

Abstract
INTRODUCTION
Since Hippocrates first described the cutaneous spreading of herpes simplex lesions, many other diseases—chickenpox, infectious mononucleosis, nasopharyngeal carcinoma, and Kaposi’s sarcoma—have been found to be associated with the nine known human herpesviruses. Among them, herpes simplex virus type 1 (HSV-1, causes cold sores), type 2 (HSV-2, causes genital herpes), and varicella-zoster virus (causes chickenpox and shingles)—which all belong to the α-herpesvirus subfamily—can establish lifelong latent infection within our peripheral nervous system.
RATIONALE
A prominent feature of these neurotropic viruses is the long-range (up to tens of centimeters) axonal retrograde transport of the DNA-containing viral capsid from nerve endings at sites of infection (such as the lips) to neuronal cell bodies at the ganglia to establish latency or, upon reactivation, anterograde transport of the progeny viral particles from the ganglia to nerve terminals, resulting in reinfection of the dermis. Capsid-associated tegument complexes (CATCs) have been demonstrated to be involved in this cytoskeleton-dependent capsid transport. Because of the large size (~1300 Å) of HSV-1 particles, it has been difficult to obtain atomic structures of the HSV-1 capsid and CATC; consequently, the structural bases underlying α-herpesviruses’ remarkable capability of long-range neuronal transport and many other aspects of its life cycle are poorly understood.
RESULTS
By using cryo–electron microscopy, we obtained an atomic model of the HSV-1 capsid with CATC, comprising multiple conformers of the capsid proteins VP5, VP19c, VP23, and VP26 and tegument proteins pUL17, pUL25, and pUL36. Crowning every capsid vertex are five copies of heteropentameric CATC. The pUL17 monomer in each CATC bridges over triplexes Ta and Tc on the capsid surface and supports a coiled-coil helix bundle of a pUL25 dimer and a pUL36 dimer, thus positioning their flexible domains for potential involvement in nuclear egress and axonal transport of the capsid. The single C-terminal helix of pUL36 resolved in the CATC links the capsid to the outer tegument and envelope: As the largest tegument protein in all herpesviruses and essential for virion formation, pUL36 has been shown to interact extensively with other tegument proteins, which in turn interact with envelope glycoproteins. Architectural similarities between herpesvirus triplex proteins and auxiliary cementing protein gpD in bacteriophage λ, in addition to the bacteriophage HK97 gp5–like folds in their major capsid proteins and structural similarities in their DNA packaging and delivery apparatuses, indicate that the commonality between bacteriophages and herpes-viruses extends to their auxiliary components. Notwithstanding this broad evolutionary conservation, comparison of HSV-1 capsid proteins with those of other herpesviruses revealed extraordinary structural diversities in the forms of domain insertion and conformation polymorphism, not only for tegument interactions but also for DNA encapsulation.
CONCLUSION
Our structure of the HSV-1 capsid with capsid-associated tegument proteins provides mechanistic insights into multiple aspects of the viral life cycle, including capsid assembly, nuclear egress, acquisition of tegument and envelope, and axonal transport in neuronal cells. The numerous molecular interactions and atomic details embodied in the structure make it a much-sought-after atlas for the search of antivirals targeting these critical steps of HSV-1 lytic replication.
Graphical abstract
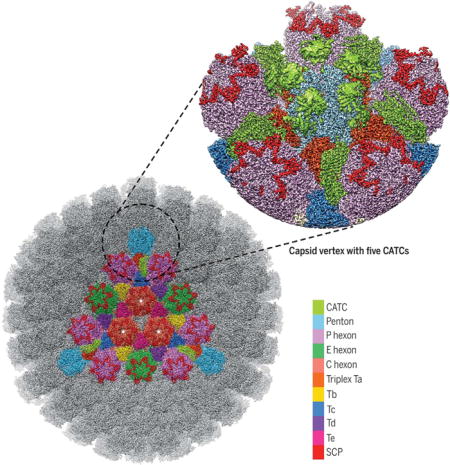
Structure of the HSV-1 capsid with capsid-associated tegument proteins Surface view of a 4.2-Å resolution map of the icosahedral capsid, with a single facet shown in color. The structure of the vertex region (magnified view) was improved to 3.5-Å resolution by subparticle refinement. P, peripentonal; C, center; E, edge; Ta to Te, heterotrimeric triplexes composed of Tri1, Tri2A, and Tri2B.
The Herpesviridae family is associated with herpes lesions at the lips (cold sores) and the genitalia as well as many other diseases—chickenpox, infectious mononucleosis, nasopharyngeal carcinoma, and Kaposi’s sarcoma (1). The nine known human herpesviruses span all three herpesvirus subfamilies—α-, β-, and γ-herpesviruses—and are grouped on the basis of their biological properties, including differences in tissue tropism. Viruses in the α-herpesvirus subfamily, including herpes simplex virus type 1 (HSV-1, causes cold sores) and type 2 (HSV-2, causes genital herpes) and varicella-zoster virus (causes chickenpox and shingles), can establish lifelong latent infections within the peripheral nervous systems of their hosts. The DNA-containing capsids of these neurotropic viruses undergo long-range (up to tens of centimeters) axonal retrograde transport from nerve endings at sites of infection (such as the lips) to neuronal cell bodies at the ganglia to establish latency. Upon reactivation, anterograde transport of the progeny viral particles from the ganglia to nerve terminals results in reinfection of the dermis. Capsid-bound tegument proteins are involved in this cytoskeleton-dependent capsid transport (2–7). Densities of tegument proteins have been visualized at the icosahedral vertices of the HSV-1 capsid by cryo– electron microscopy (cryo-EM) (8), leading to designations such as C-capsid–specific component (9), capsid vertex-specific component (10), and, more generally across different subfamilies of herpesviruses, the capsid-associated tegument complex (CATC) (11). Molecular labeling, together with the fitting of crystallographic models into subnanometer resolution cryo-EM structures, has shown that CATC from α-herpesviruses contains at least pUL25 and pUL17 (10–14) and possibly pUL36 (15–17), all of which are essential for viral propagation. However, the CATC in the nonneurotropic β-herpesvirus human cytomegalovirus (HCMV) bears neither compositional nor structural similarities to that in α-herpesviruses. The β-herpesvirus–specific tegument protein pp150 forms a spherical net securing the capsid (18–20) that is pressurized by a viral double-stranded DNA (dsDNA) genome of 235 kb, 50% larger than that of HSV-1. Capsid proteins are more conserved than tegument proteins, with herpesviruses across different subfamilies all containing homologous capsid proteins, though the level of conservation varies greatly in both sequence and function.
In the absence of atomic structures of the HSV-1 capsid and CATC, the structural bases underlying long-range neuronal transport and aspects of genome packaging are poorly understood. Because of the large size (~1300 Å) of HSV-1 particles, the progress toward an atomic description of HSV-1 has been astonishingly slow, with the highest resolution as yet being 6.8 Å (17), which is insufficient for de novo atomic modeling.
Overall structures
To obtain an atomic description of the HSV-1 capsid and its associated tegument proteins, we imaged intact HSV-1 virions by electron-counting cryo-EM (fig. S1) and obtained a three-dimensional icosahedral reconstruction at an effective resolution of 4.2 Å by combining 23,800 virion particles (Fig. 1A and fig. S2A). Like earlier reconstructions at lower resolution [for example, (17, 21)], our density map only reveals the icosahedrally ordered components of the virion, including a triangulation number (T) = 16 capsid containing pentons, 3 quasi-equivalent hexons (P, peripentonal; E, edge; and C, center), 6 quasi-equivalent triplexes (Ta to Tf), and 12 star-shaped CATC densities, each crowning a penton at the icosahedral vertex (Figs. 1, A to C, and 2A). Through a subparticle-refinement procedure, we further improved the structure of the vertex region containing CATC to 3.5-Å resolution (fig. S2, A and B). High-resolution features resolved in our cryo-EM map, such as amino acid side chains (Movies 1 and 2 and figs. S3 to S5), enabled us to build atomic models for both the capsid and CATC (Fig. 1D). The atomic models for the capsid proteins are all built de novo, including 16 copies of the major capsid protein (MCP) VP5, 10 copies of the triplex dimer protein (Tri2) VP23, 5 copies of the triplex monomer protein (Tri1) VP19c, and 15 copies of the smallest capsid protein (SCP) VP26, amounting to more than 28,000 residues in total.
Fig. 1. Cryo-EM reconstruction and atomic modeling of the HSV-1 capsid.
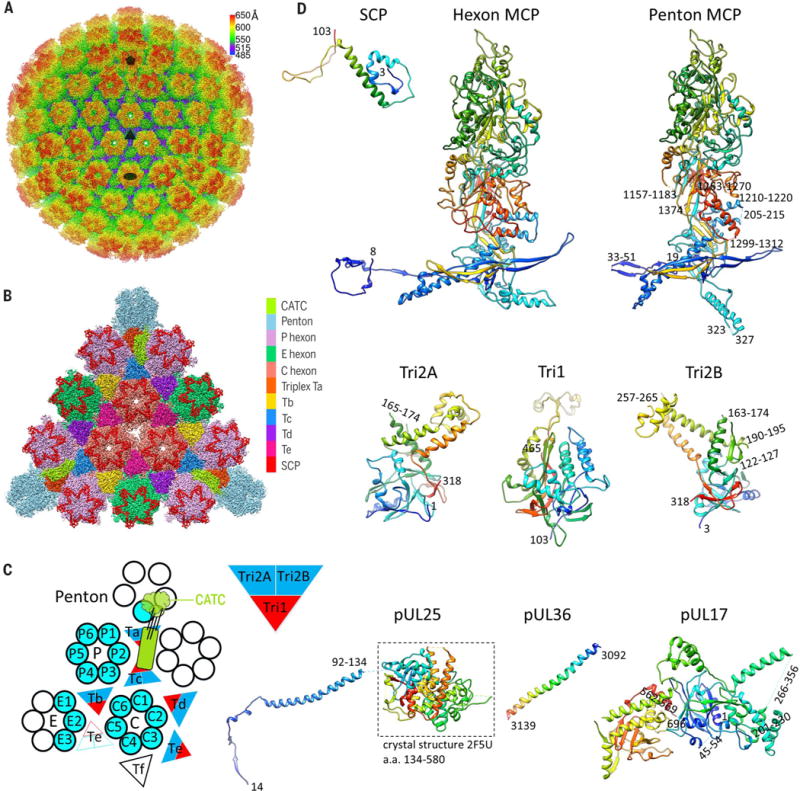
(A) Radially colored cryo-EM density map of the HSV-1 capsid viewed along a threefold axis. Fivefold, threefold, and twofold axes are denoted by a pentagon, triangle, and oval, respectively. (B) Magnified view of one facet of the icosahedral capsid with structural components differentially colored. The density of triplex Tf at the center is not shown. (C) A schematic representation of one asymmetric unit (shaded) of the capsid. An extra copy of triplex Te (unshaded) from an adjacent asymmetric unit is shown to depict that triplex pair Tb-Te has a similar configuration as Ta-Tc, thus providing a second potential binding site for the CATC (11, 17). The enlarged red and blue triangle shows the heterotrimeric nature of a triplex. a.a., amino acid. (D) Atomic models of individual capsid or tegument proteins in rainbow-colored ribbon (from blue at the N terminus to red at the C terminus). Numbers denote chain termini or flexible segments that are not modeled. a.a., amino acid.
Fig. 2. Structure of the CATC.
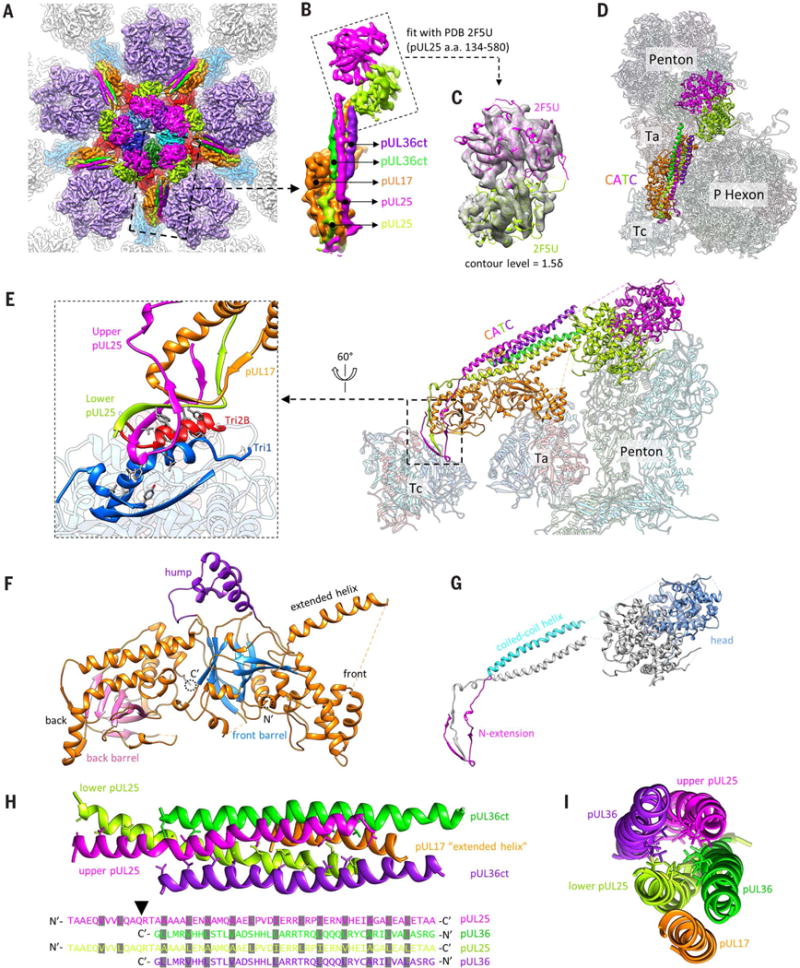
(A) Cryo-EM densities of the CATC surrounding a penton vertex. The density map was low-pass filtered to 6-Å resolution to show the flexible pUL25 head region. (B) Magnified view of a segmented-out CATC unit from the dashed square in (A), showing its composition. (C) Fitting of two copies of pUL25 C-terminal region crystal structure (PDB 2F5U) (22) into the bi-lobed head region of CATC. The contour level for the pUL25 head region in (A) to (C) was 1.5d (where d is the standard deviation) and that of the rest was 3d. (D and E) Top view (D) and side view (E) of CATC interacting with underlying capsid components, shown with atomic models. On the left in (E) is a magnified view of the interface between the CATC and triplex Tc. Hydrophobic side chains of the pUL25 dimer, Tri1, and Tri2B involved in the interactions are highlighted. (F and G) Domain organization of pUL17 (F) and pUL25 (G). (H and I) The five-helix bundle viewed from the top of the capsid (H) or along the center of the coiled coil (I). In (H), side chains are shown for hydrophobic residues in the center of the helix bundle (top), with their identities marked (grayed) in the sequence (bottom). The arrowhead in (H) points to the position in the pUL25 sequence where the hydrophobic residue distribution pattern breaks. Nʹ, N terminus; Cʹ, C terminus.
Movie 1.
Overall structure and high-resolution features of the HSV-1 capsid icosahedral reconstruction
Movie 2.
High-resolution features in the 3.5-Å resolution subparticle reconstruction of the HSV-1 capsid vertex region
Structure of the CATC
Surrounding each capsid vertex, five CATC densities form a five-pointed star crowning the pen-ton (Fig. 2A). The relative density values of CATC versus its underlying triplexes are similar (Movie 2 and fig. S2B), suggesting nearly full occupancy of CATC, in contrast to the low occupancy of CATC in the virion of γ-herpesvirus Kaposi’s sarcoma– associated herpesvirus (KSHV) (11). Each CATC contains a triplex-binding region bridging triplexes Ta and Tc, an extended five-helix bundle, and a bi-lobed head region that lies in the space between two neighboring penton MCPs (Fig. 2, B to E). The triplex-binding region and the five-helix bundle are well resolved (fig. S5), but the bi-lobed head region is not and requires low-pass filtering to 6-Å resolution for interpretation, indicating its higher level of flexibility (fig. S2B). Nonetheless, the clearly resolved secondary structures in the head region enabled reliable fitting of two copies of the crystal structure of the pUL25 C-terminal fragment [amino acids 134 to 580, Protein Data Bank (PDB) 2F5U] (22) (Fig. 2C and Movie 3), inconsistent with the assignment of one pUL25 and one pUL36 to the corresponding bi-lobed densities in a previous cryo-EM reconstruction of HSV-1 at 6.8-Å resolution (17). Our CATC model comprises these fitted dimer structures of the pUL25 head domain and de novo atomic models for the triplex-binding region and the five-helix bundle. We can model near full-length pUL17, two copies of N-terminal pUL25 (~90 amino acids), and two copies of C-terminal pUL36 (pUL36ct) (amino acids 3092 to 3139) (Fig. 1D).
Movie 3.
Fitting of two copies of the pUL25 C-terminal region crystal structure into the bi-lobed head region of CATC
The 696-amino acid–long pUL17 monomer comprises an eight-stranded β barrel at the N terminus (“front barrel”), a seven-stranded β barrel at the C terminus (“back barrel”), and a helix-rich middle segment that enwraps the β barrels to form two lobes (labeled “front” and “back”) (Fig. 2F and Movie 4). A small helical domain extends from between the two lobes (the “hump”). A helix extending from the front lobe (“extended helix,” amino acids 241 to 266) is the fifth helix of the five-helix bundle and guides the positioning of the pUL25-pUL36 helix bundle (Fig. 2, E and F). The hump on one side and the P hexon on the other further anchor and stabilize the helix bundle (Fig. 2D). Overall, the position of the CATC is constrained by triplexes Ta and Tc and the P hexon tower (Fig. 2D and fig. S6).
Movie 4.
Structure of the CATC and its interactions with the underlying capsid components
The two subunits of the pUL25 dimer in CATC differ in structural details but bear the same domain organization, consisting of an N-terminal β-strand domain (N extension, amino acids 1 to 47) that interacts with pUL17, a coiled-coil helix domain (48 to 94), and the C-terminal head domain (134 to 580) (Fig. 2G and Movie 4). The N extensions of the two pUL25 monomers intertwine with each other and with the back lobe of pUL17: Three β strands (amino acids 3 to 5, 10 to 14, 24 to 26) of the upper pUL25, two β strands (14 to 20, 26 to 28) of the lower pUL25, and two β strands (503 to 505, 510 to 512) of pUL17 together form two small β sheets (Fig. 2E, magnified view). In this way, the two differently folded N extensions of the pUL25 dimer work together to fasten the coiled-coil helix bundle to pUL17. The N extensions of the two pUL25 molecules also insert a number of hydrophobic side chains (Phe7, Leu10, Val12, Phe18, and Val20 of the upper pUL25 and Phe18 and Val20 of the lower pUL25) into two hydrophobic clefts of triplex Tc, one at the interface between Tri1 and Tri2B and the other on the surface of Tri1 (Fig. 2E, magnified view). Notably, only pUL25 residues are directly involved in triplex Tc binding, as opposed to previous suggestions that pUL17 directly interacts with both Ta and Tc—a reasonable interpretation when amino acid residues were not resolved (17).
Our identification of two copies of pUL36ct in the helix bundle of the CATC is supported by side-chain densities resolved in the 3.5-Å resolution subparticle reconstruction map (fig. S5). It is also consistent with biochemical data showing that the C-terminal 62–amino acid segment of pUL36 (also termed VP1/2) is essential for the association of pUL36 with the capsid and that this capsid association requires interaction with pUL25 in both pseudorabies virus (PRV) and HSV-1 (23, 24). In our model, the pUL36ct dimer joins the pUL25 dimer to form a four-helix coiled coil, which in turn rests on the extended helix of pUL17, giving rise to the five-helix bundle (Fig. 2, H and I, and Movie 4). Within the pUL25-pUL36ct four-helix coiled coil, hydrophobic side chains (predominantly leucine) are spaced every three or four residues in all four helices, similar to those in a leucine zipper (Fig. 2H). There is a break in this pattern of hydrophobic residues at position Gln58-Arg59 in pUL25 (arrowhead in Fig. 2H) where pUL36ct incorporates into the CATC. The bundling with pUL36ct may also stabilize the pUL25 dimer and likely the entire CATC, as previous cryo-EM reconstruction of a pUL36-deletion mutant of HSV-1 showed diminished CATC densities (16). The extensive interactions among all five subunits of CATC suggest that they likely coassemble in the nucleus and act together as a functional unit in binding the capsid, consistent with their detections in capsids purified from the host-cell nucleus (10, 25).
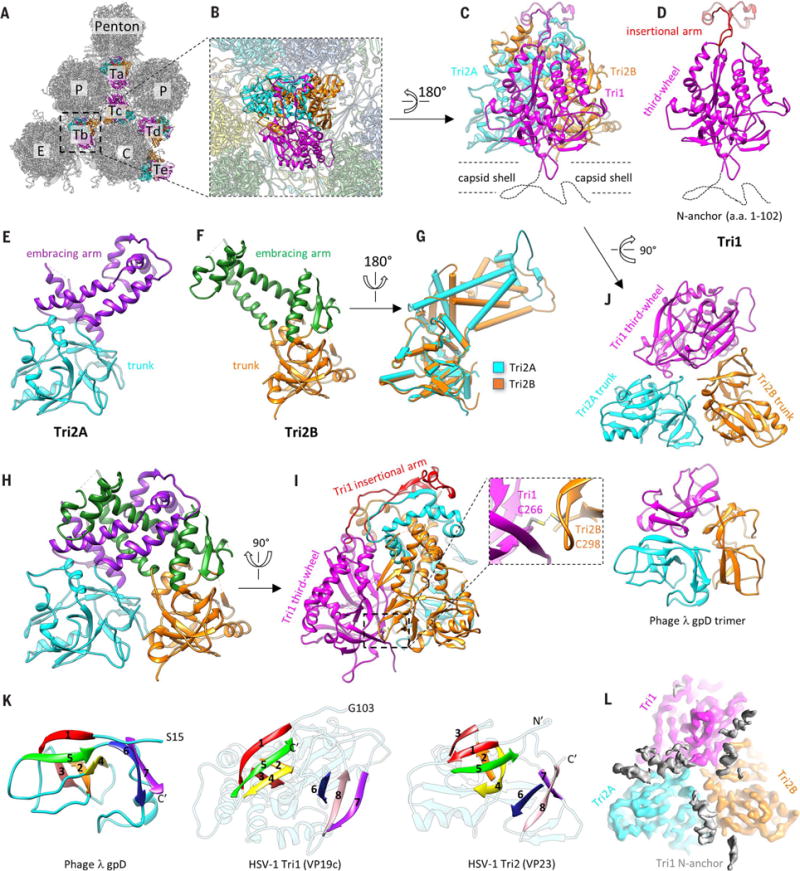
Bacteriophage-related motif and α-herpesvirus–specific features of the triplex
Protruding from the floor of the canyons between towering capsomers are six quasi-equivalent triplexes Ta to Tf (Figs. 1, B and C, and 3, A and B), with only Ta and Tc providing the platform for CATC binding (Movie 4). Each triplex is a heterotrimer consisting of one Tri1 (VP19c) and two conformers of Tri2 (VP23)—Tri2A and Tri2B (Fig. 3, C to F). The sequence of HSV-1 Tri1 protein VP19c is about 40% longer than those of the Tri1 homologs in both β- and γ-herpesviruses, whereas the Tri2 protein sequences are similar in length across the three herpesvirus subfamilies. The two Tri2 subunits in each triplex interact through their embracing-arm domains (amino acids 145 to 289) (Fig. 3H). The embracing-arm domains are structurally variable between the two subunits, whereas their trunk domains (1 to 144, 290 to 318) are constant (Fig. 3G). This Tri2A-Tri2B dimer is bound by the third-wheel domain (103 to 356, 412 to 465) of the Tri1 monomer from the side and latched at the top by a 55–amino acid insertional-arm domain (357 to 411) of Tri1 (Fig. 3I). Near the bottom of this heterotrimeric interface, an intermolecular disulfide bond formed between Cys266 of Tri1 and Cys298 of Tri2B likely further stabilizes the triplex (Fig. 3I, magnified view, and fig. S4).
Fig. 3. Structure of the triplex.
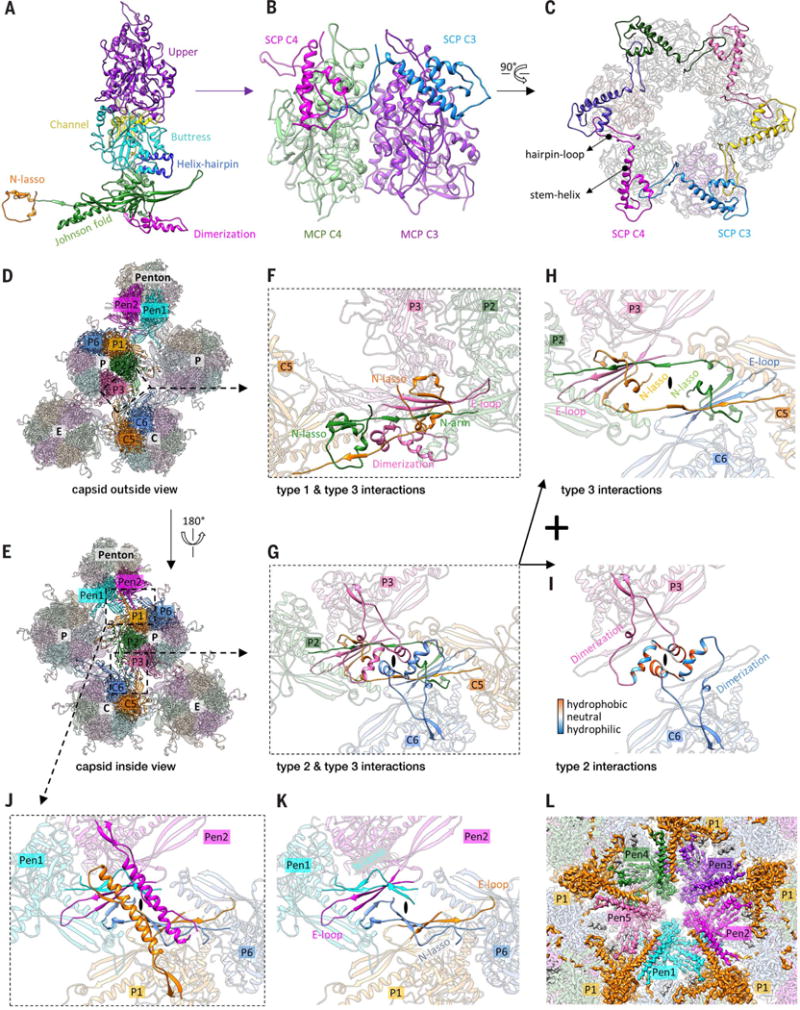
(A) Distribution of triplexes in the MCP network. (B) Magnified view of a triplex Tb as viewed from the outside of the capsid. (C to F) Detailed structures of triplex Tb (C) and its components Tri1 (D), Tri2A (E), and Tri2B (F). Dotted curves in (C) and (D) denote the N-terminal region of Tri1 that was not modeled because of its flexibility. (G) Superposition of Tri2A and Tri2B showing constant trunk domains and conformational differences in their embracing-arm domains. (H) Tri2A and Tri2B embrace each other with their embracing-arm domains to form a dimer. (I) The Tri1 insertional arm binding on top of the Tri2 dimer and the Tri1-Tri2B intermolecular disulfide bond (shown in magnified view) stabilize the triplex structure. (J) Architectural similarities between HSV-1 triplex (top) and the gpD trimer of phage λ (bottom, PDB 1C5E) (26). Both are viewed from the inside of the capsid. (K) Same topology of β strands among gpD, Tri1, and Tri2. Corresponding β strands are colored in the same way and numbered one to eight, from Nʹ to Cʹ. (L) Cryo-EM density of triplex Tb in the same orientation as (J), showing the tri-lobed density of the flexible Tri1 N anchor (gray), which penetrates through the capsid floor and anchors the entire triplex to the capsid shell, as depicted in (C).
Despite no recognizable similarity between the Tri1 (VP19c) and Tri2 (VP23) protein sequences, the third-wheel domain of Tri1 and the trunk domain of Tri2 both predominantly contain two β barrels, giving rise to the homotrimeric appearance of the triplex when viewed from the inside of the capsid (Fig. 3J). Notably, the arrangement of the two β barrels in both Tri1 and Tri2 closely resembles the homotrimeric cementing proteins of dsDNA bacteriophages, such as gpD in phage λ (Fig. 3J and fig. S7). Indeed, side-by-side comparison of the chain tracing in gpD (26), Tri1, and Tri2 reveals identical topology of their β strands (Fig. 3K). This observation suggests that HSV-1 Tri1 and Tri2 evolved from an ancestral gene shared by phage λ gpD. Perhaps a gene-duplication event in the herpesvirus ancestor led to divergent evolution of Tri1 and Tri2. For example, whereas each subunit of the gpD trimer contributes an N-terminal extension to augment a β sheet on the capsid surface (27), the two Tri2 subunits in the herpes-virus triplex do not have such a capsid-binding N-terminal extension. Instead, the Tri1 monomer inserts its N-terminal extension (N anchor) through the capsid floor and folds into a tri-lobed structure inside the capsid (Fig. 3, C, D, and L) (20, 28), anchoring the entire triplex. Selection pressure to mediate binding of divergent tegument proteins outside the capsid and to accommodate genomes of varying sizes inside the capsid could have also forced Tri1 to diverge on both its outer and inner sides. Indeed, the Tri1 insertional arm, which emanates from a topologically similar location on one of the two β barrels as that of the Tri2 embracing arm, is the most externally located and is completely missing in HCMV Tri1 (20) and KSHV Tri1 (28). Likewise, the genome-facing N anchor of HSV-1 Tri1 is substantially longer than (102 versus 44 amino acids), and not as rigid as, that of HCMV (20).
HSV-1–specific features in the SCP and MCP
Like that of HCMV or KSHV (20, 28), the HSV-1 MCP (VP5) subunit can be thought of as an extensive elaboration on a central bacteriophage HK97 gp5–like (“Johnson”) fold (amino acids 51 to 193, 232 to 298, 365 to 396, 1061 to 1113), with six additional domains, including the N lasso (1 to 50), dimerization (299 to 364), helix-hairpin (194 to 231), channel (407 to 480, 1335 to 1374), buttress (397 to 406, 1048 to 1060, 1114 to 1334) and upper (481 to 1047) domains (Fig. 4A).
Fig. 4. SCP and MCP network interactions.
(A) Domain organization in a hexon MCP. (B and C) SCP binds the upper domain of hexon MCP (B), and six copies of SCP form a gear-shaped ring crowning and stabilizing the hexon (C). (D and E) Part of the MCP network viewed from the outside (D) or the inside (E) of the capsid. Pen, penton. (F to I) Three types of network interactions among hexon MCPs. (H) and (I) show the individual structures that make up (G). The type I interaction is an intracapsomeric augmentation of β strands from adjacent MCPs [P2 and P3 in (F)] in the same capsomer.Type II and type III are intercapsomeric interactions among two pairs of MCPs [P2-P3 and C5-C6 in (G)] diagonally across the local twofold axis [ovals in (G) to (I)]. The type III interaction builds on and fortifies the type I interaction [see C5 N lasso in (F)]. The two dimerization domains joined by a type II interaction (I) also sit atop a pair of type III interactions, when looking from the inside of the capsid (G), and prevent the two N lassos [C5 and P2 N lassos in (G) and (H)] from unwinding. (J and K) Interactions between penton MCPs and the P1 and P6 hexon MCPs are different from the canonical hexon MCP network interactions [compare (J) and (G), (K) and (H)]. (L) Cryo-EM density at the penton area viewed from the inside of the capsid.The five copies of penton MCP are differently colored, whereas all P1 MCPs are colored in gold.
The upper domain of MCP (MCPud) provides the binding site for SCP (VP26) (Fig. 4B), which is the most divergent, both structurally and functionally, capsid protein across all herpesvirus subfamilies. Our atomic model of the 112–amino acid SCP monomer encompasses residues 3 to 103 and consists of a helix-rich N-terminal domain (amino acids 1 to 71) featuring the characteristic “stem helix” involved in MCPud binding (29) and the C-terminal hairpin-loop domain (72 to 112). Six SCP subunits form a gear-shaped hexameric ring crowning each hexon, with their hairpin-loop domains bridging the gaps between adjacent hexon MCP upper domains (Fig. 4, B and C). This “cross-linking” interaction of SCP resembles that of the 170–amino acid SCP-homolog ORF65 in γ-herpesvirus KSHV and thus likely has a similar function in stabilizing the hexons of the capsid (29). This is consistent with the previous observation that the VP26 null mutation resulted in decreased production of infectious virus in the nervous system of infected mice, though it had little effect in cell culture (30). Notably, the 75– amino acid SCP-homolog UL48.5 in β-herpesvirus HCMV completely lacks the C-terminal cross-linking moiety found in HSV-1 and KSHV but has evolved to stabilize the capsid by mediating the binding of a β-herpesvirus–specific tegument protein pp150 (19, 20).
As in HCMV and KSHV (20, 28), three types of MCP-MCP interactions form a hierarchical network of interactions on the HSV-1 capsid floor (Fig. 4, D and E, and Movie 5). The type I interaction is intracapsomeric β augmentation between adjacent MCPs within a capsomer, as exemplified by P2 and P3 MCPs in Fig. 4F: Two β strands in the N arm of P2, two β strands in the E loop, and one β strand in the dimerization domain of P3 together form a five-stranded β sheet. Type II and type III interactions are intercapsomeric interactions involving the dimerization and N-lasso domains, respectively. As exemplified in Fig. 4, G and H, the C5 N lasso extends and lashes around the P2 N arm and P3 E loop and the P2 N lasso extends and lashes around the C5 N arm and C6 E loop to form a pair of type III interactions related by a local twofold axis. These type III interactions are built on and likely strengthen the type I interactions, as the C5 N lasso contributes two β strands to augment the existing five-stranded β sheet from P2 and P3’s type I interaction into a seven-stranded β sheet (Fig. 4, F and H), as does the P2 N lasso to C5 and C6 (Fig. 4H). Furthermore, a type II interaction is formed by the dimerization domains of P3 and C6 pairing with one another around the local twofold axis with hydrophobic residues (Fig. 4I) and secures the pair of type III interactions among P2, P3, C5, and C6 (Fig. 4G).
Movie 5.
MCP network interactions in the capsid floor
The network interactions of the penton and its surrounding P hexons (Fig. 4J and Movie 5) are different from those of other hexons (Fig. 4G) in two ways. First, the N-terminal regions of penton MCP and P6 MCP refold and lose the canonical N-lasso structure of the hexon MCP (Fig. 4K). As a result, the penton neither lashes nor is lashed by adjacent P hexons. Second, the dimerization domain of penton MCP, as well as that of P1 MCP, refold from the helix-turn-helix structure in a canonical type II interaction (Fig. 4I) into a single long helix (Fig. 4J and fig. S8). The two resulting helices interact through a series of interdigitated leucine resides (that is, hydrophobic interactions) buried within the interface and a series of hydrogen-bonded arginine and glutamate residues lining the inner surface of the capsid (fig. S8). Five such helix pairs radiate from the penton channel and give rise to a star-shaped appearance (Fig. 4L and Movie 5) not seen in the capsids of β- (20) and γ-herpesviruses (29) and even the closely related α-herpesvirus PRV (17), thus providing an extraordinary example of structural diversification of the HSV-1 capsid.
Discussion
Our observation of architectural similarities between herpesvirus triplex proteins and auxiliary cementing protein gpD in bacteriophage λ, in addition to earlier identifications of bacteriophage HK97 gp5–like folds in their major capsid proteins (27, 31) and structural similarities in their DNA packaging and delivery apparatuses (32), indicates that the commonality between bacterio-phages and herpesviruses extends to their auxiliary components. Nonetheless, evolution seems to have driven the diversification of capsid-fortifying strategies in these viruses to accommodate differently sized genomes. Whereas a gpD-like cementing protein might have been replaced during evolution by autocatalytic chemical cross-linking of capsid proteins in HK97 (27, 33, 34), the much larger genome of herpesvirus necessitates more extensive involvement of the triplex in building a larger-sized capsid—indeed, the triplex coassembles with MCP as a major building block of the procapsid in herpesvirus (35–37), in contrast to the gpD trimer, which is an auxiliary addition to the matured phage λ capsid after genome packaging (27).
Together with the above, the model of an HSV-1 particle presented here provides not only the basis for understanding capsid assembly but also mechanistic insights into previously unexplained aspects of the viral life cycle. We show that CATC is a complex of tegument proteins pUL17, pUL25, and pUL36, which have been suggested to play multiple roles, including reinforcement of the capsid (38), retrograde transport of the incoming viral capsid during initial infection (3, 5, 7, 39, 40), ejection of the viral genome through the nuclear pore (41–43), and nuclear and cytoplasmic egress of the progeny viral particles (25, 44–49). CATC binds only the Ta-Tc triplex pair near two P hexons but not the other quasi-equivalent Tb-Te triplex pair near one C hexon and one E hexon (Fig. 1C), where the slightly different geometry of the C hexon presents a steric hindrance to CATC binding (fig. S9), as also revealed in the 6.8-Å resolution HSV-1 structure (17). This stringent selection of binding site suggests high rigidity of the CATC, which can be attributed to the characteristic of the pUL17 structure—two rigidity-promoting β barrels stuffed beneath the length-defining, helix-rich middle segment. The specificity of CATC binding might also have functional implications in ensuring the right timing for nuclear egress of capsid, a function that has been associated primarily with pUL25 for its interaction with the pUL31 component of the nuclear egress complex (44–48). In the host-cell nucleus, CATC must select mature capsids over immature ones (procapsids) for nuclear egress; the geometry of neighboring capsomers and triplexes in the spherical procapsid differs from that in the angularized mature capsid (35), preventing premature association of CATC to the procapsids. The structural constraints on CATC binding also raise the question of whether it can bind to the portal vertex. Although icosahedral reconstruction precludes knowledge of the geometry surrounding the portal, functional studies have shown an essential role of pUL25 in HSV-1 genome release (41–43), arguing for CATC’s existence at the portal vertex.
The bundling of pUL36ct with the pUL25 coiled-coil helix provides an anchoring point for the recruitment of pUL36 and, in turn, pUL36-binding tegument proteins, such as the conserved pUL37 (50, 51) and pUL48 (VP16) (52), both of which can interact with cytoplasmic tails of viral glycoproteins to facilitate virion envelopment (53–56). In this regard, pUL36 is a pivot connecting herpesvirus capsid assembly and primary tegumentation in the host-cell nucleus to its secondary tegumentation (49) and envelopment in cytoplasm, and our structure provides the first atomic view of the early steps in this cascade of events leading to the morphogenesis of herpesvirus virion. Beyond its C-terminal helix resolved here, the central one-third of pUL36 also dimerizes to form a coiled-coil helix bundle (57). Intriguingly, this structural organization of the CATC is somewhat reminiscent of cellular motor proteins, which all contain a coiled-coil helix bundle joining their cytoskeleton-binding globular head domains with their cargo-binding domains (58–60). The involvement of CATC in cytoskeleton-dependent α-herpesvirus capsid transport, through either direct or indirect interactions with cellular motor proteins (2–7, 39, 40), now opens the door for new inquiries into the remarkable ability of long-range axonal transport of these neurotropic viruses.
Materials and Methods
Virus culture and purification
Vero cells (ATCC CCL-81) were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) to 100% confluence and inoculated with HSV-1 (strain KOS, ATCC VR-1493) at a multiplicity of infection (MOI) of ~0.01. At 3 days postinfection, the culture media were collected and centrifuged at 8000g for 15 min to remove cell debris. Viral particles were pelleted by centrifuging at 80,000g for 1 hour, resuspended in phosphate-buffered saline (PBS, pH 7.4), and then purified by centrifuging through a 15 to 50% (w/v) sucrose density gradient at 80,000g for 1 hour. Two bands in the density gradient were usually identified. The lower band contained more noninfectious, enveloped particles lacking nucleocapsid and thus was discarded. The upper band was collected, diluted with PBS, and pelleted by centrifuging at 80,000g for 1 hour. For a 1-liter start volume of viral culture, the purified HSV-1 virions were resuspended in 30 μl PBS and subjected to cryo-EM sample preparation.
Cryo-EM sample preparation and data collection
To best preserve the structural integrity of the capsid-associated tegument proteins, we decided to image intact virions instead of detergent-treated ones, which would have the benefits of reduced sample thickness and better contrast as demonstrated in the study of HCMV (20). For cryo-EM sample preparation, 2.5 μl of purified HSV-1 virion was applied to a glow-discharged Quantifoil R2/1 Cu grid. The grid was manually blotted with filter paper to remove excess sample and flash-frozen in liquid ethane with a homemade plunger freezing apparatus. The frozen-hydrated grids were loaded into an FEI Titan Krios electron microscope operated at 300 kV for automated image acquisition with Leginon (61). A total of about 8000 movies were acquired with a Gatan K2 Summit direct electron detection camera operated in super-resolution electron-counting mode at a nominal magnification of ×14,000, giving a pixel size of 1.03 Å per pixel. The dose rate on the camera was set to ~8e per physical pixel per second and the total exposure time for each movie was 13 s fractionated into 26 frames with 0.5-s exposure time for each frame. Frame images in each movie were aligned and averaged to produce a single micrograph after correction of beam-induced drift using the GPU-accelerated motion correction program (62).
Image processing
Defocus values of micrographs were determined by CTFFIND3 (63) to be in the range of –1 to –3 μm. A total of 45,530 particles (1440 by 1440 pixels) were manually picked from 7356 selected micro-graphs using boxer in EMAN (64). The particle images were binned 8x, 4x, or 2x stepwise to speed up the data processing. Orientation and center parameters for each particle image were determined and refined with the common-line based IMIRS program (65, 66), and 3D reconstruction was done with GPU-accelerated eLite3D (67). In the initial step, only data lower than 30-Å resolution were used, and no CTF correction was applied. On the basis of the reported phase residue (PR) value distribution, 28,042 high-quality particles were selected out and divided into two random halves for further refinement. After convergence of refinement for both halves, an FSC curve was calculated and the resolution was determined to be 4.2 Å on the basis of the gold-standard FSC = 0.143 criterion (68). In the final step, the two halves of the data set were combined, and a total of 23,800 particles were used to calculate the final density map. A B-factor of –200 Å2 was applied to sharpen the density map for model building and structure analysis.
Local averaging and atomic model building
Quasi-equivalentcopiesof MCP C1 to C6, or triplexes Tb to Te, in an asymmetric unit were segmented out in cuboid, fitted to each other, and averaged in Chimera (69) to further improve quality of the density map and facilitate backbone tracing for atomic model building. Ab initio models were built manually in Coot (70), with the local-averaged maps by the following standard modeling procedure: First, the density was traced by the Baton mode tool in Coot; the amino acid residues were then registered to this Baton model; and the geometry of the full atomic model was then regularized manually in Coot and finally iteratively refined with Phenix real space refinement program (71). For the MCP upper domain, its existing crystal structure (PDB 1NO7) (72) was fitted into the cryo-EM density map to guide backbone tracing of this region. The atomic models were then fitted into every quasi-equivalent position in the asymmetric unit and manually adjusted to account for minor structural variations.
Because of the limited resolution of the penton area, model building for the penton MCP and the CATC was not as straightforward as for the above-described other capsid proteins, including hexon MCPs, SCPs, and triplex proteins, and additional strategies had to be used. Briefly, fitting of C1 hexon MCP model into the penton of a low-pass filtered density map indicated that only their floor regions have substantial structural differences, but their tower regions are structurally similar. The floor region of penton MCP has adequate quality for de novo modeling, and this model was combined with the model of the C1 hexon MCP tower to generate a chimera atomic model of the penton MCP.
Each CATC density contains a triplex-binding region bridging triplexes Ta and Tc, and a bi-lobed head region situated above penton MCPs, joined by a coiled-coil five-helix bundle. The triplex-binding region and the five-helix bundle are well resolved, whereas the bi-lobed head region is only discernable when low-pass filtered to 6-Å resolution. To model the triplex-binding region, which was known to be mainly occupied by a pUL17 monomer (10), we first traced its contiguous, nonbranching density segments and then used sequence-based secondary structure prediction of pUL17 as a roadmap to determine how to join these segments. Correct chain tracing of pUL17 is cross-validated by the good match of predicted secondary structures and those resolved in the density map.
To model the pUL25 of CATC, we first fitted two copies of the crystal structure of pUL25 C-terminal region (PDB 2F5U) (22) into the bi-lobed head region in the low-pass filtered map. These two fitted structures were combined with de novo models of two pUL25 N-terminal segments, each spanning a helix in the five-helix bundle and an extended loop in the triplex-binding region, to produce the full atomic model of the two pUL25 subunits. De novo modeling of the two remaining helices in the five-helix bundle as pUL36ct was straightforward in Coot, thanks to their simple structure as a contiguous helix.
In total, we built atomic models for a total of 51 unique conformers of the four capsid proteins and three tegument proteins: 15 hexon MCP (amino acids 8 to 1374), 1 penton MCP (19 to 33, 51 to 205, 215 to 323, 327 to 411, 430 to 1157, 1183 to 1210, 1220 to 1263, 1270 to 1299, 1312 to 1374), 15 hexon SCP (3 to 103), 5 Tri1 (103 to 465), 10 Tri2 (1 to 165, 174 to 318 of Tri2A; 3 to 122, 127 to 163, 174 to 190, 195 to 257, 265 to 318 of Tri2B), 1 pUL17 (1 to 45, 54 to 201, 230 to 266, 356 to 562, 569 to 696), 2 pUL25 (1 to 94 for one copy, 13 to 92 for the other copy, and each chimerized with PDB 2F5U resolving 134 to 577), and 2 pUL36 (3092 to 3139). As the last step, the refined atomic models of all individual conformers were combined and refined together in Phenix to resolve intermolecular clashes at the interface.
Subparticle refinement and model improvement
Because of the large size of the HSV-1 capsid, any overall particle deformation and the defocus gradient across the particle (that is, the Ewald sphere effect) (73) would result in considerable dampening of the high-resolution information in conventional icosahedral reconstruction by treating the particle as a whole. Both effects are the most severe in the capsid vertex region, limiting attainable resolution of the penton and the CATC. To further improve resolution of the vertex region, we applied a subparticle refinement and reconstruction procedure considering both local variations (74) and defocus gradient. Specifically, icosahedral orientation and center parameters of each particle image determined above were used to guide extraction of all vertex regions as subparticles with their defocus values adjusted according to their locations on each particle. The orientation and center parameters of these sub-particles were locally refined with Relion (75), and, by imposing C5 symmetry, we obtained a final reconstruction at 3.5-Å resolution on the basis of the gold-standard FSC = 0.143 criterion (68). Atomic models of components in the vertex region (including hexon MCPs P1, P2, and P6; penton MCP; triplex Ta; and the CATC) were fitted into the refined subparticle map, manually checked in Coot, and refined with Phenix. Overall, our models built from the 4.2-Å resolution icosahedral map match well with the improved subparticle map at 3.5-Å resolution, indicative of good quality and validity of our original models.
Supplementary Material
Acknowledgments
We thank S. Shivakoti for assistance in sample preparation, X. Zhang and Y. Liu for assistance in data processing, J. Jih for advice in modeling and movie production, L. J. Wang for assistance in figure preparation, and both L. J. Wang and S. Ye for critical reading of the manuscript.
Funding: This project was supported in part by grants from the NIH (GM071940, DE025567, and AI094386). We acknowledge the use of instruments at the Electron Imaging Center for Nanomachines [supported by the University of California, Los Angeles, and by instrumentation grants from the NIH (1S10OD018111, 1U24GM116792) and NSF (DBI-1338135, DMR-1548924)]. X.D. was supported in part by fellowships from the China Scholarship Council.
Footnotes
Data and materials availability: Coordinates and EM maps are available at the PDB and the Electron Microscopy Data Bank (EMDB), respectively, under the accession numbers PDB 6CGR, EMD-7472, and EMD-7473. All other data to support the conclusions are in the main paper or supplementary materials.
The list of author affiliations is available in the full article online.
Author contributions: Z.H.Z. and X.D. conceived the project, performed the experiment, interpreted the data, and wrote the paper.
Competing interests: None declared.
SUPPLEMENTARY MATERIALS
www.sciencemag.org/content/360/6384/eaao7298/suppl/DC1 Figs. S1 to S9
References (76, 77)
REFERENCES AND NOTES
- 1.Pellett P, Roizman B. Fields Virology, D Knipe, P Howley, Eds (Lippincott Williams and Wilkins, Philadelphia. 2013;2:1802–1822. [Google Scholar]
- 2.Smith GA, Enquist LW. Break ins and break outs: Viral interactions with the cytoskeleton of mammalian cells. Annu Rev Cell Dev Biol. 2002;18:135–161. doi: 10.1146/annurev.cellbio.18.012502.105920. [DOI] [PubMed] [Google Scholar]
- 3.Luxton GW, et al. Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc Natl Acad Sci USA. 2005;102:5832–5837. doi: 10.1073/pnas.0500803102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luxton GW, Lee JI, Haverlock-Moyns S, Schober JM, Smith GA. The pseudorabies virus VP1/2 tegument protein is required for intracellular capsid transport. J Virol. 2006;80:201–209. doi: 10.1128/JVI.80.1.201-209.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antinone SE, Smith GA. Retrograde axon transport of herpes simplex virus and pseudorabies virus: A live-cell comparative analysis. J Virol. 2010;84:1504–1512. doi: 10.1128/JVI.02029-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zaichick SV, Bohannon KP, Smith GA. Alphaherpesviruses and the cytoskeleton in neuronal infections. Viruses. 2011;3:941–981. doi: 10.3390/v3070941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zaichick SV, et al. The herpesvirus VP1/2 protein is an effector of dynein-mediated capsid transport and neuroinvasion. Cell Host Microbe. 2013;13:193–203. doi: 10.1016/j.chom.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou ZH, Chen DH, Jakana J, Rixon FJ, Chiu W. Visualization of tegument-capsid interactions and DNA in intact herpes simplex virus type 1 virions. J Virol. 1999;73:3210–3218. doi: 10.1128/jvi.73.4.3210-3218.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trus BL, et al. Allosteric signaling and a nuclear exit strategy: Binding of UL25/UL17 heterodimers to DNA-filled HSV-1 capsids. Mol Cell. 2007;26:479–489. doi: 10.1016/j.molcel.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toropova K, Huffman JB, Homa FL, Conway JF. The herpes simplex virus 1 UL17 protein is the second constituent of the capsid vertex-specific component required for DNA packaging and retention. J Virol. 2011;85:7513–7522. doi: 10.1128/JVI.00837-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dai X, Gong D, Wu TT, Sun R, Zhou ZH. Organization of capsid-associated tegument components in Kaposi’s sarcoma-associated herpesvirus. J Virol. 2014;88:12694–12702. doi: 10.1128/JVI.01509-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conway JF, et al. Labeling and localization of the herpes simplex virus capsid protein UL25 and its interaction with the two triplexes closest to the penton. J Mol Biol. 2010;397:575–586. doi: 10.1016/j.jmb.2010.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cockrell SK, Huffman JB, Toropova K, Conway JF, Homa FL. Residues of the UL25 protein of herpes simplex virus that are required for its stable interaction with capsids. J Virol. 2011;85:4875–4887. doi: 10.1128/JVI.00242-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Homa FL, et al. Structure of the pseudorabies virus capsid: Comparison with herpes simplex virus type 1 and differential binding of essential minor proteins. J Mol Biol. 2013;425:3415–3428. doi: 10.1016/j.jmb.2013.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cardone G, et al. The UL36 tegument protein of herpes simplex virus 1 has a composite binding site at the capsid vertices. J Virol. 2012;86:4058–4064. doi: 10.1128/JVI.00012-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fan WH, et al. The large tegument protein pUL36 is essential for formation of the capsid vertex-specific component at the capsid-tegument interface of herpes simplex virus 1. J Virol. 2015;89:1502–1511. doi: 10.1128/JVI.02887-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huet A, et al. Extensive subunit contacts underpin herpesvirus capsid stability and interior-to-exterior allostery. Nat Struct Mol Biol. 2016;23:531–539. doi: 10.1038/nsmb.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu X, et al. Biochemical and structural characterization of the capsid-bound tegument proteins of human cytomegalovirus. J Struct Biol. 2011;174:451–460. doi: 10.1016/j.jsb.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dai X, et al. The smallest capsid protein mediates binding of the essential tegument protein pp150 to stabilize DNA-containing capsids in human cytomegalovirus. PLOS Pathog. 2013;9:e1003525. doi: 10.1371/journal.ppat.1003525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu X, Jih J, Jiang J, Zhou ZH. Atomic structure of the human cytomegalovirus capsid with its securing tegument layer of pp150. Science. 2017;356:eaam6892. doi: 10.1126/science.aam6892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou ZH, et al. Seeing the herpesvirus capsid at 8.5 Å. Science. 2000;288:877–880. doi: 10.1126/science.288.5467.877. [DOI] [PubMed] [Google Scholar]
- 22.Bowman BR, et al. Structural characterization of the UL25 DNA-packaging protein from herpes simplex virus type 1. J Virol. 2006;80:2309–2317. doi: 10.1128/JVI.80.5.2309-2317.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JI, Luxton GW, Smith GA. Identification of an essential domain in the herpesvirus VP1/2 tegument protein: The carboxy terminus directs incorporation into capsid assemblons. J Virol. 2006;80:12086–12094. doi: 10.1128/JVI.01184-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coller KE, Lee JI, Ueda A, Smith GA. The capsid and tegument of the alphaherpesviruses are linked by an interaction between the UL25 and VP1/2 proteins. J Virol. 2007;81:11790–11797. doi: 10.1128/JVI.01113-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leelawong M, Lee JI, Smith GA. Nuclear egress of pseudorabies virus capsids is enhanced by a subspecies of the large tegument protein that is lost upon cytoplasmic maturation. J Virol. 2012;86:6303–6314. doi: 10.1128/JVI.07051-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang F, et al. Novel fold and capsid-binding properties of the λ-phage display platform protein gpD. Nat Struct Biol. 2000;7:230–237. doi: 10.1038/73347. [DOI] [PubMed] [Google Scholar]
- 27.Lander GC, et al. Bacteriophage lambda stabilization by auxiliary protein gpD: Timing, location, and mechanism of attachment determined by cryo-EM. Structure. 2008;16:1399–1406. doi: 10.1016/j.str.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dai X, et al. Structure and mutagenesis reveal essential capsid protein interactions for KSHV replication. Nature. 2018;553:521–525. doi: 10.1038/nature25438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dai X, et al. CryoEM and mutagenesis reveal that the smallest capsid protein cements and stabilizes Kaposi’s sarcoma-associated herpesvirus capsid. Proc Natl Acad Sci USA. 2015;112:E649–E656. doi: 10.1073/pnas.1420317112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Desai P, DeLuca NA, Person S. Herpes simplex virus type 1 VP26 is not essential for replication in cell culture but influences production of infectious virus in the nervous system of infected mice. Virology. 1998;247:115–124. doi: 10.1006/viro.1998.9230. [DOI] [PubMed] [Google Scholar]
- 31.Baker ML, Jiang W, Rixon FJ, Chiu W. Common ancestry of herpesviruses and tailed DNA bacteriophages. J Virol. 2005;79:14967–14970. doi: 10.1128/JVI.79.23.14967-14970.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rixon FJ, Schmid MF. Structural similarities in DNA packaging and delivery apparatuses in herpesvirus and dsDNA bacteriophages. Curr Opin Virol. 2014;5:105–110. doi: 10.1016/j.coviro.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 33.Duda RL. Protein chainmail: Catenated protein in viral capsids. Cell. 1998;94:55–60. doi: 10.1016/S0092-86740081221-0. [DOI] [PubMed] [Google Scholar]
- 34.Wikoff WR, et al. Topologically linked protein rings in the bacteriophage HK97 capsid. Science. 2000;289:2129–2133. doi: 10.1126/science.289.5487.2129. [DOI] [PubMed] [Google Scholar]
- 35.Aksyuk AA, et al. Subassemblies and asymmetry in assembly of herpes simplex virus procapsid. mBio. 2015;6:e01525–e15. doi: 10.1128/mBio.01525-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spencer JV, Newcomb WW, Thomsen DR, Homa FL, Brown JC. Assembly of the herpes simplex virus capsid: Preformed triplexes bind to the nascent capsid. J Virol. 1998;72:3944–3951. doi: 10.1128/jvi.72.5.3944-3951.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trus BL, et al. The herpes simplex virus procapsid: Structure, conformational changes upon maturation, and roles of the triplex proteins VP19c and VP23 in assembly. J Mol Biol. 1996;263:447–462. doi: 10.1016/S0022-28369680018-0. [DOI] [PubMed] [Google Scholar]
- 38.Snijder J, et al. Vertex-specific proteins pUL17 and pUL25 mechanically reinforce herpes simplex virus capsids. J Virol. 2017;91:e00123–17. doi: 10.1128/JVI.00123-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Granzow H, Klupp BG, Mettenleiter TC. Entry of pseudorabies virus: An immunogold-labeling study. J Virol. 2005;79:3200–3205. doi: 10.1128/JVI.79.5.3200-3205.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McElwee M, Beilstein F, Labetoulle M, Rixon FJ, Pasdeloup D. Dystonin/BPAG1 promotes plus-end-directed transport of herpes simplex virus 1 capsids on microtubules during entry. J Virol. 2013;87:11008–11018. doi: 10.1128/JVI.01633-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Preston VG, Murray J, Preston CM, McDougall IM, Stow ND. The UL25 gene product of herpes simplex virus type 1 is involved in uncoating of the viral genome. J Virol. 2008;82:6654–6666. doi: 10.1128/JVI.00257-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pasdeloup D, Blondel D, Isidro AL, Rixon FJ. Herpesvirus capsid association with the nuclear pore complex and viral DNA release involve the nucleoporin CAN/Nup214 and the capsid protein pUL25. J Virol. 2009;83:6610–6623. doi: 10.1128/JVI.02655-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huffman JB, et al. The C terminus of the herpes simplex virus UL25 protein is required for release of viral genomes from capsids bound to nuclear pores. J Virol. 2017;91:e00641–17. doi: 10.1128/JVI.00641-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Klupp BG, Granzow H, Keil GM, Mettenleiter TC. The capsid-associated UL25 protein of the alphaherpesvirus pseudorabies virus is nonessential for cleavage and encapsidation of genomic DNA but is required for nuclear egress of capsids. J Virol. 2006;80:6235–6246. doi: 10.1128/JVI.02662-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang K, Baines JD. Selection of HSV capsids for envelopment involves interaction between capsid surface components pUL31, pUL17, and pUL25. Proc Natl Acad Sci USA. 2011;108:14276–14281. doi: 10.1073/pnas.1108564108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leelawong M, Guo D, Smith GA. A physical link between the pseudorabies virus capsid and the nuclear egress complex. J Virol. 2011;85:11675–11684. doi: 10.1128/JVI.05614-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mettenleiter TC, Müller F, Granzow H, Klupp BG. The way out: What we know and do not know about herpesvirus nuclear egress. Cell Microbiol. 2013;15:170–178. doi: 10.1111/cmi.12044. [DOI] [PubMed] [Google Scholar]
- 48.Newcomb WW, et al. The primary enveloped virion of herpes simplex virus 1: Its role in nuclear egress. mBio. 2017;8:e00825–17. doi: 10.1128/mBio.00825-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Desai PJ. A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J Virol. 2000;74:11608–11618. doi: 10.1128/JVI.74.24.11608-11618.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klupp BG, Fuchs W, Granzow H, Nixdorf R, Mettenleiter TC. Pseudorabies virus UL36 tegument protein physically interacts with the UL37 protein. J Virol. 2002;76:3065–3071. doi: 10.1128/JVI.76.6.3065-3071.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mijatov B, Cunningham AL, Diefenbach RJ. Residues F593 and E596 of HSV-1 tegument protein pUL36 (VP1/2) mediate binding of tegument protein pUL37. Virology. 2007;368:26–31. doi: 10.1016/j.virol.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 52.Ko DH, Cunningham AL, Diefenbach RJ. The major determinant for addition of tegument protein pUL48 (VP16) to capsids in herpes simplex virus type 1 is the presence of the major tegument protein pUL36 (VP1/2) J Virol. 2010;84:1397–1405. doi: 10.1128/JVI.01721-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu Q, Courtney RJ. Chemical cross-linking of virion envelope and tegument proteins of herpes simplex virus type 1. Virology. 1994;204:590–599. doi: 10.1006/viro.1994.1573. [DOI] [PubMed] [Google Scholar]
- 54.Gross ST, Harley CA, Wilson DW. The cytoplasmic tail of herpes simplex virus glycoprotein H binds to the tegument protein VP16 in vitro and in vivo. Virology. 2003;317:1–12. doi: 10.1016/j.virol.2003.08.023. [DOI] [PubMed] [Google Scholar]
- 55.Kamen DE, Gross ST, Girvin ME, Wilson DW. Structural basis for the physiological temperature dependence of the association of VP16 with the cytoplasmic tail of herpes simplex virus glycoprotein H. J Virol. 2005;79:6134–6141. doi: 10.1128/JVI.79.10.6134-6141.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jambunathan N, et al. Herpes simplex virus 1 protein UL37 interacts with viral glycoprotein gK and membrane protein UL20 and functions in cytoplasmic virion envelopment. J Virol. 2014;88:5927–5935. doi: 10.1128/JVI.00278-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Scrima N, et al. Insights into herpesvirus tegument organization from structural analyses of the 970 central residues of HSV-1 UL36 protein. J Biol Chem. 2015;290:8820–8833. doi: 10.1074/jbc.M114.612838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Woehlke G, Schliwa M. Walking on two heads: The many talents of kinesin. Nat Rev Mol Cell Biol. 2000;1:50–58. doi: 10.1038/35036069. [DOI] [PubMed] [Google Scholar]
- 59.Hirokawa N, et al. Submolecular domains of bovine brain kinesin identified by electron microscopy and monoclonal antibody decoration. Cell. 1989;56:867–878. doi: 10.1016/0092-86748990691-0. [DOI] [PubMed] [Google Scholar]
- 60.Kull FJ, Sablin EP, Lau R, Fletterick RJ, Vale RD. Crystal structure of the kinesin motor domain reveals a structural similarity to myosin. Nature. 1996;380:550–555. doi: 10.1038/380550a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Suloway C, et al. Automated molecular microscopy: The new Leginon system. J Struct Biol. 2005;151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 62.Li X, et al. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat Methods. 2013;10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mindell JA, Grigorieff N. Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol. 2003;142:334–347. doi: 10.1016/S1047-84770300069-8. [DOI] [PubMed] [Google Scholar]
- 64.Ludtke SJ, Baldwin PR, Chiu W. EMAN: Semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 65.Liang Y, Ke EY, Zhou ZH. IMIRS: A high-resolution 3D reconstruction package integrated with a relational image database. J Struct Biol. 2002;137:292–304. doi: 10.1016/S1047-84770200014-X. [DOI] [PubMed] [Google Scholar]
- 66.Liu H, et al. Symmetry-adapted spherical harmonics method for high-resolution 3D single-particle reconstructions. J Struct Biol. 2008;161:64–73. doi: 10.1016/j.jsb.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 67.Zhang X, Zhang X, Zhou ZH. Low cost, high performance GPU computing solution for atomic resolution cryoEM single-particle reconstruction. J Struct Biol. 2010;172:400–406. doi: 10.1016/j.jsb.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rosenthal PB, Henderson R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol. 2003;333:721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 69.Pettersen EF, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 70.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bowman BR, Baker ML, Rixon FJ, Chiu W, Quiocho FA. Structure of the herpesvirus major capsid protein. EMBO J. 2003;22:757–765. doi: 10.1093/emboj/cdg086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang X, Zhou ZH. Limiting factors in atomic resolution cryo electron microscopy: No simple tricks. J Struct Biol. 2011;175:253–263. doi: 10.1016/j.jsb.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ilca SL, et al. Localized reconstruction of subunits from electron cryomicroscopy images of macromolecular complexes. Nat Commun. 2015;6:8843. doi: 10.1038/ncomms9843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Scheres SH. RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.