

Abstract
Image-based, high-throughput screening of genetic perturbations will advance both biology and biotechnology. We report a high-throughput screening method that allows diverse genotypes and corresponding phenotypes to be imaged in numerous individual cells. We achieve genotyping by introducing barcoded genetic variants into cells and using massively multiplexed FISH to measure the barcodes. We demonstrated this method by screening mutants of the fluorescent protein YFAST, yielding brighter and more photostable YFAST variants.
High-throughput screening of genetic variants or perturbations is playing an increasingly important role in advancing the understanding of biological systems and facilitating biotechnology applications. Large-scale screening is greatly facilitated by pooled, high-diversity libraries of genetic variants. Methods such as error-prone PCR1 or cloning with large pools of array-synthesized oligonucleotides2 allow pooled libraries with a large number of genetic variants to be created. However, unlike screening of individually constructed variants where the genotype is known a priori, screening of pooled libraries requires methods to measure the genotype that produced the desired phenotype, and this is typically done by selecting/enriching library members with desired phenotypes and then using sequencing to determine their corresponding genotypes. Such approaches have been used to identify protein variants with desired properties, such as fluorescent proteins with improved brightness3, reversible photoswitching4,5 and increased lifetime6–8. At the genome scale, RNAi or CRISPR-based approaches have been used in pooled library screens to measure the role of numerous genes in cellular phenotypes such as viability9 and, more recently, in the expression of the transcriptome10–12. However, many important phenotypes cannot be measured easily with existing high-throughput screening approaches. Phenotypes ranging from cellular morphology and dynamics to the intracellular organization of proteins or RNAs require high-resolution imaging to be measured. Moreover, time-lapsed imaging can also facilitate the screening of photo-physical properties of fluorescent proteins. Unfortunately, it is challenging to combine pooled library screening with high-resolution imaging because of the difficulty associated with isolating library members based on their imaged phenotype for genotyping. To enable such imaging-based screening, it is thus desirable to directly measure the genotype of individual library members in situ by imaging. Moreover, by imaging the genotypes and phenotypes of all library members, such an approach could map the full genotype-phenotype landscape.
Here we report a high-throughput, imaging-based screening method that allows the characterization of both the phenotype and genotype of individual cells in pooled libraries. In this method, we associate each genetic variant with a unique nucleic acid barcode that can be identified via multiplexed FISH imaging. We then use imaging to determine both the phenotype and the corresponding genotype of each cell. We demonstrate the power of this method by screening 20 million E. coli cells containing ~60,000 variants of a fluorescent protein YFAST13 and identifying YFAST mutants with increased brightness and photostability.
Our barcodes are comprised of a series of nucleic acid hybridization sites, each corresponding to one bit in a N-bit binary code. For each bit, we designed two different sequences (termed readout sequences), representing the values of “1” and “0”, respectively (Fig. 1a). Different barcode designs, such as ternary codes, are also possible. Because the number of unique barcodes grows exponentially with the number of bits, our barcoding scheme potentially allows the screening of millions of genetic variants with barcodes that contain just tens of hybridization sites. We randomly assigned a barcode to each genetic variant by randomly incorporating these barcodes into plasmids containing the desired genetic variants (Fig. 1b). To minimize the probability that the same barcode is assigned to multiple genetic variants, we designed a bottlenecking strategy: specifically, after generating the plasmid library containing the barcoded genetic variants, we selected a small, random subset of library members, the number of which is much smaller than the total number of possible N-bit binary barcodes. With this strategy, only a small fraction of the selected barcodes would be associated with more than one genetic variant by chance (see more details in the Supplementary Note). We then used next generation sequencing to determine which barcode was associated with each variant and removed any barcode assigned to more than one genetic variant from further analysis. An added benefit of this bottlenecking strategy is that it provides error robustness to the barcode detection, i.e. if any bit of a barcode is mis-read, it will most likely produce an invalid barcode that is not present in the library—an error that can be detected and removed.
Fig. 1. A high-throughput, image-based screening method using massively multiplexed fluorescence in situ hybridization.
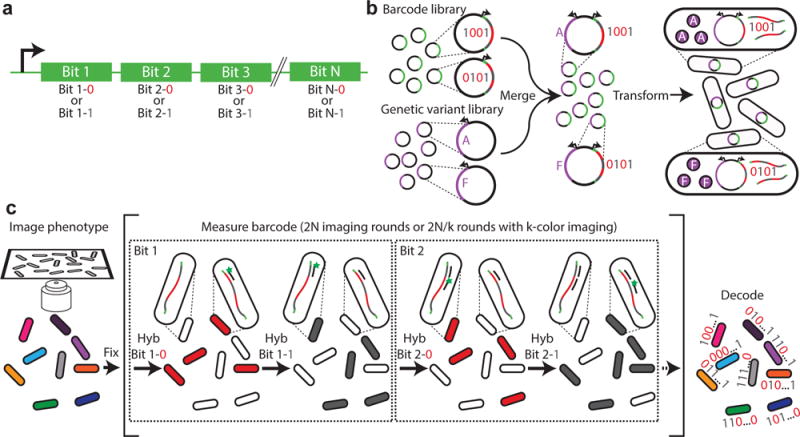
(a) Schematic depiction of a nucleic acid barcode. Each barcode consists of the concatenation of hybridization sites, each of which is associated with a different bit in a N-bit binary barcode. Each hybridization site can utilize one of two readout sequences unique to that site, with one readout sequence representing the value of “1” and another representing “0”. (b) Schematic depiction of barcoded genetic variant library construction. The library of barcodes is merged with a library of genetic variants and transformed into cells. (c) Schematic diagram of the image-based phenotype-genotype characterization. The phenotype is first imaged. Then, the cells are fixed, and multiple rounds of hybridization are used to measure the RNA barcodes expressed in the cells. During the first round, readout probe 1-0 is added and cells with barcodes that read “0” in bit 1, i.e. which contain the readout sequence 1-0, should bind to the probe and become fluorescent, whereas cells with barcodes that read “1” in bit 1 should remain dark. Once readout probe 1-0 is extinguished, readout probe 1-1 is added and the cells with barcodes that read “1” in bit 1 should become fluorescent. This process is repeated for the remaining bits, allowing the barcode of each cell, and hence the identity of the genetic variant contained in the cell, to be determined and linked to the measured phenotype of the cell.
The barcoded genetic-variant library was then incorporated into cells, where the genetic variant was expressed and the barcode sequence was transcribed into RNAs. The phenotype of each cell was then determined via imaging (Fig. 1c), and the sequence of the RNA barcode expressed within each cell was determined using a modified version of multiplexed error-robust fluorescence in situ hybridization (MERFISH)14. Specifically, we used multiple rounds of hybridization to detect the barcodes, each round probing either a single readout sequence using a complementary FISH probe (readout probe) or multiple readout sequences simultaneously using multiple readout probes linked to spectrally distinct dyes (Fig. 1c). Unlike our previous MERFISH experiments14,15, where we exploit single-molecule FISH16,17 to quantify numerous RNA species within single cells, here each individual cell expressed only one barcode RNA in high abundance, and we measured the total signal from all barcode RNA molecules within each cell. Thus, the bright signals should lead to a low error rate, and we exploited the error robustness provided by the bottlenecking strategy to detect any remaining errors.
To demonstrate the feasibility and accuracy of our method, we created a high-diversity, barcoded genetic variant library that contains ~80,000 distinct barcodes and associated these barcodes with only two genotypes—the presence or absence of a fluorescent protein, which produced simple and clear phenotypes—the presence or absence of fluorescence in cells. To this end, we first created a library of all possible 21-bit binary barcodes and inserted into this library a construct that expresses either the translational fusion of the blue fluorescent protein mTagBFP218 and the photo-switchable red fluorescent protein mMaple319 (mMaple3+) or mTagBFP2 alone (mMaple3-) (Fig. 2a). We transformed these plasmids into E. coli, bottlenecked this library to ~80,000 unique barcodes (only ~4% of the 2 million possible barcodes), and used next generation sequencing to determine which barcodes were present in the final library and their corresponding (mMaple3+ or mMaple3-) genotype. To determine the phenotypes of individual cells in the library, we imaged the cells alive, first measuring the mTagBFP2 fluorescence, then photoactivating and measuring the red mMaple3 fluorescence, and normalized the mMaple3 signal to the mTagBFP2 signal to remove differences in protein expression levels between cells. To identify the RNA barcode expressed within each cell, we fixed cells and performed MERFISH imaging with three spectrally distinct readout probes in each round of hybridization (Supplementary Table 1). In total, we screened 1.5 million E. coli cells in a ~40-hour measurement.
Fig. 2. Performance characterization of the imaged-based screening method by measuring 1.5 million cells containing 80,000 barcodes associated with two genotypes and phenotypes.
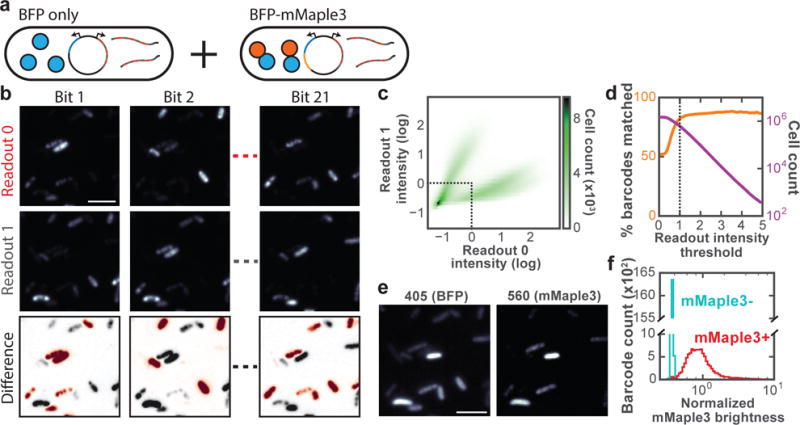
(a) Schematic diagram of the library constituents. (b) Fluorescent images for each readout from a subset of the 21 bits. The top and middle panels for each bit show the images of cells after hybridization to the readout probes corresponding to “0” (top) or “1” (middle) at this bit. The difference image (bottom) indicates whether the cell appears brighter when hybridized to readout probe “0” (red) or “1” (gray). (c) Two-dimensional histogram of normalized fluorescence intensities for readout “0” and readout “1” of bit 1 for all cells. The intensities are normalized to the median values of all cells. The dotted line depicts the threshold used for eliminating cells that appear dim in both readouts. (d) Orange: The percent of decoded barcodes that match valid barcodes present in the library as a function of the readout intensity threshold used to eliminate dim cells. Magenta: The number of cells above the readout intensity threshold. The dotted line corresponds with the threshold shown in (c). (e) Fluorescence images of mTagBFP2 and post-activation mMaple3 of the same region as (b). (f) Histograms of median mMaple3 intensity normalized to mTagBFP intensity for barcodes associated with the mMaple3-mTagBFP2 fusion gene (mMaple3+, red) and for those associated with the mTagBFP2 gene (mMaple3-, cyan). Only barcodes that were measured in at least five cells were analyzed. All scale bars are 5 μm.
As expected, for each bit, most cells were either bright when stained with the readout probe representing the value of “1” and dim when stained with the readout probe representing “0”, or vice versa (Fig. 2b, c). However, a fraction of cells appeared relatively dark in both “1” and “0” channels. We therefore used a thresholding strategy to remove these dim cells from further analysis (Fig. 2c). After thresholding, more than 600,000 of the 1.5 million measured cells remained (Fig. 2d). We then calculated the ratio of the detected “0” and “1” signals for each bit, and used this “0”-to-“1” intensity ratio to determine the barcode sequence for each cell, calling the bit value “0” or “1” if the “0”-to-“1” intensity ratio was above or below a threshold (Online Methods). Using this approach, we found that 84% of the measured barcodes matched a valid barcode that was present in the library as determined by sequencing (Fig. 2d). Among the cells that were assigned to valid barcodes, the distribution of the “0”-to-“1” intensity ratio for each bit showed two well separated populations of cells with essentially zero overlap (Supplementary Fig. 1). More stringent intensity thresholds for each bit discarded more cells without substantial improvement to the fraction of matching barcodes (Fig. 2d).
Next, we estimate our barcode misidentification rate using two different approaches. In the first approach, we exploit the knowledge that only 4% of the possible 21-bit barcodes were present in the library due to our bottlenecking strategy. Hence, 96% of the barcode reading errors generated barcodes that were not present in the library, thus not resulting in genotype misidentification; only 4% of the barcode reading errors generated valid barcodes present in the library, which would give rise to genotype misidentification. Based on the observation that 16% of the measured barcodes did not match the valid barcodes, we estimate our genotype misidentification rate to be < 1%. In the second approach, we verify our barcode measurement accuracy by taking the genotype determined from the phenotype measurement (presence and absence of mMaple3) as the ground truth and comparing this assignment to the genotype determined from the barcode measurement (Fig. 2e, f). We found that < 1% of genotype assignments disagree, which also suggests a <1% barcode misidentification rate. A more detailed description of these error estimates can be found in the Online Methods.
To demonstrate the power of our approach for screening large libraries of genetic variants, we screened for improved variants of a fluorescent protein, YFAST. YFAST is not itself fluorescent but becomes fluorescent upon binding to an exogenous, GFP-like chromophore, such as HMBR (Fig. 3a)13. We observed that YFAST exhibited complex and reversible photobleaching behavior: (1) the photobleaching of YFAST shows a biphasic behavior with one decay component much faster than the other; and (2) after the illumination was stopped, the fluorescence of YFAST rapidly recovered to a substantial extent (Supplementary Fig. 2).
Fig. 3. Screening YFAST mutants for improved brightness and photostability.
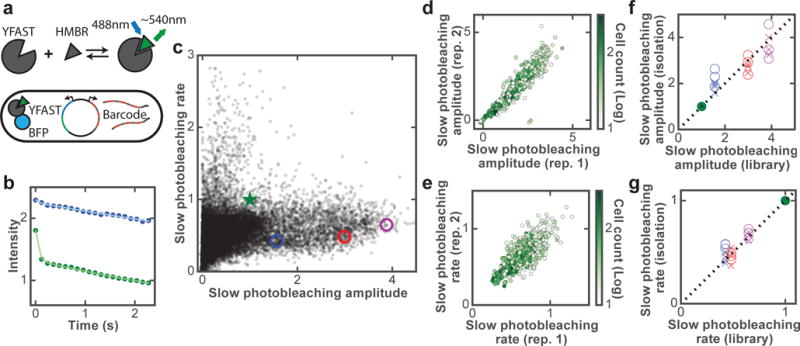
(a) Schematic of YFAST library design. (b) Photobleaching curves of the original YFAST (green circles) and a mutant (blue circles) measured from single cells in the library measurement with fits to a double exponential decay (solid lines). (c) Amplitudes and rate constants of the slow bleaching component for YFAST variants in all screened libraries. Results of the original YFAST and three selected mutants are indicated by the green star and colored circles, respectively. In the event that YFAST displays more complicated photobleaching kinetics beyond our measurement duration, the rate constants reported here should be considered the initial decay rates of the slow component. (d-e) Amplitude (d) and rate constant (e) of the slow bleaching component for two replicate library measurements containing a subset of mutants. Each point in (c-e) represents the median values of all cells corresponding to each mutant and only mutants containing at least 10 imaged cells are depicted. Furthermore, (e) only contains mutants with a slow amplitude that is at least half of that of the original YFAST. (f-g) Amplitudes (f) and rate constants (g) of the slow bleaching component for the three selected mutants (blue, red, purple) measured in isolation versus the library measurement results. Data from multiple technical replicates of isolation measurements conducted at the library-screen time resolution (120 ms; crosses) or at 4-ms time resolution (circles) are shown. In all panels, amplitudes and rate constants are normalized to those of the original YFAST (green).
We thus sought to identify YFAST mutants that are both brighter and more photostable. Specifically, because of the biphasic photobleaching behavior, we screened for mutants that exhibit a relatively large amplitude of the slow decay component as compared to the original YFAST, and preferably also with a slower decay rate of this component. We note that while brightness is a property that can be measured via simple screening methods such as FACS, screening for photobleaching kinetics requires a time-resolved measurement during screening, and, thus, would benefit from our image-based screening approach. To measure the brightness of different YFAST variants while controlling for potential variations in the expression level, we fused YFAST variants to mTagBFP2 and normalized the measured brightness of YFAST to that of mTagBFP2 for each cell (Fig. 3a). To characterize the photobleaching kinetics of YFAST variants, we measured the intensity decrease over time upon 488-nm illumination (Fig. 3b) and independently determined the background level (see Online Methods). For each mutant, we determined the two key parameters described earlier, the amplitude and rate constant of the slow bleaching component, as well as the apparent amplitude of the fast bleaching component at our 120-ms time resolution (Online Methods).
In total, we screened ~20 million cells containing ~60,000 YFAST mutants and ~160,000 barcodes (See Online Methods for the library design). We grouped cells based on the genotypes (YFAST mutants) measured and computed the median values of the three quantities mentioned above for each mutant (Fig. 3c and Supplementary Fig. 3a). We replicated the screen for a subset of the YFAST variants and found that the measured amplitudes and rate constants were reproducible between these replicate measurements (Fig. 3d, e and Supplementary Fig. 3b).
To further test the accuracy of our library measurements, we selected a few improved variants that show both larger amplitudes and slower rate constants of the slow bleaching component (Supplementary Table 2), and measured their properties in isolated clones. The results from these isolated mutant measurements are quantitatively comparable to the results that we obtained from the library screen for all three measured quantities (Fig. 3f, g and Supplementary Fig. 3c). This agreement indicates that the massive scale of parallelization in our library measurement did not cause a substantial reduction in the measurement accuracy. Because of the limited time resolution (120 ms) of our measurements, the near-zero values observed for the apparent amplitudes of the fast component for these mutants could be because the fast component was indeed diminished or because the decay rate of this component became much faster for these mutants, or both. To test these scenarios, we performed measurements of these mutants at a faster time resolution (4 ms) and found that the fast amplitudes of these mutants were indeed much smaller that of the original YFAST and, in addition, the decay rates of the fast component also became substantially faster (Supplementary Fig. 3d-f). The combined effects of these changes produced the effective elimination of the fast component at 120-ms time resolution. As expected, the amplitudes and rate constants of the slow component obtained with the increased (4-ms) time resolution still agree with the results from our library screen measurements conducted at lower time-resolution (Fig. 3f, g).
Because a standard fluorescence microscope is used for both phenotype and genotype measurements in our methods, we envision that this method can be extended, with simple adaptations, to measure a broad range of cellular phenotypes in response to a wide variety of genetic variations, ranging from mutations of single proteins to the inhibitions and activations of genes by CRISPR or RNAi. We thus expect that this high-throughput, image-based screening method can be applied broadly to improve existing properties or identify new properties of proteins and nucleic acids, as well as to decipher the roles of genes on cellular behaviors at the genomic scale.
Online Methods
Barcode library assembly
The barcode library consists of a set of plasmids, each containing a DNA barcode sequence that encodes a RNA designed to represent a single N-bit binary word. Every barcode in the library has N readout sequences, one corresponding to each bit, designed to be read out by hybridizing fluorescent probes with the complementary sequence. For each bit position, we assigned one 20-mer sequence to encode a value of “0” and another 20-mer sequence to encode a value of “1”. To increase the rate of hybridization, these encoding sequences were constructed from a three-letter nucleotide alphabet, one with only A, T, and C, in order to destabilize potential secondary structures20. The utilized sequences were drawn from those previously used for MERFISH15 with additional sequences designed using approaches described previously15. For each barcode, the bits are concatenated with a single G separating each. Although 22 bits are present in the barcode set that was constructed here, to reduce the number of hybridization rounds, experiments were conducted by reading out either 21 or 18 of the possible bits, depending on the library size.
We assembled this barcode library by ligating a mixture of short, overlapping oligonucleotides, each representing a pair of adjacent bits. For each pair of adjacent bits, there are four unique combinations of bit values (“00”, “01”, “10”, and “11”). Each corresponding sequence was synthesized as a single-stranded oligo. These oligos were then ligated to form complete, double-stranded barcodes that contain concatenated sequences of all bits with all possible bit values. For the ligation step, all oligos were mixed and diluted so that each oligo was present at a concentration of 100 nM. The mixture was phosphorylated by incubating with T4 polynucleotide kinase (16 μL oligo mixture, 2 μL T4 ligase buffer, 2 μL PNK [NEB, M0201S]) at 37 °C for 30 minutes and ligated by adding 1 μL T4 ligase (NEB, M0202S) and incubating for 1 hour at room temperature.
To prepare a plasmid library containing these barcode sequences under the control of the lpp promoter, we diluted the ligation product 10-fold and amplified it by limited-cycle PCR on a Bio-Rad CFX96 using Phusion polymerase (NEB, M0531S0) and EvaGreen (Biotium, 3100). The PCR product was run in an agarose gel, and the band of the expected length was extracted and purified (Zymo Zymoclean Gel DNA Recovery Kit, D4002). The purified product was inserted by isothermal assembly21 for 1 hour at 50 °C (NEB NEBuilder HiFi DNA Assembly Master Mix, E2621L) into a plasmid backbone fragment containing the colE1 origin, the ampicillin resistance gene, and other elements taken from the pZ series of plasmids22. The assembled plasmids were purified (Zymo DNA Clean and Concentration, D4003), eluted into 6 μL water, mixed with 10 μL of electro-competent E. coli on ice (NEB, C2986K), and electroporated using an Amaxa Nucleofector II. Immediately after electroporation, 1 mL SOC was added and the culture was incubated at 37 °C on a shaker for one hour. Subsequently, the SOC culture was diluted into 50 mL of LB (Teknova, L8000) supplemented with 0.1 mg/mL carbenicillin (ThermoFisher, 10177-012) and placed on the shaker at 37 °C overnight. The following day, the culture was miniprepped (Zymo Zyppy Plasmid Miniprep Kit, D4019), yielding the complete barcode library.
Assembling protein mutant libraries
To create a library of mutant proteins, short nucleotide sequences containing regions of the protein with the desired mutations were synthesized as complex oligonucleotide pools. To then create the desired mutant genes from these pools, we amplified the pool and its corresponding expression plasmid via limited cycle PCR and assembled these fragments using isothermal assembly21. The expression backbone was derived from the colE1 origin and the chloramphenicol resistance gene from the pZ series of plasmids22. Oligo pool synthesis is prone to deletions, which could lead to frameshift mutations that produce non-viable proteins. To remove these variants prior to measurement, the protein variants were translationally fused upstream to the chloramphenicol resistance protein. These constructs were electroporated into E. coli, as described above, and these cultures grown in the presence of chloramphenicol to select only for protein variants that did not have frame-shift mutations and which could, thus, translate competent chloramphenicol resistance. These plasmids were re-isolated via plasmid miniprep and the genetic variants extracted via PCR prior to combination with the barcode library.
Merging mutation libraries with the barcode library
To merge a mutant library with the barcode library, the corresponding halves of each plasmid library were amplified by limited-cycle PCR. Of note, the forward primer for amplifying the barcode library contained 20 random nucleotides so that each assembled plasmid contained a 20-mer unique molecular identifier (UMI)23,24. Also, the protein mutant half contained the plasmid’s replication origin (colE1) while the barcode half contained the ampicillin resistance gene ensuring that only plasmids containing both halves were competent. The two halves were assembled by isothermal assembly and transfected into electrocompetent E. coli as described earlier. After incubating in SOC for 1 hour at 37 °C, the culture was again diluted into 50 mL LB and grown until it reached an optical density at 600 nm (OD600) of ~1. To limit the possibility that a single bacterium had taken up more than one plasmid, plasmids were extracted again from this culture and reinserted at a concentration where the number of E. coli cells significantly outnumbered the number of plasmids. Specifically, 2 μL of the plasmid library at 100pg/μL was re-electroporated into 10 μL of fresh electro-competent E. coli. This culture was then grown and diluted to a concentration of ~1000 cells/μL by using the OD600 to determine the number of cells in the culture and, thus, the appropriate dilution. From the diluted culture, a volume containing the desired number of cells, and hence the desired number of unique barcode-mutant pairs, was inoculated into a new culture. This culture was incubated at 37 °C overnight and the following day it was archived for future imaging experiments by diluting 1:1 in 50% glycerol (Teknova, G1796), separating into 100 μL aliquots, and storing at −80 °C. The remaining culture was mini-prepped to use as a PCR template for constructing the barcode to genotype lookup table.
Constructing the barcode-to-genotype lookup table
Since barcodes and gene variants were assembled randomly, next generation sequencing was used to construct a look-up table that links barcodes to their corresponding gene variant. The total length of the combined sequence of the gene variant and the barcode exceeded the read length of the sequencing platform used (Illumina MiSeq). To circumvent this challenge, multiple fragments were extracted from each library, sequenced independently and grouped computationally using the UMI.
The mini-prepped libraries were prepared for sequencing by two sequential limited-cycle PCRs. The first PCR extracted the desired region while adding the sequencing priming regions, and the second PCR added multiplexing indices and the Illumina adapter sequences. Between PCRs, the product was purified in an agarose gel and the final product was gel purified prior to sequencing.
For each sequencing read, the corresponding barcode or gene variant sequence was extracted. The reads were then grouped by common UMI, and the most frequently occurring barcode and gene variant seen for each UMI was assigned to that UMI, constructing the barcode-to-gene variant lookup table for every variant in the library. Any ambiguous barcode (i.e. a barcode assigned to more than one genetic variant) was excluded from further analysis. This analysis was conducted in custom software written in Matlab.
Library design of YFAST variants
Since YFAST is a recently developed fluorescent protein, the consequences of mutating different regions of the protein are not well characterized in the literature. Hence, we began our screen by concurrently designing libraries following two distinct strategies. In the first strategy, we took a structurally naïve view and constructed a library (library type 1, LT1) that consists of mutants corresponding to all possible single amino acid substitutions, insertions, and deletions at each location within YFAST. The second strategy made use of structural information of the YFAST precursor, Photoactive Yellow Protein (PYP) (PDB: 1NWZ)25 to target residues adjacent to the chromophore (library type 2, LT2-1), introducing up to 6 amino-acid substitutions per mutant. We screened these libraries using our screening method. Since many of the mutants in LT2-1 appeared dark, we refined the selection of mutations by redesigning the oligo pool to only include those amino-acid substitutions that appeared bright with relatively high frequencies in the LT2-1 library and created another library (LT2-2) that combined these substitutions, containing up to 6 substitutions per mutant. We then screened this library with our method as well. We then created a library (library type 3, LT3) by combining mutations found to have favorable brightness and photostability (i.e. relatively large amplitude of the slow bleaching component) in LT1 with those mutations found to have favorable brightness and photostability in all LT2. Each variant in LT3 contains up to 10 mutations. We then screened LT3 and identified a mutant with 6 amino acid substitutions that is particularly photostable with a large amplitude of the slow bleaching component and a nearly eliminated the fast component at our library measurement time resolution. Next, to further improve the fluorescent properties of this mutant, we created a new library (library type 4, LT4) that contained all possible single amino acid substitution, insertion, and deletion at every residue of this mutant. Finally, based on the screening results of LT4, we created library type 5 (LT5) by splitting the entire protein sequence into 6 regions, selecting LT4 mutations with favorable brightness and photostability in each region, and creating all possible combinations of these mutations. LT5 contains 6-12 mutations per library member.
Some of the above libraries were constructed and measured concurrently while we were developing and optimizing our screening protocol. Therefore, we re-measured all of the libraries again, by mixing them into pools containing ~25,000 barcodes each. Instead of combining all libraries into a single pool and measuring a very large number of cells in a single screen over a long time, we opted to split the measurements into smaller pools and measured 1-2 million cells per experiment. Since the phenotype accuracy increases with the number of cells measured, we also included the results from the earlier measurements of individual libraries that were performed using the optimize protocol. Fig. 3 and Supplementary Fig. 3 contain results from all library measurements performed with the optimized protocol.
Phenotype and barcode imaging
Each library was prepared for imaging by thawing the 100 μL aliquot from −80 °C to room temperature and diluting into 2 mL LB supplemented with 0.1 mg/mL carbenicillin. Imaging coverslips (Bioptechs, 0420-0323-2) in 60-mm-diameter cell culture dishes were prepared by covering them in 1% polyethylenimine (Sigma-Aldrich, P3143-500ML) in water for 30 minutes followed by a single wash with phosphate buffered saline (PBS). The E. coli culture was diluted 10-fold into PBS, poured into the culture dish, and spun at 100g for 5 minutes to adhere cells to the surface.
The sample coverslip was assembled into a Bioptech’s FCS2 flow chamber. A peristaltic pump (Gilson, MINIPULS 3) pulled liquid through the chamber while three computer-controlled valves (Hamilton, MVP and HVXM 8-5) were used to select the input fluid. The sample was imaged on a custom microscope built around a Nikon Ti-U microscope body with a Nikon CFI Plan Apo Lambda 60× oil immersion objective with 1.4 NA. Illumination was provided at 405, 488, 560, 647, and 750 nm using solid-state single-mode lasers (Coherent, Obis 405nm LX 200mW; Coherent, Genesis MX488-1000; MPB Communications, 2RU-VFL-P-2000-560-B1R, MPB Communication, 2RU-VFL-P-1500-647-B1R; and MPB Communications, 2RU-VFL-P-500-750-B1R) in addition to the overhead halogen lamp for bright field illumination. The Gaussian profile from the lasers was transformed into a top-hat profile using a refractive beam shaper (Newport, GBS-AR14). The intensity of the 488-, 560-, and 647-nm lasers was controlled by an acousto-optic tunable-filter (AOTF), the 405-nm laser was modulated by a direct digital signal, and the 750-nm laser and overhead lamp were switched by mechanical shutters. The excitation illumination was separated from the emission using a custom dichroic (Chroma, zy405/488/561/647/752RP-UF1) and emission filter (Chroma, ZET405/488/461/647-656/752m). The emission was imaged onto an Andor iXon+ 888 EMCCD camera. During acquisition, the sample was translated using a motorized XY stage (Ludl, BioPrecision2) and kept in focus using a home-built autofocus system.
Phenotype measurements were conducted immediately after cells were deposited onto the coverslip, inserted into the flow chamber, and immersed in PBS. For imaging E. coli cells expressing mMaple3-mTagBFP2 fusion or mTagBFP2 alone, an image was first acquired for 1 frame with 405-nm illumination to excite mTagBFP2 at a frame rate of 8.4 Hz (120 ms), followed by illumination with 405-nm light for 30 additional frames at 8.4 Hz to photoactivate mMaple3. Then an image was acquired with 560-nm illumination for 1 frame to detect mMaple3 fluorescence. For imaging E. coli cells expressing the YFAST mutants, images were first acquired in the absence of the chromophore with 405-nm illumination for 1 frame to measure the mTagBFP2 fluorescence to determine the position of each cell followed by an image with bright-field illumination for alignment between multiple imaging rounds. Then 10 μM of the chromophore HMBR (synthesized as described previously13) in PBS was flowed over the cells and a fluorescence image was acquired with 488-nm illumination for 1 frame to measure YFAST intensity, 405-nm illumination for 1 frame to measure mTagBFP2 intensity, and a bright-field image was acquired again for alignment, followed by at least 20 frames at 8.4 Hz with constant 488-nm illumination to measure the decrease in intensity upon photobleaching. Since 8.4 Hz is the full field frame rate of the camera that we used, increasing the time resolution would require imaging a smaller field of view per frame and hence a reduction in the measurement throughput. Images were acquired at thousands of locations in the sample, each corresponding to a ~200×200 μm2 field-of-view. All fields were imaged prior to the addition of the chromophore to determine the position of each cell, and then after the chromophore was added, all of the subsequent exposure sequence described above was completed at each field prior to moving to the next. The illumination intensities at the back-focal plane used in these experiments were 1 W/cm2, 3 W/cm2, and 10 W/cm2 for the 405-nm, 488-nm, and 561-nm lasers, respectively. Following the phenotype measurement, the cells were fixed by incubation for 30 minutes in a mixture of methanol and acetone at a 4:1 ratio for fast hybridization to RNA26. To prevent salts from precipitating and clogging the flow system, water was flowed before and after the fixation mixture. Once fixed, the cells were washed in 2× Saline Sodium Chloride (SSC) and hybridizations for MERFISH imaging were started.
To determine the RNA barcode expressed within each cell, we performed multiple rounds of hybridizations. For each hybridization round, the sample was incubated for 30 minutes in hybridization buffer [2×SSC; 5% w/v dextran sulfate (EMD Millipore, 3730-100ML), 5% w/v ethylene carbonate (Sigma-Aldrich, E26258-500G), 0.05% w/v yeast tRNA, and 0.1% v/v Murine RNase inhibitor (NEB, M0314L)] with a mixture of readout probes labeled with either ATTO565, Cy5, or Alexa750 (Bio-Synthesis Inc) each at a concentration of 10 nM. In the readout probes, the dyes were linked to the oligonucleotides through a disulfide bond15. Then, the hybridization buffer was replaced by an oxygen-scavenging buffer for imaging27 [2×SSC; 50 mM TrisHCl pH 8, 10% w/v glucose (Sigma-Aldrich, G8270), 2 mM Trolox (Sigma-Aldrich, 238813), 0.5 mg/mL glucose oxidase (Sigma-Aldrich, G2133), and 40 μg/mL catalase (Sigma-Aldrich, C100-500mg)]. Each position in the flow cell was imaged with 750-, 647-, and 560-nm illumination from longest to shortest wavelength followed by bright-field illumination for alignment before continuing to the next location. Following the imaging of all regions, the disulfide bonds linking the dyes to the oligonucleotides in the readout probes were cleaved by incubating the sample in 50 mM tris (2-carboxyethyl)phosphine (TCEP; Sigma-Aldrich, 646547-10X1ML) in 2×SSC for 15 minutes. The sample was then rinsed in 2×SSC and the next hybridization round started. For each round of hybridization, three readout probes with spectrally discernable dyes (ATTO565, Cy5, and Alexa750) were hybridized simultaneously as described above (see Supplementary Table 1). Altogether, with 14 hybridization rounds, all 42 readouts corresponding to 21 bits were measured in 40 hours. For smaller libraries, the imaging area was reduced, and the number of hybridization rounds was decreased to 12 (for 18-bit readout), reducing the measurement time to 22 hours.
Image analysis
To correct for residual illumination variations across the camera, a flat-field correction was performed as follows. Every image was divided by the mean intensity image for all images with the given illumination color. Then, the images for different rounds corresponding to the same region were aligned using the image acquired under bright field illumination by up-sampled cross-correlation, creating a normalized image stack of all images at each position in the flow chamber. If the radial power spectral density of any given bright field image did not contain sufficient high frequency power, the image was designated as out-of-focus and all images for the corresponding region were excluded from further analysis.
To extract cell intensities, the edges of each cell were detected using the Canny edge detection algorithm on the image acquired with 405-nm illumination for mTagBFP2 imaging. The edges that formed closed boundaries were filled in and closed regions of pixels were extracted. If a given closed pixel region had a filled area of more than 20 pixels and the ratio of the filled area to the area of the convex hull was greater than 0.9, it was classified as a cell. To increase the cell detection efficiency, the detected cells were then removed from the binary image, the image was dilated, filled, and eroded and cells were extracted again. This allowed cells where gaps exist in the detected edges to still be detected. For each cell, the mean intensity was extracted for the corresponding pixels in every image.
From the cell intensities, the phenotypes and barcodes were calculated. For each measured readout sequence, the measured intensity was normalized by subtracting the minimum and dividing by the median signal observed for that readout sequence across all cells. To determine whether a barcode contained a “1” or a “0” at each bit, the measured intensities of the “1” readout sequence and the “0” readout sequence for that bit were compared. Specifically, a threshold was selected on the ratio of these two values, called the “0”-to-“1” intensity ratio. If the “0”-to-“1” intensity ratio was above the threshold, the bit was called as a “0”. Otherwise, the bit was called as a “1”. Because the “1” and “0” readout sequences were measured in different hybridization rounds and we observed variation in staining quality between rounds, it was necessary to optimize this threshold for each bit individually. This optimization was performed by randomly selecting 150 barcodes (a training set) from the set of known barcodes that were determined to be present in the library by sequencing. An initial set of thresholds was selected and the fraction of cells matching these barcodes was determined. The threshold for each bit was then varied independently to identify the threshold set that maximizes this fraction. This optimized threshold set was then used for determining the bit values for all cells.
Once the barcode was determined for each cell, cells were grouped by barcode and the median of the various phenotype values was computed to determine the measured phenotype for the genotype corresponding to that barcode. For the mMaple3 measurement, the normalized brightness was determined from the ratio of the mMaple3 intensity under 560-nm illumination to the mTagBFP2 intensity under 405-nm illumination, as discussed above. For YFAST measurements, the normalized intensity was determined by the ratio of the YFAST fluorescence intensities under 488-nm illumination in the presence of the YFAST chromophore HMBR to the mTagBFP2 fluorescence intensities under 405-nm illumination. To account for the fluorescence background present in E. coli upon 488-nm illumination, the background was independently determined and subtracted before calculating the fluorescence ratio. The background was estimated by calculating the median intensity of all cells upon 488-nm illumination predicted to contain a non-fluorescent YFAST mutant. Specifically, cells, grouped by barcode, were assigned to the non-fluorescent population if the Pearson correlation coefficient between the fluorescence intensity measured under 488-nm illumination (YFAST channel) and those measured under 405-nm illumination (mTagBFP2 channel) for the grouped cells fell below a threshold of 0.2. Since the YFAST variant is translationally fused to mTagBFP2, when the two intensities are uncorrelated, it suggests that the number of YFAST proteins in the cells does not affect the brightness of the cell and hence the YFAST associated with that barcode should be dark.
Our initial high-time resolution (4-ms) measurements of the original YFAST variant revealed a biphasic decay of fluorescence with time (Supplementary Fig. 2). To quantify this behavior, we fit the background-subtracted photobleaching curve, , to the sum of two exponentials:
where and represent the amplitude and decay rate constant for the fast photobleaching component and and represent the corresponding values for the slow photobleaching component. These fits of the original YFAST showed that the decay rate constants for the fast and slow components were ~10 s−1and ~0.1 s−1, respectively, under our illumination intensity.
This double-exponential decay function was also used to characterize our library screen measurements. However, to increase the throughput of our screens, we utilized the full imaging frame of our camera, which required the use of a slower frame rate (8.4 Hz, ~120 ms). This frame rate was comparable to the decay rate observed for the fast component of the original YFAST variant; thus, we did not anticipate that the rate constant associated with the fast component would be well constrained by this double-exponential fit. To address this problem, we initially fixed the rate constant of the fast component to the value determined from the original YFAST and allowed the other three parameters to vary in the fit. The time resolution of our library measurements was much higher than the decay time constant of the slow component; thus, the parameters associated with the slow component, and B, were well constrained by this fit—a point confirmed by our observation that and B did not change appreciable (by <0.5%) when we varied the fixed value of over a wide range or let also be a fitting parameter. Furthermore, we anticipate that the time resolution, 120 ms, and duration, 2.5 s, of our library measurements, plus the independent determination of the background level (discussed above), should allow and B to be determined reliably. Though, we cannot rule out the possibility that beyond our measurement duration, YFAST displays a more complicated photobleaching kinetics with more decay rate constants, in which case, our reported rate constant B for the slow component should be considered the initial decay rate of this component. To estimate the fast component amplitude, , we utilized the well constrained value of the slow component amplitude, . Specifically, we calculated from the difference of the initial brightness of each variant ( ) and the fit value for the slow component amplitude, . Because of the limited time resolution of our library screen, we did not extract the rate constant of the fast bleaching component, and we note that the apparent amplitude that we determined for the fast bleaching component is likely to systematically underestimate this amplitude. Nonetheless, it should still provide useful information for future imaging experiments using the YFAST variants at ~100 ms or slower time resolution.
The reported values for the slow component amplitude and decay rate are normalized to the corresponding values measured for the original YFAST, unless otherwise mentioned. The fast photobleaching component amplitude was not normalized in this fashion but rather was reported as the fraction of the total brightness, i.e. [ ], which we termed the fractional fast photobleaching amplitude.
This analysis was conducted in custom software written in Python.
Estimating barcode misidentification rate
We estimate our barcode misidentification rate using two different approaches. In the first approach, we estimate our error rate in barcode identification using the observation that 16% of the measured barcodes did not match the valid barcodes present in the library. We note that there are two types of errors. If the measurement error produces an invalid barcode that was not present in the library (type I error), this barcode read out error would be detected, and hence this type of error would not affect our accuracy in genotype identification. If, however, the measurement error produces a valid barcode that was present in the library (type II error), this error would not be detected and hence would cause a genotype misidentification. We recognize that the frequency of type I error occurrence is the product of the frequency of barcode error occurrence and the fraction of all possible 21-bit binary barcodes that are not present in the library. Since the frequency of type I error occurrence was measured to be 16% and 96% of all possible barcodes were not present in the library, the frequency of barcode error occurrence should be 16.7%. The frequency of type II error occurrence, which is the product of the frequency of barcode error occurrence (16.7%) and the fraction of all possible barcodes that are present in the library (4%), should then be only 0.67%. Hence our genotype misidentification rate was < 1%. This low error rate illustrates the benefit of our barcode bottlenecking strategy.
In the second approach, we verify our barcode measurement accuracy by taking the genotype determined from the phenotype measurement (presence and absence of mMaple3) as the ground truth and comparing this assignment to the genotype determined from the barcode measurement. To determine the phenotype of each cell, we normalized the mMaple3 fluorescence intensity of the cell to the mTagBFP2 fluorescence intensity to remove differences in protein expression levels. We then calculated the median of the normalized brightness for all cells assigned to the same barcode and constructed a histogram of this normalized mMaple3 brightness for all barcodes associated with the mMaple3+ genotype as well as a histogram for all barcodes associated with the mMaple3- genotype. As expected, these two histograms are well separated with only a very small overlap (Fig. 2f). Next, we determined the fraction of barcodes that were misidentified by assuming that the overlap between the two histograms were solely due to barcode misidentification. To this end, we set a threshold based on the intersection point of the two histograms and assigned all cells with normalized mMaple3 brightness larger (or smaller) than this threshold as having a mMaple3+ (or mMaple3-) genotype. We then compared this genotype assignment to the genotype assignment based on the measured barcode and found that < 1% of genotype assignments disagree, which also suggests a <1% barcode misidentification rate. We note that this error rate is likely an overestimate since in addition to barcode misidentification, the natural spread in the intensity distribution of cells in each group should also contribute to the overlap of these distributions.
Code, data and protocol availability
The Python code used for image analysis and the Matlab code for sequence analysis is available at github.com/ZhuangLab as well as in the Supplementary Software. The data that support the findings of this study are available from the corresponding authors upon request. A detailed step-by-step protocol is accessible via Protocol Exchange28.
A summary of the experimental design and software availability can be found in the Life Sciences Reporting Summary.
Supplementary Material
Acknowledgments
We thank H. Babcock for instrumentation advice and many readers for participating in the discussion of the preprint of this paper on bioRxiv (doi.org/10.1101/143966). This work was supported in part by the NIH. X.Z. is an HHMI investigator.
Footnotes
Author contributions
G.E., J.R.M., and X.Z. conceived the study and designed the experiments. G.E. performed experiments and analyzed data. G.E., J.R.M., and X.Z. interpreted the data and wrote the manuscript.
Competing financial interests
G.E., J.R.M., and X.Z. are inventors on a patent applied for by Harvard University that covers the screening method described here.
References
- 1.Cadwell RC, Joyce GF. Randomization of genes by PCR mutagenesis. PCR Methods Appl. 1992;2:28–33. doi: 10.1101/gr.2.1.28. [DOI] [PubMed] [Google Scholar]
- 2.Kosuri S, Church GM. Large-scale de novo DNA synthesis: technologies and applications. Nat Methods. 2014;11:499–507. doi: 10.1038/nmeth.2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang J, Campbell RE, Ting AY, Tsien RY. Creating new fluorescent probes for cell biology. Nat Rev Mol Cell Biol. 2002;3:906–918. doi: 10.1038/nrm976. [DOI] [PubMed] [Google Scholar]
- 4.Grotjohann T, et al. Diffraction-unlimited all-optical imaging and writing with a photochromic GFP. Nature. 2011;478:204–208. doi: 10.1038/nature10497. [DOI] [PubMed] [Google Scholar]
- 5.Brakemann T, et al. A reversibly photoswitchable GFP-like protein with fluorescence excitation decoupled from switching. Nat Biotechnol. 2011;29:942–947. doi: 10.1038/nbt.1952. [DOI] [PubMed] [Google Scholar]
- 6.Shaner NC, et al. Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat Methods. 2008;5:545–551. doi: 10.1038/nmeth.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davis LM, Lubbeck JL, Dean KM, Palmer AE, Jimenez R. Microfluidic cell sorter for use in developing red fluorescent proteins with improved photostability. Lab Chip. 2013;13:2320. doi: 10.1039/c3lc50191d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dean KM, et al. Microfluidics-based selection of red-fluorescent proteins with decreased rates of photobleaching. Integr Biol. 2015;7:263–273. doi: 10.1039/c4ib00251b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shalem O, Sanjana NE, Zhang F. High-throughput functional genomics using CRISPR-Cas9. Nat Rev Genet. 2015;16:299–311. doi: 10.1038/nrg3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dixit A, et al. Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell. 2016;167:1853–1866.e17. doi: 10.1016/j.cell.2016.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adamson B, et al. A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell. 2016;167:1867–1882.e21. doi: 10.1016/j.cell.2016.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jaitin DA, et al. Dissecting Immune Circuits by Linking CRISPR-Pooled Screens with Single-Cell RNA-Seq. Cell. 2016;167:1883–1896.e15. doi: 10.1016/j.cell.2016.11.039. [DOI] [PubMed] [Google Scholar]
- 13.Plamont MA, et al. Small fluorescence-activating and absorption-shifting tag for tunable protein imaging in vivo. Proc Natl Acad Sci U S A. 2016;113:497–502. doi: 10.1073/pnas.1513094113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen KH, Boettiger AN, Moffitt JR, Wang S, Zhuang X. Spatially resolved, highly multiplexed RNA profiling in single cells. Science (80-) 2015;348:aaa6090–aaa6090. doi: 10.1126/science.aaa6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moffitt JR, et al. High-throughput single-cell gene-expression profiling with multiplexed error-robust fluorescence in situ hybridization. Proc Natl Acad Sci. 2016;113:11046–11051. doi: 10.1073/pnas.1612826113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Femino AM. Visualization of single RNA transcripts in situ. Science (80-) 1998;280:585–590. doi: 10.1126/science.280.5363.585. [DOI] [PubMed] [Google Scholar]
- 17.Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods. 2008;5:877–879. doi: 10.1038/nmeth.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Subach OM, Cranfill PJ, Davidson MW, Verkhusha VV. An enhanced monomeric blue fluorescent protein with the high chemical stability of the chromophore. PLoS One. 2011;6:e28674. doi: 10.1371/journal.pone.0028674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang S, Moffitt JR, Dempsey GT, Xie XS, Zhuang X. Characterization and development of photoactivatable fluorescent proteins for single-molecule-based superresolution imaging. Proc Natl Acad Sci. 2014;111:8452–8457. doi: 10.1073/pnas.1406593111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Online Methods References
- 20.Zhang Z, Revyakin A, Grimm JB, Lavis LD, Tjian R. Single-molecule tracking of the transcription cycle by sub-second RNA detection. Elife. 2014;3:e01775. doi: 10.7554/eLife.01775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gibson DG, et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 22.Lutz R, Bujard H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 1997;25:1203–10. doi: 10.1093/nar/25.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kivioja T, et al. Counting absolute numbers of molecules using unique molecular identifiers. Nat Methods. 2011;9:72–74. doi: 10.1038/nmeth.1778. [DOI] [PubMed] [Google Scholar]
- 24.Shiroguchi K, Jia TZ, Sims PA, Xie XS. Digital RNA sequencing minimizes sequence-dependent bias and amplification noise with optimized single-molecule barcodes. Proc Natl Acad Sci. 2012;109:1347–1352. doi: 10.1073/pnas.1118018109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Getzoff ED, Gutwin KN, Genick UK. Anticipatory active-site motions and chromophore distortion prime photoreceptor PYP for light activation. Nat Struct Biol. 2003;10:663–668. doi: 10.1038/nsb958. [DOI] [PubMed] [Google Scholar]
- 26.Shaffer SM, Wu MT, Levesque MJ, Raj A. Turbo FISH: A method for rapid single molecule RNA FISH. PLoS One. 2013;8:e75120. doi: 10.1371/journal.pone.0075120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rasnik I, McKinney SA, Ha T. Nonblinking and long-lasting single-molecule fluorescence imaging. Nat Methods. 2006;3:891–893. doi: 10.1038/nmeth934. [DOI] [PubMed] [Google Scholar]
- 28.Emanuel G, Moffitt JR, Zhuang X. Protocol Exchange. doi: 10.1038/protex.2017.109. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.