

Abstract
Brain-derived neurotrophic factor (BDNF) is identified as a potent neuroprotective and neuroregenerative agent for many neurological diseases. Regrettably, its delivery to the brain is hampered by poor serum stability and rapid brain clearance. Here, a novel nanoformulation is reported composed of a bio-compatible polymer, poly(ethylene glycol)-b-poly(L-glutamic acid) (PEG-PLE), that hosts the BDNF molecule in a nanoscale complex, termed here Nano-BDNF. Upon simple mixture, Nano-BDNF spontaneously forms uniform spherical particles with a core–shell structure. Molecular dynamics simulations suggest that binding between BDNF and PEG-PLE is mediated through electrostatic coupling as well as transient hydrogen bonding. The formation of Nano-BDNF complex stabilizes BDNF and protects it from nonspecific binding with common proteins in the body fluid, while allowing it to associate with its receptors. Following intranasal administration, the nanoformulation improves BDNF delivery throughout the brain and displays a more preferable regional distribution pattern than the native protein. Furthermore, intranasally delivered Nano-BDNF results in superior neuroprotective effects in the mouse brain with lipopolysaccharides-induced inflammation, indicating promise for further evaluation of this agent for the therapy of neurologic diseases.
Keywords: central nervous system, drug delivery, intranasal delivery, neurotrophic factors, polyion complexes
1. Introduction
Nanoformulations of therapeutic protein should enable efficient loading and retention of the protein in a nanocarrier, be stable in biological milieu, and ensure rapid release of a functionally active protein to interact with its receptor at the site of action. No current nanoparticle formulation has simultaneously achieved these goals because strong entrapment of the protein in a nanoparticle simultaneously restricts its availability to receptors. Current formulations solve this dilemma by releasing the protein over a period of time, after which the released protein immediately loses protection, often resulting in aggregation, activity loss or immunogenicity. Here we demonstrate that the brain-derived neurotrophic factor (BDNF) self-assembles with an anionic block copolymer into nanosized complexes, stabilized by cooperative electrostatic interactions and H-bonds. While the complexes remain stable in the presence of salt and serum proteins, active BDNF is readily abstracted from them upon interaction with its specific receptors. As a result, nanoformulated BDNF (Nano-BDNF) is as active as the free neurotrophin in activating its receptor and downstream signaling. The seamless transition between the “preservation” and “action” states of BDNF minimizes the potential side effects caused by nonspecific release. Nano-BDNF shows profoundly improved delivery to the brain and neuroprotective effects in vivo.
BDNF promotes neuronal survival and modulates long-term potentiation of synaptic transmission, which implicates this neurotrophin as a prime therapeutic candidate for a host of neurological disorders.[1] However, despite the ability of BDNF to cross the blood-brain barrier (BBB), it has a short serum half-life and is rapidly cleared from the brain, which severely limits the brain bioavailability of this neurotrophin after peripheral administration.[2] To bypass the BBB and minimize exposure to peripheral tissues, some proteins can be delivered more directly to the central nervous system (CNS) using the intranasal to brain (INB) route.[3] In this approach, the drug is administered into the nasal cavity to the level of the olfactory nerve/cribriform plate where it can travel up along the olfactory and/or trigeminal nerves to reach the CNS.[4] However, BDNF delivered via INB route is primarily found in the olfactory bulb and trigeminal nerve and shows little penetration to the therapeutically targeted brain regions, such as cortex or hippocampus.[5] The poor intranasal-to-brain bioavailability of BDNF can be attributed to its cationic surface charges and extensive binding to a variety of polysaccharides, resulting in its entrapment and degradation within the negatively charged and sialic-acid-rich nasal mucosa.[6] Although nasal adsorption of proteins can be ameliorated by PEGylation,[7] this approach requires covalent modification of BDNF with poly(ethylene-glycol) (PEG) chains, which can potentially hinder interaction of the neurotrophin with its receptor and therefore entails tedious separation of the more active populations.[8] In this study, we propose a new approach for noncovalent, spontaneous, and agile nanoformulation of BDNF by simply mixing it with PEG-poly(L-glutamic acid) (PEG-PLE) in an aqueous solution. Driven by electrostatic interactions and hydrogen bonding, BDNF and PEG-PLE form stable nanoparticles that are negatively charged and coated by noncovalently attached PEG chains. We present compelling data on the stability of these nanoparticles in physiological conditions, the unaffected activity of encapsulated BDNF, the improved nasal to brain delivery and the decreased brain clearance of BDNF in the Nano-BDNF formulation, which is novel and practically significant.
2. Results
2.1. A Simple and Straightforward Nanoformulation
Consistent with prior reports,[9] native BDNF forms large and highly heterogeneous aggregates with an effective diameter (Deff) of about 600 nm and a polydispersity index (PDI) greater than 0.4 (Figure 1a,b). Therefore, it cannot be used as an injectable pharmaceutical agent without proper formulation. Our approach addresses this problem by simply mixing BDNF and PEG-PLE in aqueous solutions without sonication or other extensive agitation. This resulted in spontaneous self-assembly of BDNF/PEG-PLE complexes that we will further refer to as “Nano-BDNF”. The properties of such complexes depend on the composition index of the mixture Z−/+, defined here as the molar ratio of the total number of glutamic acid residues of PEG-PLE to the combined number of lysine and arginine residues of BDNF. The Nano-BDNF particles were nearly monodispersed (PDI = 0.026–0.115) at the charge ratios (Z−/+) from 0.5 to 5 (Figure 1a,b). The particle size increased from 191 to 246 nm as Z−/+ changed from 0.5 to 3 and then remained essentially constant. At Z−/+ = 10, the polydispersity increased (PDI = 0.214), but it was diminished when we used a microfluidics mixer to mix the components (Figures S1 and S2, Supporting Information). Transmission electron microscopy (TEM) (Figure 1) and atomic force microscopy (AFM) (Figure 1d) suggested that Nano-BDNF consists primarily of smaller roundish shaped particles, while BDNF formed irregularly shaped aggregates.
Figure 1.
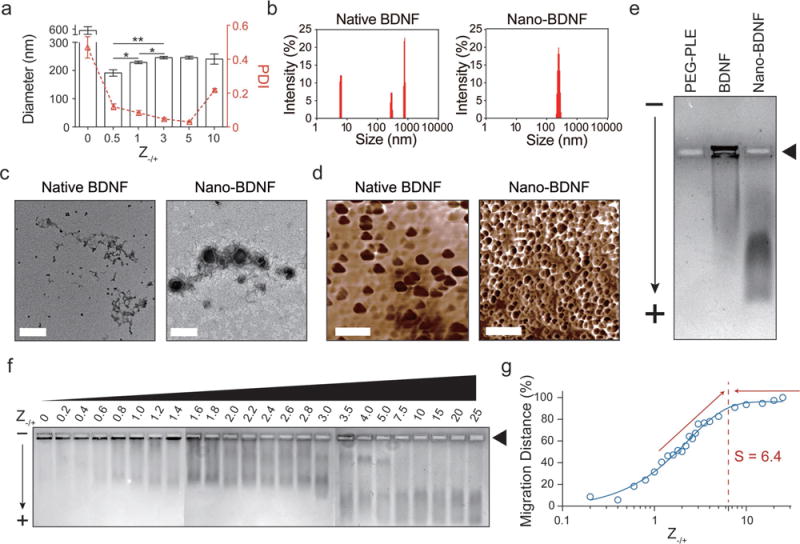
Characterization of Nano-BDNF’s physicochemical features and determination of its minimum saturation Z−/+ value. a) Effective hydrodynamic diameters of native BDNF and Nano-BDNF samples prepared at different Z−/+ values were measured with DLS. The Z-averaged diameter was plotted to the left Y-axis, and the PDI was plotted to the right Y-axis (n = 5). Statistical significance is defined as P < 0.05 and indicated by *, p < 0.05; **, p < 0.01. b) Representative intensity-size graphs of native BDNF and Nano-BDNF (Z−/+ = 5). c) Representative TEM images of native BDNF and Nano-BDNF (Z−/+ = 10) prepared in 10 × 10−3 M phosphate buffer, pH = 7.4 (scale bar = 200 nm). d) Representative AFM phase diagrams of native BDNF and Nano-BDNF (Z−/+ = 10) in 10 × 10−3 M phosphate buffer, pH = 7.4 (scale bar = 500 nm). e) The electrophoretic mobility of PEG-PLE, native BDNF, and Nano-BDNF (Z−/+ = 100) was determined in 0.5% agarose gel (pore size ≈ 500 nm)[43] at 80 V for 1 h. Samples were loaded in the rectangular wells indicated by the black triangle. The “−” and “+” signs denote cathode and anode, respectively. The black arrow indicates direction of electron flow. Commassie Blue staining was performed to visualize BDNF, since it has minimal staining of free PEG-PLE copolymer. Despite the positive charge on BDNF at the pH of the running buffer, a small fraction of BDNF was always observed to migrate toward the anode, possibly due to binding and subsequent charge-conversion by free agarose oligomers and/or polymers that are not completely immobilized in the gel. f) The electrophoretic mobility of Nano-BDNF prepared at different Z−/+ values were evaluated and presented in the same way as described in (e). Lanes 1–24 represent samples with different Z−/+ values from 0 to 25. g) To quantify the migration distance of BDNF, we located the center of mass of the BDNF smear in each lane of (f) and measured its distance from the loading well of that lane. The distances were then quantified and normalized with the value obtained at Z−/+ = 25 being 100% and plotted against their Z−/+ values on a logarithmic scale. A sigmoidal curve was fitted to the plot, and the minimum saturation Z−/+ value “S” was determined to be about 6.4 by elongating and intersecting the two linear portions of the curve on the graph.
2.2. Formation of Overcharged Nano-BDNF Species
Polyion complexes form as a result of electrostatic interactions between the reacting polyions.[10] In this regard, one can monitor formation of such complexes by the changes of the electrophoretic mobility of the reacting species in horizontal agarose gel electrophoresis (HAGE). Although naked BDNF is highly positively charged, it does not migrate in agarose gels due to strong binding with the agarose matrix (Figure 1e).[11] In contrast, Nano-BDNF migrates much further toward the anode suggesting that PEG-PLE prevented BDNF from binding with agarose. It also suggests that the positive charges of BDNF are neutralized by PEG-PLE and the complex particles acquired a net negative charge (Figure 1e). Notably, albeit the same amount of BDNF was loaded in every well in HAGE experiments, the Nano-BDNF bands always appeared fainter than the native BDNF bands after Coomassie Blue staining. This can be explained by the binding of Coomassie Blue dye to basic/aromatic amino acids on the protein,[12] both of which can be attenuated by encapsulation of the protein in a negatively charged polyion complex with a hydrophilic corona.
As the ratio of PEG-PLE to BDNF increases, we observed progressive incorporation of BDNF in the complex (Figure 1f). Several bands corresponding to different complex species emerged as the proportion of PEG-PLE increases, which suggested dispro-portionation, but eventually only one type of the complex species is observed. Surprisingly, the “saturation” and complete incorporation of BDNF in the complex is achieved at a high excess of PEG-PLE. By plotting the weighted distance of the complex migration in the gel against the charge ratio, the saturation Z−/+ in this experiment was determined to be 6.4 (Figure 1g). This value is further supported by an isothermal titration calorimetry (ITC) experiment where the exothermic reaction between BDNF and PEG-PLE was recorded until Z−/+ reached 6.8 (Figure S3, Supporting Information). Therefore, a considerable excess of PEG-PLE appears to have been incorporated in the complex. Hence, the complex becomes strongly negatively charged due to the excess of bound polyanion as was evident from the HAGE experiment.
2.3. Molecular Dynamics (MD) Simulation of the Complex Formation
Overcharging (incorporation of an excess of polyion) is common for polyion complexes like Nano-BDNF when interacting species can aggregate due to hydrophobic or other molecular interactions.[13] However, the charge excess of PEG-PLE (six to sevenfold) incorporated in Nano-BDNF seems to be unusually high compared to similar complexes of other proteins and nucleic acids. If one takes into account the length of the PLE—50 repeating units (r.u.), then each PLE chain in the block copolymer on average would interact with only seven to eight amino groups of the BDNF. Such a small number of ion pairs would be comparable with the minimum number needed for polyion coupling.[14] Consequently, such complexes would not be very stable (especially in the presence of low-molecular-mass salt). To better understand possible molecular interactions between the reacting species, we carried out MD simulations. In this approach, we simulated single PEG-PLE chains and a BDNF dimer for 100 ns using four starting seed positions (Figure S4A, Supporting Information). We observe association and binding in every simulation experiment, although the binding at any given residue is transient (Figure S4, Supporting Information). The interaction interface consists of many residues in BDNF and PEG-PLE that come into close contact and is indicative of hydrogen bonding. We used HBplus, a program designed to identify and analyze hydrogen bonds,[15] to determine if hydrogen bonds were formed. Thus, we located residues with high frequency of interaction and H-bond formation with the glutamic acid residues of PEG-PLE (Figure 2a,b and Table S1, Supporting Information). BDNF is a compact globular protein consisting of ≈70% ß-strands and 20% ß-turns.[16] Our simulations suggest that the binding sites are located primarily in the highly positively charged regions of the BDNF molecule, which are also characterized by high β-turn/random coil contents (Figure S5, Supporting Information, and Figure 2b,c). Furthermore, due to the distance between the cationic patches (Figure S5, Supporting Information), a single PEG-PLE chain is unable to saturate both sites present in the BDNF dimer. Hence, we simulated higher molar ratios of PEG-PLE to BDNF (Figure S6, Supporting Information) and found that the maximum number of PEG-PLE chains to form Nano-BDNF with a single BDNF dimer is 3, which is equivalent to Z−/+ of 3.4 and lower than the saturation Z−/+ value we determined earlier. We note that while the MD simulation serves well to locate the binding sites on BDNF, it does not capture the full complexity of the polyion complexes where more than one BDNF molecule participates in the interaction simultaneously as is evident from their size. Therefore, it appears that the combined effect of the crowding behavior of self-assembly and H-bond formation resulted in significant further overcharging of Nano-BDNF complexes.
Figure 2.
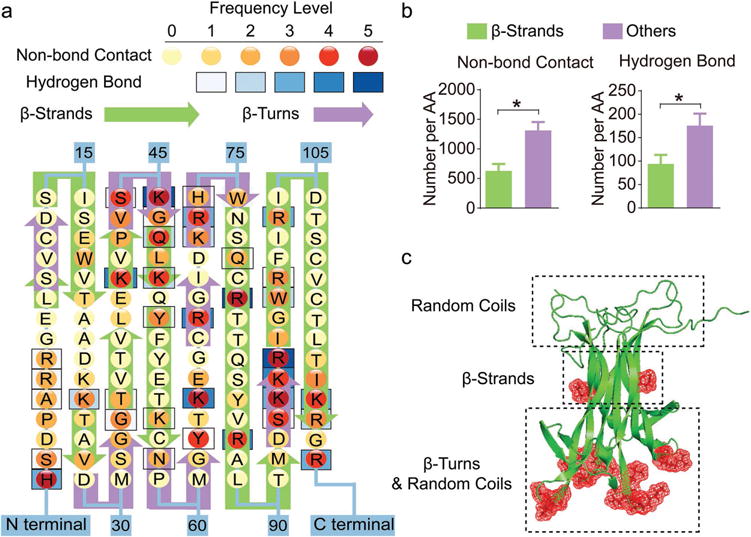
Binding sites with PEG-PLE primarily locate in regions that are rich in random coil and β-turn structures of BDNF. a) Frequency map of BDNF-polymer interactions on each amino acid of BDNF. The frequency levels of hydrogen bonds and nonbonding contacts are determined separately based on their total counts (summarized in Table S1, Supporting Information) in the trajectories obtained from four independent simulations with one BDNF dimer and one PEG-PLE chain. b) Quantification of BDNF–polymer interaction occurring on β-strands versus other structural domains. The result is presented as the number of occurrences averaged by the number of amino acids (denoted by AA in the figure) forming each structural domain. “*” denotes statistical significance between two groups (P < 0.05). c) Cartoon visualization of a BDNF dimer molecule with illustrations of predominant structural domains at each part of the protein. The red meshes represent amino acids that are actively involved in the binding between BDNF and its receptor, TrkB.
2.4. Nano-BDNF Complexes are Stable in the Presence of Salt and Proteins
Polyion complexes dissociate upon an increase in ionic strength, which usually proceeds in a narrow range of concentration with a low-molecular-mass electrolyte.[17] This range depends on the structure of the reacting species as well as the type of electrolyte, and normally does not exceed 0.3–0.7 M NaCl for most biologically relevant systems. Quite surprisingly, depending on the charge ratio, Nano-BDNF displayed two distinct types of behavior. The complexes prepared at low charge ratios (Z−/+ ≤ 3) appeared to disintegrate at 0.15 M NaCl (Figure 3a,b). In contrast, complexes prepared at Z−/+ ≥ 5 remain at least partially stable as observed by the presence of the fraction of Nano-BDNF migrating in the gel even in 1.05 M NaCl (Figure 3c–e). This fraction features lower migration velocity at high ionic strength, possibly due to partial detachment of BDNF from the complex and subsequent osmotic swelling of the remaining species, which is typical for this type of systems.[13d] Remarkably, at a very large excess of PEG-PLE (Z−/+ = 100), the amount of free BDNF that was stripped from the complex greatly diminished, suggesting an easy approach to prepare stable Nano-BDNF (Figure 3e). Altogether, the behavior involving differential stability of polyion complexes to ionic strength depending on the PEG-PLE excess is unusual. It could, however, be explained by the transition from a regular polyion complex formed at a lower Z−/+ ratio to the formation of a cooperative H-bond complex along with aggregation and structural rearrangement at high Z−/+ ratios where the complex saturates.
Figure 3.
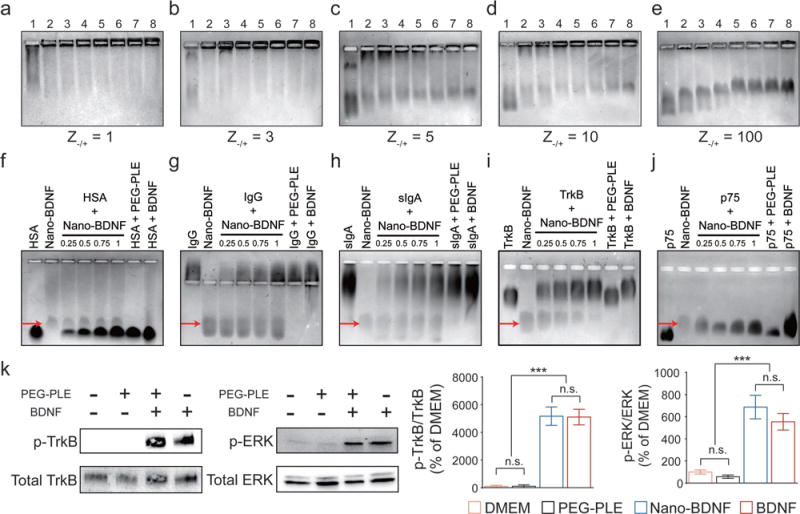
Nano-BDNF prevents BDNF from binding with nasal defensive proteins, while readily releasing active BDNF upon incubation with the BDNF receptors, TrkB and p75NTR. a–e) Nano-BDNF samples of different Z−/+ values were prepared in NaCl solutions before horizontal agarose gel electrophoresis. The concentration of NaCl in the Nano-BDNF samples gradually increased from lane 1 to lane 8 (0, 0.15, 0.30, 0.45, 0.60, 0.75, 0.90, and 1.05 M, respectively). The amount of free BDNF in each experimental condition can be estimated by the intensities of staining at the edge of the respective loading well. f–j) The nasal mucosal proteins f) HSA, g) IgG, h) sIgA, and BDNF receptors i) TrkB, j) p75NTR, were incubated with Nano-BDNF (Z−/+ = 200) in lactated ringer’s solution (LR) for 30 min before subjected to horizontal agarose gel electrophoresis. From left to right, lane 1: Competitor; lane 2: Nano-BDNF; lane 3: Competitor incubated with Nano-BDNF (molar ratio 0.25: 1); lane 4: Competitor incubated with Nano-BDNF (molar ratio 0.5: 1); lane 5: Competitor incubated with Nano-BDNF (molar ratio 0.75: 1); lane 6: Competitor incubated with Nano-BDNF (molar ratio 1:1); lane 7: Competitor incubated with PEG-PLE (same amounts as in lane 6); lane 8: Competitor incubated with native BDNF (molar ratio 1:1). Red arrows indicate the location of Nano-BDNF bands in each graph. k) Western blots showing the level of TrkB receptor phosphorylation at the site of Tyrosine 515 as well as the level of ERK phosphorylation in vitro after treatment with (from left to right) dulbecco’s modified eagle medium (DMEM), PEG-PLE, Nano-BDNF, or BDNF for 5 min at 37 °C (n = 6). Quantification of band density was presented by the ratio of phosphorylated TrkB over total TrkB. Data were analyzed with unpaired Student’s t-test between each two groups; *** denotes p < 0.001 and n.s. denotes no significant difference.
Aside from stability at physiological ionic strengths, polyion complexes for biomedical use need to be stable in the presence of natural polyelectrolytes present in biological fluids that can disintegrate the complex via a polyion interchange reaction.[17b] Therefore, we evaluated the integrity of Nano-BDNF using HAGE after incubation with proteins commonly found in body fluids, including human serum albumin (HSA), serum immunoglobulin G (IgG), and secretory immunoglobulin A (sIgA). Surprisingly, the negatively charged HSA showed no competency to disintegrate Nano-BDNF (Figure 3f), Similarly, serum IgG and sIgA do not compromise the integrity of Nano-BDNF as evidenced in Figure 3g,h. Stability in the presence of proteins may be explained by stabilization of the Nano-BDNF by a cooperative system of H-bonds as well as steric hindrance of the PEG corona, both of which can facilitate intravenous and intranasal delivery of our formulation.
2.5. Nano-BDNF Releases Active BDNF without Compromising Its Intrinsic Stability
The challenge of the use of nanoparticulate carriers is to maintain integrity of BDNF and release it uncompromised to the relevant receptor, tropomyosin receptor kinase B (TrkB) in the brain. In this context, we characterized the interaction of TrkB and Nano-BDNF by incubating the soluble, recombinant extracellular domain of TrkB with BDNF and Nano-BDNF, and then performing HAGE experiments. Incubation of native BDNF and TrkB yields a single species that migrates as a distinct band (Figure 3i). When mixed with Nano-BDNF, TrkB associates in a 1:1 stoichiometry with the BDNF dimer, thus causing the band containing Nano-BDNF to proportionally decrease in intensity. Formation of a TrkB-BDNF complex was further evidenced by impeded migration of the TrkB band in the agarose gel, which suggests charge neutralization (Figure 3i). Similar effects were observed upon mixing Nano-BDNF with a low-affinity BDNF receptor, p75NTR (Figure 3j). Therefore, both the high- and low-affinity receptors displace PEG-PLE and abstract BDNF from the Nano-BDNF complex, while HSA, serum IgG, and sIgA at the same molar ratios are not able to do so.
Some previous reports indicated that interaction with poly-electrolytes could destabilize protein cargos.[18] Hence, we analyzed the protein stability in two distinct assays to determine how the stability and functionality of BDNF were affected. We assayed the melting temperatures (Tm) of BDNF and Nano-BDNF using a fluorometric dye (which responds to environmental changes, namely, hydrophobicity). Here we show that native BDNF has a Tm value of ≈83 °C (Figure S7A, Supporting Information), consistent with previous reports.[19] We demonstrate that encapsulation in Nano-BDNF yields a slightly higher stability compared to naked BDNF (≈0.5 °C) at Z−/+ of 5 and 10 (Figure S7B, Supporting Information).
In order to determine if functionality was maintained as well, we incubated BDNF and Nano-BDNF with a genetically engineered NIH 3T3 cell line[20] which stably expresses TrkB without producing BDNF. Activation of TrkB was tested using a phosphospecific antibody to detect phosphorylation in the TrkB kinase domain. TrkB phosphorylation is observed at the same level when stimulated with BDNF and Nano-BDNF, indicating that the vehicle does not inhibit activity or association (Figure 3k). We also observed phosphorylated extracellular signal regulated kinase (ERK), a downstream target kinase,[21] at similar levels after TrkB stimulation with BDNF and Nano-BDNF (Figure 3k).
2.6. Nano-BDNF Enhanced the Delivery of BDNF to Different Brain Regions and Was Neuroprotective against Lipopolysaccharides (LPS)-Induced Neuroinflammation after INB Administration
We next performed an in vivo pharmacokinetic study to evaluate the ability of this formulation to deliver BDNF to the brain after INB administration, which is known to bypass the BBB and can potentially deliver therapeutic proteins to a wide range of brain regions.[4] Indeed, in most of the brain regions, both native and nanoformulated 125I-BDNF was readily detected as soon as 5 min after INB administration (Figure 4a). Within 30 min after INB administration, the level of Nano-BDNF accumulation in the brain was significantly higher than that of the native BDNF in all brain regions we examined, except for midbrain where there was a trend for the increase. The areas under the curve (AUC) from 0 to 30 min suggest that Nano-BDNF increases the delivery of the neurotrophin in all brain regions, most notably in the olfactory bulb (≈6.7 times), hippocampus (≈9.9 times), and brainstem (≈4.0 times) (Figure 4b and Table S2, Supporting Information). The overall increase for the whole brain was also significant albeit less dramatic (≈3.5 times) (Table S2, Supporting Information). By dividing the AUC for different brain regions we further concluded that, compared with native BDNF, the relative brain regional distribution pattern of BDNF after treatment with Nano-BDNF was very different. Specifically, Nano-BDNF was more preferable for distribution to the hippocampus, as compared with olfactory bulb, hypothalamus and the rest of the brain (Figure S8, Supporting Information).
Figure 4.
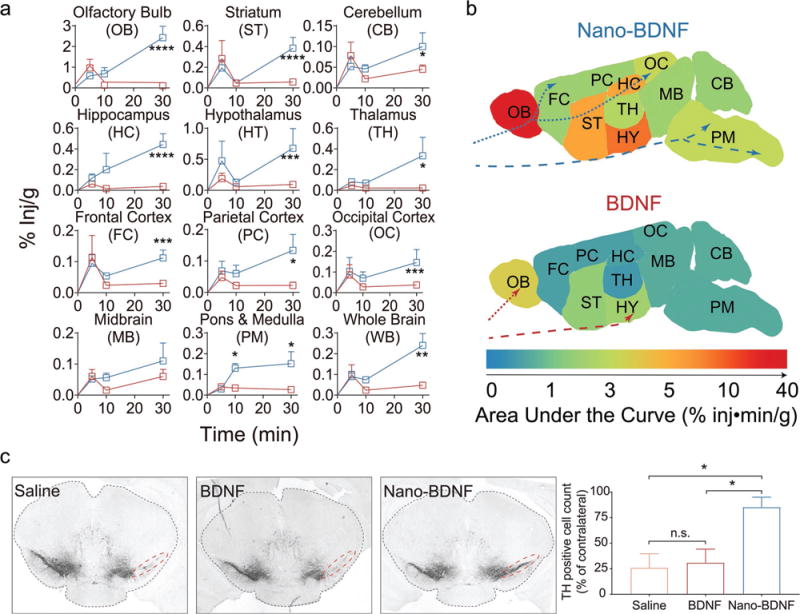
Nano-BDNF significantly improves concentration of therapeutic BDNF in different regions of the brain and proves neuroprotective in a rodent neuroinflammation model. a) Significant differences in BDNF concentration were detected in multiple brain regions over a period of 30 min following INB administration of either Nano-BDNF (blue curve) or native BDNF (red curve). b) Heat maps demonstrating the area under the curve (AUC) of BDNF or Nano-BDNF detected in different brain regions over 30 min after INB administration. The traversing trajectories of Nano-BDNF (blue) and native BDNF (red) are shown for both the olfactory pathway (dotted lines) and the trigeminal pathway (dashed lines) based on descriptions in the literature.[4] c) Coronal brain sections at 2.7 ± 0.1 mm posterior bregma, where dopaminergic neuronal loss was observed in ipsilateral substantia nigra induced by intrastriatal LPS injection. Lesions were observed in animals (n = 3) treated with saline (left lane) as well as in those treated with native BDNF (middle lane). In the animals treated with Nano-BDNF, there was significantly less dopaminergic neuronal loss compared to the other two groups. Quantification of TH-positive cells in the SN area (indicated by red dotted lines) of each brain after treatment was performed as described in the Experimental Section. The numbers are normalized as % of TH-positive cells in the SN area of the contralateral hemisphere on the same brain. Data were analyzed with unpaired Student’s t-test between each two groups; * denotes p < 0.05, ** denotes p < 0.01, *** denotes p < 0.001, **** denotes p < 0.0001, and n.s. denotes no significant difference.
Based on these observations, we further tested in vivo neuroprotective effects of Nano-BDNF using a model of acute neuroinflammation induced via intrastriatal LPS injection (Figure 4c).[22] In animals treated with saline, we observed a clear loss of dopaminergic neurons in the ipsilateral substantia nigra (SN) at 2.7 ± 0.1 mm posterior Bregma. Animals treated with intra-nasal administration of BDNF showed little or no attenuation of dopaminergic neuronal loss. In contrast, animals treated with Nano-BDNF suffered significantly less dopaminergic neuronal loss induced by LPS injection. Hence, Nano-BDNF demonstrated a neuroprotective effect against LPS induced acute neuroinflammation.
We note that the amount of Nano-BDNF detected in the serum after intranasal administration was lower than native BDNF (Table S2, Supporting Information). Analysis of the peripheral distribution of Nano-BDNF suggests that within 30 min following INB delivery, Nano-BDNF was detected in liver, spleen, kidney, lung, and heart—albeit at levels less than 0.5% inj g−1. Marginally higher amounts of the INB delivered Nano-BDNF were found in the esophagus, trachea, and stomach but these amounts still were in the range of 1%–2% inj g−1 tissue (Figure S9, Supporting Information) and were likely due to limitations of our INB procedure in a mouse (“post nasal drip”). In addition, literature as well as our own studies suggest the existence of a brain-clearance process for native BDNF.[2] Impressively, we observed a much slower brain-clearance rate of Nano-BDNF (t1/2 = 167 min) from brain to blood when it was directly injected to cerebral ventricle (ICV) relative to native BDNF (t1/2 = 25 min, Figure 5). This observation may help explain why, despite the increased accumulation of Nano-BDNF in the brain, its release into the serum was reduced.
Figure 5.
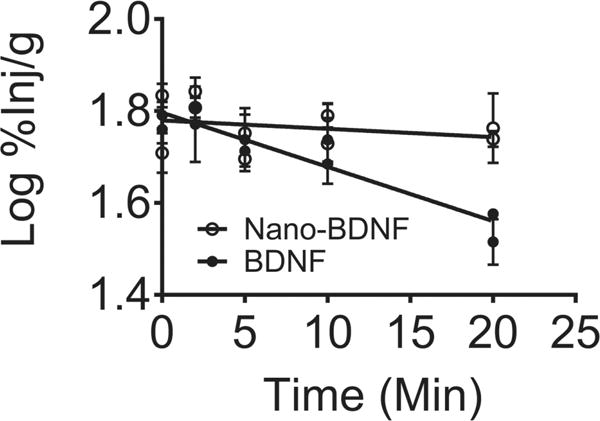
Nano-BDNF inhibits BDNF efflux from the brain to the blood. Brain efflux curves of 125I-BDNF or 125I-Nano-BDNF after ICV injection showing the rate of efflux, as indicated by the slopes of the lines were −0.012 ± 0.0014 (r2 = 0.89, n = 26) for BDNF and −0.0018 ± 0.0023 (r2 = 0.07, n = 30) for Nano-BDNF, respectively. Both slopes were significantly nonzero and differences between the slopes are extremely significant.
The serum clearance and brain uptake of Nano-BDNF after intravenous (IV) administration is shown in Figure S10 in the Supporting Information. Consistent with ICV data, IV administered Nano-BDNF displays decreased brain efflux and greater overall brain uptake than the native BDNF. Comparing the Brain/Serum ratios for IV (≈30–40 μL g−1) and INB (≈1000– 3000 L g−1) of Nano-BDNF, we conclude that the blood to brain route could explain only ≈1%–4% of total brain uptake of Nano-BDNF after nasal administration. Therefore, the increases in BDNF concentration in the brain after intranasal administration of Nano-BDNF are due to the improved delivery of the nanoformulated neurotrophin along the nose-to-brain pathway.
3. Discussion
We present a novel nanoformulation of BDNF that has immediate translational potential. As a protein delivery system, Nano-BDNF remains stable at high salt concentrations, protects BDNF from nonspecific binding with serum proteins, and is as active as the native neurotrophin in activating its receptor, TrkB and the downstream signaling in the cells. Most current nanoparticulate formulations of BDNF (poly(D,L-lactide-co-glycolide) microspheres,[23] nanoporous PLE particles,[24] agarose scaffolds)[25] control the release of the peptide temporally, i.e., release it over a period of time. In contrast, Nano-BDNF releases BDNF in a “spatially controlled” manner. That is, the release of BDNF is triggered by the presence of its target receptors that can abstract BDNF from the complex. Computational molecular modeling suggests that both the PLE chain of the PEG-PLE block copolymer and the receptor interact with approximately the same sites in the BDNF molecule (Figure 2c), with the receptor apparently having higher affinity and thereby displacing the copolymer from the complex. Indeed, the apparent dissociation constant (Kd) of Nano-BDNF is around 1.8 × 10−6 M as measured in the ITC experiment (Figure S5, Supporting Information). This number is ≈1000 times higher than the reported Kd value of BDNF binding to cell surface-bond TrkB,[26] which not only is consistent with our HAGE result but also explains the demonstrated Nano-BDNF activity in vitro and in vivo. While cooperative polyion displacement has been well known for complexes of synthetic polyelectrolytes and those of DNA,[13a] this is to the best of our knowledge the first example of such a process in which a polyion is competitively displaced from a complex with a compact globular protein ligand by a receptor that becomes activated during the process. This is a novel and in our view very useful concept in nanomedicine.
Many prior studies attempted to deliver native proteins and peptides to the brain through the INB route in their native form, but the outcomes varied from protein to protein. For example, intranasal delivery of insulin has shown promise in Alzheimer’s disease (AD) patients,[27] yet the same approach did not appear to work for BDNF which was primarily restricted to the olfactory bulb and the trigeminal nerve after INB administration.[6] Surprisingly, very little work has been done to improve BDNF delivery to the brain via this pathway. To the best of our knowledge, there are currently only two such reports. Vaka et al.[28] utilize 0.25% w/v chitosan as a permeability enhancer to disrupt the nasal mucosa barrier, and Chen et al.[29] applied focused ultrasound to induce contractions in the perivascular space of localized brain regions. To this end, our work is the first in this field to improve BDNF delivery with a nanoformulation.
Our data also indicate low accumulation level and restricted regional distribution pattern of native BDNF that are consistent with previous reports.[6,30] The nanoformulation not only increased the amount of BDNF delivered but also significantly altered its distribution pattern. Specifically, Nano-BDNF increased the proportion of BDNF in the brainstem and hippocampus, both being important brain regions widely involved in different CNS disease pathologies. Although neither PEG nor PLE are known nasal absorption enhancers, there is a possibility that the presence of PEG-PLE itself could enhance BDNF delivery to the brain by modulating the composition, viscosity, and permeability of nasal mucosa. However, disruption of the mucosal barrier alone would not explain the altered brain-regional distribution of BDNF.
The passage of therapeutic proteins from nose to brain is known to associate with two pathways: the peripheral olfactory system pathway (transporting to the olfactory bulb and rostral brain regions) and the trigeminal nerve pathway (transporting to the brain stem and caudal brain regions).[4] The distribution pattern of native BDNF in the brain suggests insufficient entry along either pathway, whereas Nano-BDNF appears to have penetrated into much deeper brain regions (Figure 4b). This can be a result from the combined effect of several factors. First, Nano-BDNF is able to stabilize the neurotrophin and minimize its interaction with nasal proteins during its transportation in the nasal tissue, as evidenced in Figure 3a–j. Notably, HSA, sIgA, and serum IgG constitute 15%, 15%, and 1%–5% of total proteins in the human nasal lavage fluid, respectively.[31] Second, the 5 KDa-PEG-coated and negatively charged nanoformulation of BDNF is expected to reduce its binding to the brain extracellular matrix and substantially increase its diffusion rates in the brain parenchyma.[32] Additionally, higher brain accumulation of Nano-BDNF could also be related to the observed decreased brain clearance of the neurotrophin, as compared with native BDNF, which appears to be cleared from the brain in conjunction with the reabsorption of the cerebrospinal fluid (CSF).[2] It is known that INB administered proteins are subject to bulk flow within the perivascular space of cerebral blood vessels where CSF plays an important role in their clearance.[5,33] A decreased clearance of the nanoformulated and PEG-coated neurotrophin may be due to its higher diffusion rates in the brain compared to native BDNF.[32] This accelerates their diffusion out of the paravascular space and distribution into the brain parenchyma to a rate that is faster than the turnover rate of CSF clearance and may reflect a general phenomenon resulting in improved brain accumulation of other INB administered proteins delivered by brain-penetrating nanoformulations.
One interesting point that remains unclear in this study is whether BDNF is transported to its final destination in the brain in the form of Nano-BDNF complexes, or is it released somewhere along the way and then transported in a free form. Based on the altered brain regional distribution observed with Nano-BDNF, if the BDNF was released, it would occur only after the complex distribution from the paravascular space into the brain parenchyma. Besides, the data on the complex stability in vitro argue against premature release of the BDNF from the complex. Nevertheless, this point warrants further investigation of the integrity of Nano-BDNF along the nose-to-brain pathway as well as how exactly the INB delivery outcomes are dependent on the size, charge, and surface properties of these complexes.
The block copolymer chemistry employed in the Nano-BDNF formulation contains two biocompatible materials, PEG and PLE, both of which have been approved by the U.S. Food & Drug Administration (PEG as an inactive ingredient in pharmaceutical preparations, and PLE as a Generally Recognized as Safe food additive).[34] Relatively low distribution of Nano-BDNF to the blood and peripheral organs should also decrease the risk of side effects associated with systemic exposure to Nano-BDNF. In the present study, the indication of the neuroprotective effect of Nano-BDNF in the brain has been provided using an LPS-induced acute neuroinflammation model. Taken together, our data represent a compelling rationale for advancing the evaluation of Nano-BDNF for treatment for CNS disorders such as stroke,[35] Rett syndrome,[36] and Alzheimer’s disease.[37]
The major limitation of this study is using the mice model to evaluate delivery efficiency. Although a similar nose-to-brain pathway exists in both human and rodents,[38] the relative surface area of the nasal cavity (surface area/volume) in mice is substantially higher than that in humans.[39] Therefore, a direct translation of data obtained in mice would overestimate the INB delivery efficiency of the same formulation in humans.
4. Experimental Section
Preparation and Characterization of Nano-BDNF
Nano-BDNF was prepared by mixing BDNF and PEG-PLE in an aqueous solution. Briefly, a concentrated PEG-PLE solution was added drop-by-drop to a concentrated BDNF solution with gentle vortex at room temperature (RT). The mixture was then diluted with either 10 × 10−3 M phosphate buffer (pH = 7.4, PB) or LR to desired concentrations, and gently vortexed for another 30 s. Complexes were allowed to incubate at RT for 30 min before use in experiments. The charge ratio (Z−/+) was calculated as the ratio of the total number of carboxylate groups in PEG-PLE (50) to that of arginine and lysine residues in BDNF (44, based on the sequence data provided by the manufacturer). To measure hydrodynamic size with dynamic light scattering (DLS), Nano-BDNF was prepared at various Z−/+ in LR and analyzed at 25 °C using a Zetasizer Nano ZS (Malvern Instruments Ltd., UK) with a scattering angle of 173°. TEM samples were deposited on thin layers of carbon films coated on copper grids and dried for 5 min. Positive staining was performed for 10 s with 2% uranyl acetate, which was then removed using filter paper. All samples were observed under a JEOL 100CX II transmission electron microscope set at 100 kV. Images were acquired with a digital imaging system. AFM samples were prepared by depositing 5 μL of aqueous dispersion of 0.01 mg mL−1 native BDNF or Nano-BDNF (Z−/+ = 10) on the surface of a glass slide followed by air drying for 15 min. A MFP3D system (Asylum Research, Santa Barbara, CA) operated in tapping mode was utilized to image the particles.
HAGE
Samples containing 2 μg of BDNF were electrophoresed through a 0.5% agarose gel at 80 V for 1 h in Tris/Glycine running buffer and then stained using Bio-Safe Coomassie Stain solution (Bio-Rad, Hercules, CA). Images were obtained using the FluorChem E series imager (ProteinSimple, San Jose, CA) with a 100 ms exposure time. The center of mass as determined by optical density was obtained using ImageJ (NIH) and defined as the migration distance of the sample loaded in that lane.
Radioactive Labeling of BDNF
125I-BDNF and 125I-Nano-BDNF were produced using a previously described Chloramine T method.[40] Briefly, 5 μg of BDNF was mixed with 0.5 mCi Na125I in a final volume of 45 μL sodium PB (0.25 M, pH 7.5). Five μL of freshly prepared 2 μg μL−1 chloramine-T solution in PB was then added to the mixture and incubated for 1 min at RT. 125I-BDNF was purified from the mixture with Illustra NAP™-5 columns (GE Healthcare, Piscataway, NJ), and fractions were collected in Eppendorf tubes pretreated with either 1% bovine serum albumine (BSA) in lactated Ringer’s solution (1% BSA-LR, for collecting 125I-BDNF) or 0.1% PEG-PLE solution in LR (for collecting 125I-Nano-BDNF) to prevent adsorption and the radioactivity of each fraction was counted in a PerkinElmer γ-counter. The integrity of 125I-BDNF was determined by trichloroacetic acid (TCA) precipitation. Briefly, 1 μL of each fraction was added to 0.5 mL of 1% BSA-LR and then precipitated in 0.5 mL of 30% TCA followed by centrifugation at 5400 g for 10 min at 4 °C. The radioactivity in the supernatant and pellet were counted with γ-counter and the values used to calculate the integrity of 125I-BDNF. Samples containing more than 100 000 count per minute (cpm) μL−1 of radioactivity and precipitate more than 95% in TCA were used for animal studies.
Cell Culture and Western Blot
NIH 3T3 cells expressing TrkB were cultured in GlutaMAX DMEM supplemented with calf serum (10%), G418 (100 μg mL−1), and penicillin-streptomycin (100–100 μg mL−1) and were maintained at 37 °C in a humidified CO2 (5%) incubator. Half a million cells were seeded 24 h before treatment. DMEM, 500 ng mL−1 of BDNF, Nano-BDNF (Z−/+ = 946), or equivalent amount of PEG-PLE polymer in the Nano-BDNF formulation were incubated with the cells for 5 min at 37 °C in serum-free DMEM then moved to ice for 20 min and lysed using radioimmunoprecipitation assay buffer with proteinase and phosphatase inhibitors. Western blot analysis was conducted as reported previously[41] with modifications. Briefly, 10 μg total protein from the cell lysates was loaded in each well of 7.5% Mini-PROTEAN® TGX™ precast gels and run at 150 V for 1 h. Proteins were then transferred to 0.45 μm polyvinylidene difluoride membranes at 500 mA current for 1 h. The membranes were then blocked in either 5% BSA (for detection of phosphorylated TrkB and ERK) or 5% nonfat dry milk (for detection of total TrkB and ERK) for 30 min at RT followed by an overnight incubation at 4 °C with primary antibody. Bands were visualized using horseradish peroxidase (HRP)-conjugated secondary antibodies after incubation with SuperSignal West Pic Chemiluminescent Substrate for 1 min. Chemiluminescent signal was recorded by a FluorChem E series imager and quantified by Image J software (National Institute of Health (NIH), Bethesda, MD) using densitometry analysis.
Animals
Charles River Laboratories supplied 8-week-old male CD-1 mice (24–28 g) and 11–12-week-old male C57/B6 mice (20–22 g). Animals were housed and humanely handled in accordance with the Principles of Animal Care outlined by National Institutes of Health. They were allowed free access to food and water and were maintained under temperature, humidity, and light-controlled conditions. Institutional Animal Care and Use Committees (IACUC) of the University of North Carolina at Chapel Hill approved all experiments involving animal subjects.
INB Administration and Pharmacokinetic Study
INB administration of 125I-BDNF or 125I-Nano-BDNF was performed by pipetting 10 μL of sample solution (total radioactivity is 1 000 000 cpm, Z−/+ = 1100, PEG-PLE concentration is 0.91 mg mL−1) with a thin tip advanced 5 mm into the left nare of the mice preanesthetized with an intraperitoneal injection of 0.2 mL of 40% urethane. The abdomen and rib cage of the mice were opened and venous blood was collected by cardiac puncture at each time point (5, 10, and 30 min) after INB administration. Next, 20 mL of phosphate-buffered saline (PBS) was perfused through the left ventricle of the heart. Mice were quickly decapitated and different brain regions were immediately dissected on ice and weighed. Serum samples were obtained by centrifugation of whole blood at 5000 g for 10 min. Peripheral organs were also collected and weighed. Levels of radioactivity in mice serum, brain regions, and peripheral organs were measured with a γ-counter.
BDNF Efflux Measurement
To measure the rate of BDNF efflux from the brain, the mouse scalp was removed and a hole was made into the skull and 1 μL LR containing 500 000 cpm of 125I-BDNF or 125I-Nano-BDNF was slowly injected into the lateral ventricle of the brain. Mice were decapitated at 2, 5, 10, and 20 min and the whole brain was removed and weighed. The level of 125I-BDNF available for transport at t = 0 was estimated in mice that had been overdosed with urethane for 20 min and had received an injection of 125I-BDNF 10 min earlier. To determine the residual levels of radioactivity in the brain, samples were counted with a γ-counter.
Neuroinflammation Model and Treatment Regimen
Striatal neuroinflammation was induced in mice as previously reported with slight modifications.[37] Mice were anesthetized with isoflurane (5% for induction and 1%–2% for maintenance) and immobilized using a stereotaxic frame (Stoelting Co., Wood Dale, IL). The skull was exposed and two burr holes were drilled above the striatum of the same hemisphere. The Dura mater was delicately pierced to introduce the injection needle. The coordinates for the injection sites from Bregma in mm were: anterior/posterior (AP) +1.2, medial/lateral (ML) −1.5, dorsal/ventral (DV) −3.5, and AP −0.3, ML −2.5, and DV −3.2. At each injection sites, 1 μL of LPS at 10 μg μL−1 was slowly injected at a rate of about 0.5 μL min−1 using a 10 μL Hamilton syringe. After injection, the needles were left in place for an additional 5 min to minimize LPS backflow.
Multiple intranasal doses of BDNF (5 μg per animal), Nano-BDNF (5 μg BDNF/animal, Z+/− = 100), or vehicle (0.9% saline) were administered to randomly grouped mice (n = 3) with striatal neuroinflammation. The doses were given 20 min prior, 4–8 h after, and 23 h after stereotaxic LPS injection. The mice were euthanized 24 h after LPS injection, perfused intracardially with 20 mL ice-cold PBS followed by 20 mL of 4% paraformaldehyde.
Immunohistochemistry
The mice brain was immediately collected after perfusion and postfixed for 24 h in 4% paraformaldehyde at 4 °C and transferred to 30% sucrose in PBS until the brain tissues sank to the bottom of the solution (a proximately 72 h process). The brain tissue was snap frozen with isopentane/dry ice and cut into 30 μm thick coronal sections containing the SN on a cryostat. The sections were then transferred to and stored in Tris Buffered Saline (TBS) with 0.1% sodium azide.
In order to investigate the neuroprotective effect of the nanoformulation, dopaminergic neurons in SN were stained with tyrosine hydroxylase (TH) antibody (MilliporeSigma, Billerica, MA) as previously described.[38] Briefly, the free-floating brain sections were incubated with anti-TH antibody diluted 1:2000 in TBS containing 2% goat serum for 48 h at 4 °C. The sections were then rinsed in TBS and incubated with secondary antibody, biotinylated goat-antirabbit IgG (Vector Laboratories, Burlingame, CA), diluted 1:400 in TBS containing 2% goat serum followed by another rinse in TBS. Immunoreactive cells were visualized using the ABC HRP Kit (Vector Laboratories). The sections were finally mounted on glass slides and cover-slipped with Permount (Fisher Scientific) for examination under a Nikon microscope. The outlines of the SN area were determined in the TH stained sections by the distribution of the dopaminergic neurons and by referencing well-established landmarks.[42] Within the SN area of each brain hemisphere, an investigator who was blind to treatment groups quantified the numbers of TH-positive cells with clearly visualized cell bodies.
Supplementary Material
Acknowledgments
The authors thank Dr. Michael Jay and Dr. Zibo Li (UNC-CH) for sharing and helping with gamma counters in radioactivity assays; Dr. Ashutosh Tripathy (Macromolecular Interactions Facility, UNC-CH) for fruitful discussions and help with ITC experiment; Dr. Wallace Ambrose and Dr. Amar S. Kumbhar (Chapel Hill Analytical and Nanofabrication Laboratory, UNC-CH) for help and trainings with TEM and AFM; Dr. Min Zheng (UNC-CH) for the help with the protein thermal shift assay; Zhenhao Bao (School of Business, University of Connecticut) for help and instructions on MD data processing; Dr. Glenn Walker (Joint UNC/NC State Department of Biomedical Engineering) for advice and help with microfluidic device fabrication. NIH 3T3 cell line stably expressing human TrkB receptor was kindly provided by Dr. David Kaplan (Department of Molecular Genetics, University of Toronto, Canada). This research was supported in parts by the National Institutes of Health Grant No. R21NS08815202 and the HeART Award from the International Rett Syndrome Foundation. J.M.F. was supported in part by NIH Grant No. 5T32GM008570-22.
Footnotes
The ORCID identification number(s) for the author(s) of this article can be found under https://doi.org/10.1002/adfm.201703982.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
A.V.K. is a founder of Neuro10−9 Pharma Inc. a startup company that is developing CNS therapeutics and holds patents to various nanoformulations. Other authors declare no other competing financial interest, except for the patent(s) filed by their institution on behalf of several co-authors (A.V.K., X.Y., and Y.J.).
Contributor Information
Dr. Yuhang Jiang, Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy, University of North Carolina, Chapel Hill, NC 27599-7362, USA; Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina, Chapel Hill, NC 27599-7362, USA.
James M. Fay, Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina, Chapel Hill, NC 27599-7362, USA Department of Biochemistry and Biophysics, School of Medicine, University of North Carolina, Chapel Hill, NC 27599-7260, USA.
Dr. Chi-Duen Poon, Research Computer Center, University of North Carolina, Chapel Hill, NC 27599, USA
Natasha Vinod, Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina, Chapel Hill, NC 27599-7362, USA; Joint UNC/NC State Department of Biomedical Engineering, University of North Carolina, Chapel Hill, NC 27599-7575, USA.
Yuling Zhao, Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy, University of North Carolina, Chapel Hill, NC 27599-7362, USA; Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina, Chapel Hill, NC 27599-7362, USA.
Kristin Bullock, Geriatric Research Education and Clinical Center, Veterans Affairs Puget Sound Health Care System, Seattle, WA 98108, USA; Division of Gerontology and Geriatric Medicine, Department of Medicine, University of Washington School of Medicine, Seattle, WA 98108, USA.
Dr. Si Qin, Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy, University of North Carolina, Chapel Hill, NC 27599-7362, USA Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina, Chapel Hill, NC 27599-7362, USA.
Prof. Devika S Manickam, Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy, University of North Carolina, Chapel Hill, NC 27599-7362, USA; Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina, Chapel Hill, NC 27599-7362, USA.
Dr. Xiang Yi, Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy, University of North Carolina, Chapel Hill, NC 27599-7362, USA Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina, Chapel Hill, NC 27599-7362, USA.
Prof. William A. Banks, Geriatric Research Education and Clinical Center, Veterans Affairs Puget Sound Health Care System, Seattle, WA 98108, USA Division of Gerontology and Geriatric Medicine, Department of Medicine, University of Washington School of Medicine, Seattle, WA 98108, USA.
Prof. Alexander V. Kabanov, Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy, University of North Carolina, Chapel Hill, NC 27599-7362, USA; Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina, Chapel Hill, NC 27599-7362, USA; Joint UNC/NC State Department of Biomedical Engineering, University of North Carolina, Chapel Hill, NC 27599-7575, USA; Laboratory for Chemical Design of Bionanomaterials, Faculty of Chemistry, M.V. Lomonosov Moscow State University, Moscow 117234, Russia.
References
- 1.Zhao H, Alam A, San CY, Eguchi S, Chen Q, Lian Q, Ma D. Brain Res. 2017;1665:1. doi: 10.1016/j.brainres.2017.03.029. [DOI] [PubMed] [Google Scholar]
- 2.Pan W, Banks WA, Fasold MB, Bluth J, Kastin AJ. Neuropharmacology. 1998;37:1553. doi: 10.1016/s0028-3908(98)00141-5. [DOI] [PubMed] [Google Scholar]
- 3.Yi X, Manickam DS, Brynskikh A, Kabanov AV. J Controlled Release. 2014;190:637. doi: 10.1016/j.jconrel.2014.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lochhead JJ, Thorne RG. Adv Drug Delivery Rev. 2012;64:614. doi: 10.1016/j.addr.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 5.Lochhead JJ, Wolak DJ, Pizzo ME, Thorne RG. J Cereb Blood Flow Metab. 2015;35:371. doi: 10.1038/jcbfm.2014.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alcalá-Barraza SR, Lee MS, Hanson LR, McDonald AA, Frey WH, McLoon LK. J Drug Targeting. 2010;18:179. doi: 10.3109/10611860903318134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ali N, Mattsson K, Rissler J, Karlsson HM, Svensson CR, Gudmundsson A, Lindh CH, Jonsson BA, Cedervall T, Karedal M. Nanotoxicology. 2016;10:226. doi: 10.3109/17435390.2015.1048324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soderquist RG, Milligan ED, Sloane EM, Harrison JA, Douvas KK, Potter JM, Hughes TS, Chavez RA, Johnson K, Watkins LR, Mahoney MJ. J Biomed Mater Res A. 2009;91a:719. doi: 10.1002/jbm.a.32254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burns ML, Malott TM, Metcalf KJ, Hackel BJ, Chan JR, Shusta EV. Appl Environ Microbiol. 2014;80:5732. doi: 10.1128/AEM.01466-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aleksandr BZ, Viktor AK. Russ Chem Rev. 1982;51:833. [Google Scholar]
- 11.Kanato Y, Kitajima K, Sato C. Glycobiology. 2008;18:1044. doi: 10.1093/glycob/cwn084. [DOI] [PubMed] [Google Scholar]
- 12.Compton SJ, Jones CG. Anal Biochem. 1985;151:369. doi: 10.1016/0003-2697(85)90190-3. [DOI] [PubMed] [Google Scholar]
- 13.a) Kabanov AV, Kabanov VA. ChemInform. 1995;26 https://doi.org/10.1002/chin.199528318. [Google Scholar]; b) Holappa S, Kantonen L, Andersson T, Winnik F, Tenhu H. Langmuir. 2005;21:11431. doi: 10.1021/la051866r. [DOI] [PubMed] [Google Scholar]; c) Bakeev KN, Izumrudov VA, Kuchanov SI, Zezin AB, Kabanov VA. Macromolecules. 1992;25:4249. [Google Scholar]; d) Zintchenko A, Zintchenko A, Rother G, Dautzenberg H. Langmuir. 2003;19:2507. [Google Scholar]
- 14.Papisov IM, Litmanovich AA. Polym Sci USSR. 1977;19:830. [Google Scholar]
- 15.McDonald IK, Thornton JM. J Mol Biol. 1994;238:777. doi: 10.1006/jmbi.1994.1334. [DOI] [PubMed] [Google Scholar]
- 16.Robinson RC, Radziejewski C, Stuart DI, Jones EY. Biochemistry. 1995;34:4139. doi: 10.1021/bi00013a001. [DOI] [PubMed] [Google Scholar]
- 17.a) Dautzenberg H. Macromolecules. 1997;30:7810. [Google Scholar]; b) Kabanov VA. Usp Khim. 2005;74:5. [Google Scholar]; c) Y. Jiang, The University of North Carolina at Chapel Hill, 2016.
- 18.a) Zhang H, Saiani A, Guenet JM, Curtis R. Macromol Symp. 2007;251:25. [Google Scholar]; b) Cousin F, Gummel J, Ung D, Boué F. Langmuir. 2005;21:9675. doi: 10.1021/la0510174. [DOI] [PubMed] [Google Scholar]; c) Stogov SV, Muronetz VI, Izumrudov VA. Dokl Biochem Biophys. 2009;427:187. doi: 10.1134/s160767290904005x. [DOI] [PubMed] [Google Scholar]
- 19.Callahan WJ, Narhi LO, Kosky AA, Treuheit MJ. Pharm Res. 2001;18:261. doi: 10.1023/a:1011034425896. [DOI] [PubMed] [Google Scholar]
- 20.Fryer RH, Kaplan DR, Kromer LF. Exp Neurol. 1997;148:616. doi: 10.1006/exnr.1997.6699. [DOI] [PubMed] [Google Scholar]
- 21.Nakamura T, Sanokawa R, Sasaki Y, Ayusawa D. Oncogene. 1996;13:1111. [PubMed] [Google Scholar]
- 22.Hunter RL, Hunter RL, Cheng B, Choi DY, Liu M. J Neurosci Res. 2009;87:1913. doi: 10.1002/jnr.22012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bertram JP, Rauch MF, Chang K, Lavik EB. Pharm Res. 2010;27:82. doi: 10.1007/s11095-009-0009-x. [DOI] [PubMed] [Google Scholar]
- 24.Tan J, Wang Y, Yip X, Glynn F. Adv Mater. 2012;24:3362. doi: 10.1002/adma.201200634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mehrotra S, Lynam D, Maloney R, Pawelec KM. Adv Funct Mater. 2010;20:247. doi: 10.1002/adfm.200901172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soppet D, Escandon E, Maragos J, Middlemas DS, Raid SW, Blair J, Burton LE, Stanton BR, Kaplan DR, Hunter T, Nikolics K, Parade LF. Cell. 1991;65:895. doi: 10.1016/0092-8674(91)90396-g. [DOI] [PubMed] [Google Scholar]
- 27.Craft S, Baker LD, Montine TJ, Minoshima S, Watson GS, Claxton A, Arbuckle M, Callaghan M, Tsai E, Plymate SR, Green PS, Leverenz J, Cross D, Gerton B. Arch Neurol. 2012;69:29. doi: 10.1001/archneurol.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vaka SR, Murthy SN, Balaji A, Repka MA. Pharm Res. 2012;29:441. doi: 10.1007/s11095-011-0572-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen H, Yang GZX, Getachew H, Acosta C, Sanchez CS, Konofagou EE. Sci Rep. 2016;6:28599. doi: 10.1038/srep28599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vaka SRK, Murthy SN, Balaji A, Repka MA. Pharm Res. 2012;29:441. doi: 10.1007/s11095-011-0572-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Casado B, Pannell LK, Iadarola P, Baraniuk JN. Proteomics. 2005;5:2949. doi: 10.1002/pmic.200401172. [DOI] [PubMed] [Google Scholar]
- 32.Nance EA, Woodworth GF, Sailor KA, Shih TY, Xu Q, Swaminathan G, Xiang D, Eberhart C, Hanes J. Sci Transl Med. 2012;4:149ra119. doi: 10.1126/scitranslmed.3003594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, Nagelhus EA, Nedergaard M. Sci Transl Med. 2012;4:147ra111. doi: 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Efremenko EN, Lyagin IV, Klyachko NL, Bronich T, Zavyalova NV, Jiang Y, Kabanov AV. J Controlled Release. 2017;247:175. doi: 10.1016/j.jconrel.2016.12.037. [DOI] [PubMed] [Google Scholar]
- 35.Harris NM, Ritzel R, Mancini NS, Jiang Y, Yi X, Manickam DS, Banks WA, Kabanov AV, McCullough LD, Verma R. Pharm, Biochem Behav. 2016;150–151:48. doi: 10.1016/j.pbb.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kline DD, Ogier M, Kunze DL, Katz DM. J Neurosci. 2010;30:5303. doi: 10.1523/JNEUROSCI.5503-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, Wang L, Blesch A, Kim A, Conner JM, Rockenstein E, Chao MV, Koo EH, Geschwind D, Masliah E, Chiba AA, Tuszynski MH. Nat Med. 2009;15:331. doi: 10.1038/nm.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peterson A, Bansal A, Hofman F, Chen TC, Zada G. J Neuro-Oncol. 2014;116:437. doi: 10.1007/s11060-013-1346-5. [DOI] [PubMed] [Google Scholar]
- 39.Ruigrok MJR, de Lange ECM. AAPS J. 2015;17:493. doi: 10.1208/s12248-015-9724-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.a) Jiang Y, Brynskikh AM, D SM, Kabanov AV. J Controlled Release. 2015;213:36. doi: 10.1016/j.jconrel.2015.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jiang Y, Arounleut P, Rheiner S, Bae Y, Kabanov AV, Milligan C, Manickam DS. J Controlled Release. 2016;231:38. doi: 10.1016/j.jconrel.2016.02.038. [DOI] [PubMed] [Google Scholar]
- 41.a) Ma Y, Wang J, Gao J, Yang H, Wang Y, Manithody C, Li J, Rezaie AR. Thrombosis Haemostasis. 2015;113:338. doi: 10.1160/TH14-04-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xu S, Spencer CM, Munger J. J Virol. 2015;89:6406. doi: 10.1128/JVI.03671-14. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Natarajan G, Perriotte-Olson C, Bhinderwala F, Powers R, Desouza CV, Talmon GA, Yuhang J, Zimmerman MC, Kabanov AV, Saraswathi V. Transl Res. 2017;188:10. doi: 10.1016/j.trsl.2017.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bentivoglio M, Bloom FE, Björklund A, Dunnett SB, Hökfelt T, Kaczmarek L, Kuhar MJ, Ottersen OP, Owman C, Quirion R, Robertson HA, Steinbusch HWM, Storm-Mathisen J, Swanson LW, Tohyama M, Vente JD, Vincent SR. Handbook of Chemical Neuroanatomy. Vol. 2. Elsevier; Amsterdam, Netherlands: 1983. [Google Scholar]
- 43.Janaky N, Jun-Ying X, Xiang-Yang L. J Phys: Conf Ser. 2006;28:83. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.