

Abstract
Diabetes mellitus (DM) complications affect patients and cause varying damage. Skin ulcers exhibit difficulties in wound healing, and the regulatory basis for this remains unclear. High glucose concentration (HG) was utilized to mimic DM in cultured cells. Reverse transcription-quantitative polymerase chain reaction, western blotting and fluorescence dye analyses were performed to analyze the effects of hedgehog signaling in regulation of HG or diabetes in fibroblasts. HG-stress suppressed hedgehog-signaling gene expression, whereas the apoptosis and inflammatory response markers, Caspase-3 and plasminogen activator inhibitor-1 (PAI1), respectively, were induced. In addition, HG-stress inhibited the fibroblast proliferation rate. In parallel, treatment with Sonic hedgehog (Shh), an activator of hedgehog signaling, together with HG eliminated effects of HG on expression of hedgehog-signaling genes, Caspase-3 and PAI1, and rescued the cell proliferation rate in fibroblasts. In addition, Shh application activated c-Jun N-terminal kinase (JNK), which was inhibited by HG stress. sp600125, a JNK specific inhibitor, treatment inhibited the effect of Shh on fibroblast proliferation and hedgehog-signaling marker gene expression. Furthermore, zinc finger protein Gli1 (Gli1) overexpression partially eliminated the effect of HG and sp600125 on fibroblast proliferation, and reduced HG-induced ROS generation in fibroblasts. Together, these results indicate that HG stress inhibits hedgehog signaling, and Shh-JNK-Gli1 pathway positively regulates HG-induced damage on fibroblasts.
Keywords: hedgehog signalling, high-glucose, skin, fibroblasts, damage
Introduction
Diabetes mellitus (DM) is a severe metabolic disease prevalent in a large number of patients worldwide (~5.38%) and is characterized by high concentrations of blood sugar (1). The challenges in DM therapy arise from complications that affect several organs, such as the heart, kidney and skin: Many patients suffer from a delay in skin wound and ulcer repair (2); and higher sugar content in blood vessels blocks angiogenesis, which is important for supplying fresh blood for the repair of damaged tissue (3).
Repair of skin ulcers is a complicated process that requires recruitment of cells such as keratinocytes, platelets, endothelial cells, fibroblasts, and macrophages. Fibroblast migration and proliferation, angiogenesis, and wound contraction, together with collagen deposition and remodeling are the key processes in wound healing. In the dermal skin layer, extracellular matrix (ECM) synthesis is the largest component of the skin generation process (4). Fibroblasts initiate ECM production and remodeling, which further forms granulation tissue (5,6). Levels of cellular reactive oxygen species (ROS) are modified to sense environmental changes, and they function as secondary messengers in regulation of diverse biological processes including cell proliferation, differentiation, and maturation (7). Fibroblasts isolated from patients with DM are typically large and widely spread, compared with age-matched fibroblasts from healthy people, and exhibit abnormal endoplasmic reticulum, higher number of vesicular particles, and altered microtubule arrays. Therefore, high glucose concentration (HG) in vivo induces normal protein maturation, cellular protein trafficking and protein secretion in diabetic ulcer fibroblasts (8,9). In addition, it has been demonstrated that cell proliferation was defective and subsequent production of extracellular matrix protein was affected in diabetic ulcer fibroblasts (8). HG was reported to inhibit c-Jun N-terminal kinase (JNK) activity, resulting in a delay in fibroblast migration (10). Previous transcriptome analysis using HG-stimulated fibroblasts has identified changes in the expression of numerous genes associated with diverse pathways including Wnt, inflammatory response, and hedgehog signaling (11).
Hedgehog signaling has been widely studied due to its significance in development and disease regulation (12). Hedgehog was first observed as a secreted protein that requires specific positional identity in embryonic segments of Drosophila melanogaster (13). The three mammalian hedgehog genes, Sonic hedgehog (Shh), Desert hedgehog and Indian hedgehog, are known for their importance in the patterning of developing tissue and biological structures (13). Notably, inhibition of hedgehog signaling either by loss of a gene or by suppression of expression severely affects development of tissues and organs, leading to skeletal malformation, craniofacial defects, polydactyly and holoprosencephaly (14). Furthermore, abnormal activation of hedgehog signaling is associated with most basal cell carcinomas, and some medulloblastomas and rhabdomyosarcomas (12,15–17). Under normal conditions, the plasma membrane-localized Patched1 (Ptch1) interacts with Smoothened (SMO) to maintain hedgehog signaling in an inactive or ‘off’ state. Following Shh secretion, it interacts with and inactivates Ptch1, resulting in the activation of SMO (16,18). SMO further triggers downstream gene transcription through the zinc finger protein Gli (Gli)-Kruppel family of transcription factors, controlling cell differentiation, proliferation, and survival (18). However, the role of hedgehog signaling in the process of fibroblast repair under DM conditions is currently unknown.
In the present study, the effects of hedgehog signaling on HG-damaged skin fibroblasts were analyzed via Shh stimulation, and the expression levels of hedgehog signaling, apoptosis and inflammatory response markers were determined. Cell proliferation rates were also analyzed. Additionally, JNK activity was analyzed following HG and Shh treatment, and cell proliferation was analyzed following Shh and JNK inhibitor supplementation together with overexpression of Gli1, which is a key transcriptional activator of hedgehog signaling (19). The findings of the present study may be useful for skin ulcer therapy for patients with DM.
Materials and methods
Fibroblast culture
Human foreskin samples were obtained from 6 male patients (age, 23–35 years), undergoing foreskin circumcision, from January 2017 to March 2017 at the Department of Dermatology, The First Affiliated Hospital of Xinxiang Medical University (Xinxiang, China). The present study was approved by the Ethics Committee of Xinxiang Medical University, and written informed consent was obtained from all patients. The human foreskin fibroblast isolation and proliferation were performed as described previously (10). Fibroblasts were divided into three groups and treated with HG (35 mM), sp600126 (100 nM; cat. no. S5567; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany), and Shh (100 ng/ml; cat. no. S0191; Sigma-Aldrich; Merck KGaA), respectively, for 3 h at 37°C. The low glucose treated group was untilized as the control.
Cell proliferation assay
Cell proliferation activity was examined following the aforementioned treatment, using a Cell Counting Kit-8 (Dojindo Molecular Technologies, Inc., Kumamoto, Japan) according to the manufacturer's protocol. Briefly, 2×103 fibroblasts were seeded in 96 well plates and respectively treated with HG (35 mM) and Shh (100 ng/ml), and cell density was analyzed as previously reported (10).
RNA isolation and reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
RNA was isolated from fibroblasts that were either untreated or treated with HG (35 mM) and Shh (100 ng/ml; cat no. ab23327; Abcam, Cambridge, MA, USA). Fibroblasts were washed twice with ice-cold PBS and 1 ml TRIzol (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) was subsequently added to each 3.5-cm diameter dish to lyse the cells. Following treatment with RQ1 DNase (Promega Corporation, Madison, WI, USA), 1 µg RNA was used to synthesize cDNA using the GoScript Reverse Transcription kit (Reverse Transcription System, Promega) following the manufacturer's instructions. A SYBR Green Master Mix (Bio-Rad Laboratories, Inc., Hercules, CA, USA) was used to perform qPCR on an Illumina Eco 3.0 system (Illumina, Inc., San Diego, CA, USA). The PCR reaction was performed in a 20 µl reaction mixture containing 10 µl SYBR Green Master Mix, 4 µl primers (2 µl each of 2.5 µM forward and reverse primer), 3 µl cDNA template and 3 µl double-distilled H2O. A typical reaction consisted of an initial denaturation at 95°C for 3 min, followed by 40 cycles of denaturation for 30 sec at 95°C, annealing for 30 sec at 60°C, and extension at 72°C for 30 sec, followed by a final extension at 72°C for 7 min. The transcription levels were normalized against those of GAPDH using the 2−ΔΔCq method (20). Primers used in RT-qPCR analysis are listed in Table I.
Table I.
Reverse transcription-quantitative polymerase chain reaction primer sequences.
| Primer | Sequence (5′-3′) |
|---|---|
| Caspase-3 F | TGATGATGACATGGCGTGTC |
| Caspase-3 R | GTTGCCACCTTTCGGTTAAC |
| PAI1 F | GAGACTGAAGTCGACCTCAG |
| PAI1 R | CTGTCCATGATGATCTCCTC |
| GAPDH F | GACCTGCCGTCTAGAAAAAC |
| GAPDH R | CTGTAGCCAAATTCGTTGTC |
| SMO F | ACCTATGCCTGGCACACTTC |
| SMO R | AGGAAGTAGCCTCCCACGAT |
| Ptch1 F | CAAACTCCTGGTGCAAACCG |
| Ptch1 R | CCGGGATTCTCAGCCTTGTT |
| Gli1 F | CCAGAGTTCAAGAGCCTGG |
| Gli1 R | CCTCGCTCCATAAGGCTCAG |
F, forward; R, reverse; PAI1, plasminogen activator inhibitor-1; SMO, Smoothened; Ptch1, Patched1; Gli1, zinc finger protein Gli1.
Western blotting
Total protein was extracted using cell lysis buffer according to the manufacturer's protocol (cat. no. P0013; Beyotime Institute of Biotechnology, Haimen, China) and the concentration was determined using a BCA assay. Total protein (20 µg) from each sample was electrophoresed using 10% SDS-PAGE. Following separation, the proteins were transferred to Immobilon-P Transfer membranes (EMD Millipore, Billerica, MA, USA), which were blocked in blocking solution (1X TBS, 5% skimmed milk and 0.05% Tween-20) for 2 h at room temperature and thereafter probed with primary antibodies at 25°C for 2 h. The following primary antibodies were used: Anti-phosphorylated (p)-stress-activated protein kinase (SAPK)/JNK (Thr183/Tyr185) antibody (1:1,000; cat. no. 4668; Cell Signaling Technology, Inc., Danvers, MA, US), anti-SAPK/JNK (1:1,000; cat. no. 9252; Cell Signaling Technology, Inc.) and anti-GAPDH antibody (1:2,000, cat. no. ab9484; Abcam). Following incubation with the primary antibodies, the membranes were washed with PBS and incubated with corresponding secondary antibodies for 1 h at room temperature. The secondary antibodies used were anti-mouse or anti-rabbit horseradish peroxidase-linked secondary antibody (1:2,000; cat. no. 7074; Cell Signaling Technology, Inc.). Samples were visualized using a Beyo ECL Plus kit (cat. no. P0018; Beyotime Institute of Biotechnology).
Overexpression of Gli1
To overexpress Gli1 in fibroblasts, the Gli1 open reading frame (ORF) sequence (Gli1; GenBank accession no. NP_005260; National Center for Biotechnology Information; https://www.ncbi.nlm.nih.gov/) was artificially synthesized by Sangon Biotech Co., Ltd. (Shanghai, China). The Gli1 ORF was then cloned into the pcDNA3 expression vector (Invitrogen; Thermo Fisher Scientific, Inc.). A total of 2 µg pcDNA3-Gli1 or pcDNA3 empty vector was transfected into human foreskin primary fibroblasts using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) following the manufacturer's instructions. RNA was extracted from transfected cells following 48 h, and the Gli1 expression level was determined via quantitative RT-qPCR under the same parameters as aforementioned, but with the following primers: Gli1, F, 5′-TTTCCCATTACCCCCAGCCCTCTC-3′; R, 5′-GAAGCCCTATTTGCCCCCACTACA-3′ and GAPDH, F, 5′-GTCATCATCTCTGCCCCCTCTGCT-3′; R, 5′-GACGCCTGCTTCACCACCTTCTTG-3′.
ROS measurement
To visualize cellular ROS levels 2′,7′-dichlorofluorescein diacetate (DCFH-DA) dye was used (21). Fibroblasts were cultured in low glucose DMEM (LG, 5.5 mM; cat. no. 11885092; Thermo Fisher Scientific, Inc.) or HG (35 mM; cat. no. 11965118; Thermo Fisher Scientific, Inc.), and Gli1 overexpressed cells were treated with HG at 37°C for 6 h. Fibroblasts were incubated with 10 µM DCFH-DA for 30 min at 37°C in the dark and fluorescence intensity was analyzed using a confocal microscope at a magnification of ×200 (excitation at 488 nm, emission at 530 nm) within 15 min.
Statistical analysis
Statistical differences were analyzed using the Prism 5 software package (Version 5.0; GraphPad Software, Inc., La Jolla, CA, USA). Data are presented as the mean ± standard deviation and three independent repeats were performed for each experiment. Significant differences between two groups were calculated using Student's t-test, and one-way analysis of variance was performed to analyze differences between more than two groups with a Turkey post hoc test. P<0.05 was considered to indicate a statistically significant difference.
Results
HG inhibits cell proliferation and hedgehog signaling in skin fibroblasts
Diabetes is a severe metabolic disease that generates diverse complications threatening patient health. To understand the mechanism of diabetes, HG is typically used to mimic the disease. Previous studies have observed that HG mediated stress altered the expression of many genes including hedgehog signaling-related genes in human fibroblast primary cells (11). To analyze the effect of HG on hedgehog signaling, key genes associated the pathway were examined for HG-mediated changes in expression. Initially, HG concentration was optimized for the relevant experimental conditions prior to testing gene expression. Previous research has identified that HG concentrations >35 mM significantly inhibit cell proliferation (11). In the present study, it was confirmed that 35 mM HG stress significantly reduced fibroblast proliferation, in comparison with LG fibroblasts (Fig. 1A). Furthermore, cell damage via HG was examined at the molecular level by evaluating expression levels of Caspase-3 (apoptosis marker) and plasminogen activator inhibitor-1 (PAI1; inflammatory response marker). RT-qPCR results demonstrated that HG treatment significantly increased Caspase-3 and PAI1 levels (Fig. 1B). Subsequently, the expression of key hedgehog signaling-related genes (SMO, Ptch1, and Gli1) was analyzed. RT-qPCR data indicate that HG suppressed the expression of all three genes (Fig. 1B).
Figure 1.
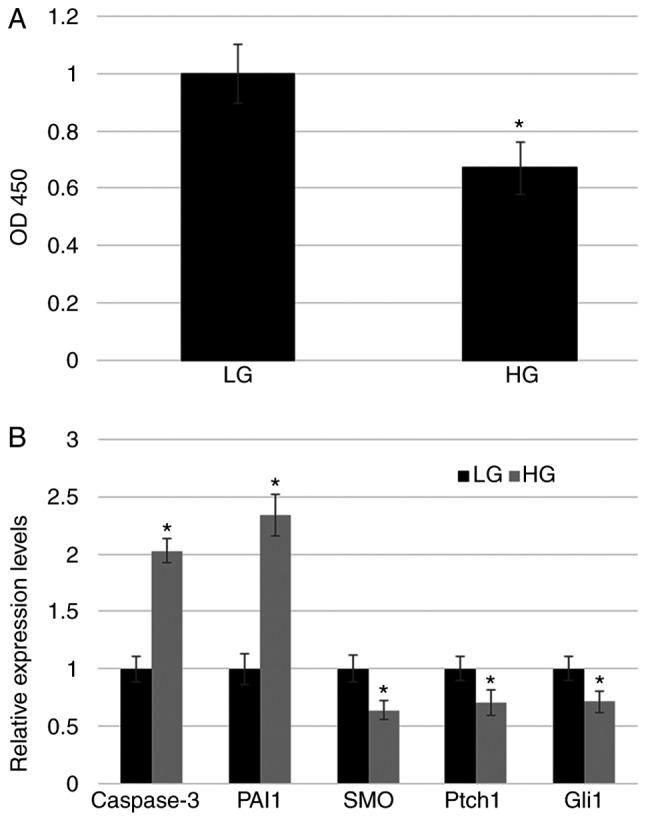
Fibroblast proliferation rate and marker gene expression upon HG stress. (A) Cell proliferation was analyzed using a cell counting kit-8 assay. (B) Expression of apoptosis marker (Caspase-3), inflammatory response marker (PAI1), and hedgehog signaling-related markers (SMO, Ptch1 and Gli1) was monitored in LG- and HG-treated fibroblasts with reverse transcription-quantitative polymerase chain reaction. *P<0.05 vs. LG. LG, low glucose concentration (5.5 mM); HG, high glucose concentration (35 mM); PAI1, plasminogen activator inhibitor-1; SMO, Smoothened; Ptch1, Patched1; Gli1, zinc finger protein Gli1; OD, optical density.
Shh treatment ameliorates the effect of HG on fibroblasts
As HG suppressed hedgehog signaling and cell proliferation, hedgehog signaling was activated via treatment with Shh in the culture medium, and cell proliferation and hedgehog gene expression were analyzed. Cell proliferation was activated by Shh treatment in LG medium and significantly inhibited by HG stress. Furthermore, addition of Shh with HG ameliorated the effect of HG on cell proliferation, and cells exhibited similar proliferation as cells grown under LG (Fig. 2A). The effects of Shh on gene expression were subsequently analyzed. Shh treatment induced Gli1 expression in LG growth medium, whereas HG significantly suppressed Gli1 expression. Treatment with Shh and HG eliminated the effect of HG on Gli1 expression, resulting in similar gene expression as that observed in cells grown under LG (Fig. 2B).
Figure 2.
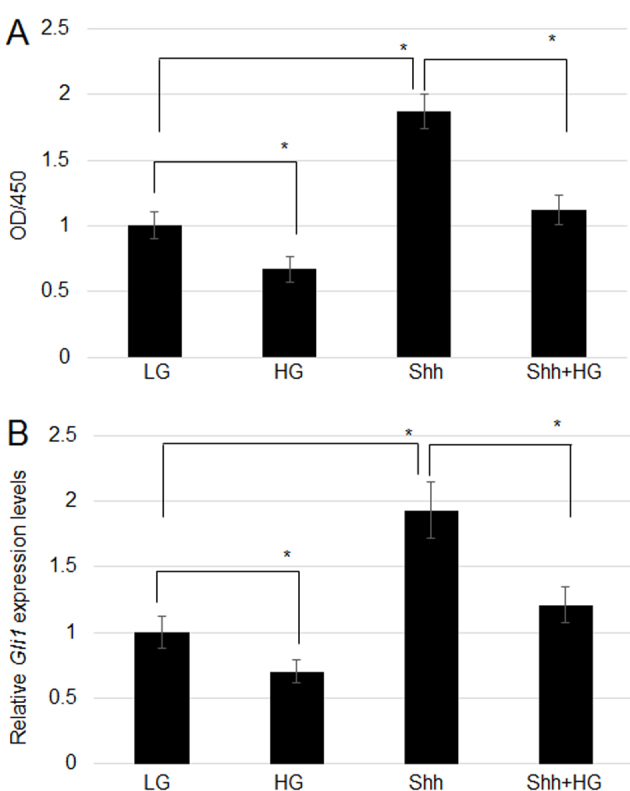
Effect of Shh on cell proliferation and gene expression. (A) Fibroblasts were treated with Shh (an inducer of hedgehog signaling), HG and HG+Shh, and cell proliferation was examined after 72 h. (B) Gli1 expression level was tested following Shh, HG, and HG+Shh treatment. Significant differences between groups are indicated with *P<0.05, HG and Shh vs. LG; Shh+HG vs. Shh. Shh, Sonic hedgehog; HG, high glucose concentration (35 mM); Gli1, zinc finger protein Gli1; OD, optical density; LG, low glucose concentration (5.5 mM).
JNK is associated with Shh-dependent cell proliferation and hedgehog signaling-related gene regulation
JNK modulates HG-mediated signaling in fibroblasts (10). It is known that HG inhibits JNK activity via suppression of p-JNK levels, without affecting total JNK (t-JNK) levels (10). Shh treatment induced a marked increase in the level of p-JNK (p-JNK) in both LG- and HG-treated cells (Fig. 3A). However, Shh, HG, and HG+Shh treatments did not markedly affect t-JNK levels (Fig. 3A). Additionally, JNK function during Shh-induced cell proliferation was examined. Compared with the LG group, treatment with sp600125, a JNK-specific inhibitor, significantly inhibited cell proliferation, whereas Shh treatment significantly promoted cell proliferation. Furthermore, sp600125 treatment significantly inhibited Shh-induced cell proliferation (Fig. 3B). As JNK has a role downstream of Shh during cell proliferation, the role of JNK in hedgehog signaling-related gene expression was further explored. Effects of sp600126 treatment in LG-grown cells on SMO, Ptch1, and Gli1 transcript levels were analyzed, indicating that inhibition of JNK via sp600125 significantly suppressed SMO, Ptch1, and Gli1 expression (Fig. 3C).
Figure 3.
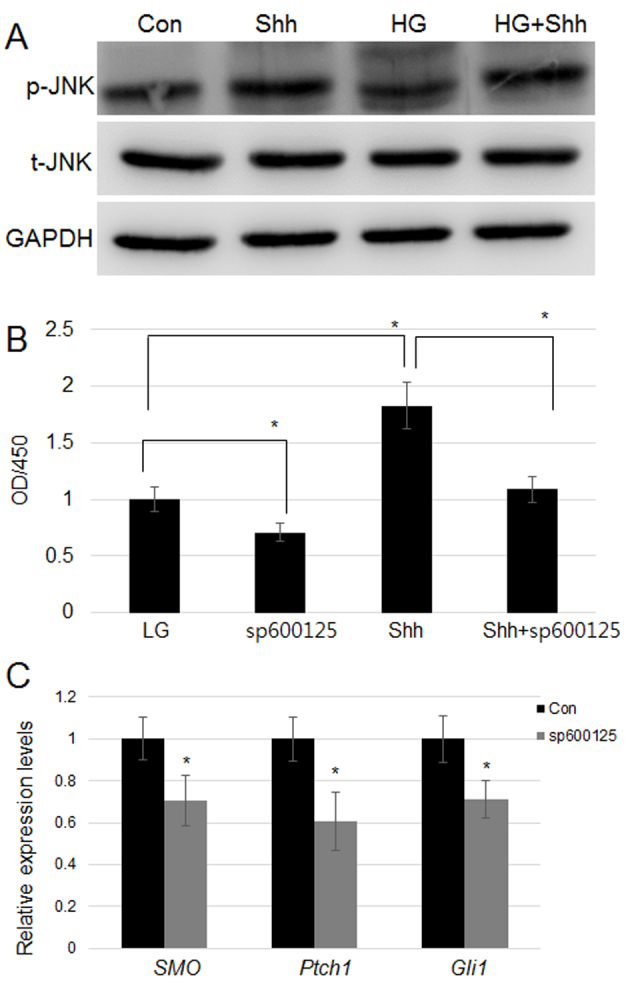
Effect of Shh on JNK activity, and of JNK inhibition on cell proliferation and hedgehog signaling-related gene expression. (A) Fibroblasts were treated with Shh, HG and HG+Shh, and p-JNK and total t-JNK levels were measured using western blotting. GAPDH was utilized as an internal control. (B) Fibroblasts were treated with Shh, sp600125, and HG+sp600125, and cell proliferation was monitored. Significant differences are marked with *P<0.05, sp60012 and Shh vs. LG; Shh+sp60012 vs. Shh. (C) SMO, Ptch1, and Gli1 expression was analyzed following treatment with sp600125. *P<0.05 vs. con. Shh, Sonic hedgehog, JNK, c-Jun N-terminal kinase; HG, high glucose concentration (35 mM); con, low glucose as the control group; p, phosphorylated; t, total; SMO, Smoothened; Ptch1, Patched1; Gli1, zinc finger protein Gli1; OD, optical density.
Overexpression of Gli1 partially eliminates HG and JNK inhibitor action
Gli1 expression was suppressed in fibroblasts upon HG and sp100125 treatment (Figs. 1 and 3). Therefore, the function of Gli1 was analyzed by overexpressing Gli1 in human fibroblasts. Following sequencing, the Lipofectamine 2000 system was used to transfect 2 µg pcDNA3-Gli1 or pcDNA3 empty plasmid into human foreskin primary fibroblasts. RT-qPCR results demonstrated that Gli1 expression was significantly increased in the Gli1-overexpressing cells than in cells transfected with empty vector (Fig. 4A). Compared with empty vector transformation, Gli1-overexpressing cells also exhibited a significant increase in cell proliferation (Fig. 4B). Furthermore, RT-qPCR analysis of Gli1-overexpressing cells treated with HG and sp600125 indicated that HG and sp600126 treatment did not significantly affect Gli1 expression levels (Fig. 4A). In addition, Gli1-overexpressing cells treated with sp600126 retained their increased proliferation rate compared with control cells (Fig. 4B), indicating that Gli1 regulates fibroblast proliferation downstream of HG and JNK signaling.
Figure 4.
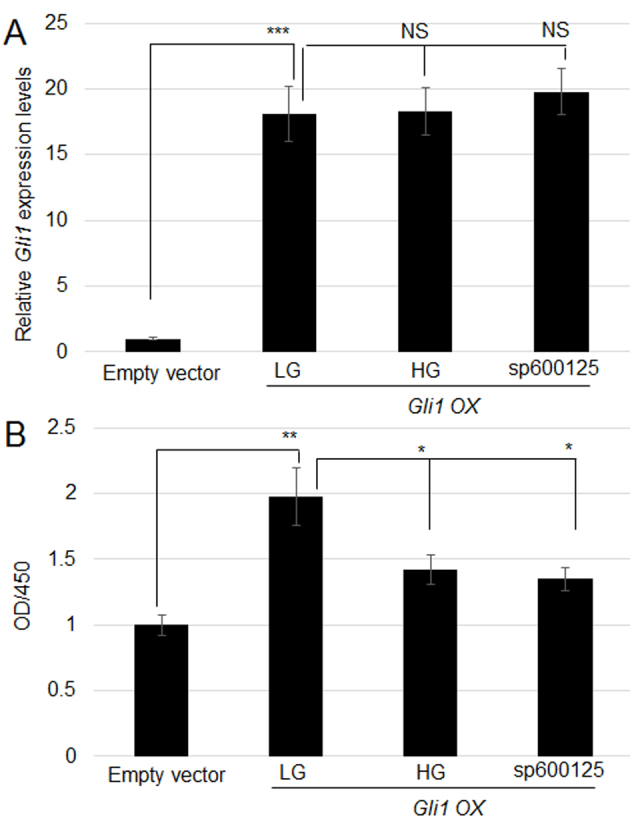
Effect of Gli1 overexpression on HG and sp600125-mediated inhibition of cell proliferation. (A) Gli1 was overexpressed in human fibroblasts, and its expression was analyzed with reverse transcription-quantitative polymerase chain reaction. Gli1-overexpressing cells treated with HG and sp600125, or without any treatment were compared. pcDNA3 empty vector-transfected cells were used as a control. ***P<0.001, LG vs. Empty vector; NS, No significance. (B) Cell proliferation was monitored in the pcDNA3 empty vector-transfected cells, Gli1-overexpressing cells, and sp600125- or HG-treated Gli1-overexpressing cells. Significant differences are indicated with **P<0.01, LG vs. Empty vector; *P<0.05, HG and sp600125 vs. LG. Gli1, zinc finger protein Gli1; HG, high glucose concentration (35 mM); LG, low glucose concentration (5.5 mM); OD, optical density; OX, overexpression.
Activation of hedgehog signaling reduces HG-induced ROS generation
Previous reports have demonstrated that HG-induced ROS production via inhibition of JNK and ROS levels is closely associated with wound repair (10,22). To further analyze the association between hedgehog signaling and HG-induced ROS production, ROS levels were detected in fibroblasts using DCFH-DA, which is a ROS indicator. LG-grown cells were used as a control and were not observed to notably accumulate ROS. However, HG-treated cells showed significantly increased ROS accumulation, compared with LG. To analyze the effects of hedgehog signaling on HG-induced ROS generation, Gli1-overexpressing cells were treated with HG, which resulted in significantly lower ROS levels (Fig. 5A and 5B). These findings suggested that Gli1 overexpression reduced HG-induced ROS production.
Figure 5.
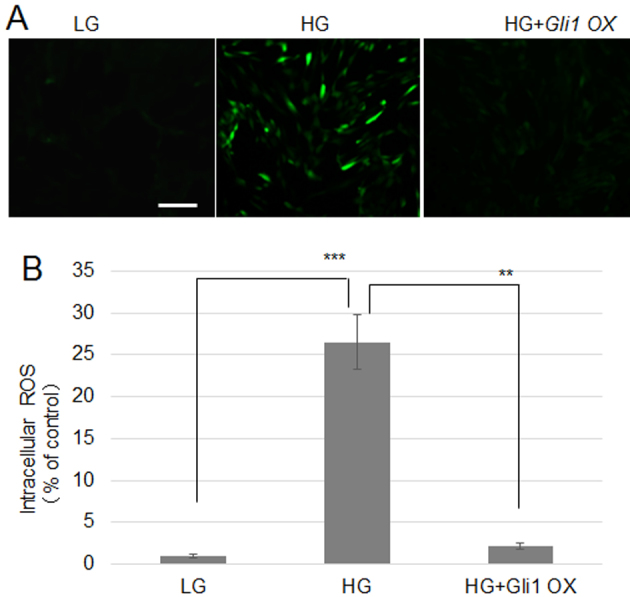
Shh treatment or Gli1 overexpression reduces cellular ROS levels in fibroblasts. (A) Cells were incubated for 1 h in LG, HG, or HG+Shh. Cellular ROS levels were visualized using 2′,7′-dichlorofluorescein diacetate. Scale bar=100 µm. (B) Fluorescence levels from 10 independent cells in each treatment group were quantified (n=10). Significant differences are shown with ***P<0.001, HG vs. LG; **P<0.01, HG+Gli1 OX vs. HG. Shh, Sonic hedgehog; Gli1, zinc finger protein Gli1; ROS, reactive oxygen species; LG, low glucose concentration (5.5 mM); HG, high glucose concentration (35 mM); OX, overexpression.
Discussion
Skin ulcers induced by DM are difficult to repair and afflict patients worldwide. Concentration of HG in in vitro fibroblast experiments is typically higher than the glucose levels in physiological levels of glucose in diabetic patients (>11.1 mM); 25–50 mM HG has been used for DM study in previous reports (10,23,24). In the present study, 35 mM HG was used to perform the experiments. Skin wound repair involves multiple steps including cell proliferation, and requires the coordination of multiple cell layers. Fibroblasts in the skin have an important role during wound repair. Abnormal activation of Ras-related C3 botulinum toxin substrate 1 and suppression of JNK were previously observed to be associated with wound repair in HG-stressed skin fibroblasts (10,25). In addition, transcriptome analysis has revealed that several biological pathways are altered in HG-damaged fibroblasts, including Wnt, hedgehog, and nuclear factor-κB signaling (11). However, to the best of our knowledge, the regulatory basis of hedgehog signaling during HG damage in fibroblasts has not been reported.
In the present study, it was observed that HG inhibited cell proliferation and suppressed the expression of key hedgehog signaling-related genes including SMO, Ptch1, and Gli1. Treatment with Shh, an activator of hedgehog signaling, was used to analyze the role of hedgehog in HG-damaged fibroblasts. Shh treatment activated cell proliferation in LG and HG growth conditions, and Gli1 expression was also induced by Shh in both LG- and HG-grown cells. Notably, cell proliferation ability and Gli1 expression were similar in LG and HG+Shh conditions, suggesting that hedgehog may be a target of HG-mediated signaling. To further examine the association of hedgehog signaling in the HG-induced damage of fibroblasts, JNK activity was analyzed in HG- and Shh-stimulated cells. The present results demonstrated that JNK phosphorylation was activated by Shh and suppressed by HG. Treatment with Shh and HG induced an increase in JNK phosphorylation. In addition, sp600125, a JNK specific inhibitor, was used, and cell proliferation was analyzed, which indicated that sp600125 inhibited JNK activity, consequently inhibiting cell proliferation, whereas Shh treatment partially rescued these effects. These results indicate that JNK positively regulates cell proliferation downstream of HG and Shh signaling. As a relatively lower concentration of sp600125 may not completely suppress JNK activity, Shh treatment may be able to partially rescue the inhibited cell proliferation. As Shh stimulation activated JNK activity, the effect of altered JNK function on hedgehog signaling was further explored. sp600125 was added to fibroblasts in LG medium and SMO, Ptch1, and Gli1 expression was evaluated. The data indicate that JNK positively regulates the expression of key hedgehog signaling-related genes and suggest that hedgehog signaling is activated via Shh, which binds the receptor to activate downstream signaling, and that JNK is downstream of hedgehog activated by Shh stimuli. However, JNK also regulates expression of SMO, Ptch1, and Gli1. These three genes were induced by Shh stimulation and suppressed by HG stress, implying that JNK may be a key downstream factor through which HG and hedgehog signaling regulate expression of SMO, Ptch1, and Gli1. Furthermore, Gli1 overexpression eliminated HG- and sp600125-mediated effects on cell proliferation, further suggesting that Gli1 function is downstream of HG and JNK signaling.
Previously, it was demonstrated that HG-represses JNK signaling to increase ROS production in human fibroblasts. A previous study (10) determined that as Shh treatment activates JNK activity, Gli1 overexpression reduced impact of treatment with a JNK inhibitor. Therefore, the ROS production in fibroblasts and in skin from diabetic rats was analyzed. HG or diabetes highly induced ROS generation in fibroblasts and rat skin, and the activation of hedgehog signaling via Shh treatment and Gli1 overexpression in diabetic rat skin and HG-stimulated fibroblasts, respectively, reduced ROS production. These data suggested that activation of hedgehog signaling reduces the effect of HG, possibly via inhibition of ROS overproduction (10). Further analysis is required to elucidate the associations between ROS generation and hedgehog signaling in human fibroblasts. Although the linkage between Gil1-JNK-ROS was identified in the present study; however the regulation of this process was not determined. Further study may be required to assess how HG stress inhibits hedgehog signaling.
The findings of the present study are unique, to the best of our knowledge, and provide important insights that may help elucidate the regulatory basis of skin ulcers induced by diabetes. Elucidating the mechanism of hedgehog signaling activation to protect fibroblasts from HG damage may provide a molecular target for clinical treatment.
Acknowledgements
Not applicable.
Funding
The present study was supported by National Natural Science Foundation of China (grant no. 81602132), and grants from the Department of Science and Technology of Henan Province (grant nos. 172102310584 and 182102310242), the Education Department of Henan Province (grant nos. 201610472017 and 201610472040). The present study was also supported by Institute of Precision Medicine Taihang Young Scholar Foundation of Xinxiang Medical University, Doctoral Scientific Research Foundation of Xinxiang Medical University (grant nos. XYBSKYZZ201513 and 201512).
Availability of data and materials
All data generated or analyzed during this study are included in this paublished paper.
Authors' contributions
NS and HW coordinated the present study and wrote the manuscript; TG, JQ, JY, YQ, QC, YZ, YC, QH and XM performed and analyzed the experiments; and TS and ZF analyzed and reviewed the data. All authors contributed to the interpretation of data, manuscript revision and critical discussion.
Ethics approval and consent to participate
The present study was approved by the Ethics Committee of Xinxiang Medical University (Xinxiang, China), and written informed consent was obtained from all patients.
Consent for publication
Written informed consent was obtained from all patients.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Shi Y, Hu FB. The global implications of diabetes and cancer. Lancet. 2014;383:1947–1948. doi: 10.1016/S0140-6736(14)60886-2. [DOI] [PubMed] [Google Scholar]
- 2.Yach D, Stuckler D, Brownell KD. Epidemiologic and economic consequences of the global epidemics of obesity and diabetes. Nature Med. 2006;12:62–66. doi: 10.1038/nm0106-62. [DOI] [PubMed] [Google Scholar]
- 3.Braiman-Wiksman L, Solomonik I, Spira R, Tennenbaum T. Novel insights into wound healing sequence of events. Toxicol Pathol. 2007;35:767–779. doi: 10.1080/01926230701584189. [DOI] [PubMed] [Google Scholar]
- 4.Brem H, Tomic-Canic M. Cellular and molecular basis of wound healing in diabetes. J Clin Invest. 2007;117:1219–1222. doi: 10.1172/JCI32169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martin P. Wound healing-aiming for perfect skin regeneration. Science. 1997;276:75–81. doi: 10.1126/science.276.5309.75. [DOI] [PubMed] [Google Scholar]
- 6.Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: Sparking the development of diabetic vascular injury. Circulation. 2006;114:597–605. doi: 10.1161/CIRCULATIONAHA.106.621854. [DOI] [PubMed] [Google Scholar]
- 7.Obayashi K, Akamatsu H, Okano Y, Matsunaga K, Masaki H. Exogenous nitric oxide enhances the synthesis of type I collagen and heat shock protein 47 by normal human dermal fibroblasts. J Dermatol Sci. 2006;41:121–126. doi: 10.1016/j.jdermsci.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 8.Loots MA, Lamme EN, Mekkes JR, Bos JD, Middelkoop E. Cultured fibroblasts from chronic diabetic wounds on the lower extremity (non-insulin-dependent diabetes mellitus) show disturbed proliferation. Arch Dermatol Res. 1999;291:93–99. doi: 10.1007/s004030050389. [DOI] [PubMed] [Google Scholar]
- 9.Rowe DW, Starman BJ, Fujimoto WY, Williams RH. Abnormalities in proliferation and protein synthesis in skin fibroblast cultures from patients with diabetes mellitus. Diabetes. 1977;26:284–290. doi: 10.2337/diab.26.4.284. [DOI] [PubMed] [Google Scholar]
- 10.Xuan YH, Huang BB, Tian HS, Chi LS, Duan YM, Wang X, Zhu ZX, Cai WH, Zhu YT, Wei TM, et al. High-glucose inhibits human fibroblast cell migration in wound healing via repression of bFGF-regulating JNK phosphorylation. PLoS One. 2014;9:e108182. doi: 10.1371/journal.pone.0108182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pang L, Wang Y, Zheng M, Wang Q, Lin H, Zhang L, Wu L. Transcriptomic study of high glucose effects on human skin fibroblast cells. Mol Med Rep. 2016;13:2627–2634. doi: 10.3892/mmr.2016.4822. [DOI] [PubMed] [Google Scholar]
- 12.Bushman W. In Prostate Cancer. Springer; 2007. Hedgehog Signaling in Development and Cancer; pp. 107–118. [DOI] [Google Scholar]
- 13.Taipale J, Beachy PA. The Hedgehog and Wnt signalling pathways in cancer. Nature. 2001;411:349–354. doi: 10.1038/35077219. [DOI] [PubMed] [Google Scholar]
- 14.Jenkins D. Hedgehog signaling in development and disease. Encyclopedia Cell Biol. 2016;3:76–85. doi: 10.1016/B978-0-12-394447-4.30007-4. [DOI] [Google Scholar]
- 15.Beauchamp EM, Ringer L, Bulut G, Sajwan KP, Hall MD, Lee YC, Peaceman D, Ozdemirli M, Rodriguez O, Macdonald TJ, et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J Clin Invest. 2011;121:148–160. doi: 10.1172/JCI42874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang K, Pan L, Che X, Cui D, Li C. Sonic Hedgehog/GLI1 signaling pathway inhibition restricts cell migration and invasion in human gliomas. Neurol Res. 2010;32:975–980. doi: 10.1179/016164110X12681290831360. [DOI] [PubMed] [Google Scholar]
- 17.Huangfu D, Anderson KV. Signaling from Smo to Ci/Gli: Conservation and divergence of Hedgehog pathways from Drosophila to vertebrates. Development. 2006;133:3–14. doi: 10.1242/dev.02169. [DOI] [PubMed] [Google Scholar]
- 18.Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317:372–376. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- 19.Takebe N, Hunsberger S, Yang SX. Expression of Gli1 in the hedgehog signaling pathway and breast cancer recurrence. Chin J Cancer Res. 2012;24:257–258. doi: 10.1007/s11670-012-0260-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 21.Walker SJ, Worst TJ, Vrana KE. Semiquantitative real-time PCR for analysis of mRNA levels. Methods Mol Med. 2003;79:211–227. doi: 10.1385/1-59259-358-5:211. [DOI] [PubMed] [Google Scholar]
- 22.Tao L, Li X, Zhang L, Tian J, Li X, Sun X, Li X, Jiang L, Zhang X, Chen J. Protective effect of tetrahydroxystilbene glucoside on 6-OHDA-induced apoptosis in PC12 cells through the ROS-NO pathway. PLoS One. 2011;6:e26055. doi: 10.1371/journal.pone.0026055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu ZX, Cai WH, Wang T, Ye HB, Zhu YT, Chi LS, Duan YM, Sun CC, Xuan YH, Jin LT. bFGF-regulating MAKPs are involved in high-glucose-mediated ROS production and delay of vascular endothelial cell migration. PLoS One. 2015;10:e0144495. doi: 10.1371/journal.pone.0144495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lamers ML, Almeida ME, Vicente-Manzanares M, Horwitz AF, Santos MF. High glucose-mediated oxidative stress impairs cell migration. PLoS One. 2011;6:e22865. doi: 10.1371/journal.pone.0022865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu P, Wang Z, Sun X, Chen X, Zeng S, Chen L, Li S. Hydrogen-rich medium protects human skin fibroblasts from high glucose or mannitol induced oxidative damage. Biochem Biophys Res Commun. 2011;409:350–355. doi: 10.1016/j.bbrc.2011.05.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this paublished paper.