

Abstract
During the last two decades, the pharmaceutical industry has progressed from detecting small molecules to designing biologic-based therapeutics. Amino acid-based drugs are a group of biologic-based therapeutics that can effectively combat the diseases caused by drug resistance or molecular deficiency. Computational techniques play a key role to design and develop the amino acid-based therapeutics such as proteins, peptides and peptidomimetics. In this study, it was attempted to discuss the various elements for computational design of amino acid-based therapeutics. Protein design seeks to identify the properties of amino acid sequences that fold to predetermined structures with desirable structural and functional characteristics. Peptide drugs occupy a middle space between proteins and small molecules and it is hoped that they can target “undruggable” intracellular protein–protein interactions. Peptidomimetics, the compounds that mimic the biologic characteristics of peptides, present refined pharmacokinetic properties compared to the original peptides. Here, the elaborated techniques that are developed to characterize the amino acid sequences consistent with a specific structure and allow protein design are discussed. Moreover, the key principles and recent advances in currently introduced computational techniques for rational peptide design are spotlighted. The most advanced computational techniques developed to design novel peptidomimetics are also summarized.
Keywords: protein-based drugs, in silico designing, protein, peptide, peptidomimetics
Introduction
Different diseases may be caused by pathogens or malfunctioning organs, and using therapeutic agents to heal them has an old recorded history. Small molecules are conventional therapeutic candidates that can be easily synthesized and administered. However, many of these small molecules are not specific to their targets and may lead to side effects.1 Moreover, a number of diseases are caused due to deficiency in a specific protein or enzyme. Thus, they can be treated using biologically based therapies that are able to recognize a specific target within crowded cells.2 Under the biologic conditions, some macromolecules such as proteins and peptides are optimized to recognize specific targets.3 Therefore, they can override the shortcomings of small molecules.3 Recently, pharmaceutical scientists have shown interest in engineering amino acid-based therapeutics such as proteins, peptides and peptidomimetics.4–6
Theoretical and experimental techniques can predict the structure and folding of amino acid sequences and provide an insight into how structure and function are encoded in the sequence. Such predictions may be valuable to interpret genomic information and many life processes. Moreover, engineering of novel proteins or redesigning the existing proteins has opened the ways to achieve novel biologic macromolecules with desirable therapeutic functions.7 Protein sequences comprise tens to thousands of amino acids. Besides, the backbone and side chain degrees of freedom lead to a large number of configurations for a single amino acid sequence. Protein design techniques give minimal frustration through precise identification of sequences and their characteristics.8–11 Considering energy landscape theory, the adequately minimal frustration in natural proteins occurs when their native state is adequately low in energy.7 The de novo design of a sequence is difficult because there are huge numbers of possible sequences: 20N for N-residue proteins with only 20 natural amino acids.12
Peptide design should incorporate computational approaches. It can benefit from searching the more advanced fields used for small molecules and protein design.13 However, the straightforward adoption of computational approaches employed to small-molecule and protein design has not be accepted as a reasonable solution to the peptide design problem.14–16 In the peptide drug design, the conformational space accessible to peptides challenges the small-molecule computational approaches. Besides, the necessity for nonstandard amino acids and various cyclization chemistries challenges the available tools for protein modeling.13 Furthermore, the aggregation of peptide drugs during production or storage can be an unavoidable problem in the peptide design procedure. Rational design of a peptide ligand is also challenging because of the elusive affinity and intrinsic flexibility of peptides.17 Peptide-focused in silico methods have been increasingly developed to make testable predictions and refine design hypotheses. Consequently, the peptide-focused approaches decrease the chemical spaces of theoretical peptides to more acceptable focused “drug-like” spaces and reduce the problems associated with aggregation and flexibility.13,18 For the discussions that follow, peptides can be defined as relatively small (2–30 residues) polymers of amino acids.18
In physiological conditions, several problems such as degradation by specific or nonspecific peptidases may limit the clinical application of natural peptides.19 Moreover, the promiscuity of peptides for their receptors emerges from high degrees of conformational flexibility that can cause undesirable side effects.20 Besides, some properties of therapeutic peptides, such as high molecular mass and low chemical stability, can result in a weak pharmacokinetic profile. Therefore, peptidomimetic design can be a valuable solution to circumvent some of undesirable properties of therapeutic peptides.21,22
In the biologic environment, peptidomimetics can mimic the biologic activity of parent peptides with the advantages of improving both pharmacokinetic and pharmacodynamic properties including bioavailability, selectivity, efficacy and stability. A wide range of peptidomimetics have been introduced, such as those isolated as natural products,23 synthesized from novel scaffolds,24 designed based on X-ray crystallographic data25 and predicted to mimic the biologic manner of natural peptides.26
Using hierarchical strategies, it is possible to change a peptide into mimic derivatives with lower undesirable properties of the origin peptide.27 Over the past 10 years, computational methods have been developed to discover peptidomimetics.28 In a part of this review, novel computational methods introduced for peptidomimetic design have been summarized.
Peptidomimetics can be categorized as follows: peptide backbone mimetics (Type 1), functional mimetics (Type 2) and topographical mimetics (Type 3).29 The first generation of peptidomimetics (Type 1) mimics the local topography of amide bond. It includes amide bond isosteres,30 pyrrolinones31 or short fragments of secondary structure, such as beta-turns.32 Such mimetics generally match the peptide backbone atom-for-atom, and comprise chemical groups that also mimic the functionality of the natural side chains of amino acids. A number of prosperous instances of Type 1 peptidomimetics have been reported.33
The second type of peptidomimetics is described as functional mimetics or Type 2 mimetics, which include small, non-peptide compounds that are able to identify the biologic targets of their parent peptide.34 At first, they were assumed to be conservative structural analogs of parent peptides. However, using site-directed mutagenesis, their binding sites to biologic targets were investigated. The results indicated that Type 2 peptidomimetics routinely bind to protein sites that are different from those selected by the original peptide.35 Therefore, Type 2 mimetics maintain the ability to interfere with the peptide–protein interaction process without the necessity to mimic the structure of the natural peptide.28
Type 3 peptidomimetics reveal the best conception of peptidomimetics. They consist of the necessary chemical groups that act as topographical mimetics and contain novel chemical scaffolds that are unrelated to natural peptides.36
Here, theoretical and computational techniques to design proteins, peptides and peptidomimetics are reviewed. However, the current review does not deeply highlight the computational aspects of amino acid-based therapeutic design, but only discusses the methods used to design the mentioned therapeutics. Figure 1 summarizes the key concepts presented in this study.
Figure 1.

A schematic summary of the key concepts presented in this review.
As some examples, the structures of Aldesleukin, Leuprolide and Spaglumic acid, important amino acid-based therapeutics approved by the US Food and Drug Administration (FDA), are shown in Figure 2A–C. The X-ray crystallographic structures of Aldesleukin (PDB ID: 1M47; Figure 2A) and Leuprolide (PDB ID: 1YY2; Figure 2B) were obtained from the Protein Data Bank (PDB; http://www.rcsb.org/) and visualized by PyMol tool. The structure of Spaglumic acid was retrieved (in MOL format) from PubChem database (https://pubchem.ncbi.nlm.nih.gov/) with the PubChem ID 188803 (Figure 2C) and visualized using PyMol. Aldesleukin, a lymphokine, is a recombinant protein used to treat adults with metastatic renal cell carcinoma (https://www.drugbank.ca/drugs/DB00041). Leuprolide, a synthetic nine-residue peptide analog of gonadotropin releasing hormone, is used to treat advanced prostate cancer (https://www.drugbank.ca/drugs/DB00007). Spaglumic acid is used in allergic conditions such as allergic conjunctivitis. The drug belongs to a class of peptidomimetics known as hybrid peptides. Hybrid peptides contain at least two dissimilar types of amino acids (alpha, beta, gamma or delta) linked to each other via a peptide bond (https://www.drugbank.ca/drugs/DB08835).
Figure 2.

The structures of three important amino acid-based therapeutics approved by the FDA.
Notes: (A) Aldesleukin (PDB ID: 1M47), a recombinant lymphokine, has been used for treatment of adults with metastatic renal cell carcinoma. (B) Leuprolide (PDB ID: 1YY2) is a synthetic nine-residue peptide analog of gonadotropin releasing hormone used to treat advanced prostate cancer. (C) Spaglumic acid (PubChem ID: 188803), a peptidomimetic, is used in allergic conditions such as allergic conjunctivitis. The structures of the drugs were visualized via PyMol.
Abbreviations: FDA, US Food and Drug Administration; PDB, Protein Data Bank.
In the current study, all FDA-approved therapeutics (in 2018) were retrieved from DrugBank (https://www.drugbank. ca/biotech_drugs) and an analysis was conducted to compare their percentages. Protein-based therapies, gene or nucleic acid-based therapies, vaccines, allergenics and cell transplant therapies made up 8.05%, 0.17%, 2.64%, 16.20% and 0.14% of total approved therapeutics, respectively. Small-molecule drugs made up 72.76% of the approved therapeutics (Figure 3).
Figure 3.
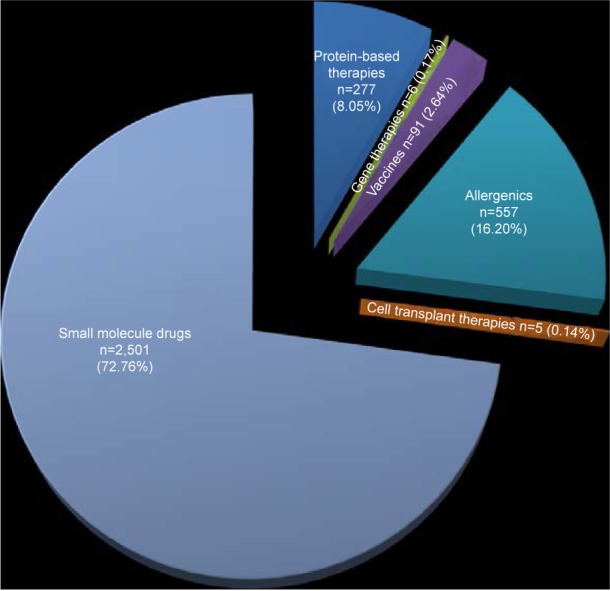
A summary of the FDA-approved small- and large-molecule therapeutics.
Notes: Number and percentage of FDA-approved therapeutics (in 2018) is shown inside the pie diagram. Protein-based therapies: 8.05% (n=277), gene and nucleic acid-based therapies: 0.17% (n=6), vaccines: 2.64% (n=91), allergenics: 16.20% (n=557), cell transplant therapies: 0.14% (n=5), small-molecule drugs: 72.76% (n=2,501).
Abbreviation: FDA, US Food and Drug Administration.
Methods and tools for computational designing of therapeutic proteins
Computational designing of proteins can be classified as follows: 1) template-based designing in which three-dimensional (3D) structure of a predefined template is adapted to design a sequence and 2) de novo designing in which the amino acids’ arrangement is changed to generate both sequence and 3D structure of a completely novel protein.3
Template-based designing
The problem of predicting the fold of an unknown sequence could be solved by utilizing templates. Since the fold is unaltered, the backbone atoms are directly located on this framework.3 Moreover, to generate a functional protein, the side chains that can effectively stabilize the structure are added to the backbone.37,38 Routine concerns and methods for template-based protein design are reviewed below.
Searching process
Selecting the template (scaffold) protein
The template (also named as scaffold protein) contains a group of backbone atom coordinates. The coordinates can be retrieved from an available X-ray crystal structure or cautiously from a nuclear magnetic resonance (NMR) structure.39 Fixing the backbone decreases the computational complication, but it may inhibit the main chain modifications to adjust sequence alternation.7 Backbone flexibility can generate designed functionalities over the protein’s normal function. The backbone flexibility is introduced through incorporating other closely associated conformations to an existing structure.40–42 Recently, new functionalities were effectively introduced into the TIM-barrel topology.43 This fold has been detected as one of the most shared structures in 21 distinct protein superfamilies.44
Sequence search and characterization
In a design procedure, a protein sequence is selected such that it meets the energetic and geometric constraints established by the chosen fold. Sequence search techniques sample different sequences and estimate their energies to gain the one owing the minimum energy.3
In order to identify the sequences subject to an objective function or a specific energy, a diverse strategies including optimization and probabilistic approaches have been developed.45 Optimization processes may recognize candidate sequences using stochastic or deterministic methods.45 Probabilistic approaches focus on characterizing the sequence space probabilistically.
Deterministic methods: To achieve a sequence folded into a global minimum energy conformation, deterministic methods search the whole sequence space and identify the global optima.3,7 These methods include dead-end elimination (DEE),46 self-consistent mean field,47 graph decomposition and linear programming.48 Stochastic algorithms search the sequence space in an exploratory manner.3 These algorithms include Monte Carlo algorithms (simulated annealing),49 graph search methods50 and genetic algorithms.51 Some of the most commonly used methods are discussed below.
DEE has been considered as a thorough search algorithm. To find and remove sequence-rotameric positions that are not portions of the global minimum energy conformation, DEE compares two amino acid rotamers and removes the one with greater interaction energy.52 Interaction energies are computed for each rotamer of the test amino acid, along with all rotamers of every other amino acid.3 The situation is repetitively examined for total amino acid states as well as their rotamers until it no longer holds true.52,53 Expanding the sequence length increases the combinatorial complication of DEE exponentially. Therefore, to design sequences of 30 amino acids or larger, application of DEE may be restricted.54 Details of the theorem are explained elsewhere.3,7
Stochastic search algorithms: As mentioned before, deterministic approaches are perfect to design proteins with small sizes, but show the applied disadvantages with extension of sequence size. Stochastic or heuristic methods are valuable to design large proteins.3 The most widely used method for protein design includes Monte Carlo sampling.3,7
Monte Carlo method samples positions of complicated proteins in a way related to a selected probability distribution such as Boltzmann distribution. Boltzmann distribution specially weighs low-energy configurations. The Monte Carlo algorithm performs iterative series of calculations. At the primary step of each search, a partially accidental test sequence is generated, and its energy is calculated via a physical potential. During the primary step, both rotamer state and amino acid identity are adjusted and an efficient temperature controls the probable energy alterations. In the next step, named simulated annealing, the temperature gradually decreases and permits favorable sampling of lower-energy configurations.55 Multiple independent calculations are carried out to converge the system to a global minimum.3,7 For more explanation about the theorems and details of the formulation of the probability distribution and weights, readers are referred to study previous reports.3,7
Probabilistic approach: Probabilistic approaches are frequently employed when thorough information is not accessible for protein design. In a probabilistic approach, site-specific amino acid probabilities may be utilized, rather than particular sequences. The procedure is partially motivated by the uncertainties to find sequences consistent with a specific structure. Briefly, the backbone atoms are fixed or greatly constrained, side chain conformations are discretely handled, energy functions are estimated and solvation is handled by simple models.7 However, in order to offer valuable sequence information for design experiments and to find structurally significant amino acids, probabilistic techniques leverage structural characteristics of interatomic interactions.7
Generally, Monte Carlo methods give a probabilistic sampling of sequences.49,55 In addition, an entropy-based formalism has been defined to predict amino acid probabilities for a certain backbone structure.56,57 The method employs concepts from statistical thermodynamics to assess the site-specific probabilities. To address the whole space of existing compositions, the theory is not restricted by the computational enumeration and sampling. Large protein structures with >100 variable residues can be supplied simply.7
Sampling sequence space to generate conformations
The chemical variability of a sequence and the number of various amino acids permitted at each position are defined as “degrees of freedom for each amino acid”. Moreover, each of the 20 natural residues search the whole sequence space.58 To decrease the degrees of freedom for each amino acid and searching the sequence space, diverse approaches such as hydrophobic patterning have been proposed.58 Monomers can be used to probe a protein structure59 and improve its function,60 other than the naturally occurring amino acids.61
Sampling of side chain conformational space to form conformations
Side chain conformations are typically consistent with the energy minima of molecular potentials and can be obtained from a structural database.62 Rotamer statuses are related to the repeatedly detected values of dihedral angles in the side chain of each amino acid. For example, the simplest amino acids including alanine and glycine have only one rotamer status, while the bigger amino acids have >80 diverse rotamer statuses.62
A variety of rotamer libraries including backbone- dependent, secondary structure-dependent and backbone-independent libraries have been developed for protein design.62,63 By using a rotamer library, one can discretize a meaningful state space to decrease the computational difficulty. Rotamer libraries can be extended beyond the 20 natural amino acids. The effective rotamers can model cofactors, ligands, water and posttranslational modifications. For example, to improve the modeling of protein–protein interactions and model water within proteins interiors, the structurally definite water molecules can be inserted as a solvated rotamer library.61
Scoring functions (energy functions)
Energy functions have been employed to quantify sequence–structure compatibilities.64 They include linear associations of hydrogen bonds made by backbone atoms, repulsion among atoms, hydrophobic attraction among non-polar groups and electrostatic interactions among sequential neighbors.65 The sequence of a protein is selected so that it can adjust the energetic and geometric constraints enforced by the favorite fold. Constraints typically contain several intramolecular interactions such as van der Waals, hydrophobic, polar and electrostatic interactions, as well as hydrogen bonds. Generally, by using a scoring function, it is possible that energetic contributions of the mentioned parameters are taken into account.3,7,65
De novo design: designing the sequence and 3D structure
Through assembly of proteins fragments66,67 or secondary-structure elements,68,69 novel structures can be modeled de novo. In the design procedures, the backbone coordinates are generally constrained.
Summary and important findings of some proteins designed using computational approach including a retroaldol enzyme,43 the Kemp elimination enzyme,70 a novel βαβ protein,71 a redesigned procarboxypeptidase,72 a novel α/β protein structure and the TOP773 are shown in Table 1.
Table 1.
Summary and important findings of case studies in protein design field
| Designed protein | Description | Main conclusion(s) | References |
|---|---|---|---|
| A retroaldol enzyme | Template-based design (using TIM-barrel template) | Designs that utilized an explicit water molecule to mediate proton shuffling were notably more favorable than those involving charged side chain networks | 43 |
| Kemp elimination enzyme | Template-based design (using TIM-barrel template) with measured rate enhancements of up to 105 and multiple turnovers | Designs were approved to have close to atomic accuracy. The results demonstrated the power of combining computational protein design with directed evolution for generating novel enzymes | 70 |
| A novel βαβ protein | De novo design included a β-sheet forming a tight core with the helix | A stand-alone βαβ motif was de novo designed with a stable tertiary structure The designed small protein may provide a model system for a protein-folding study | 71 |
| The redesign of a procarboxypeptidase | Computational protein design protocol RosettaDesign was used to completely redesign the sequence of the activation domain of human procarboxypeptidase A2 | Yielding a highly stable and fast-folding antiparallel dimer | 72 |
| The design of a novel α/β protein structure, TOP7 | A general computational strategy iterated between sequence design and structure prediction was used to design a 93-residue α/β protein named Top7 with a novel sequence and topology | Top7 was experimentally found to be folded and extremely stable, and the X-ray crystal structure of Top7 was similar to the design model | 73 |
Peptide design
Methods and tools
Peptide design methods have been categorized as ligand- and target-based design methods. In the ligand-based designing procedure, information derived from peptides is used to design novel therapeutic peptides. In the target-based method, information derived from target proteins is specifically utilized. Typically, a hybrid approach including both ligand- and target-based design is utilized.13
Ligand-based peptide design
The ligand-based design has been classified as follows: 1) sequence-based, 2) property-based and 3) conformation-based design.
Sequence-based approach uses the information of conserved regions and analyzes the multiple sequence alignments. This method is directed by the hypothesis that conserved regions are functionally and structurally significant.13 Computational tools allow the ligand-based peptide design, although they lag behind bioinformatics strategies developed for protein designing.13 Recently, using a method based on a PAM250 matrix, the relationship between a series of 35 collagen peptides and antiangiogenic activity including proliferation, migration and adhesion was analyzed.74 The PAM250 matrix captured information of mutation rates among all pairs of amino acids. Based on the results, regions at the C and N termini of the peptides were detected to be significant for an ideal activity and suggested as two distinct binding sites. The approach showed the potential worth of the sequence-based peptide design.74 In another report, a computational platform called SARvision was developed to support sequence-based design. SARvision signifies an important step for peptide sequence/activity relationship (SAR) analysis. Moreover, it pools the improved visualization abilities with advanced sequence/activity analysis.75
Compared to small molecules, property-based design methods for peptides are in the early stages of development. In a recent study, the ΔG decomposition per residue and the physicochemical characteristics of amino acids, such as hydrophilicity, hydrophobicity and volume, were used to model peptide binding to targets of interest.76,77 Finally, a model was built to estimate peptide ΔG values for binding to the class I major histocompatibility complex (MHC) protein HLA-A*0201.78 Furthermore, in a wide range of studies, antimicrobial peptides were successfully analyzed by using the property-based approach.79 For example, a machine-learning method was employed to design novel antimicrobial peptides.80 The victory of the property-based methods with antimicrobial peptides may be explained by the fact that the desired biologic activity of membrane disruption is relatively nonspecific.13
In the case of conformation-based peptide design, computational techniques were developed to predict the conformational ensembles or structure of peptides and analyze the SARs.81,82 PEP-FOLD is an online tool used to predict the 3D structures of peptides of length 9–36 residues.81 A remarkable suggestion from the data is that PEP-FOLD seems to solve the conformational sampling problem.13,81
In order to search conformational spaces of a peptide, long timescale molecular dynamic simulations have been employed.83,84 Besides, quantum mechanical calculations are promising to address the scoring deficiency in the peptide conformational examination.85 Apparently, to affect the peptide design processes positively, improving the major theoretical and technical issues is necessary before such computationally sophisticated and costly procedures.
Conformation of a peptide may be modeled to generate a 3D pharmacophore hypothesis. A certain pharmacophore hypothesis is useful to determine the ADME/Tox activities or particular potencies of a peptide.86 For example, screening of a peptide library was jointed to generate a pharmacophore hypothesis to identify potent agonists of melanocortin-4 receptor isoforms. A combinatorial tetrapeptide library was screened, and SAR and ligand-derived pharmacophore templates were generated. The pharmacophore hypothesis was proposed to allow continuous attempts in the rational design of melanocortin receptor molecules.86
Target-based peptide design
Compared to ligand-based peptide design, target-based design appears to be in a more improved level.13 Target-based design is initiated with the computer-aided survey of a ligand-bound or unbound protein target to recognize its potential binding sites, prospective specificity surfaces and other pharmacologic activity elements. The phase is generally followed by an in silico design phase where computational methods perform, refine and evaluate peptide design ideas. Some recently developed computational methods for target-based peptide design are reviewed below.
Structure survey
Recently, an increase in the number of protein–peptide 3D structures deposited in the PDB has assisted to search the molecular mechanism and structural basis of peptide recognition and binding.87 Information of crystal structures of protein–peptide complexes can improve our knowledge of the chemical forces involved in the binding and special modes of binding. Dynamic data of the complexes can be partially extracted from the solution NMR structures deposited in the PDB. To record the structures and functions of various protein–peptide complexes, the experimentally resolved structure data were gathered, annotated and analyzed, and several distinctive databases such as PepX,88 PepBind89 and peptidDB were generated.90 The PepX database, derived from the PDB, comprises unique protein–peptide interface collections.88 The PepBind database contains 4,986 protein–peptide complex structures from the PDB.89 PeptidDB is a curated database of 103 protein–peptide complexes.90
The abundance of the structural information specifically on monomeric proteins could be gathered to design protein–peptide interactions with no requirement for their sequence homology.91
Protein–peptide docking
Precise docking of a highly flexible peptide is a major challenge.18 Traditional docking protocols, such as AutoDock, Vina92,93 and MOE-Dock,94 developed for docking of small molecules, were also used to dock a peptide to a protein receptor. However, comparative studies revealed that these techniques would face failure if the docked peptides were >3 residues long.95 Therefore, development of peptide-focused docking protocols is very important.96 Other protein–protein docking tools such as z-dock and Hex have been used for the computational peptide design in some studies.96 Below, details of recently developed peptide-focused docking approaches are discussed.
First, heuristic evolution procedures were applied to search the large conformational space of linear peptides before the binding.97 However, these procedures were not efficient and their use was limited.18 Then, a scheme based on conformational sampling became common in the peptide docking. Besides, several illustrative approaches were proposed to balance between the accuracy and efficacy of the flexible peptide docking. In this aspect, DocScheme,98 DynaDock99 and pepspec100 were integrated to online user-friendly interfaces and introduced.
Recently, PepCrawler101 and FlexPepDock102 were developed as the peptide docking tools.18 It is reported that FlexPepDock102 has sub-angstrom accuracy in reproducing the crystal structures of protein–peptide complexes.103 All of the FlexPepDock-based methods assume previous information about the peptide-binding site.13
AnchorDock, a recently described algorithm, allows powerful blind docking calculations through relaxing the constraint.104 The program predicts anchoring origins on a protein surface. Following recognition of the anchoring origins, an assumed peptide conformation is refined using an anchor-constrained molecular dynamic process.105
HADDOCK, a well-known protein–protein docking tool, has been recently expanded to run the flexible peptide–protein docking.105 To handle a docking procedure, HADDOCK uses ambiguous interaction restraints based on the experimental information about intermolecular interactions. This rigid body peptide docking is followed through a flexible-simulated annealing process. The novel HADDOCK strategy initiates docking computations from an ensemble of three dissimilar peptide conformations (eg, α-helix, extended and polyproline-II) that are high informative inputs.105
CABS-dock is a recently introduced protein–peptide docking tool and runs a primary docking procedure whose outcomes can be refined by other tools such as FlexPepDock.106 In the primary phase of the procedure, random conformations of a peptide are predicted and located around the protein target of interest. The process is followed by replica exchange Monte Carlo dynamics. Subsequently, 10 models are selected for the last optimization using the Modeller tool to gain accurate scoring and ranking poses.13,106
GalaxyPepDock was developed to use experimentally resolved protein–peptide structures for running the template-based docking pooled by flexible energy-based optimization.107
Atomistic simulation
Atomistic Monte Carlo and molecular dynamics simulations are accurate, but they are meticulous techniques to investigate peptide–protein binding interactions. These techniques can also detect the thermodynamic profile and trajectory included in protein–peptide identification. These methods predict the association among conformations of a peptide in solution or protein.108 In a study, in order to describe the binding of a decapeptide to the cognate SH3 receptor, a long-term molecular dynamic simulation was used and a two-state model was built.109 In the first step, a relatively quick diffusion phase, nonspecific encounter complexes were generated and stabilized by using electrostatic energy. The secondary step was a slow modification phase, in which the water molecules were emptied out from the space between the peptide ligand and the receptor.109 In another report, by using Monte Carlo method, the mentioned two-state model was verified to trace some oligopeptide routes for binding to various PDZ (Post synaptic density protein, Drosophila disc large tumor suppressor, and Zonula occludens-1 protein) domains.110
The affinity of BH3 peptides to Bcl-2 protein was investigated, and results showed the higher affinity of bound peptides occurred when the corresponding peptides were in a lower degree of disorder in unbound states and vice versa.111 These results showed that the highly structured peptides could increase their affinity through reducing the entropic loss associated with the binding. Overall, in addition to the electrostatic and hydrophobic forces, protein–peptide interactions can be affected by the entropic effect and conformational flexibility that could be willingly examined with atomistic simulations.111
Very recently, using a fast molecular dynamics simulation, the energetic and dynamic features of protein–peptide interactions were studied. In most cases, the native binding sites and native-like postures of protein–peptide complexes were recapitulated. Additional investigation showed that insertion of motility and flexibility in the simulation could meaningfully advance the correctness of protein–peptide binding prediction.112
Peptide affinity prediction
Most features of computational peptide design are based on the accuracy and efficacy of affinity prediction. Hence, the fast and reliable prediction of peptide–protein affinity is significant for rational peptide design.18 In this aspect, two categories of prediction algorithms including sequence- and structure-based approaches were developed. The sequence-based method uses the information derived from primary polypeptide sequences to approximate and evaluate the standards of the binding affinity. The structure-based process takes the information derived from 3D structures of protein–peptide complexes to predict the binding affinity.113
At the sequence level, the quantitative structure–activity relationships (QSARs) have been widely utilized to forecast the binding affinity of peptides and conclude the biologic function.114 To model the statistical correlation between sequence patterns and biologic activities of experimentally assessed peptides, machine-learning methods such as partial least squares (PLS), artificial neural networks (ANN) and support vector machine (SVM) have been used. The obtained correlations have been used to infer experimentally undetermined peptides.115
The relationship between the biologic activity and molecular structure is an important issue in biology and biochemistry. QSAR is a well-established method employed in pharmaceutical chemistry and has become a standard tool for drug discovery. However, the predictive capacity of QSAR techniques is generally weaker than statistics-based approaches. Therefore, a combination of the QSAR method with a statistic-based technique may bring out the best in each other and can be a trend in future developments of drug discovery.114
At the structural level, numerous reports on affinity prediction have addressed the MHC-binding peptides. Plentiful MHC–peptide complex structure records have been deposited in the PDB.116
The significance of domain-peptide recognition has been recently illustrated in the metabolic pathway and cell signaling.117 To predict the protein–peptide binding potency, a number of strict theories were suggested based on the potential free energy perturbation. The theories computed the alteration of free energies upon the interaction between phosphor-tyrosine-tetra-peptide (pYEEI) and human Lck SH2 domain.118 Furthermore, to obtain a deep insight into the structural and energetic aspects of peptide recognition by the SH3 domain, a number of molecular modeling experiments such as homology modeling, molecular docking and mechanism dynamics were used.119 Peptide array strategies confirmed that some peptide candidates may be potent binders of the Abl SH3 domain.120 Very recently, an approach including quantum mechanics/molecular mechanics, semi-empirical Poisson–Boltzmann/surface area and empirical conformational free energy analysis was developed to quantitatively illustrate the energetic contributions involved in the affinity losing of PDZ domain and OppA protein to their peptide ligands.121,122
De novo peptide design
Recently, in order to de novo target-based peptide design, two remarkable methodologies including the VitAL method and an approach developed by Bhattacherjee and Wallin were introduced. The VitAL method pools verterbi algorithm with AutoDock to design peptides for the binding sites of a target.123 The “Bhattacherjee and Wallin” approach explores both peptide sequence and conformational space around a protein target at the same time.124 This approach was tested on three dissimilar peptide–protein domains to assess its ability.13
A brief list of the existing computational resources employed in peptide design is presented in Table 2.
Table 2.
A brief list of available computational resources employed in the peptide design
| Resource | Description | References |
|---|---|---|
| TumorHoPe | Tumor-homing peptide database | Kapoor et al144 |
| Brainpep | Blood–brain barrier peptide database | Volpe145 |
| SARvision | Peptide bioinformatics | Hansen et al75 |
| PEP-FOLD | Peptide structure prediction | Thevenet et al81 |
| PepX | Unique protein–peptide structural clusters | Vanhee et al146 |
| PepBind | PDB-derived protein–peptide structures | Das et al89 |
| PeptiDB | Survey of protein–peptide interactions | London et al90 |
| FlexPepDock | Protein–peptide docking | London et al147 |
| AnchorDock | Protein–peptide docking | Ben-Shimon and Niv104 |
| HADDOCK | Protein–peptide docking | Trellet et al105 |
| CABS-dock | Protein–peptide docking | Kurcinski et al106 |
| GalaxyPepDock | Protein–peptide docking | Lee et al107 |
| DocScheme | Protein–peptide docking | Niv and Weinstein98 |
| DynaDock | Protein–peptide docking | Antes99 |
| Pepspec | Protein–peptide docking | King and Bradley100 |
| PepCrawler | Protein–peptide docking | Donsky and Wolfson101 |
| pDOCK | Protein–peptide docking | Khan and Ranganathan148 |
| ACCLUSTER | Peptide-binding site prediction | Yan and Zou149 |
| ACCLUSTER+BriX | Peptide-binding site prediction | Verschueren et al150 |
| PEP-SiteFinder | Peptide-binding site prediction | Saladin et al151 |
| VitAL | De novo peptide design | Unal et al152 |
Abbreviation: PDB, Protein Data Bank.
In silico peptidomimetics design
In recent years, some computational methods have been proposed to design peptidomimetics. These methods can be classified based on their specificity to translate peptides to peptidomimetics.28 To select the best method, awareness about the structure of peptide–protein complexes is important.28,96 Herein, recently introduced methods for computer-aided design of peptidomimetics are presented.
De novo design method
GrowMol is a combinatorial algorithm employed in the peptidomimetics design. GrowMol searches a variety of probable ligands for the binding sites of a target protein125 and produces molecules with the chemical and steric complementarity for the 3D structure of binding sites.
This method was used to generate peptidomimetic inhibitors of thermolysin, HIV protease and pepsin. By using the X-ray crystal structures of pepstatin–pepsin complexes, GrowMol predicted therapeutic peptidomimetics against the aspartic proteases. The algorithm created some cyclic inhibitors bridging the side chains of cysteine residues in the Pl and P3 inhibitor subsites. The binding modes were checked using X-ray crystallography.125,126
LUDI is another interesting software referring to the de novo methodology.127 By using natural and non-natural amino acids as building blocks, the software designed peptidomimetics against renin, thermolysin and elastase.127 Conformational flexibility of each novel peptidomimetic was searched through sampling the multiple conformers of each amino acid.127
Peptide-driven pharmacophoric method
Peptide-driven pharmacophoric hypothesis is the most perceptive computational technique discovered in the peptidomimetics design. The method is especially useful when the X-ray structures of protein–protein complexes exist.28 The main idea is to adapt the hot spot concept into the associated pharmacophoric feature concept. With a pharmacophore-based virtual screening process, this strategy can determine novel type 3 mimetics.128 In fact, the side chains of each amino acid can be simply categorized based on the conventional pharmacophoric characteristics, such as hydrogen bond donors and acceptors, aromatic ring and charged and hydrophobic centers.
For example, in a report, pharmacophore model directed synthesis of the non-peptide analogs of a cationic antimicrobial peptide identified an anti-staphylococcal activity.129 To make a pharmacophore hypothesis, a model of RNA III-inhibiting peptide (RIP), a well-known heptapeptide inhibitor of the staphylococcal pathogenesis, was utilized. Through the virtual screening of 300,000 commercially available small molecules based on the RIP-based pharmacophore, Hamamelitannin was discovered as a non-peptide mimetic of RIP. Hamamelitannin is a tannin derivate extracted from Hamamelis virginiana.28,129
In another study, two rounds of in silico screening were performed to discover potential peptidomimetics able to mimic a cyclic peptide (cyclo-[CPFVKTQLC]) that is known to bind the anb3 integrin receptor.130 At the end of the process, the most potent representatives were at least 2,000 times better than the original cyclopeptide (around 2 mM).130
In a prosperous instance, virtual screening was done by using multi-conformational forms of a large commercial library. A target-based pharmacophoric model mapped the CD4-binding site on HIV-1 gp120. The pharmacophore hypothesis was made based on a homology model of the protein cavity. In a cell-based assay, two of the top scoring molecules were detected as micromolar inhibitors of HIV-1 replication.131
The pharmacophore-based screening was used to find the novel Alzheimer’s therapeutics as mimetics of neurotrophins.132 The therapeutic utilization of neurotrophins might be restricted because of several deficiencies such as its reduced central nervous system penetration, decreased stability and potency to enhance neuronal death through interaction with the p75NTR receptor. The mimetism of particular nerve growth factor domains could inhibit neuronal death. Peptidomimetics of the loop 1 and loop 4 domains of nerve growth factor can prevent neuronal death induced by p75NTR-dependent and Trk-related signaling.132
In another study, a full-computational pharmacophore-based approach assessed the FDA-approved drugs as valuable candidates to inhibit protein–protein interactions.133 Peptide structures were designated in terms of pharmacophores and searched against the FDA-approved drugs to detect same molecules. The top ranking drug matches contained several nuclear receptor ligands and matched allosterically to the binding site on the target protein. The top ranking drug matches were docked to the peptide-binding site. The majority of the top-ranking matches presented a negative free energy change upon binding that was comparable to the standard peptide.133
Geometry similarity method
Geometry similarity methods create a geometric similarity between non-peptide templates and peptide patches. In a study, the SuperMimic tool was developed to recognize peptide mimetics.134 In the program, a complex library of peptidomimetics composed of several protein structure libraries has been deposited. Moreover, SuperMimic includes the D-peptides, synthetic components (reported as beta-turn or gamma-turn mimetics) and peptidomimetic ligands obtained from the PDB.134 In the program, the searching process allows scanning a library of small molecules that mimic the tertiary structure of a query peptide followed by scanning of a protein library where a query for small molecule can adopt into the backbone.28,134
Sequence-based method
Recently, a method has been developed to rank peptide compound matches that are limited to short linear motifs in proteins and compounds with amino acid substituents.135 The algorithm allows mapping the side chain-like substituents on every compound of a large chemical library. The complete molecule can be signified by a short sequence, and each fragment in the molecule can be represented as a distinct letter abbreviation.28 A cross-search between the PubChem database (about 5.4 million molecules) and a non-redundant collection of 11,488 peptides obtained from PDB demonstrated that the algorithm can be useful for high-throughput measurements.28 To recognize a true positive, the method explored identified protein motifs against the National Cancer Institute Developmental Therapeutic Program compound database.135
In another study, the Similarity of Amino Acid Motifs to Compounds web server was developed to ease screening of identified motif structures against bioactive compound databases.136 The methodology was reported to be efficient since the compound databases were preprocessed to maximize the accessible data, and the necessary input data was minimal.136 In Similarity of Amino Acid Motifs to Compounds, motif matching can be full or partial that may decrease or enhance the number of potential mimetics, respectively. Using a novel search algorithm, the web service can perform a fast screening of known or putative motifs against ready compound libraries. The classified results can be examined by linking to appropriate databases.28,136
Fragment-based method
Replacement with Partial Ligand Alternatives through Computational Enrichment is a fragment-based approach.137
By using structures of peptide-bound proteins as design anchors, the program can computationally find a non-peptide mimetic for specific determinants of known peptide ligands.137
Hybrid peptide-driven shape and pharmacophoric method
Development and application of strategies for pharmacophore modeling indicate that the medicinal chemistry community has broadly accepted the intuitive nature of the pharmacophore concept. Besides, shape complementarity has been identified as a significant element in the molecular identification between ligands and their targets.28 In virtual screening efforts, using the pharmacophore- and shape-based techniques distinctly may increase the rate of false-positive results.128 Therefore, incorporating both pharmacophore- and shape-matching techniques into one program can potentially diminish the rate of false positives.128
Recently, to discover novel peptidomimetics, a web-oriented virtual screening tool named pepMMsMIMIC138 was developed to pool the conventional pharmacophore matching with shape complementarity. A library of 17 million conformers were extracted from 3.9 million commercially available chemicals and gathered in the MMsINC database. The database was used as a skeleton to develop pepMMsMIMIC.139 In the pepMMsMIMIC interface, the 3D structure of a protein-bound peptide is used as an input. Then, chemical structures able to mimic the pharmacophore and shape similarity of the original peptide are proposed to involve in the protein–protein recognition.139
A list of in silico methods used to design potential peptidomimetics along with their strengths and weaknesses is presented in Table 3.
Table 3.
A list of the in silico methods utilized to design potential peptidomimetics, along with their strengths and weaknesses
| Methods | Description | Strengths | Weaknesses |
|---|---|---|---|
| De novo design | When the structure of the host protein is only available | Generation of highly diverse candidates | Does not allow large-scale screening of virtual libraries |
| Receptor-based pharmacophore hypothesis | When the structure of the host protein is only available | Appropriate to a large-scale virtual screening campaign | 3D pharmacophoric hypothesis is needed; protein atom-type parameterization is required |
| Conventional hot spots-based pharmacophore hypothesis | When the protein–protein complex structure is available | Suitable to a large-scale virtual screening campaign | 3D pharmacophoric hypothesis is required; protein atom-type parameterization is necessary |
| Sequence based | Method is used to rank peptide–compound matches that is limited to short linear motifs in proteins and compounds involving amino acid substituents | Useful for high-throughput screening as a prefiltering tool | Limited validation of current Methods Restricted to small linear motifs |
| Geometry similarity | When the structure of the guest peptide is only available | Efficiency in identification of similarities between peptide patches and non-peptide templates | Current implementation is limited to small libraries; similarity search is restricted to backbone features |
| Fragment based | When the structure of the guest peptide is only available | – | Limited fragment library |
Abbreviation: 3D, three dimensional.
Conclusion
Overall, design and development of therapeutics are tedious, expensive and time-consuming procedures. Therefore, using modern approaches including computer-aided design methods can lessen the examination phase, price and failure of therapeutics discovery. Computational methods used to design amino acid-based therapeutics can increase the range of available biotherapeutics. Benefiting from the dramatic advance in bioinformatics, computational tools can be used to find and develop therapeutic proteins, peptides and peptidomimetics.140,141 Moreover, using the computational tools decrease the cost of therapeutics development, from concept to market, by up to 50%.140
However, in the computational protein designing, there are some challenges such as our inadequate knowledge of folding and physical forces that stabilize protein structures. Moreover, sequences and local structures have many degrees of freedom that can complicate the sequence search. Therefore, there is a requirement for effective methods to find sequences related to a particular structure and measure essential protein folding criteria.
Overall, in silico design of amino acid-based therapeutics includes many challenges that should be removed to improve the overall performance of the design processes. For example, although structure determination of all disease-related proteins through crystallography and NMR is a laborious task, it is necessary to gather much structural information of peptide–protein interactions. Besides, development of vigorous algorithms to calculate protein–protein binding energies is essential. The estimation of binding constant between two macromolecules with an appropriate speed–accuracy tradeoff needs millisecond scale molecular dynamics. Moreover, understanding of both protein–protein and protein–peptidomimetics recognition processes in a molecular level can be improved using higher accurate force fields such as quantum mechanical polarizable force.
In recent years, there are growing examples on the approval of monoclonal antibodies (therapeutic antibodies) by the FDA for treatment of various diseases. This important area of amino acid-based therapeutics has been covered in more depth elsewhere.142,143 For more explanation about the theorems and details of antibody informatics for drug discovery as well as the computer-aided antibody design, readers are referred to study previous reports.142,143
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Mócsai A, Kovács L, Gergely P. What is the future of targeted therapy in rheumatology: biologics or small molecules? BMC Med. 2014;12:43. doi: 10.1186/1741-7015-12-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leader B, Baca QJ, Golan DE. Protein therapeutics: a summary and pharmacological classification. Nat Rev Drug Discov. 2008;7:21–39. doi: 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- 3.Roy A, Nair S, Sen N, Soni N, Madhusudhan MS. In silico methods for design of biological therapeutics. Methods. 2017;131:33–65. doi: 10.1016/j.ymeth.2017.09.008. [DOI] [PubMed] [Google Scholar]
- 4.Farhadi T, Hashemian SMR. Constructing novel chimeric DNA vaccine against Salmonella enterica based on SopB and GroEL proteins: an in silico approach. Int J Pharm Investig. 2017:1–17. [Google Scholar]
- 5.Farhadi T, Nezafat N, Ghasemi Y. In silico phylogenetic analysis of Vibrio cholera isolates based on three housekeeping genes. Int J Comput Biol Drug Des. 2015a;8(1):62–74. doi: 10.1504/IJCBDD.2015.068789. [DOI] [PubMed] [Google Scholar]
- 6.Farhadi T, Nezafat N, Ghasemi Y, Karimi Z, Hemmati S, Erfani N. Designing of complex multi-epitope peptide vaccine based on Omps of Klebsiella pneumoniae: an in silico approach. Int J Pept Res Ther. 2015a;21(3):325–341. [Google Scholar]
- 7.Samish I, MacDermaid CM, Perez-Aguilar JM, Saven JG. Theoretical and computational protein design annu. Rev Phys Chem. 2011;62:129–149. doi: 10.1146/annurev-physchem-032210-103509. [DOI] [PubMed] [Google Scholar]
- 8.Angamuthu K, Piramanayagam S. Evaluation of in silico protein secondary structure prediction methods by employing statistical techniques. Biomed Biotechnol Res J. 2017;1:29–36. [Google Scholar]
- 9.Wankhade G, Kamble S, Deshmukh S, Jena L, Waghmare P, Harinath BC. Inhibition of mycobacterial CYP125 enzyme by sesamin and β-sitosterol: an in silico and in vitro study. Biomed Biotechnol Res J. 2017;1:49–54. [Google Scholar]
- 10.Onuchic JN, Luthey-Schulten Z, Wolynes PG. Theory of protein folding: the energy landscape perspective. Annu Rev Phys Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 11.Onuchic JN, Wolynes PG, Luthey-Schulten Z, Socci ND. Toward an outline of the topography of a realistic protein-folding funnel. Proc Natl Acad Sci U S A. 1995;92:3626–3630. doi: 10.1073/pnas.92.8.3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chino M, Maglio O, Nastri F, Pavone V, DeGrado WF, Lombardi A. Artificial diiron enzymes with a de novo designed four-helix bundle structure. Eur J Inorg Chem. 2015:3371–3390. doi: 10.1002/ejic.201500470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diller DJ, Swanson J, Bayden AS, Jarosinski M, Audie J. Rational, computer-enabled peptide drug design: principles, methods, applications and future directions. Future Med Chem. 2015;7(16):2173–2193. doi: 10.4155/fmc.15.142. [DOI] [PubMed] [Google Scholar]
- 14.Rentzsch R, Renard BY. Docking small peptides remains a great challenge: an assessment using AutoDock Vina. Brief Bioinform. 2015;16(6):1045–1056. doi: 10.1093/bib/bbv008. [DOI] [PubMed] [Google Scholar]
- 15.Pike DH, Nanda V. Empirical estimation of local dielectric constants: toward atomistic design of collagen mimetic peptides. Biopolymers. 2015;104(4):360–370. doi: 10.1002/bip.22644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Audie J, Swanson J. Recent work in the development and application of protein-peptide docking. Future Med Chem. 2012;4(12):1619–1644. doi: 10.4155/fmc.12.99. [DOI] [PubMed] [Google Scholar]
- 17.Bradbury J. Rational design of peptide drugs: avoiding aggregation. Drug Discov Today. 2005;10:1208–1209. doi: 10.1016/S1359-6446(05)03603-2. [DOI] [PubMed] [Google Scholar]
- 18.Zhou P, Wang C, Ren Y, Yang C, Tian F. Computational peptidology: a new and promising approach to therapeutic peptide design. Curr Med Chem. 2013;20:1985–1996. doi: 10.2174/0929867311320150005. [DOI] [PubMed] [Google Scholar]
- 19.Ong ZY, Wiradharm N, Yang YY. Strategies employed in the design and optimization of synthetic antimicrobial peptide amphiphiles with enhanced therapeutic potentials. Adv Drug Deliv Rev. 2014;78:28–45. doi: 10.1016/j.addr.2014.10.013. [DOI] [PubMed] [Google Scholar]
- 20.Góngora-Benítez M, Tulla-Puche J, Albericio F. Multifaceted roles of disulfide bonds. Peptides as therapeutics. Chem Rev. 2014;114(2):901–926. doi: 10.1021/cr400031z. [DOI] [PubMed] [Google Scholar]
- 21.Vagner J, Qu H, Hruby VJ. Peptidomimetics, a synthetic tool of drug discovery. Curr Opin Chem Biol. 2008;12(3):292–296. doi: 10.1016/j.cbpa.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watkins AM. An in silico pipeline for the design of peptidomimetic protein-protein interaction inhibitors (Order 10188557) 2016. [Accessed October 12, 2017]. Available from ProQuest Dissertations & Theses A&I; ProQuest Dissertations & Theses Global. (1845861691). Available from: https://search.proquest.com/docview/1845861691?accountid=42543.
- 23.Newman DJ, Cragg GM. Natural products as sources of new drugs over the last 25 years. Nat Prod. 2007;70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- 24.Isidro-Llobet A, Murillo T, Bello P, et al. Diversity-oriented synthesis of macrocyclic peptidomimetics. Proc Natl Acad Sci U S A. 2011;108(17):6793–6798. doi: 10.1073/pnas.1015267108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghosh AK, Xi K, Grum-Tokars V, et al. Structure-based design, synthesis, and biological evaluation of peptidomimetic SARS-CoV 3CLpro inhibitors. Bioorg Med Chem Lett. 2007;17:5876–5880. doi: 10.1016/j.bmcl.2007.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abell A, editor. Advances in Amino Acid Mimetics and Peptidomimetics. Vol. 1. New York: JAI Press, Inc; 1997. p. 167. [Google Scholar]
- 27.Marshall GR. A hierarchical approach to peptidomimetic design. Tetrahedron. 1993;49:3547–3558. [Google Scholar]
- 28.Floris M, Moro S. Mimicking Peptides… In Silico. Mol Inf. 2012;31:12–20. doi: 10.1002/minf.201100093. [DOI] [PubMed] [Google Scholar]
- 29.Ripka AS, Rich DH. Peptidomimetic design. Curr Opin Chem Biol. 1998;2:441–452. doi: 10.1016/s1367-5931(98)80119-1. [DOI] [PubMed] [Google Scholar]
- 30.Agdeppa ED. Rational design for peptide drugs. J Nucl Med. 2006;47(12):22N–24N. [Google Scholar]
- 31.Shah A, Barathi B, Nair A, Rajam C. Peptidomimetics as a cutting edge tool for advanced healthcare. Int J Pharm Sci Res. 2017;8(12):4992–5000. [Google Scholar]
- 32.Seebach D, Gardiner J. β-Peptidic Peptidomimetics. Acc Chem Res. 2008;41:1366–1375. doi: 10.1021/ar700263g. [DOI] [PubMed] [Google Scholar]
- 33.Gautier A, Pitrat D, Hasserodt J. An unusual functional group interaction and its potential to reproduce steric and electrostatic features of the transition states of peptidolysis. Bioorg Med Chem. 2006;14:3835–3847. doi: 10.1016/j.bmc.2006.01.031. [DOI] [PubMed] [Google Scholar]
- 34.Alig L, Edenhofer A, Hadvary P, et al. Low molecular weight, non-peptide fibrinogen receptor antagonists. J Med Chem. 1992;35:4393–4407. doi: 10.1021/jm00101a017. [DOI] [PubMed] [Google Scholar]
- 35.Longo FM, Xie Y, Massa SM. Neurotrophin small molecule mimetics: candidate therapeutic agents for neurological disorders. Curr Med Chem. 2005;5:29–41. [Google Scholar]
- 36.Hruby VJ, Li G, Haskell-Luevano C, Shenderovich M. Design of peptides, proteins, and peptidomimetics in chi space. Biopolymers. 1997;43:219–266. doi: 10.1002/(SICI)1097-0282(1997)43:3<219::AID-BIP3>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 37.Pabo C, Molecular technology Designing proteins and peptides. Nature. 1983;301:200. doi: 10.1038/301200a0. [DOI] [PubMed] [Google Scholar]
- 38.Drexler KE. Molecular engineering: an approach to the development of general capabilities for molecular manipulation. Proc Natl Acad Sci U S A. 1981;78:5275–5278. doi: 10.1073/pnas.78.9.5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schneider M, Fu XR, Keating AE. X-ray versus NMR structures as templates for computational protein design. Proteins. 2009;77:97–110. doi: 10.1002/prot.22421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harbury PB, Plecs JJ, Tidor B, Alber T, Kim PS. High-resolution protein design with backbone freedom. Science. 1998;282:1462–1467. doi: 10.1126/science.282.5393.1462. [DOI] [PubMed] [Google Scholar]
- 41.Humphris EL, Kortemme T. Prediction of protein-protein interface sequence diversity using flexible backbone computational protein design. Structure. 2008;16:1777–1788. doi: 10.1016/j.str.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 42.Mandell DJ, Kortemme T. Backbone flexibility in computational protein design. Curr Opin Biotechnol. 2009;20:420–428. doi: 10.1016/j.copbio.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 43.Jiang L, Althoff EA, Clemente FR, et al. De novo computational design of retro-aldol enzymes. Science. 2008;319:1387–1391. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagano N, Orengo CA, Thornton JM. One fold with many functions: the evolutionary relationships between TIM barrel families based on their sequences, structures and functions. J Mol Biol. 2002;321:741–765. doi: 10.1016/s0022-2836(02)00649-6. [DOI] [PubMed] [Google Scholar]
- 45.Samish I. Search and sampling in structural bioinformatics. In: Gu J, Bourne PE, editors. Structural Bioinformatics. New York: Wiley; 2009. pp. 207–235. [Google Scholar]
- 46.Desmet J, De Maeyer M, Hazes B, Lasters I. The dead-end elimination theorem and its use in protein side-chain positioning. Nature. 1992;356:539–542. doi: 10.1038/356539a0. [DOI] [PubMed] [Google Scholar]
- 47.Koehl P, Delarue M. Application of a self-consistent mean field theory to predict protein sidechains conformation and estimate their conformational entropy. J Mol Biol. 1994;239:249–275. doi: 10.1006/jmbi.1994.1366. [DOI] [PubMed] [Google Scholar]
- 48.Grigoryan G, Reinke AW, Keating AE. Design of protein-interaction specificity gives selective bZIP-binding peptides. Nature. 2009;458:859–864. doi: 10.1038/nature07885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang X, Saven JG. Computational methods for protein design and protein sequence variability: biased Monte Carlo and replica exchange. Chem Phys Lett. 2005;401:205–210. [Google Scholar]
- 50.Leach AR, Lemon AP. Exploring the conformational space of protein side chains using dead-end elimination and the A* algorithm. Proteins. 1998;33:227–239. doi: 10.1002/(sici)1097-0134(19981101)33:2<227::aid-prot7>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 51.Desjarlais JR, Handel TM. Side-chain and backbone flexibility in protein core design. J Mol Biol. 1999;290:305–318. doi: 10.1006/jmbi.1999.2866. [DOI] [PubMed] [Google Scholar]
- 52.LuCore SD, Litman JM, Powers KT, Gao S, Lynn AM, Tollefson WTA, et al. Dead-end elimination with a polarizable force field repacks PCNA structures. Biophysical Journal. 2015;109(4):816–826. doi: 10.1016/j.bpj.2015.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krivov GG, Shapovalov MV, Dunbrack RL., Jr Improved prediction of protein side-chain conformations with SCWRL4. Proteins. 2009;77:778–795. doi: 10.1002/prot.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Voigt CA, Gordon DB, Mayo SL. Trading accuracy for speed: a quantitative comparison of search algorithms in protein sequence design. J Mol Biol. 2000;299:789–803. doi: 10.1006/jmbi.2000.3758. [DOI] [PubMed] [Google Scholar]
- 55.Zou J, Saven JG. Using self-consistent fields to bias Monte Carlo methods with applications to designing and sampling protein sequences. J Chem Phys. 2003;118:3843–3854. [Google Scholar]
- 56.Calhoun JR, Kono H, Lahr S, Wang W, DeGrado WF, Saven JG. Computational design and characterization of a monomeric helical dinuclear metalloprotein. J Mol Biol. 2003;334:1101–1115. doi: 10.1016/j.jmb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 57.Zou JM, Saven JG. Statistical theory of combinatorial libraries of folding proteins: energetic discrimination of a target structure. J Mol Biol. 2000;296:281–294. doi: 10.1006/jmbi.1999.3426. [DOI] [PubMed] [Google Scholar]
- 58.Marshall SA, Mayo SL. Achieving stability and conformational specificity in designed proteins via binary patterning. J Mol Biol. 2001;305:619–631. doi: 10.1006/jmbi.2000.4319. [DOI] [PubMed] [Google Scholar]
- 59.Serrano AL, Troxler T, Tucker MJ, Gai F. Photophysics of a fluorescent non-natural amino acid: p-cyanophenylalanine. Chem Phys Lett. 2010;487:303–306. doi: 10.1016/j.cplett.2010.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chin JW, Cropp TA, Anderson JC, Mukherji M, Zhang Z, Schultz PG. An expanded eukaryotic genetic code. Science. 2003;301:964–967. doi: 10.1126/science.1084772. [DOI] [PubMed] [Google Scholar]
- 61.Jiang L, Kuhlman B, Kortemme T, Baker D. A “solvated rotamer” approach to modeling watermediated hydrogen bonds at protein-protein interfaces. Proteins. 2005;58:893–904. doi: 10.1002/prot.20347. [DOI] [PubMed] [Google Scholar]
- 62.Dunbrack RL., Jr Rotamer libraries in the 21st century. Curr Opin Struct Biol. 2002;12:431–440. doi: 10.1016/s0959-440x(02)00344-5. [DOI] [PubMed] [Google Scholar]
- 63.Peterson RW, Dutton PL, Wand AJ. Improved side-chain prediction accuracy using an ab initio potential energy function and a very large rotamer library. Protein Sci. 2004;13:735–751. doi: 10.1110/ps.03250104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Boas FE, Harbury PB. Potential energy functions for protein design. Curr Opin Struct Biol. 2007;17:199–204. doi: 10.1016/j.sbi.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 65.Jin WZ, Kambara O, Sasakawa H, Tamura A, Takada S. De novo design of foldable proteins with smooth folding funnel: automated negative design and experimental verification. Structure. 2003;11:581–590. doi: 10.1016/s0969-2126(03)00075-3. [DOI] [PubMed] [Google Scholar]
- 66.Simons KT, Kooperberg C, Huang E, Baker D. Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. J Mol Biol. 1997;268:209–225. doi: 10.1006/jmbi.1997.0959. [DOI] [PubMed] [Google Scholar]
- 67.Tsai CJ, Zheng J, Aleman C, Nussinov R. Structure by design: from single proteins and their building blocks to nanostructures. Trends Biotechnol. 2006;24:449–454. doi: 10.1016/j.tibtech.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 68.Cochran FV, Wu SP, Wang W, et al. Computational de novo design and characterization of a four-helix bundle protein that selectively binds a nonbiological cofactor. J Am Chem Soc. 2005;127:1346–1347. doi: 10.1021/ja044129a. [DOI] [PubMed] [Google Scholar]
- 69.McAllister KA, Zou HL, Cochran FV, et al. Using α-helical coiled coils to design nanostructured metalloporphyrin arrays. J Am Chem Soc. 2008;130:11921–11927. doi: 10.1021/ja800697g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rothlisberger D, Khersonsky O, Wollacott AM, et al. Kemp elimination catalysts by computational enzyme design. Nature. 2008;453:190–195. doi: 10.1038/nature06879. [DOI] [PubMed] [Google Scholar]
- 71.Liang HH, Chen H, Fan KQ, et al. De novo design of a βαβ motif. Angew Chem Int Ed Engl. 2009;48:3301–3303. doi: 10.1002/anie.200805476. [DOI] [PubMed] [Google Scholar]
- 72.Dantas G, Corrent C, Reichow SL, et al. High-resolution structural and thermodynamic analysis of extreme stabilization of human procarboxypeptidase by computational protein design. J Mol Biol. 2007;366:1209–1221. doi: 10.1016/j.jmb.2006.11.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302:1364–1368. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 74.Rivera CG, Rosca EV, Pandey NB, Koskimaki JE, Bader JS, Popel AS. Novel peptide-specific quantitative structure activity relationship (QSAR) analysis applied to collagen IV peptides with antiangiogenic activity. J Med Chem. 2011;54(19):6492–6500. doi: 10.1021/jm200114f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hansen MR, Villar HO, Feyfant E. Development of an informatics platform for therapeutic protein and peptide analytics. J Chem Inform Model. 2013;53(10):2774–2779. doi: 10.1021/ci400333x. [DOI] [PubMed] [Google Scholar]
- 76.Du QS, Ma Y, Xie NZ, Huang RB. Two-level QSAR network (2L-QSAR) for peptide inhibitor design based on amino acid properties and sequence positions. SAR QSAR Environ Res. 2014;25(10):837–851. doi: 10.1080/1062936X.2014.959049. [DOI] [PubMed] [Google Scholar]
- 77.Du QS, Xie NZ, Huang RB. Recent development of peptide drugs and advance on theory and methodology of peptide inhibitor design. Med Chem. 2015;11(3):235–247. doi: 10.2174/1573406411666141229163355. [DOI] [PubMed] [Google Scholar]
- 78.Du QS, Wei YT, Pang ZW, Chou KC, Huang RB. Predicting the affinity of epitope-peptides with class I MHC molecule HLA-A*0201: an application of amino acid-based peptide prediction. Protein Eng Des Sel. 2007;20(9):417–423. doi: 10.1093/protein/gzm036. [DOI] [PubMed] [Google Scholar]
- 79.Bhonsle JB, Clark T, Bartolotti L, Hicks RP. A brief overview of antimicrobial peptides containing unnatural amino acids and ligand-based approaches for peptide ligands. Curr Top Med Chem. 2013;13(24):3205–3224. doi: 10.2174/15680266113136660226. [DOI] [PubMed] [Google Scholar]
- 80.Giguere S, Laviolette F, Marchand M, et al. Machine learning assisted design of highly active peptides for drug discovery. PLoS Comput Biol. 2015;11(4):e1004074. doi: 10.1371/journal.pcbi.1004074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Thevenet P, Shen Y, Maupetit J, Guyon F, Derreumaux P, Tuffery P. PEP-FOLD: an updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res. 2012;40:W288–W293. doi: 10.1093/nar/gks419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Beaufays J, Lins L, Thomas A, Brasseur R. In silico predictions of 3D structures of linear and cyclic peptides with natural and non-proteinogenic residues. J Peptide Sci. 2012;18(1):17–24. doi: 10.1002/psc.1410. [DOI] [PubMed] [Google Scholar]
- 83.Klepeis JL, Lindorff-Larsen K, Dror RO, Shaw DE. Long-timescale molecular dynamics simulations of protein structure and function. Curr Opin Struct Biol. 2009;19(2):120–127. doi: 10.1016/j.sbi.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 84.Lindorff-Larsen K, Piana S, Dror RO, Shaw DE. How fastfolding proteins fold. Science. 2011;334(6055):517–520. doi: 10.1126/science.1208351. [DOI] [PubMed] [Google Scholar]
- 85.Improta R, Vitagliano L, Esposito L. Bond distances in polypeptide backbones depend on the local conformation. Acta Crystallogr D Biol Crystallogr. 2015;71(Pt 6):1272–1283. doi: 10.1107/S1399004715005507. [DOI] [PubMed] [Google Scholar]
- 86.Haslach EM, Huang H, Dirain M, et al. Identification of tetrapeptides from a mixture based positional scanning library that can restore nM full agonist function of the L106P, I69T, I102S, A219V, C271Y, and C271R human melanocortin-4 polymorphic receptors (hMC4Rs) J Med Chem. 2014;57(11):4615–4628. doi: 10.1021/jm500064t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Berman HM, Westbrook J, Feng Z, et al. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Verschueren E, Vanhee P, van der Sloot AM, Serrano L, Rousseau F, Schymkowitz J. Protein design with fragment databases. Curr Opin Struct Biol. 2011;21(4):452–459. doi: 10.1016/j.sbi.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 89.Das AA, Sharma OP, Kumar MS, Krishna R, Mathur PP. PepBind: a comprehensive database and computational tool for analysis of protein-peptide interactions. Genomics Proteomics Bioinform. 2013;11(4):241–246. doi: 10.1016/j.gpb.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.London N, Movshovitz-Attias D, Schueler-Furman O. The structural basis of peptide-protein binding strategies. Structure. 2010;18(2):188–199. doi: 10.1016/j.str.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 91.Vanhee P, Stricher F, Baeten L, et al. Protein-peptide interactions adopt the same structural motifs as monomeric protein folds. Structure. 2009;17:1128–1136. doi: 10.1016/j.str.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 92.Ciemny MP, Kurcinski M, Kozak KJ, Kolinski A, Kmiecik S. Highly Flexible Protein-Peptide Docking Using CABS-Dock. In: Schueler-Furman O, London N, editors. Modeling Peptide-Protein Interactions Methods in Molecular Biology. Vol. 1561. New York, NY: Humana Press; 2017. [DOI] [PubMed] [Google Scholar]
- 93.Farhadi T, Fakharian A, Ovchinnikov RS. Virtual screening for potential inhibitors of CTX-M-15 protein of Klebsiella pneumoniae. Interdisciplin Sci. 2017 doi: 10.1007/s12539-017-0222-y. [DOI] [PubMed] [Google Scholar]
- 94.Yagi Y, Terada K, Noma T, Ikebukuro K, Sode K. In silico panning for a non-competitive peptide inhibitor. BMC Bioinformatics. 2007;8:11. doi: 10.1186/1471-2105-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kellenberger E, Rodrigo J, Muller P, Rognan D. Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins. 2004;57:225–242. doi: 10.1002/prot.20149. [DOI] [PubMed] [Google Scholar]
- 96.Farhadi T. In silico designing of peptide inhibitors against pregnane X receptor: the novel candidates to control drug metabolism. Int J Pept Res Ther. 2017:1–12. [Google Scholar]
- 97.Desmet J, Wilson IA, Joniau M, de Maeyer M, Lasters I. Computation of the binding of fully flexible peptides to proteins with flexible side chains. FASEB J. 1997;11:164–172. doi: 10.1096/fasebj.11.2.9039959. [DOI] [PubMed] [Google Scholar]
- 98.Niv MY, Weinstein H. A flexible docking procedure for the exploration of peptide binding selectivity to known structures and homology models of PDZ domains. J Am Chem Soc. 2005;127:14072–14079. doi: 10.1021/ja054195s. [DOI] [PubMed] [Google Scholar]
- 99.Antes I. DynaDock: a new molecular dynamics-based algorithm for protein-peptide docking including receptor flexibility. Proteins. 2010;78(5):1084–1104. doi: 10.1002/prot.22629. [DOI] [PubMed] [Google Scholar]
- 100.King CA, Bradley P. Structure-based prediction of proteinpeptide specificity in Rosetta. Proteins. 2010;78:3437–3449. doi: 10.1002/prot.22851. [DOI] [PubMed] [Google Scholar]
- 101.Donsky E, Wolfson HJ. PepCrawler: a fast RRT-based algorithm for high-resolution refinement and binding-affinity estimation of peptide inhibitors. Bioinformatics. 2011;27:2836–2842. doi: 10.1093/bioinformatics/btr498. [DOI] [PubMed] [Google Scholar]
- 102.Raveh B, London N, Zimmerman L, Schueler-Furman O. Rosetta FlexPepDock ab-initio: simultaneous folding, docking and refinement of peptides onto their receptors. PLoS ONE. 2011;6(4):e18934. doi: 10.1371/journal.pone.0018934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Raveh B, London N, Schueler-Furman O. Sub-angstrom modeling of complexes between flexible peptides and globular proteins. Proteins. 2010;78:2029–2040. doi: 10.1002/prot.22716. [DOI] [PubMed] [Google Scholar]
- 104.Ben-Shimon A, Niv MY. AnchorDock: blind and flexible anchor-driven peptide docking. Structure. 2015;23(5):929–940. doi: 10.1016/j.str.2015.03.010. [DOI] [PubMed] [Google Scholar]
- 105.Trellet M, Melquiond AS, Bonvin AM. A unified conformational selection and induced fit approach to proteinpeptide docking. PLoS One. 2013;8(3):e58769. doi: 10.1371/journal.pone.0058769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kurcinski M, Jamroz M, Blaszczyk M, Kolinski A, Kmiecik S. CABS-dock web server for the flexible docking of peptides to proteins without prior knowledge of the binding site. Nucleic Acids Res. 2015;43(W1):W419–W424. doi: 10.1093/nar/gkv456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lee H, Heo L, Lee MS, Seok C. GalaxyPepDock: a proteinpeptide docking tool based on interaction similarity and energy optimization. Nucleic Acids Res. 2015;43(W1):W431–W435. doi: 10.1093/nar/gkv495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Voelz VA, Shell MS, Dill KA. Predicting peptide structures in native proteins from physical simulations of fragments. PLoS Comput Biol. 2009;5:e1000281. doi: 10.1371/journal.pcbi.1000281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ahmad M, Gu W, Helms V. Mechanism of fast peptide recognition by SH3 domains. Angew Chem Int Ed. 2008;47:7626–7630. doi: 10.1002/anie.200801856. [DOI] [PubMed] [Google Scholar]
- 110.Staneva I, Wallin S. Binding free energy landscape of domain peptide interactions. PLoS Comput Biol. 2011;7:e1002131. doi: 10.1371/journal.pcbi.1002131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lama D, Sankararamakrishnan R. Molecular dynamics simulations of pro-apoptotic BH3 peptide helices in aqueous medium: relationship between helix stability and their binding affinities to the anti-apoptotic protein Bcl-XL. J Comput Aided Mol Des. 2011;25:413–426. doi: 10.1007/s10822-011-9428-y. [DOI] [PubMed] [Google Scholar]
- 112.Dagliyan O, Proctor EA, D’Auria KM, Ding F, Dokholyan NV. Structural and dynamic determinants of protein-peptide recognition. Structure. 2011;19:1837–1845. doi: 10.1016/j.str.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhou P, Tian F, Wu Y, Li Z, Shang Z. Quantitative sequenceactivity model (QSAM): applying QSAR strategy to model and predict bio-activity and function of peptides, proteins and nucleic acids. Curr Comput Aided Drug Des. 2008;4:311–321. [Google Scholar]
- 114.Du QS, Huang RB, Chou KC. Recent advances in QSAR and their applications in predicting the activities of chemical molecules, peptides and proteins for drug design. Curr Protein Pept Sci. 2008;9:248–260. doi: 10.2174/138920308784534005. [DOI] [PubMed] [Google Scholar]
- 115.Zhou P, Tian F, Lv F, Shang Z. Comprehensive comparison of eight statistical modelling methods used in quantitative structure retention relationship studies for liquid chromatographic retention times of peptides generated by protease digestion of the Escherichia coli proteome. J Chromatogr A. 2009;1216:3107–3116. doi: 10.1016/j.chroma.2009.01.086. [DOI] [PubMed] [Google Scholar]
- 116.Lafuente EM, Reche PA. Prediction of MHC-peptide binding: a systematic and comprehensive overview. Curr Pharm Des. 2009;15:3209–3220. doi: 10.2174/138161209789105162. [DOI] [PubMed] [Google Scholar]
- 117.Reimand J, Hui S, Jain S, Law B, Bader GD. Domain mediated protein interaction prediction: from genome to network. FEBS Lett. 2012;586:2751–2763. doi: 10.1016/j.febslet.2012.04.027. [DOI] [PubMed] [Google Scholar]
- 118.Woo HJ, Roux B. Calculation of absolute protein-ligand binding free energy from computer simulations. Proc Natl Acad Sci U S A. 2005;102:6825–6830. doi: 10.1073/pnas.0409005102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hou T, McLaughlin W, Lu B, Chen K, Wang W. Prediction of binding affinities between the human amphiphysin-1 SH3 domain and its peptide ligands using homology modeling, molecular dynamics and molecular field analysis. J Proteome Res. 2006;5:32–43. doi: 10.1021/pr0502267. [DOI] [PubMed] [Google Scholar]
- 120.Hou T, Xu Z, Zhang W, et al. Characterization of domain-peptide interaction interface: a generic structure-based model to decipher the binding specificity of SH3 domains. Mol Cell Proteomics. 2009;8:639–649. doi: 10.1074/mcp.M800450-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tian F, Yang L, Lv F, Luo X, Pan Y. Why OppA protein can bind sequence-independent peptides? A combination of QM/MM, PB/SA, and structure-based QSAR analyses. Amino Acids. 2011;40:493–503. doi: 10.1007/s00726-010-0661-9. [DOI] [PubMed] [Google Scholar]
- 122.Tian F, Lv Y, Zhou P, Yang L. Characterization of PDZ domain-peptide interactions using an integrated protocol of QM/MM, PB/SA, and CFEA analyses. J Comput Aided Mol Des. 2011;25:947–958. doi: 10.1007/s10822-011-9474-5. [DOI] [PubMed] [Google Scholar]
- 123.Vanhee P, van der Sloot AM, Verschueren E, Serrano L, Rousseau F, Schymkowitz J. Computational design of peptide ligands. Trends in Biotechnology. 2011;29(5):231–239. doi: 10.1016/j.tibtech.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 124.Bhattacherjee A, Wallin S. Exploring protein-peptide binding specificity through computational peptide screening. PLoS Comput Biol. 2013;9(10):e1003277. doi: 10.1371/journal.pcbi.1003277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bohacek RS, McMartin CJ. Multiple highly diverse structures complementary to enzyme binding sites: results of extensive application of a de novo design method incorporating combinatorial growth. J Am Chem Soc. 1994;116:5560–5571. [Google Scholar]
- 126.Rich DH, Bohacek RS, Dales NA, Glunz P, Ripka AS. Transformation of peptides into non-peptides. Synthesis of computer-generated enzyme inhibitors. Chimia. 1997;51:45–47. [Google Scholar]
- 127.Bohm HJ. Towards the automatic design of synthetically accessible protein ligands: peptides, amides and peptidomimetics. J Comp Aided Mol Des. 1996;10:265–272. doi: 10.1007/BF00124496. [DOI] [PubMed] [Google Scholar]
- 128.Lower M, Proschak E. Structure-based pharmacophores for virtual screening. Mol Inf. 2011;5:398–404. doi: 10.1002/minf.201100007. [DOI] [PubMed] [Google Scholar]
- 129.Hansen T, Alst T, Havelkova M, Strom MB. Antimicrobial activity of small β-peptidomimetics based on the pharmacophore model of short cationic antimicrobial peptides. J Med Chem. 2010;53:595–606. doi: 10.1021/jm901052r. [DOI] [PubMed] [Google Scholar]
- 130.Hall PR, Leitao A, Ye C, et al. Small molecule inhibitors of hantavirus infection. Bioorg Med Chem Lett. 2010;20:7085–7091. doi: 10.1016/j.bmcl.2010.09.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Caporuscio F, Tafi A, Gonzlez E, Manetti F, Est JA, Botta M. A dynamic target-based pharmacophoric model mapping the CD4 binding site on HIV-1 gp120 to identify new inhibitors of gp120–CD4 protein–protein interactions. Bioorg Med Chem Lett. 2009;19:6087–6091. doi: 10.1016/j.bmcl.2009.09.029. [DOI] [PubMed] [Google Scholar]
- 132.Massa SM, Xie Y, Longo FM. Alzheimer’s therapeutics. J Mol Neurosci. 2003;20:323–326. doi: 10.1385/JMN:20:3:323. [DOI] [PubMed] [Google Scholar]
- 133.Parthasarathi L, Casey F, Stein A, Aloy P, Shields DC. Approved drug mimics of short peptide ligands from protein interaction motifs. J Chem Inf Model. 2008;48:1943–1948. doi: 10.1021/ci800174c. [DOI] [PubMed] [Google Scholar]
- 134.Goede A, Michalsky E, Schmidt U, Preissner R. SuperMimic–Fitting peptide mimetics into protein structures. BMC Bioinformatics. 2006;10:7–11. doi: 10.1186/1471-2105-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Baran I, Varekova RS, Parthasarathi L, Suchomel S, Casey F, Shields DC. Identification of potential small molecule peptidomimetics similar to motifs in proteins. J Chem Inf Model. 2007;47:464–474. doi: 10.1021/ci600404q. [DOI] [PubMed] [Google Scholar]
- 136.Casey FP, Davey NE, Baran I, Varekova RS, Shields DC. Web server to identify similarity of amino acid motifs to compounds (SAAMCO) J Chem Inf Model. 2008;48:1524–1529. doi: 10.1021/ci8000474. [DOI] [PubMed] [Google Scholar]
- 137.Andrews MJ, Kontopidis G, McInnes C, et al. REPLACE: a strategy for iterative design of cyclin-binding groove inhibitors. ChemBioChem. 2006;7:1909–1915. doi: 10.1002/cbic.200600189. [DOI] [PubMed] [Google Scholar]
- 138.Floris M, Masciocchi J, Fanton M, Moro S. Swimming into peptido-mimetic chemical space using pepMMsMIMIC. Nucleic Acids Res. 2011;39:W261–W269. doi: 10.1093/nar/gkr287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Masciocchi J, Frau G, Fanton M, et al. MMsINC: a large-scale chemoinformatics database. Nucleic Acids Res. 2009;37:D284–D290. doi: 10.1093/nar/gkn727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ou-Yang S, Lu J, Kong X, Liang Z, Luo C, Jiang H. Computational drug discovery. Acta Pharmacol Sin. 2012;33:1131–1140. doi: 10.1038/aps.2012.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zurdo J. Developability assessment as an early de-risking tool for biopharmaceutical development. Pharm Bioprocess. 2013;1:29–50. [Google Scholar]
- 142.Shirai H, Prades C, Vita R, et al. Antibody informatics for drug discovery. Biochim Biophys Acta. 2014;1844:2002–2015. doi: 10.1016/j.bbapap.2014.07.006. [DOI] [PubMed] [Google Scholar]
- 143.Kuroda D, Shirai H, Jacobson MP, Nakamura H. Computer-aided antibody design. Protein Eng Des Sel. 2012;25(10):507–521. doi: 10.1093/protein/gzs024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kapoor P, Singh H, Gautam A, Chaudhary K, Kumar R, Raghava GP. TumorHoPe: a database of tumor homing peptides. PLoS One. 2012;7(4):e35187. doi: 10.1371/journal.pone.0035187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Volpe DA. Drug-permeability and transporter assays in Caco-2 and MDCK cell lines. Future Med Chem. 2011;3(16):2063–2077. doi: 10.4155/fmc.11.149. [DOI] [PubMed] [Google Scholar]
- 146.Vanhee P, Reumers J, Stricher F, et al. PepX: a structural database of non-redundant protein-peptide complexes. Nucleic Acids Res. 2010;38:D545–D551. doi: 10.1093/nar/gkp893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.London N, Raveh B, Cohen E, Fathi G, Schueler-Furman O. Rosetta FlexPepDock web server – high resolution modeling of peptide-protein interactions. Nucleic Acids Res. 2011;39:W249–W253. doi: 10.1093/nar/gkr431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Khan JM, Ranganathan S. pDOCK: a new technique for rapid and accurate docking of peptide ligands to major histocompatibility complexes. Immun Res. 2010;6:S2. doi: 10.1186/1745-7580-6-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yan C, Zou X. Predicting peptide binding sites on protein surfaces by clustering chemical interactions. J Comput Chem. 2015;36(1):49–61. doi: 10.1002/jcc.23771. [DOI] [PubMed] [Google Scholar]
- 150.Verschueren E, Vanhee P, Rousseau F, Schymkowitz J, Serrano L. Protein-peptide complex prediction through fragment interaction patterns. Structure. 2013;21(5):789–797. doi: 10.1016/j.str.2013.02.023. [DOI] [PubMed] [Google Scholar]
- 151.Saladin A, Rey J, Thevenet P, Zacharias M, Moroy G, Tuffery P. PEP-SiteFinder: a tool for the blind identification of peptide binding sites on protein surfaces. Nucleic Acids Res. 2014;42:W221–W226. doi: 10.1093/nar/gku404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Unal EB, Gursoy A, Erman B. VitAL: Viterbi algorithm for de novo peptide design. PLoS One. 2010;5(6):e10926. doi: 10.1371/journal.pone.0010926. [DOI] [PMC free article] [PubMed] [Google Scholar]