

Abstract
The aim of the present study was to identify the effect of heme oxygenase (HO)-1 gene on cerebral ischemia-reperfusion injury. Sprague-Dawley rats were divided randomly into four groups: Sham group, vehicle group, empty adenovirus vector (Ad) group and recombinant HO-1 adenovirus (Ad-HO-1) transfection group. Rats in the vehicle, Ad and Ad-HO-1 groups were respectively injected with saline, Ad or Ad-HO-1 for 3 days prior to cerebral ischemia-reperfusion injury. Subsequently, the middle cerebral artery occlusion method was used to establish the model of cerebral ischemia-reperfusion injury. Following the assessment of neurological function, rats were sacrificed, and the infarction volume and apoptotic index in rat brains were measured. Furthermore, the protein expression levels of HO-1 in brain tissues were detected using western blot analysis. Results indicated that the neurological score of the Ad-HO-1 group was significantly increased compared with the Ad or vehicle groups, respectively (P<0.001). The volume of cerebral infarction and the index score of neuronal apoptosis in the vehicle and Ad groups was significantly increased compared with the Ad-HO-1 group (P<0.01). The death of neuronal cells following cerebral ischemia-reperfusion injury reduced remarkably induced by over-expression of HO-1. These findings suggest a neuroprotective role of HO-1 against brain injury induced by transient cerebral ischemia-reperfusion injury.
Keywords: heme oxygenase, cerebral ischemia, middle cerebral artery occlusion adenovirus, rat
Introduction
The research on ischemic preconditioning (IPC) has demonstrated that IPC-induced brain tolerance may be early or late, with the early phase ranging between min and h after the onset of IPC, and the late phase ranging between h and days after the onset of IPC (1). Furthermore, IPC activates several protective molecular pathways, including protein kinase C, mitogen-activated protein kinase and phosphatidylinositol 3-kinase at different time points (1). A previous study has indicated that, between 12 and 24 h after IPC, the expression of heat shock protein heme oxygenase (HO)-1 was activated in newborn rat brains, and upregulation of HO-1 expression remained for 7 days (2). A previous study also indicated that HO-1 was able to partially mediate the neuroprotective roles on IPC (3).
It is known that the rate-limiting enzyme HO is able to degrade heme in vivo and subsequently antagonize stress reactions, resulting in a protective effect against cell damage (4). The pro-oxidant heme may be subjected to oxidative degradation into the potent antioxidants carbon monoxide, biliverdin, and free iron by HO (5). Subsequently, biliverdin reductase is converted rapidly from biliverdin to bilirubin (4–6). There are two isoenzymes of HO, namely HO-1 and HO-2 (6). HO-2 gene knockout reduces oxidative stress in nerve cell damage (6). Conversely, HO-1 knockout mice are more sensitive to ischemia-reperfusion injury; however induced HO-1 overexpression may promote a protective effect (7). Furthermore, HO-1 upregulation may provide protection against damage induced by cold ischemia (7). These findings suggest that promoting the activity of HO-1, may have a potential cytoprotective effect against cell damage. In HO-1 transgenic rats, overexpression of HO-1 has been demonstrated to attenuate ischemic stroke damage induced by middle cerebral artery occlusion (MCAO) (8). It has been indicated that the activity of HO-1 with pharmacological stimulation may be a novel therapeutic target for the treatment of ischemic injury (8–10).
Gene transfer was used in the present study to investigate stroke by transducing genes into neurons (11). There are alternative approaches for expressing transgenes of interest in adult animals, and then investigating their effects on cerebral ischemia-reperfusion injury (11). One viral gene transfer method involves the use of recombinant adeno-associated viral vectors, which may deliver transgenes into neurons through a number of delivery pathways, including intramuscular (12), subcutaneous, intraneural (13), intrathecal (14,15) and intraspinal applications (16,17), direct injections into dorsal root ganglia (18), and application of virus accompanied with herpes simplex virus locality (19).
The objectives of the present study included the following: i) Identification of the role of HO-1 gene in cerebral ischemia-reperfusion injury based on the transfection method; ii) confirmation of the high expression of the HO-1 gene and its anti-apoptotic effects; and iii) to illuminate the cellular or physiological effect of recombinant adenovirus bearing HO-1 (Ad-HO-1) against ischemia-reperfusion injury of the brain, which may have a protective role against stroke or neurological disorders associated with stroke.
Materials and methods
Adenovirus vector production
The adenovirus-bacterial recombination system (AdEasy) was supplied by Dr Chen Jing from Thomas Jefferson University (Philadelphia, PA, USA), including the expression vectors pGEM-3ZF (+) and pAdEasy-1, which were labeled with green fluorescent protein (GFP). The rat HO-1 gene was constructed into adenoviral vectors in the AdEasy Vector System as described previously (20). The spleens were taken from 6 3-month-old female Wistar rats (weight, 250±25 g) and stored at −80°C. The rats were maintained within a specific pathogen free facility with a constant humidity (55±5%) and temperature (22±2°C) in a 12 h light/dark cycle with free access to food and water. Total RNA was extracted from the rats spleen cells using an RNeasy Total RNA Isolation kit (Qiagen, Inc., Valencia, CA, USA). Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) was performed and the products were used for the gene amplification of HO-1, and subsequently cloned into pAdEasy-1 vectors. The RNA (2 µg per reaction) was reverse transcribed using avian myeloblastosis virus reverse transcriptase (Promega Corporation, Madison, WI, USA) at 42°C for 70 min in the presence of 250 µM dNTPs, avian myeloblastosis reverse transcription 5X reaction buffer (250 mM Tris-HCl, 250 mM KCl, 50 mM MgCl2, 50 mM DTT and 2.5 mM spermidine; all Promega Corporation) and 1 µM oligo-p (dT)15 primer (Roche Molecular Diagnostics, Pleasanton, CA, USA). PCR amplification reactions were performed in a total volume of 20 µl containing 1.5 U Taq DNA polymerase, 2 µl 10X PCR buffer (100 mM Tris-HCl, 500 mM KCl, 15 mM MgCl2), 0.4 µl 10 mM dNTP (all Promega Corporation) and ~100 ng DNA template on a GeneAmp® PCR System 9700 (Roche Molecular Diagnostics). The thermocycling conditions were as follows: Predenaturing at 94°C for 5 min, followed by 32 cycles of denaturing at 94°C for 45 sec, annealing at the temperature appropriate for each pair of primers for 45 sec and extending at 72°C for 45 sec with a final extension at 72°C for 10 min. According to the published GeneBank sequence of rat HO-1 (NM_010442) (ncbi.nlm.nih.gov/nuccore/NM_010442.2?report=genbank), the upstream primer sequence was 5′-ATGGAGCGTCCACAGCTCGAC-3′ and the downstream primer sequence was 5′-GGGCCAACACTGCATTTACAT-3′. The HO-1 gene was synthesized by Shanghai Sangon Biological Engineering Technology & Services Co., Ltd. (Shanghai, China). Prior to use, assessments for viral concentration and titer of the pAdEasy-1 viral vectors were conducted. The concentration of virus [plaque-forming unit (pfu)/ml] was measured (21). The 50% tissue culture infection dosage (TCID50) method was used to measure virus titers. The cells were seeded into clear, flat-bottomed 96-well plates at 105 cells in 100 µl of DMEM containing 10% fetal calf serum (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA), 1% 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 1% L-glutamine, and 1% penicillin/streptomycin. The cells were added to all wells except column 10, which was left blank to separate the infected wells and the uninfected control wells. Prior to the assay an additional 80 µl of maintenance media with DMEM containing 2% FCS was added to all wells. A total of 20 µl neat or diluted virus was added to each well in the first column of the plate. The virus was then serially diluted (1:10, 20 µl into 180 µl) across the plate from columns 1 to 9 with pipette tips changed between each column. Column 10 was left empty, and columns 11 and 12 contained media only and served as controls for uninfected cells. The plates were then incubated at 37°C with 5% CO2 for 6 days. On the 6th day, 3 µl (1.5%) Neutral Red solution (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was added to every well containing cells to aid visualization and the plates were returned to the incubator for an additional 24 h. On the 7th day postinoculation, the cells were fixed with 100 µl 10% formal-saline (prepared by diluting 37% formaldehyde with PBS, pH 7.4). Following fixation for a minimum of 30 min at room temperature, all liquid was removed from the plate and each well was examined for cytopathic effect. For each dilution, the number of wells that were positive for CPE was scored. A Reed and Muench calculation was then performed to determine the 50% infectious dose, which was then scaled up to give a count of the TCID50 per ml (21). The viral titers of Ad-GFP and Ad-HO-1 were 2.0×1011 pfu/ml and 2.5×1011 pfu/ml, respectively. Briefly, the vector plasmids and ViraPower Package mixture (containing the pLP1, pLP2 and pLP/VSVG plasmids) were cotransfected into 293T cells and cultured at 37°C. At 48 h post transfection the viruses were harvested and concentrated in PBS by ultracentrifugation (for 16 h at 9,000 × g at 4°C) to 1.25×109 transducing units/ml. The virus titer kit was purchased from MellGen Laboratories, Inc. (Surrey, BC, Canada) and used according to the manufacturer's protocol (21).
Animals and adenovirus infection
Based on the National Institutes of Health Guidelines on Use of Laboratory Animals, all experiments were conducted and approved by the Second Hospital of Shanxi Medical University Committee on Animal Care (Shanxi, China).
Male Sprague-Dawley (SD) rats (n=48; weight, 240–280 g; age, 3–6 months) were purchased from the Experimental Animal Center of the Fourth Military Medical University (Xi'an, China). The rats were maintained in a specific pathogen free facility with a constant humidity (55±5%) and temperature (22±2°C) and a 12 h light/dark cycle with free access to food and water. Rats were randomly divided into 4 groups (n=6/group): Sham group, vehicle group, empty adenovirus vector (Ad) group and Ad-HO-1 transfection group. Rats in the vehicle, Ad and Ad-HO-1 groups were respectively injected with 20 µl saline, saline containing 1.25 µl empty vector adenovirus or saline containing 1 µl recombinant HO-1 adenovirus via the right lateral ventricle once daily for 3 days prior to cerebral ischemia-reperfusion injury induction. The injection site was referred to as the Paxinos and Watson rat brain stereotactic atlas (10).
Transient focal cerebral ischemia-reperfusion injury models
Rats in the vehicle group, Ad group and Ad-HO-1 group were anesthetized (3% isoflurane for induction and 1% for maintenance) and the transient focal cerebral ischemia-reperfusion injury models were induced via the MCAO method and intraluminal suture technique (22,23). The rats in the sham group did not receive MACO or intraluminal suture, however a sham operation was performed. Blood flow was reduced to 87–90% in the ipsilateral parietal cortex, which was considered as confirmation of successful occlusion. During the experiment, a laser-Doppler flowmeter (Probe 407-1; Perimed AB, Järfälla, Sweden) was used for all groups to monitor the blood flow at a maintained body temperature (37.0±0.5°C). Following occlusion for 90 min, blood flow was restored for 90 min to the ischemic region of brain and the filament was removed from lumen. All rats recovered from anesthesia prior to being returned to their cages.
Detection of GFP
Following a 24-h recovery period from MCAO, rat MCAO models were anesthetized terminally with sodium pentobarbital (60 mg/kg; intraperitoneal; Sigma-Aldrich; Merck KGaA) and saline was used for transcardial perfusion. Rat brains were harvested, and sliced into 5 coronal sections (2-mm thick) from the frontal pole, fixed in 4% formalin overnight at 30°C and stained with 0.5% w/v triphenyltetrazolium chloride (TTC) dissolved in PBS for 15 min at room temperature. Infarct areas of each slice were analyzed using the SigmaScan Pro 5 Image Analysis software (Systat Software, Inc., Point Richmond, CA, USA). Direct GFP signals were digitally photographed using an Olympus BX51 fluorescence microscope (magnification, ×200; Olympus Corporation, Tokyo, Japan). A total of 5 fields of view were randomly selected in each slice, 100 cells were randomly selected in each, and the number of fluorescent cells were counted. Subsequently, the efficiency of transfection was calculated as follows: Efficiency of transfection=number of positively stained cells/total number of cells ×100%, which was automatically calculated and exported to Image-ProPlus 6.0 (Media Cybernetics, Inc., Rockville, MD, USA) and GraphPad Prism 5.0 (GraphPad Software, Inc., La Jolla, CA, USA) for further analysis.
Western blot analysis
Total protein was extracted from cortices of SD rats in all groups using a radioimmunoprecipitation lysis buffer (Thermo Fisher Scientific, Inc.). Protein concentration was determined using the BCA Protein Assay kit (Pierce; Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol. Equal amounts of protein (50 µg/lane) were resolved by 10% SDS-PAGE and transferred to a polyvinylidene difluoride membrane (EMD Millipore, Billerica, MA, USA). The western blot assays were performed with a Mini-PROTEAN®TGX™ system (Bio-Rad Laboratories, Inc., Hercules, CA, US). A 5% skimmed milk powder in Tris-buffered saline with Tween-20 blocking solution was used for 1 h at room temperature. The membranes were incubated with β-actin (cat. no. RB-9010-R7; 1:4,000; Invitrogen; Thermo Fisher Scientific, Inc.) and anti-HO-1 (cat. no. sc-120745; 1:3,000; Santa Cruz Biotechnology, Inc., Dallas, TX, USA) primary antibodies overnight at 4°C. The membranes were incubated with horseradish peroxidase-conjugated anti-mouse immunoglobulin G secondary antibodies (cat. no. 7076; 1:5,000; Cell Signaling Technology, Inc., Danvers, MA, USA) at room temperature for 1 h. The protein bands were detected using the enhanced chemiluminescent method with ECL™ Western Blotting Detection reagents (GE Healthcare, Chicago, IL, USA) and subsequent autoradiography. The protein levels of the bands were normalized against β-actin.
Assessment of neurological deficit score (NDS)
Following a 24-h recovery period from MCAO, the NDSs of rats were examined as described previously (18) with a modified scoring system. The modified scoring system was adopted to determine the neurological functional deficit at 4 h after the MCAO. Neurological deficit score (NDS; 0–10% normal, 100% maximum deficit). The modified neurological severity score (mNSS), a composite of motor, sensory, reflex and balance tests was used to test the sensorimotor deficit. The mNSS was graded on a scale of 0–18. A normal score was 0 and the maximal deficit score was 18, 1 point was awarded for the inability to perform the task or the lack of a tested reflex. (0, no neurological deficits; 1, failure to extend right forepaw fully; 2, circling to right; 3, falling to right; 4, did not walk spontaneously and has depressed levels of consciousness).
Volume of infarct regions
At 24 h following MCAO establishment, the tissues of rat brains were sectioned as described above. Six samples from each group were used in each experiment. The volume of infarct regions was assessed using TTC staining in the dark overnight at 30°C, and images of each section were captured using an Olympus BX51 fluorescence microscope (magnification, ×200). The infarct areas of each slice were analyzed using the SigmaScan™ Pro V5.0 Image Analysis software (Systat Software, Inc., San Jose, CA, USA). The injured tissues remained unstained by TTC, as staining did not occur in areas where neuronal loss was observed. Therefore, the unstained areas (infarct tissues) were examined and analyzed with Photoshop software 7.0 (Adobe Systems, Inc., San Jose, CA, USA). The sections were used to determine the volumes of infarct region. For the ipsilateral hemisphere, the infarct regions of each brain were calculated in mm3, which was corrected for swelling in the same brain with translation on the percentage of contralateral hemisphere as described previously (24).
Terminal deoxynucleotidyl-transferase-mediated dUTP nick end (TUNEL) staining
At 24 h following MCAO, brain tissue sections were fixed in 4% formalin overnight at 30°C then stained with TUNEL (Roche Diagnostics GmbH; Mannheim, Germany) at 37°C for 1 h. The incorporated digoxigenin-conjugated nucleotides were detected using a horseradish peroxidase-conjugated anti-digoxigenin antibodies and 3,3′-diaminobenzidine. The dehydrated sections were cleared in xylene, mounted with Canada balsam and enclosed with coverslips. The sections of brain tissues (n=6 per group) in contralateral hemisphere and five fields of view were randomly selected for counting and NDSs were determined. Subsequently, brain tissue sections were dewaxed and rehydrated at 60°C for 2 h. These slides were incubated for 30 min at room temperature in a 20 µg/ml DNase-free proteinase K and rat terminal deoxynucleotidyl-transferase (both Sigma-Aldrich; Merck KGaA) reaction mix. Following rinsing with PBS 3 times for 15 min each time, these sections were treated with streptavidin horseradish peroxidase (1:200; Roche Diagnostics GmbH) at 37°C for 30 min and positive signals were enhanced with diaminobenzidine (1:200; Roche Diagnostics GmbH) at room temperature for 30 min. The tissue slide was digitally photographed using an Olympus BX51 fluorescence microscope (magnification, ×400). The apoptotic index (positively stained cells count/total number of cells ×100%) was automatically calculated and exported to GraphPad Prism 5.0 for further analysis.
Activation of caspase-3
The neuronal activity of caspase-3 was examined using a Caspase-3 Activity Assay kit (cat. no. CST 5723S; Cell Signaling Technology Inc.) according to the manufacturer's protocol. Brain tissues of the total suspension weighing 3 g were homogenized for 2 min in Mcllwain's buffer, then the homogenates were centrifuged at 3,000 g for 5 min at room temperature and the supernatants were collected.
Statistical analysis
The normally distributed continuous variables were indicated as the mean ± standard deviation. Statistical analysis was performed using GraphPad Prism 5.0. Between groups, the Student's t-test (for two group comparisons) or one-way analysis of variance with Kruskal-Wallis post hoc tests (for multiple group comparisons) were used to analyze and compare quantitative data. The data that were abnormally distributed were analyzed between groups with Kruskal-Wallis analysis of variance method. SPSS software (version 18.0; SPSS, Inc., Chicago, IL, USA) was used to analyze these statistical data. P<0.05 was considered to indicate a statistically significant difference.
Results
Overexpression of HO-1 protects against transient cerebral ischemia-reperfusion injury
MCAO establishment was considered successful when the blood flow of the rat models was decreased to 80–90% of the baseline level in the core area. Following MCAO establishment, the cerebral blood flow was not significantly different in the model groups treated with vehicle or Ad (data not shown). The total volume of infarction and infarction area of the representative brain sections at 24 h following ischemia-reperfusion injury establishment in each group are indicated in Fig. 1. The total volume of infarction in the Ad-HO-1 group was 27.31±2.913%, which was significantly decreased compared with those in the vehicle (60.36±6.750%; P<0.01) and Ad groups (61.80±7.544%; P<0.01).
Figure 1.
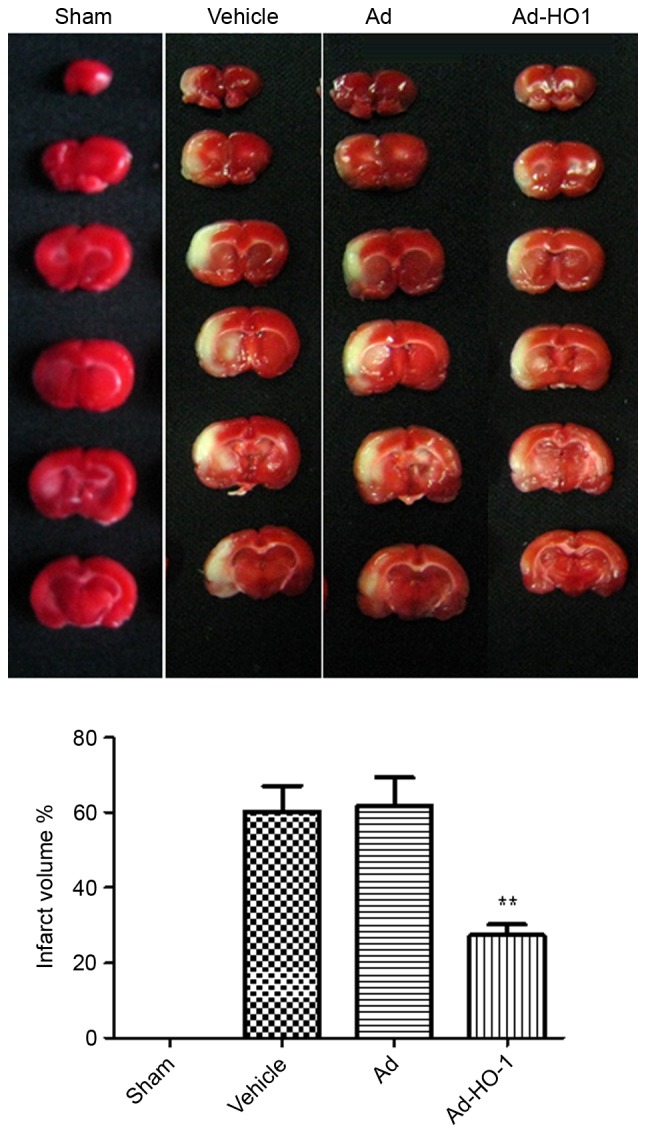
Overexpression of HO-1 attenuated infarct volume in the ipsilateral cortex following MCAO. Just after 2 h of MCAO, the blood flow was decreased to 80–90% of the baseline level in the core area, which classified the model as successful. Following 24 h of MCAO, the cerebral blood flow was not significantly different between the two model groups (vehicle and Ad groups). The total volume of infarction and infarction area of representative brain sections in each group were indicated at 24 h following ischemia. Compared with the sham group, volume of cerebral infarction in V and Ad groups were significantly increased. Over-expression of HO-1significantly attenuated infarct volume in the ipsilateral cortex. Data are presented as the mean ± standard deviation (repeats, n=6). **P<0.01 vs. vehicle and Ad groups. MCAO, middle cerebral artery occlusion; Ad, empty adenovirus vector; HO-1, heme oxygenase-1; Ad-HO-1, recombinant HO-1 adenovirus.
Overexpression of HO-1 with recombinant adenovirus reverses post-ischemic neuronal apoptosis induced by secondary brain damage in transient cerebral ischemia-reperfusion injury
In sham and vehicle groups, limited GFP was detected (Fig. 2A and B, respectively). Conversely, expression of GFP was prevalent in the brain tissue (hippocampus, striatum, cortex penumbra) when transfected with Ad and Ad-HO-1 (Fig. 2C-E). The expression levels of HO-1 were assessed in each group using western blot analysis (Fig. 3A). In HO gene-treated rats, HO-1 expression levels were increased significantly compared with the vehicle and Ad groups (P<0.05; Fig. 3B). Furthermore, compared with the sham group, transient cerebral ischemia-reperfusion injury induced in the vehicle and Ad groups exhibited a significant increase in apoptotic index score at 24 h following MCAO (P<0.01; Fig. 3C). However, overexpression of HO-1 significantly reversed this score (18.28±0.18%) compared with the vehicle (57.68±2.11%) and Ad (58.22±1.96%) groups with transient cerebral ischemia-reperfusion injury (P<0.05).
Figure 2.

Expression of green fluorescent protein. No green fluorescent protein was detected in the (A) sham and (B) vehicle groups. However, significant expression of green fluorescent protein (transfection rate was 34.5±3.4%) was indicated in the (C) Ad (36.31±2.21) and (D) Ad-HO-1 groups (27.31±1.38) (P<0.01) at 24 h after ischemia-reperfusion injury was established (magnification, ×400). (E) Quantification of fluorescence. **P<0.01 vs. vehicle and Ad groups. Ad, empty adenovirus vector group; HO-1, heme oxygenase-1; Ad-HO-1, recombinant HO-1 adenovirus.
Figure 3.

Overexpression of HO-1 via recombinant adenovirus transfection ameliorated post-ischemic neuronal apoptosis induced by secondary brain damage in transient cerebral ischemia-reperfusion injury. (A and B) Transfection of HO-1 gene caused significantly increased HO-1 protein expression levels in the cerebral cortex of rats compared with that in the vehicle and Ad groups. Furthermore, overexpression of HO-1 significantly enhanced the anti-apoptotic effect as determined by (C) terminal deoxynucleotidyl-transferase-mediated dUTP nick end staining and (D) caspase-3 activity. Data are presented as the mean ± standard deviation (repeats, n=6). *P<0.05 vs. vehicle and Ad-HO-1 groups and **P<0.01 vs. sham group. Ad, empty adenovirus vector group; HO-1, heme oxygenase-1; Ad-HO-1, recombinant HO-1 adenovirus.
The role of the caspase-dependent apoptosis pathway in neuronal cell apoptosis was assessed based on caspase-3 activity. Neuronal apoptosis induced by ischemia was further examined following transfection with the HO-1 gene to identify its specific role. The vehicle and Ad groups were significantly increased compared with the sham group (P<0.01). Caspase-3 activation in rat brains induced by cerebral ischemia-reperfusion injury was significantly reduced following treatment with HO-1 gene compared with the vehicle and Ad group (P<0.05; Fig. 3D). In the sham group, the percentage of detected TUNEL-positive cells was 0.1% (Fig. 4). Results demonstrated that detectable neuronal apoptosis was not induced by surgical procedure in the sham group. Conversely, in the tissues from brains of ischemic-reperfused rats, the nuclei with TUNEL-positive stain were prevalent. However, the number of TUNEL-positive cells was significantly reduced following treatment with HO-1 gene (18.83±1.82) compared with the Ad group (59.33±2.95; P<0.001) (Fig. 4). These findings suggest that HO-1 gene overexpression may have an inhibitive role in post-ischemic neuronal apoptosis.
Figure 4.

TUNEL stain. In the brain tissues of the sham group, the percentage of positive cells stained with TUNEL was 0.1% (magnification, ×400). Conversely, in the tissues from brains of ischemic-reperfused rats, TUNEL-positive nuclei were prevalent. However, the number of TUNEL-positive cells was markedly reduced following treatment with Ad-HO-1. (A) Sham group, (B) Vehicle group, (C) Ad group and (D) Ad-HO-1 transfection group. (E) The results were quantified. ***P<0.001 vs. the vehicle group and ###P<0.001 vs. the Ad group. TUNEL, terminal deoxynucleotidyl-transferase-mediated dUTP nick end staining, Ad, empty adenovirus vector; HO-1, heme oxygenase-1; Ad-HO-1, recombinant HO-1 adenovirus.
Overexpression of HO-1 with recombinant adenovirus reverses behavioral impairments induced by secondary brain damage in transient cerebral ischemia-reperfusion injury
Transient cerebral ischemia-reperfusion damage markedly reduced the NDS (Fig. 5) at 24 h following MCAO. The neurological deficit scores were significantly reduced in the MCAO groups (Vehicle and Ad group) compared with the sham group (P<0.01). Overexpression of HO-1 significantly ameliorated the NDS (12.45±0.18%) compared with the vehicle (9.82±0.17%) and Ad (9.96±0.09%) groups (P<0.001; Fig. 5). These findings suggest that overexpression of the HO-1 gene has a potential protective role against secondary brain damage of the cerebral cortex induced by transient cerebral ischemia-reperfusion injury, and may restore neurological/behavioral impairment.
Figure 5.

Overexpression of HO-1 via recombinant adenovirus transfection reversed behavioral impairments induced by secondary brain damage in transient cerebral ischemia-reperfusion injury. Transient cerebral ischemia-reperfusion damage reduced the neurological score at 24 h following middle cerebral artery occlusion. Overexpression of HO-1 significantly attenuated the neurological deficit score compared with transient cerebral ischemia injury groups (vehicle and Ad groups). Data are presented as the mean ± standard deviation (repeats, n=6). **P<0.01 vs. the sham group; ***P<0.001 vs. vehicle; ###P<0.001 vs. the Ad group. Ad, empty adenovirus vector; HO-1, heme oxygenase-1; Ad-HO-1, recombinant HO-1 adenovirus.
Discussion
Globally, stroke is one of the major causes of long-term disability and death (25). Stroke is associated with notable socioeconomic and clinical implications; furthermore, there is an urgent requirement for effective therapies to treat stroke. In practice, stroke is a pathological process involving multiple factors that are typically associated with oxidative stress, inflammation, calcium overload, brain edema and cell apoptosis (26).
TIAs (transient ischemic attacks) are known as brief ischemic episodes and have been studied for over two decades (27,28). Tolerance to stroke may be induced by TIAs through elevating the safety threshold of human brain tissue (29). The potential molecular mechanisms of neuroprotection induced by TIAs were unclear (29). No clinically effective therapies are currently available for treating stroke, despite a number of studies based on experimental neuroprotective compounds and animal models that have demonstrated promising results (30,31). Investigation into the potential molecular mechanisms associated with TIAs may aid the identification of the novel targets for the prevention and treatment for stroke (32).
HO-1 has a crucial role in the normal function of neurons (33). In normal rats, the expression of HO-1 is located at the neuronal nuclei of Purkinje or hindbrain cells in the cerebellum (34). Furthermore, heat shock proteins, trauma, bleeding and hypoxia may induce the expression of HO-1 (35). HO broadly participates in the functional regulation of the heart, brain, lungs, liver, kidneys and other tissue cells associated with anti-oxidative stress injury (36). Previous results have demonstrated that the increase in HO expression was able to protect neuronal cells against ischemia through anti-oxidative stress (37), anti-apoptosis (38) and inhibition of the inflammatory response (39), thereby improving the cerebral microcirculation blood flow, stabilizing the cell membrane and mitochondrial membrane, and reducing brain edema (40). IPC induces the expression of HO-1, which has an important role in mediating the protection against transient and permanent brain ischemia (41). Additionally, following permanent ischemia, HO-1 may be the critical factor in improving the NDS and reducing infarct volume of the brain (42). In summary, HO-1 is the key element in the protection against stroke, particularly in the tolerance to stroke induced by IPC (3).
Conventional therapeutic strategies, which are administered via a range of methods, including orally, subcutaneously, intramuscularly and intravenously, are subject to different degrees of blood-brain barrier obstacles, which makes the outcome of an effective therapeutic response a challenge. As a novel therapeutic strategy, brain stereotactic injection of exogenous gene therapy for brain disease has received a lot of interest in recent years (43).
In the present study, the recombinant adenovirus vector of HO-1 was transfected into rats on the right lateral ventricle and the HO-1 gene was injected directly into the brain. The protein expression levels of HO-1 were continuously detected for 3 days following transfection, and were significantly increased in the cortex compared with the Ad group. However, protein expression levels of HO-1 were decreased in the Ad group transfected with adenovirus vector compared with the Ad-HO-1 group, which demonstrated that the injection method of intracerebroventricular administration was feasible and effective.
The present results indicated that intraventricular injection with Ad-HO-1 is an effective method to enhance the expression of HO-1 and reverse brain injury induced by transient focal cerebral ischemia-reperfusion injury in rats. Furthermore, the present findings suggested that this approach may significantly inhibit neuronal apoptosis and attenuate neurological behavioral deficits. The results also demonstrated that rats that received MCAO and pretreatment with HO-1 gene exhibited a significant decrease in infarct volume compared with rats that were subjected to MCAO and treated with vehicle or Ad. Furthermore, the present findings indicated that intraventricular injection with the HO-1 gene significantly induced the expression of HO-1, which improved neurological behavioral. This intervention method may lead notable clinical benefits through decreasing the volume of infarct at a later time point (at 24 h after reperfusion).
Similar to reports in previous studies (44,45), the present study demonstrated that the expression of HO-1 increased significantly in focal ischemic brains, which indicated that HO-1 may be a potential sensor for therapy in vitro. In the present study, a common adenovirus vector was used for the control, which did not affect the expression level of HO-1. However, in the Ad-HO-1 group, expression was significantly increased. These results demonstrated that the protective effects originated from the successful expression of HO-1 introduced by transfection compared with endogenous HO-1.
In conclusion, the HO-1 gene carried by adenovirus may be effectively transfected into the brain tissue and expressed stably in vivo. The programmed cell death in neuronal cells following cerebral ischemia-reperfusion injury may be significantly reversed by overexpression of HO-1. The results of the present study suggested that HO-1 was able to mediate the effective reduction of infarct volume, and provide neuroprotection against transient cerebral ischemia-reperfusion induced brain damage.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science Foundation of China (grant no. 30600524).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
XL performed all experiments analyzed and interpreted the data. RG, WH and ZS performed some of the experiments. GW, LW and YX were major contributors in writing the manuscript and contributed to the conception and design of the study. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The study was approved by The Second Hospital of Shanxi Medical University Committee on Animal Care (Shanxi, China).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Steiger HJ, Hanggi D. Ischaemic preconditioning of the brain, mechanisms and applications. Acta Neurochir (Wien) 2007;149:1–10. doi: 10.1007/s00701-006-1057-1. [DOI] [PubMed] [Google Scholar]
- 2.Bergeron M, Ferriero DM, Vreman HJ, Stevenson DK, Sharp FR. Hypoxia-ischemia, but not hypoxia alone, induces the expression of heme oxygenase-1 (HSP32) in newborn rat brain. J Cereb Blood Flow Metab. 1997;17:647–658. doi: 10.1097/00004647-199706000-00006. [DOI] [PubMed] [Google Scholar]
- 3.Zeynalov E, Shah ZA, Li RC, Doré S. Heme oxygenase 1 is associated with ischemic preconditioning-induced protection against brain ischemia. Neurobiol Dis. 2009;35:264–269. doi: 10.1016/j.nbd.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poss KD, Tonegawa S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc Natl Acad Sci USA. 1997;94:10919–10924. doi: 10.1073/pnas.94.20.10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol Rev. 2006;86:583–650. doi: 10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- 6.Tenhunen R, Marver HS, Schmid R. The enzymatic catabolism of hemoglobin: Stimulation of microsomal heme oxygenase by hemin. J Lab Clin Med. 1970;75:410–421. [PubMed] [Google Scholar]
- 7.Kato H, Amersi F, Buelow R, Melinek J, Coito AJ, Ke B, Busuttil RW, Kupiec-Weglinski JW. Heme oxygenase-1 overexpression protects rat livers from ischemia/reperfusion injury with extended cold preservation. Am J Transplant. 2001;1:121–128. doi: 10.1034/j.1600-6143.2001.10205.x. [DOI] [PubMed] [Google Scholar]
- 8.Panahian N, Yoshiura M, Maines MD. Overexpression of heme oxygenase-1 is neuroprotective in a model of permanent middle cerebral artery occlusion in transgenic rats. J Neurochem. 1999;72:1187–1203. doi: 10.1111/j.1471-4159.1999.721187.x. [DOI] [PubMed] [Google Scholar]
- 9.Shen C, Cheng W, Yu P, Wang L, Zhou L, Zeng L, Yang Q. Resveratrol pretreatment attenuates injury and promotes proliferation of neural stem cells following oxygen-glucose deprivation/reoxygenation by upregulating the expression of Nrf2, HO-1 and NQO1 in vitro. Mol Med Rep. 2016;14:3646–3654. doi: 10.3892/mmr.2016.5670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ryu MJ, Chung HS. Fucoidan reduces oxidative stress by regulating the gene expression of HO-1 and SOD-1 through the Nrf2/ERK signaling pathway in HaCaT cells. Mol Med Rep. 2016;14:3255–3260. doi: 10.3892/mmr.2016.5623. [DOI] [PubMed] [Google Scholar]
- 11.Tulsulkar J, Glueck B, Hinds TD, Jr, Shah ZA. Ginkgo biloba extract prevents female mice from ischemic brain damage and the mechanism is independent of the HO1/Wnt pathway. Transl Stroke Res. 2016;7:120–131. doi: 10.1007/s12975-015-0433-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sugiura S, Kitagawa K, Tanaka S, Todo K, Omura-Matsuoka E, Sasaki T, Mabuchi T, Matsushita K, Yagita Y, Hori M. Adenovirus-mediated gene transfer of heparin-binding epidermal growth factor-like growth factor enhances neurogenesis and angiogenesis after focal cerebral ischemia in rats. Stroke. 2005;36:859–864. doi: 10.1161/01.STR.0000158905.22871.95. [DOI] [PubMed] [Google Scholar]
- 13.Shen F, Fan Y, Su H. Adeno-associated viral vector-mediated hypoxia-regulated VEGF gene transfer promotes angiogenesis following focal cerebral ischemia in rats. Gene Ther. 2008;15:30–39. doi: 10.1038/sj.gt.3303048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paxinos G, Watson CR, Emson PC. AChE-stained horizontal sections of the rat brain in stereotaxic coordinates. J Neurosci Methods. 1980;3:129–149. doi: 10.1016/0165-0270(80)90021-7. [DOI] [PubMed] [Google Scholar]
- 15.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.STR.20.1.84. [DOI] [PubMed] [Google Scholar]
- 16.Pantoni L, Bartolini L, Pracucci G, Inzitari D. Interrater agreement on a simple neurological score in rats. Stroke. 1998;29:871–872. doi: 10.1161/01.STR.29.4.871. [DOI] [PubMed] [Google Scholar]
- 17.Amantea D, Nappi G, Bernardi G, Bagetta G, Corasaniti MT. Post-ischemic brain damage: Pathophysiology and role of inflammatory mediators. FEBS J. 2009;276:13–26. doi: 10.1111/j.1742-4658.2008.06766.x. [DOI] [PubMed] [Google Scholar]
- 18.Maines MD. Heme oxygenase: Function, multiplicity, regulatory mechanisms and clinical applications. FASEB J. 1998;2:2557–2568. doi: 10.1096/fasebj.2.10.3290025. [DOI] [PubMed] [Google Scholar]
- 19.Regan RF, Guo Y, Kumar N. Heme oxygenase-1 induction protects murine cortical astrocytes from hemoglobin toxicity. Neurosci Lett. 2000;282:1–4. doi: 10.1016/S0304-3940(00)00817-X. [DOI] [PubMed] [Google Scholar]
- 20.He TC, Zhou S, Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sodt AJ, Venable RM, Lyman E, Pastor RW. Nonadditive compositional curvature energetics of lipid bilayers. Phys Rev Lett. 2016;117:138104. doi: 10.1103/PhysRevLett.117.138104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shah ZA, Namiranian K, Klaus J, Kibler K, Doré S. Use of an optimized transient occlusion of the middle cerebral artery protocol for the mouse stroke model. J Stroke Cerebrovasc Dis. 2006;15:133–138. doi: 10.1016/j.jstrokecerebrovasdis.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 23.Zeynalov E, Nemoto M, Hurn PD, Koehler RC, Bhardwaj A. Neuroprotective effect of selective kappa opioid receptor agonist is gender specific and linked to reduced neuronal nitric oxide. J Cereb Blood Flow Metab. 2006;26:414–420. doi: 10.1038/sj.jcbfm.9600196. [DOI] [PubMed] [Google Scholar]
- 24.Dziennis S, Qin J, Shi L, Wang RK. Macro-to-micro cortical vascular imaging underlies regional differences in ischemic brain. Sci Rep. 2015;5:10051. doi: 10.1038/srep10051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang L, Yin CY, Wu HY, Tian BB, Zhu Y, Luo CX, Zhu DY. (+)-Borneol is neuroprotective against permanent cerebral ischemia in rats by suppressing production of proinflammatory cytokines. J Biomed Res. 2017;31:306–314. doi: 10.7555/JBR.31.20160138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Voelker R. Reducing stroke disability. JAMA. 2016;316:1538. doi: 10.1001/jama.2016.14561. [DOI] [PubMed] [Google Scholar]
- 27.Phan TG, Srikanth V, Cadilhac DA, Grimley R, Donnan GA, Anderson CS. Better outcomes for hospitalized patients with TIA when in stroke units: An observational study. Neurology. 2016;87:1745–1746. doi: 10.1212/WNL.0000000000003277. [DOI] [PubMed] [Google Scholar]
- 28.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.CIR.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 29.Wegener S, Gottschalk B, Jovanovic V, Knab R, Fiebach JB, Schellinger PD, Kucinski T, Jungehülsing GJ, Brunecker P, Müller B, et al. Transient ischemic attacks before ischemic stroke: Preconditioning the human brain? A multicenter magnetic resonance imaging study. Stroke. 2004;35:616–621. doi: 10.1161/01.STR.0000115767.17923.6A. [DOI] [PubMed] [Google Scholar]
- 30.Lip GY, Kongnakorn T, Phatak H, Kuznik A, Lanitis T, Liu LZ, Iloeje U, Hernandez L, Dorian P. Cost-effectiveness of apixaban versus other new oral anticoagulants for stroke prevention in atrial fibrillation. Clin Ther. 2014;36:192–210. doi: 10.1016/j.clinthera.2013.12.011. [DOI] [PubMed] [Google Scholar]
- 31.Cherry MG, Greenhalgh J, Osipenko L, Venkatachalam M, Boland A, Dundar Y, Marsh K, Dickson R, Rees DC. The clinical effectiveness and cost-effectiveness of primary stroke prevention in children with sickle cell disease: A systematic review and economic evaluation. Health Technol Assess. 2012;16:1–129. doi: 10.3310/hta16430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilson D, Charidimou A, Ambler G, Fox ZV, Gregoire S, Rayson P, Imaizumi T, Fluri F, Naka H, Horstmann S, et al. Recurrent stroke risk and cerebral microbleed burden in ischemic stroke and TIA: A meta-analysis. Neurology. 2016;87:1501–1510. doi: 10.1212/WNL.0000000000003183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Doan KV, Kinyua AW, Yang DJ, Ko CM, Moh SH, Shong KE, Kim H, Park SK, Kim DH, Kim I, et al. FoxO1 in dopaminergic neurons regulates energy homeostasis and targets tyrosine hydroxylase. Nat Commun. 2016;7:12733. doi: 10.1038/ncomms12733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li W, Jiang D, Li Q, Yao S, Sun X, Yang Y, Meng Z, Liu W. Lipopolysaccharide-induced preconditioning protects against traumatic spinal cord injury by upregulating Nrf2 expression in rats. Life Sci. 2016;162:14–20. doi: 10.1016/j.lfs.2016.08.008. [DOI] [PubMed] [Google Scholar]
- 35.Lin R, Cai J, Kostuk EW, Rosenwasser R, Iacovitti L. Fumarate modulates the immune/inflammatory response and rescues nerve cells and neurological function after stroke in rats. J Neuroinflammation. 2016;13:269. doi: 10.1186/s12974-016-0733-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hawrylycz M, Anastassiou C, Arkhipov A, Berg J, Buice M, Cain N, Gouwens NW, Gratiy S, Iy Leszl-Ishiguro M, Horvath B, Johnson RA, Johnson FK, Lenzsér G, Hermán P, Horváth EM, Benyó Z. Influence of the heme-oxygenase pathway on cerebrocortical blood flow. Neuroreport. 2007;18:1193–1197. doi: 10.1097/WNR.0b013e3282202459. [DOI] [PubMed] [Google Scholar]
- 37.Chen K, Gunter K, Maines MD. Neurons overexpressing heme oxygenase-1 resist oxidative stress-mediated cell death. J Neurochem. 2000;75:304–313. doi: 10.1046/j.1471-4159.2000.0750304.x. [DOI] [PubMed] [Google Scholar]
- 38.Leszl-Ishiguro M, Horvath B, Johnson RA, Johnson FK, Lenzsér G, Hermán P, et al. Influence of the heme-oxygenase pathway on cerebrocortical blood flow. Neuroreport. 2007;18:1193–1197. doi: 10.1097/WNR.0b013e3282202459. [DOI] [PubMed] [Google Scholar]
- 39.Zhen-Wei X, Jian-Le S, Qi Q, Wen-Wei Z, Xue-Hong Z, Zi-Li Z. Heme oxygenase-1 improves the survival of discordant cardiac xenograft through its anti-inflammatory and anti-apoptotic effects. Pediatr Transplant. 2007;11:850–859. doi: 10.1111/j.1399-3046.2007.00701.x. [DOI] [PubMed] [Google Scholar]
- 40.Jumnongprakhon P, Govitrapong P, Tocharus C, Tocharus J. Melatonin promotes blood-brain barrier integrity in methamphetamine-induced inflammation in primary rat brain microvascular endothelial cells. Brain Res. 2016;1646:182–192. doi: 10.1016/j.brainres.2016.05.049. [DOI] [PubMed] [Google Scholar]
- 41.Zhang X, Xiao Z, Yao J, Zhao G, Fa X, Niu J. Participation of protein kinase C in the activation of Nrf2 signaling by ischemic preconditioning in the isolated rabbit heart. Mol Cell Biochem. 2013;372:169–179. doi: 10.1007/s11010-012-1458-9. [DOI] [PubMed] [Google Scholar]
- 42.Mohammadi E, Bigdeli MR. Effects of preconditioning with normobaric heperoxia on Na+/Ca2+ exchanger in the rat brain. Neuroscience. 2013;237:277–284. doi: 10.1016/j.neuroscience.2013.01.064. [DOI] [PubMed] [Google Scholar]
- 43.Gowing G, Svendsen S, Svendsen CN. Ex vivo gene therapy for the treatment of neurological disorders. Prog Brain Res. 2017;230:99–132. doi: 10.1016/bs.pbr.2016.11.003. [DOI] [PubMed] [Google Scholar]
- 44.Yen TL, Chen RJ, Jayakumar T, Lu WJ, Hsieh CY, Hsu MJ, Yang CH, Chang CC, Lin YK, Lin KH, Sheu JR. Andrographolide stimulates p38 mitogen-activated protein kinase-nuclear factor erythroid-2-related factor 2-heme oxygenase 1 signaling in primary cerebral endothelial cells for definite protection against ischemic stroke in rats. Transl Res. 2016;170:57–72. doi: 10.1016/j.trsl.2015.12.002. [DOI] [PubMed] [Google Scholar]
- 45.Xue F, Huang JW, Ding PY, Zang HG, Kou ZJ, Li T, Fan J, Peng ZW, Yan WJ. Nrf2/antioxidant defense pathway is involved in the neuroprotective effects of Sirt1 against focal cerebral ischemia in rats after hyperbaric oxygen preconditioning. Behav Brain Res. 2016;309:1–8. doi: 10.1016/j.bbr.2016.04.045. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.