

Abstract
To uncover the genes associated with the development of esophageal squamous cell carcinoma (ESCC), an ESCC microarray dataset was used to identify genes differentially expressed between ESCC and normal control tissues. The dataset GSE17351 was downloaded from the Gene Expression Omnibus, containing 5 tumor esophageal mucosa samples and 5 adjacent normal esophageal mucosa samples from 5 male patients with ESCC. The differentially expressed genes (DEGs) were identified using the Linear Models for Microarray Data R package. Then, a co-expression network was constructed using the Weighted Correlation Network Analysis (WGCNA) package, and co-expression network modules were obtained with a hierarchical clustering algorithm. Additionally, functional enrichment analyses for DEGs in the top 2 modules with the highest significance were respectively conducted using the WGCNA package and the cluster Profiler package. In total, 487 upregulated and 468 downregulated DEGs were identified. A total of 24 modules were obtained from the co-expression network, and the top 2 modules with the highest significance, designated as ‘blue4’ and ‘magenta’, were further analyzed. In the module blue4, DEGs were significantly enriched in a number of Gene Ontology terms, including ‘spindle organization’ [e.g., ubiquitin conjugating enzyme E2 C (UBE2C) and SAC3 domain containing 1] and ‘cell cycle process’ [e.g., UBE2C, minichromosome maintenance complex component 6 (MCM6) and cell division cycle 20 (CDC20)]. Furthermore, a number of DEGs (e.g., UBE2C, CDC20 and MCM6) were enriched in the ‘cell cycle’ and ‘ubiquitin mediated proteolysis’ pathways. In the module ‘magenta’, a number of DEGs [e.g., transferrin receptor (TFRC) and TEA domain transcription factor 4 (TEAD4)] were enriched in the primary metabolic process and intracellular membrane-bounded organelle. Additionally, 308 upregulated genes and 215 downregulated genes were differentially expressed in the same pattern in another dataset, GSE20347, including UBE2C, CDC20, MCM6, TFRC, TEAD4, protein phosphatase 1 regulatory subunit 3C and MAL, T-cell differentiation protein. These DEGs may function in the development of ESCC.
Keywords: esophageal squamous cell carcinoma, differentially expressed gene, co-expression network, module
Introduction
Esophageal squamous cell carcinoma (ESCC) is one of histologic subtypes of esophageal cancer, and it occurs at a relatively high frequency in China, with five-year survival rates of 14% in Hong Kong (1). However, the molecular mechanism underlying the development of ESCC remains poorly understood.
In recent years, substantial advances have been made in ESCC research. Signal transducer and activator of transcription 3 (STAT3) has been demonstrated to be upregulated by the β-catenin/T cell factor pathway in ESCC (2). STAT3β expression is significantly associated with a shorter survival time for patients with ESCC, and it may suppress the oncogenic effects of STAT3α in ESCC cell lines (3). Furthermore, xerophilusin B can induce the G2/M cell cycle arrest and apoptosis of ESCC cells (4). A previous study identified that extracellular matrix protein 1b is downregulated in ESCC compared with normal esophageal tissues, and that it served a potential suppressive function in tumorigenesis and metastasis (1). Additionally, plasma matrix metalloproteinase 1 was observed to be highly expressed in ESCC compared with normal esophageal tissues, and it may have contributed to the detection and survival prediction of ESCC (5). However, the pathogenesis of ESCC is not yet completely characterized.
In 2010, Lee et al (6) performed gene expression profiling to investigate target genes for hypoxia-inducible factor (HIF) in the esophageal tumor microenvironment; the study identified a number of HIF target genes, including prostaglandin E synthase, cyclooxygenase 2 and insulin-like growth factor binding protein-3. However, the co-expression networks and functional enrichment analysis of the differentially expressed genes (DEGs) in the ESCC samples were not investigated. In the present study, the microarray dataset of GSE17351 produced by Lee et al (6) was analyzed to identify DEGs in ESCC samples. Following this, the Weighted Correlation Network Analysis (WGCNA), a systems biology method for identifying the correlation between genes across microarray samples (7), was used to analyze co-expression networks for the upregulated DEGs. Subsequently, gene ontology (GO) and pathway enrichment analyses were performed for the DEGs in the two most significant network modules. Additionally, the DEGs were validated using another gene expression dataset, GSE20347 (8) from the Gene Expression Omnibus (GEO). These results may contribute to an improved understanding of the etiology of ESCC.
Materials and methods
Affymetrix microarray data
The GSE17351 microarray expression profile (6), based on the platform of the GPL570 [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array (Affymetrix; Thermo Fisher Scientific, Inc., Waltham, MA, USA), was downloaded from the GEO (http://www.ncbi.nlm.nih.gov/geo/) database. The dataset contains 5 tumor esophageal mucosa samples and 5 adjacent normal esophageal mucosa samples from 5 male patients with ESCC, with an age range of 51–76 years. All esophageal tissues were originally obtained through surgery at the Okayama University Hospital, Kitano Hospital and the Hospital of the University of Pennsylvania through the Cooperative Human Tissue Network (6).
Data preprocessing
The expression values of all probes in each sample were reduced to a single value by determining the mean expression value via the aggregate function method (9). Missing data were assigned using the k-nearest neighbor method (10). Quantile normalization for complete data was performed using the preprocess Core package in Bioconductor (11). When numerous probes were mapped to one gene, the mid-value of the data was defined as the expression level of the gene. However, when numerous genes were mapped by one probe, this probe was considered to lack specificity, and was removed from the analysis.
Identification of DEGs
The Linear Models for Microarray Data package of Bioconductor (12) was used to identify genes that were significantly differentially expressed in ESCC samples. The raw P-value was adjusted using the Benjamin and Hochberg method (13), and a |log2 fold change (FC)|>0.585 and P<0.05 were selected as the cut-off criteria.
Construction of co-expression networks and identification of co-expression network modules
The WGCNA package of R (7) was used to analyze the co-expression network for DEGs, and the co-expression networks were visualized using Cytoscape (Version 3.2.0) (14). The weighting coefficient β was set to 25. The adjacency matrix power method (15) was used to transfer matrixes to weighted co-expression networks.
Co-expression network modules were obtained using a hierarchical clustering algorithm (16). The number of genes in each module was at least 30. Then, the significant modules were identified using correlation coefficient and network significance methods included in the WGCNA package. Gene significance (GS) measure was defined as a function GS, and a module significance (MS) measure as a mean of the GS in the module. A larger MS value indicated a greater association of a module with ESCC.
GO and pathway enrichment analyses for DEGs in the significant modules
The WGCNA package was used to obtain significant GO terms for DEGs in the significant modules, and the cluster Profiler package of R (17) was used to perform Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis for DEGs in modules. P<0.05 was selected as the cut-off criterion.
Data validation of the DEGs
The gene expression dataset GSE20347 (8) from GEO was used to validate the expression of the identified DEGs. The dataset included the data from 17 micro-dissected ESCC tumor tissues and 17 matched normal adjacent tissues from patients with ESCC. The data were with the GPL571 [HG-U133A_2] Affymetrix Human Genome U133A 2.0 Array platform (Affymetrix; Thermo Fisher Scientific, Inc.).
Data preprocessing and DEG identification were performed with the same methods as for the data in GSE17351. The overlapping upregulated and downregulated DEGs between GSE20347 and GSE17351 were identified and illustrated using an online Venn diagram plotter tool (https://omics.pnl.gov/software/venn-diagram-plotter). The overlapping DEGs were considered to be preliminarily validated by the GSE20347 dataset.
Results
Identification of DEGs
Following data preprocessing, a total of 6,899 genes in 10 samples were excluded. Based on the cut-off criteria, 955 DEGs were identified, including 487 upregulated and 468 downregulated DEGs.
Analysis of co-expression network modules
A total of 24 co-expression network modules were identified. Of these 24, the modules ‘blue4’ and ‘magenta’ were the most significantly associated with ESCC (Fig. 1).
Figure 1.

Relative strength of the association of co-expressed network modules with esophageal squamous cell carcinoma. The x-axis displays the names of the modules.
Construction of co-expression subnetworks for the blue4 and magenta modules
The top 30 genes with the highest connectivity degree in the modules blue4 and magenta were selected for the construction of co-expression subnetworks. In the co-expression subnetworks of the modules blue4 (Fig. 2) and magenta (Fig. 3), there were 435 gene interactions per module.
Figure 2.

Interaction network for the co-expression module ‘blue4’. The size of the nodes is proportional to the value of the |log2fold change|; gray nodes represent the downregulated genes and white nodes represent the upregulated genes. The width of the lines is proportional to the co-expression coefficient.
Figure 3.

Interaction network for the co-expression module ‘magenta’. The size of the nodes is proportional to the value of |log2fold change|; gray nodes represent the downregulated genes and white nodes represent the upregulated genes. The width of lines is proportional to the co-expression coefficient.
It was observed that a number of DEGs [including cysteine rich secretory protein 3 (CRISP3), neural EGFL like 2 (NELL2), MYB proto-oncogene like 2 (MYBL2) and plasminogen activator, urokinase (PLAU)] in the co-expression subnetwork of the module blue4 and a number of DEGs [including MAL, protein phosphatase 1 regulatory subunit 3C (PPP1R3C), cytochrome P450 family 3 subfamily A member 5 (CYP3A5) and biglycan (BGN)] in the co-expression subnetwork of the module magenta had a particularly high |log2FC|, indicating that these genes may have a greater extent of association with ESCC.
Enrichment analyses for the DEGs in the co-expression subnetworks of the modules blue4 and magenta
According to GO enrichment analysis, DEGs in the co-expression subnetwork of the module blue4 were predominantly enriched in GO terms regarding cell division, including ‘spindle organization’ [e.g., ubiquitin conjugating enzyme E2 C (UBE2C) and SAC3 domain containing 1 (SAC3D1)], ‘cell cycle process’ [e.g., UBE2C, minichromosome maintenance complex component 6 (MCM6) and cell division cycle 20 (CDC20)], ‘protein binding’ (e.g., UBE2C, MCM6 and CDC20) and ‘anaphase-promoting complex’ (APC; e.g., UBE2C and CDC20; Table I). In addition, two KEGG pathways were significantly enriched, including ‘cell cycle’ (CDC20 and MCM6) and ‘ubiquitin mediated proteolysis’ (e.g., UBE2C and CDC20; Table II).
Table I.
Top 5 most significantly enriched GO terms for the 30 differentially expressed genes with the highest connectivity degree in the module ‘blue4’ in BP and CC, and all significantly enriched terms in MF.
| Category | ID | Term | P-value | Count | Genes |
|---|---|---|---|---|---|
| BP | GO:0007051 | Spindle organization | 0.000091 | 5 | UBE2C, SAC3D1, MYBL2, AURKB, NCOR1 |
| BP | GO:0051225 | Spindle assembly | 0.000131 | 4 | SAC3D1, MYBL2, AURKB, NCOR1 |
| BP | GO:0022402 | Cell cycle process | 0.000602 | 10 | CDKN3, UBE2C, SAC3D1, MCM6, MYBL2, KAT2B, AURKB, NOLC1, NCOR1, CDC20 |
| BP | GO:0000280 | Nuclear division | 0.001058 | 6 | UBE2C, SAC3D1, MYBL2, AURKB, NOLC1, CDC20 |
| BP | GO:0007067 | Mitosis | 0.001058 | 6 | UBE2C, SAC3D1, MYBL2, AURKB, NOLC1, CDC20 |
| MF | GO:0005515 | Protein binding | 0.004683 | 22 | CDKN3, GPNMB, ADRM1, UBE2C, LAMB3, MCM6, NELL2, PLAU, NDRG2, CDC20… |
| MF | GO:0005488 | Binding | 0.032021 | 26 | GNE, CDKN3, GPNMB, ADRM1, UBE2C, MCM6, NELL2, PLAU, NDRG2, CDC20… |
| CC | GO:0005680 | Anaphase-promoting complex | 0.013075 | 2 | UBE2C, CDC20 |
| CC | GO:0031974 | Membrane-enclosed lumen | 0.013075 | 12 | UBE2C, CRAT, CACYBP, HNRNPA2B1, MCM6, NUDT1, CCDC86, KAT2B, AURKB, CDC20… |
| CC | GO:0005819 | Spindle | 0.013075 | 4 | SAC3D1, AURKB, NCOR1, CDC20 |
| CC | GO:0000152 | Nuclear ubiquitin ligase complex | 0.013075 | 2 | UBE2C, CDC20 |
| CC | GO:0044427 | Chromosomal part | 0.018472 | 5 | MYBL2, KAT2B, AURKB, H2AFV, NCOR1 |
GO, Gene Ontology; BP, biological process; CC, cellular component; MF, molecular function.
Table II.
Enriched pathways for differentially expressed genes in the module ‘blue4’.
| ID | Description | P-value | Count | Genes |
|---|---|---|---|---|
| hsa04110 | Cell cycle | 0.022640607 | 2 | CDC20, MCM6 |
| hsa04120 | Ubiquitin mediated proteolysis | 0.026418251 | 2 | UBE2C, CDC20 |
DEGs in the co-expression subnetwork of the magenta module were mainly enriched in GO terms associated with metabolism, including ‘energy derivation by oxidation of organic compounds’ e.g., adenylate cyclase 3 (ADCY3), electron transfer flavoprotein dehydrogenase (ETFDH) and glucosidase α, acid (GAA), ‘primary metabolic process’ [e.g., CYP3A5, TEA domain transcription factor 4 (TEAD4) and transferrin receptor (TFRC)], and ‘intracellular membrane-bounded organelle’ (e.g., TEAD4 and TFRC; Table III). However, no KEGG pathways were significantly enriched in the DEGs of the co-expression subnetwork in the magenta module.
Table III.
Top 5 most enriched GO terms for differentially expressed genes in the ‘magenta’ module in BP and CC.
| Category | ID | Term | P-value | Count | Genes |
|---|---|---|---|---|---|
| BP | GO:0015980 | Energy derivation by oxidation of organic compounds | 0.018735 | 5 | ADCY3, ETFDH, GAA, ATP5O, PPP1R3C |
| BP | GO:0055114 | Oxidation-reduction process | 0.018735 | 6 | ADCY3, CYP3A5, ETFDH, GAA, ATP5O, PPP1R3C |
| BP | GO:0006091 | Generation of precursor metabolites and energy | 0.027604 | 5 | ADCY3, ETFDH, GAA, ATP5O, PPP1R3C |
| BP | GO:0044238 | Primary metabolic process | 0.027604 | 23 | CNPPD1, HSPE1, ATP5O, PPP1R3C, PRSS8, SNAPC3, TEAD4, TFRC, ZNF426, FBXO11… |
| BP | GO:0021680 | Cerebellar Purkinje cell layer development | 0.032836 | 2 | AARS, RORA |
| CC | GO:0044429 | Mitochondrial part | 0.022333 | 6 | MTERFD2, ETFDH, GATM, HSPE1, ATP5O, PINK1 |
| CC | GO:0043231 | Intracellular membrane-bounded organelle | 0.022333 | 23 | PSMA1, RORA, BGN, PINK1, SNAPC3, TEAD4, TFRC, EZR, ZNF426, FBXO11… |
| CC | GO:0043227 | Membrane-bounded organelle | 0.022333 | 23 | PSMA1, RORA, BGN, PINK1, SNAPC3, TEAD4, TFRC, EZR, ZNF426, FBXO11… |
| CC | GO:0031974 | Membrane-enclosed lumen | 0.023484 | 11 | MTERFD2, ETFDH, GATM, HSPE1, PSMA1, RORA, BGN, SNAPC3, TEAD4, FBXO11… |
| CC | GO:0044444 | Cytoplasmic part | 0.033213 | 18 | RNF11, HSPE1, MAL, ATP5O, BCAP29, PSMA1, BGN, PINK1, TFRC, EZR… |
GO, Gene Ontology; BP, biological process; CC, cellular component.
Validation of DEGs
In total, 308 upregulated genes (e.g., UBE2C, CDC20, MCM6, TFRC and TEAD4) and 215 downregulated genes (e.g., PPP1R3C and MAL) were identified as overlapping between the GSE17351 and GSE20347 datasets (Fig. 4).
Figure 4.
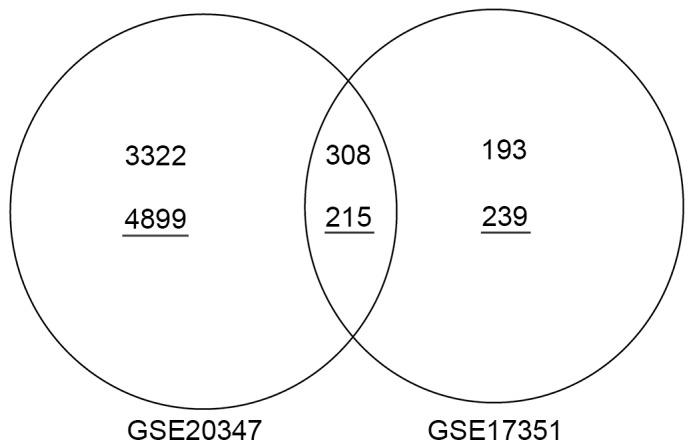
Venn diagram of the number of overlapping up- and downregulated genes between the datasets GSE17351 and GSE20347. The underlined numbers represent the number of downregulated genes; the numbers without an underline represent the number of upregulated genes.
Discussion
In the present study, a total of 955 DEGs were identified, including 487 upregulated and 468 downregulated DEGs. According to WGCNA analysis, two significant co-expression network modules, blue4 and magenta, were identified. The GO and KEGG pathway enrichment analyses for the top 30 DEGs with the highest connectivity degrees in the module blue4 revealed that a number of DEGs were associated with the ‘cell cycle’ and ‘ubiquitin mediated proteolysis’ KEGG pathways, including UBE2C, CDC20 and MCM6.
UBE2C encodes a member of the E2 ubiquitin-conjugating enzyme family, which is involved in protein ubiquitination (18). UBE2C is associated with cell cycle progression and checkpoint control, as it modulates the degradation of short-lived proteins (19). A previous study reported that UBE2C expression was elevated in 73% (11 of 15) of esophageal adenocarcinoma samples relative to Barrett's metaplasia, and the transfection of UBE2C small interfering (si)RNA induced the inhibition of cell proliferation and a distortion in cell cycle distribution (20). Furthermore, key roles of UBE2C have been demonstrated in other cancer types, including hepatocellular carcinoma (21), cervical carcinoma (22), non-small cell lung cancer (23) and pancreatic ductal adenocarcinoma (24). UBE2C and CDC20 were identified as being associated with the term ‘anaphase-promoting complex’ in the present study. The APC serves a crucial role in modulating cell cycle progression via forming two functionally distinct E3 ubiquitin ligase subcomplexes, APCCdc20 and APCCadherin 1 (25). The inhibition of CDC20 by siRNA may induce G2/M cell cycle arrest and suppress cell growth; CDC20 is negatively regulated by p53 (26).
Accumulating evidence has demonstrated that CDC20 serves a notable function in the development and progression of human cancer (27). Thus, UBE2C and CDC20 may be associated with ESCC. MCM6 is essential for the initiation of eukaryotic genome replication. A previous study reported that MCM6 is potentially associated with the lymph node metastasis of ESCC (28), indicating the crucial role of MCM6 in ESCC. In the network, UBE2C, CDC20 and MCM6 were all connected with CRISP3, which had a high |log2FC|. The low expression of CRISP3 in ESCC compared with normal tissue was identified in a previous study (29), consistent with the result of the present study. The DNA copy number loss of CRISP3 has been demonstrated in oral squamous cell carcinoma. Thus, it may be speculated that CRISP3 is associated with the carcinogenesis of ESCC. Furthermore, NELL2 was observed to have a higher |log2FC| in the co-expression subnetwork of the module blue4 in the present study; it was coexpressed with UBE2C, CDC20, MCM6 and CRISP3. A homolog of NELL2, it has been demonstrated that the promoter hypermethylation of NELL1 is higher in ESCC than in normal esophagus tissues, and that it is associated with poor prognosis in early-stage esophageal adenocarcinoma (30). Hence, it may be speculated that NELL2 may function in the occurrence of ESCC.
In the co-expression subnetwork of the magenta module, the upregulated TFRC and TEAD4, in addition to the downregulated PPP1R3C, MAL and CYP3A5 were observed to have the highest |log2FC|. A previous study reported that the elevated expression of TFRC was associated with the distant metastasis of ESCC, and patients with positive results for TFRC mRNA expression have a notably worse prognosis (31). Additionally, TEAD4 is a member of the transcriptional enhancer factor (TEA) family of transcription factors (32). TEAD and its coactivators may co-activate gene transcription, and it is pivotal for physiologically important processes including cell proliferation, cell differentiation and stem cell maintenance (32). One coactivator of TEAD, Yes-associated protein, is reported to be overexpressed in primary ESCC tumors (33). PPP1R3C catalyzes reversible protein phosphorylation, which is important in a range of cellular activities. Downregulated PPP1R3C was previously observed to be associated with lymph node metastasis in ESCC (34) and was identified as potentially contributing to the development of ESCC (35). In the present study, TFRC, TEAD4 and PPP1R3C were enriched in ‘primary metabolic processes’. Iron metabolism was identified to be altered in esophageal adenocarcinoma, and the overexpression of TFRC was associated with increased iron deposition in esophageal adenocarcinoma (36). Thus, TFRC, TEAD4 and PPP1R3C may serve important roles in the development of ESCC.
Downregulated MAL in ESCC has been identified in previous studies (37,38), which is consistent with the results of the present study. In esophageal cancer, MAL is able to suppress esophageal cancer cell motility, invasion and tumorigenicity and promote apoptosis via the Fas pathway (39), indicating a potential role for MAL in esophageal cancer. Additionally, genetic polymorphisms of CYP3A5 in combination with the sulfotransferase family 1A member 1 *2/*2 genotype are associated with an increased risk of esophageal cancer (40), suggesting a crucial role for CYP3A5 in esophageal cancer.
In the present study, DEGs were also validated using another ESCC gene expression dataset, GSE20347. A total of 308 upregulated genes and 215 downregulated genes were differentially expressed in the same pattern in GSE20347, including the aforementioned upregulated genes UBE2C, CDC20, MCM6, TFRC and TEAD4, in addition to the downregulated genes PPP1R3C and MAL. The validation results further indicate that these genes may serve crucial functions in the progression of ESCC.
However, the present study has a number of limitations. Potential microRNAs and transcription factors targeting the identified DEGs should have been identified, and these predictions should have been validated by experiments. In further studies, investigation of the etiology of ESCC will be performed in depth.
In conclusion, in the present study, 487 upregulated and 468 downregulated DEGs were identified. A number of DEGs (e.g., UBE2C, CDC20 and MCM6) enriched in ‘cell cycle’ and ‘ubiquitin mediated proteolysis’, those (e.g., TFRC and TEAD4) enriched in ‘primary metabolic process’ and ‘intracellular membrane-bounded organelle’, and a number of others (e.g., CRISP3, NELL2, PPP1R3C, MAL and CYP3A5), may be important in the initiation and development of ESCC.
Acknowledgements
Not applicable.
Funding
No funding received.
Availability of data and materials
Not applicable.
Author's contributions
XW conceived and designed the research and drafted the manuscript. GL and QL acquired the data, analyzed and interpreted the data and performed statistical analysis. CG conceived and designed the research and revised the manuscript to present important, intellectual content.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Yu VZ, Ko JM, Law S, Wang LD, Lung ML. Abstract 1158: Differential expression and functional impact of the alternatively spliced transcripts of extracellular matrix protein 1 in esophageal squamous cell carcinoma. Cancer Res. 2016;76:1158. doi: 10.1158/1538-7445.AM2016-1158. [DOI] [Google Scholar]
- 2.Yan S, Zhou C, Zhang W, Zhang G, Zhao X, Yang S, Wang Y, Lu N, Zhu H, Xu N. β-catenin/TCF pathway upregulates STAT3 expression in human esophageal squamous cell carcinoma. Cancer Lett. 2008;271:85–97. doi: 10.1016/j.canlet.2008.05.035. [DOI] [PubMed] [Google Scholar]
- 3.Zhang H, Lai R, Li E, Xu L. Abstract 3507: STAT3beta suppresses tumorigenesis via modulating the phosphorylation dynamics and transcription activity of STAT3alpha in esophageal squamous cell carcinoma. Cancer Res. 2014;74(19 Suppl):S3507–S3507. doi: 10.1158/1538-7445.AM2014-3507. [DOI] [Google Scholar]
- 4.Yao R, Chen Z, Zhou C, Luo M, Shi X, Li J, Gao Y, Zhou F, Pu J, Sun H, He J. Xerophilusin B induces cell cycle arrest and apoptosis in esophageal squamous cell carcinoma cells and does not cause toxicity in nude mice. J Nat Prod. 2015;78:10–16. doi: 10.1021/np500429w. [DOI] [PubMed] [Google Scholar]
- 5.Chen YK, Tung CW, Lee JY, Hung YC, Lee CH, Chou SH, Lin HS, Wu MT, Wu IC. Plasma matrix metalloproteinase 1 improves the detection and survival prediction of esophageal squamous cell carcinoma. Sci Rep. 2016;6:30057. doi: 10.1038/srep30057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee JJ, Natsuizaka M, Ohashi S, Wong GS, Takaoka M, Michaylira CZ, Budo D, Tobias JW, Kanai M, Shirakawa Y, et al. Hypoxia activates the cyclooxygenase-2-prostaglandin E synthase axis. Carcinogenesis. 2010;31:427–434. doi: 10.1093/carcin/bgp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Langfelder P, Horvath S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu N, Clifford RJ, Yang HH, Wang C, Goldstein AM, Ding T, Taylor PR, Lee MP. Genome wide analysis of DNA copy number neutral loss of heterozygosity (CNNLOH) and its relation to gene expression in esophageal squamous cell carcinoma. BMC Genomics. 2010;11:576. doi: 10.1186/1471-2164-11-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li X. An aggregate function method for nonlinear programming. Sci In China (A) 1991;34:1467–1473. [Google Scholar]
- 10.Altman NS. An introduction to kernel and nearest-neighbor nonparametric regression. American Statist. 1992;46:175–185. doi: 10.1080/00031305.1992.10475879. [DOI] [Google Scholar]
- 11.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 12.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 13.Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J Royal Stat Soc (Methodological) 1995;57:289–300. [Google Scholar]
- 14.Kohl M, Wiese S, Warscheid B. Cytoscape: Software for visualization and analysis of biological networks. Methods Mol Biol. 2011;696:291–303. doi: 10.1007/978-1-60761-987-1_18. [DOI] [PubMed] [Google Scholar]
- 15.Rücker G, Rücker C. On using the adjacency matrix power method for perception of symmetry and for isomorphism testing of highly intricate graphs. J Chem Inf Comput Sci. 1991;31:123–126. doi: 10.1021/ci00001a022. [DOI] [PubMed] [Google Scholar]
- 16.Wang J, Li M, Chen J, Pan Y. A fast hierarchical clustering algorithm for functional modules discovery in protein interaction networks. IEEE/ACM Trans Comput Biol Bioinform. 2011;8:607–620. doi: 10.1109/TCBB.2010.75. [DOI] [PubMed] [Google Scholar]
- 17.Nepusz T, Yu H, Paccanaro A. Detecting overlapping protein complexes in protein-protein interaction networks. Nat Methods. 2012;9:471–472. doi: 10.1038/nmeth.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scheffner M, Nuber U, Huibregtse JM. Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature. 1995;373:81–83. doi: 10.1038/373081a0. [DOI] [PubMed] [Google Scholar]
- 19.Jesenberger V, Jentsch S. Deadly encounter: Ubiquitin meets apoptosis. Nat Rev Mol Cell Biol. 2002;3:112–121. doi: 10.1038/nrm731. [DOI] [PubMed] [Google Scholar]
- 20.Lin J, Raoof DA, Wang Z, Lin MY, Thomas DG, Greenson JK, Giordano TJ, Orringer MB, Chang AC, Beer DG, Lin L. Expression and effect of inhibition of the ubiquitin-conjugating enzyme E2C on esophageal adenocarcinoma. Neoplasia. 2006;8:1062–1071. doi: 10.1593/neo.05832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ieta K, Ojima E, Tanaka F, Nakamura Y, Haraguchi N, Mimori K, Inoue H, Kuwano H, Mori M. Identification of overexpressed genes in hepatocellular carcinoma, with special reference to ubiquitin-conjugating enzyme E2C gene expression. Int J Cancer. 2007;121:33–38. doi: 10.1002/ijc.22605. [DOI] [PubMed] [Google Scholar]
- 22.Rajkumar T, Sabitha K, Vijayalakshmi N, Shirley S, Bose MV, Gopal G, Selvaluxmy G. Identification and validation of genes involved in cervical tumourigenesis. BMC Cancer. 2011;11:80. doi: 10.1186/1471-2407-11-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao L, Jiang L, Wang L, He J, Yu H, Sun G, Chen J, Xiu Q, Li B. UbcH10 expression provides a useful tool for the prognosis and treatment of non-small cell lung cancer. J Cancer Res Clin Oncol. 2012;138:1951–1961. doi: 10.1007/s00432-012-1275-2. [DOI] [PubMed] [Google Scholar]
- 24.Zhao ZK, Wu WG, Chen L, Dong P, Gu J, Mu JS, Yang JH, Liu YB. Expression of UbcH10 in pancreatic ductal adenocarcinoma and its correlation with prognosis. Tumor Biol. 2013;34:1473–1477. doi: 10.1007/s13277-013-0671-9. [DOI] [PubMed] [Google Scholar]
- 25.McLean JR, Chaix D, Ohi MD, Gould KL. State of the APC/C: Organization, function, and structure. Crit Rev Biochem Mol Biol. 2011;46:118–136. doi: 10.3109/10409238.2010.541420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kidokoro T, Tanikawa C, Furukawa Y, Katagiri T, Nakamura Y, Matsuda K. CDC20, a potential cancer therapeutic target, is negatively regulated by p53. Oncogene. 2008;27:1562–1571. doi: 10.1038/sj.onc.1210799. [DOI] [PubMed] [Google Scholar]
- 27.Smolders L, Teodoro JG. Targeting the anaphase promoting complex: Common pathways for viral infection and cancer therapy. Expert Opin Ther Targets. 2011;15:767–780. doi: 10.1517/14728222.2011.558008. [DOI] [PubMed] [Google Scholar]
- 28.Yamabuki T, Daigo Y, Kato T, Hayama S, Tsunoda T, Miyamoto M, Ito T, Fujita M, Hosokawa M, Kondo S, Nakamura Y. Genome-wide gene expression profile analysis of esophageal squamous cell carcinomas. Int J Oncol. 2006;28:1375–1384. [PubMed] [Google Scholar]
- 29.Zinovyeva MV, Monastyrskaya GS, Kopantzev EP, Vinogradova TV, Kostina MB, Sass AV, Filyukova OB, Uspenskaya NY, Sukhikh GT, Sverdlov ED. Identification of some human genes oppositely regulated during esophageal squamous cell carcinoma formation and human embryonic esophagus development. Dis Esophagus. 2010;23:260–270. doi: 10.1111/j.1442-2050.2009.01008.x. [DOI] [PubMed] [Google Scholar]
- 30.Jin Z, Mori Y, Yang J, Sato F, Ito T, Cheng Y, Paun B, Hamilton JP, Kan T, Olaru A, et al. Hypermethylation of the nel-like 1 gene is a common and early event and is associated with poor prognosis in early-stage esophageal adenocarcinoma. Oncogene. 2007;26:6332–6340. doi: 10.1038/sj.onc.1210461. [DOI] [PubMed] [Google Scholar]
- 31.Wada S, Noguchi T, Takeno S, Kawahara K. PIK3CA and TFRC located in 3q are new prognostic factors in esophageal squamous cell carcinoma. Ann Surg Oncol. 2006;13:961–966. doi: 10.1245/ASO.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 32.Pobbati AV, Hong W. Emerging roles of TEAD transcription factors and its coactivators in cancers. Cancer Biol Ther. 2013;14:390–398. doi: 10.4161/cbt.23788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muramatsu T, Imoto I, Matsui T, Kozaki K, Haruki S, Sudol M, Shimada Y, Tsuda H, Kawano T, Inazawa J. YAP is a candidate oncogene for esophageal squamous cell carcinoma. Carcinogenesis. 2011;32:389–398. doi: 10.1093/carcin/bgq254. [DOI] [PubMed] [Google Scholar]
- 34.Uchikado Y, Inoue H, Haraguchi N, Mimori K, Natsugoe S, Okumura H, Aikou T, Mori M. Gene expression profiling of lymph node metastasis by oligomicroarray analysis using laser microdissection in esophageal squamous cell carcinoma. Int J Oncol. 2006;29:1337–1347. [PubMed] [Google Scholar]
- 35.Shen Y, Tantai J, Zhao H. Ranking candidate genes of esophageal squamous cell carcinomas based on differentially expressed genes and the topological properties of the co-expression network. Eur J Med Res. 2014;19:52. doi: 10.1186/s40001-014-0052-x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36.Boult J, Roberts K, Brookes MJ, Hughes S, Bury JP, Cross SS, Anderson GJ, Spychal R, Iqbal T, Tselepis C. Overexpression of cellular iron import proteins is associated with malignant progression of esophageal adenocarcinoma. Clin Cancer Res. 2008;14:379–387. doi: 10.1158/1078-0432.CCR-07-1054. [DOI] [PubMed] [Google Scholar]
- 37.Hu YC, Lam KY, Law S, Wong J, Srivastava G. Identification of differentially expressed genes in esophageal squamous cell carcinoma (ESCC) by cDNA expression array: Overexpression of Fra-1, Neogenin, Id-1, and CDC25B genes in ESCC. Clin Cancer Res. 2001;7:2213–2221. [PubMed] [Google Scholar]
- 38.Luo A, Kong J, Hu G, Liew CC, Xiong M, Wang X, Ji J, Wang T, Zhi H, Wu M, Liu Z. Discovery of Ca2+-relevant and differentiation-associated genes downregulated in esophageal squamous cell carcinoma using cDNA microarray. Oncogene. 2004;23:1291–1299. doi: 10.1038/sj.onc.1207218. [DOI] [PubMed] [Google Scholar]
- 39.Mimori K, Shiraishi T, Mashino K, Sonoda H, Yamashita K, Yoshinaga K, Masuda T, Utsunomiya T, Alonso MA, Inoue H, Mori M. MAL gene expression in esophageal cancer suppresses motility, invasion and tumorigenicity and enhances apoptosis through the Fas pathway. Oncogene. 2003;22:3463–3471. doi: 10.1038/sj.onc.1206378. [DOI] [PubMed] [Google Scholar]
- 40.Dandara C, Li DP, Walther G, Parker MI. Gene-environment interaction: The role of SULT1A1 and CYP3A5 polymorphisms as risk modifiers for squamous cell carcinoma of the oesophagus. Carcinogenesis. 2006;27:791–797. doi: 10.1093/carcin/bgi257. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.