: 10.1038/19992. [DOI] [PubMed] [Google Scholar]
- Sadanandam A, Lyssiotis CA, Homicsko K, Collisson EA, Gibb WJ, Wullschleger S, Ostos LCG, Lannon WA, Grotzinger C, Del Rio M, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med. 2013;19:619–625. doi: 10.1038/nm.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansam CL, Wells KS, Emeson RB. Modulation of RNA editing by functional nucleolar sequestration of ADAR2. Proc Natl Acad Sci USA. 2003;100:14018–14023. doi: 10.1073/pnas.2336131100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- Stalder L, Heusermann W, Sokol L, Trojer D, Wirz J, Hean J, Fritzsche A, Aeschimann F, Pfanzagl V, Basselet P, et al. The rough endo-plasmatic reticulum is a central nucleation site of siRNA-mediated RNA silencing. EMBO J. 2013;32:1115–1127. doi: 10.1038/emboj.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, Schwille P, Brügger B, Simons M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science. 2008;319:1244–1247. doi: 10.1126/science.1153124. [DOI] [PubMed] [Google Scholar]
- Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, Waldvogel B, Vannier C, Darling D, zur Hausen A, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11:1487–1495. doi: 10.1038/ncb1998. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.