

Abstract
Adequate nutrition is critical for human brain development, which depends particularly upon glucose. The adult human brain accounts for 20–25% of total body resting glucose consumption, and studies indicate that the developing brain requires an even greater percentage of glucose. Here we critically review the currently available data on glucose requirements for early childhood brain development. Implications of these findings are then discussed in the context of childhood malnutrition and future areas of investigation.
Three to five key words not used in title: FDG-PET, malnutrition, glycolysis
Introduction
The human brain is highly reliant on glucose for fuel and growth. An adult human brain at rest consumes approximately 20–25% of its resting total body glucose consumption rate (1,2). This is nearly double that required by our nearest evolutionary cousin, the chimpanzee (3,4). Presumably cooking and other dietary, gastrointestinal and/or metabolic adaptations have enabled humans to maintain the caloric needs of their brains (4–6). However, what is often not mentioned in these analyses are the heightened costs of the developing brain. Here we critically review studies that measure the glucose requirements of the early developing brain in humans.
Tracer-based studies of glucose consumption in the developing human brain
There are few studies on human brain glucose use during development due to the ethical limitations of performing invasive and radiotracer-related studies in healthy normal children. Thus, studies to date have largely been based on children being evaluated for intermittent epilepsy, suspected perinatal hypoxic-ischemic event, or suspected Sturge-Weber syndrome, who were otherwise neurologically normal at the time of the PET study. Chugani and colleagues reported their quantitative PET studies in such children, first in 1987 (7,8). This landmark study presented data in 29 children selected from over 100, whom the authors felt were as close to healthy and neurologically normal as reasonably possible. The children ranged in age from five days to approximately 15 years of age; more than half were taking anticonvulsants (most commonly carbamazepine or phenobarbital) for seizure prevention; and all were reported to be neurologically normal at the time of the PET scan and on follow-up. Five of the 29 were scanned to assess for Sturge-Weber syndrome due to a facial birthmark, and brain PET findings in all 5 of these children were normal. It should be noted that this study derived an arterial input function, which is necessary to quantitate cerebral metabolic rate for glucose (CMRGlc), from ‘arterialized’ venous sampling, which is a method that had been previously validated and is more acceptable to perform in children (7,8). Also, this study employed a model that includes an a priori constant (‘lumped constant’) that had been previously derived from studies in young adults. The authors reported that CMRGlc during the first two years of life was similar to that in young adults, which is typically between 23–25 μmol/100g/min (8,9). The CMRGlc values in children then rose over the next few years to approximately double the adult value around 3–5 years of age, where it remained for several years and then gradually fell over subsequent years to average adult values (8).
The study by Chugani and colleagues included only three infants in the first three months of life. Kinnala et al. chose 20 PET studies in neonates and infants, ranging between 32.7 and 60.3 weeks postconceptional age, who were being evaluated for suspected hypoxic ischemic injury or neonatal hypoglycemia, but were then found to be neurologically normal at further evaluation and subsequent follow-up (10). Two infants were studied twice. Studies were performed during postprandial sleep without sedation. To quantitate CMRGlc, time-activity curves from the left ventricle of the heart and venous samples were used to estimate the input function. This group used a slightly higher lumped constant in their quantitative model as compared to the study by Chugani et al., which would be expected to result in slightly lower estimates. Nonetheless, they reported similar CMRGlc measurements for infants greater than 50 weeks postconceptional age. For premature and term-equivalent neonates, the CMRGlc was found to be lower than that seen in infants, approximately half of the whole brain CMRGlc achieved at 5–6 months of age. Powers et al. studied another six preterm infants born between 25 and 34 weeks estimated gestational age and within one week of birth, with similar results to those found by Kinnala and colleagues in their preterm infants (11).
Table 1 summarizes the quantitative measurements of CMRGlc to date in (nearly) healthy children. An outstanding question in these studies is whether quantitative methods utilizing FDG-PET to estimate CMRGlc are adequate in children. Though there is no evidence to suggest otherwise, these tracer-based studies rely on model estimates, such as the lumped constant, which relates FDG-PET-based estimates of whole brain CMRGlc to Kety-Schmidtbased measurements that were derived in young adults (8,10). Measurements in neonates are particularly suspect as these studies were often done in the setting of clinical suspicion for hypoxic-ischemic injury or neonatal hypoglycemia (10,11). Also, the neonates were typically asleep during the study, which may lower brain metabolism relative to the awake state. Thus, current data may have underestimated CMRGlc in this age group.
TABLE 1.
Quantitative estimates of CMRGlu in children.
| Age | n | CMRGlu (μmol/100g/min) | Reference |
|---|---|---|---|
| Preterm | 6 | 8.8 | Powers et al. (8) |
| Preterm | 8 | 5.5 | Kinnala et al. (7) |
| Term-2m | 8 | 7.3 | Kinnala et al. (7) |
| 2m-6m | 6 | 16.5 | Kinnala et al. (7) |
| 0-1y | 7 | 20.4 | Chugani et al. (5) |
| 1y-2y | 4 | 27.4 | Chugani et al. (5) |
| 3y-8y | 12 | 48.1 | Chugani et al. (5) |
| 9y-15y | 6 | 39.2 | Chugani et al. (5) |
| 19y-30y | 7 | 24.3 | Chugani et al. (5) |
| 21y-28y | 8 | 23.3 | Madsen et al. (6) |
Quantitative assessments of brain glucose use in older children have not been adequately replicated. One study of brain glucose metabolism in autistic children also reported on 3 control children and found a similar gray matter CMRGlc value as that reported by Chugani et al. (12). Another study, reported in 2014, obtained PET images of the brain in over 100 children (five months to twenty-three years) with non-CNS involving malignancies prior to treatment (13). This authors reported many of the same regional changes in brain glucose utilization identified by Chugani and colleagues. For example, normalized peak glucose use in medial frontal regions occurred later than in the occipital lobe. Unfortunately, this study was non-quantitative and therefore cannot provide accurate estimates of the glucose requirements during brain development.
Complementary studies on brain metabolism during development
Complementary studies investigating cerebral blood flow have found similar two-fold increases during early childhood as compared to values in adults (14). Brain oxygen use also rises in children, though not as highly as glucose use, suggesting that a proportion of the increase in brain glucose use during prepubescent childhood likely supplies non-oxidative pathways, i.e., aerobic glycolysis (14).
Aerobic glycolysis is a key metabolic feature of proliferating/growing tissues in general, including cancer and immune cells (15). Aerobic glycolysis might, in fact, represent one of the principals reasons for increased glucose requirement in the developing brain, due to the various biosynthetic processes required for synaptic development and myelination (14,16,17). As discussed further below, aerobic glycolysis might have an important role during the nutritional rehabilitation of malnourished children.
Studies of synaptic and, more recently, spine-density changes during the course of brain development demonstrate that synaptic and spine-density changes closely mirror temporal changes in CMRGlc (18–20). This suggests that the rapid rise in CMRGlc seen soon after birth reflects the rapid synaptic proliferation occurring in the brain at this time, and that, conversely, the gradual fall in CMRGlc after early childhood occurs largely due to synaptic pruning (18). It should be noted that so-called synaptic ‘pruning’ likely reflects a period of ongoing synaptic proliferation, now matched by higher rates of synaptic elimination, rather than a ‘switch’ from proliferation to elimination (21).
Recent advances in genomic technologies have led to large scale studies of human brain gene expression across the lifespan (22). Expression rates for somatic genes associated with mitochondrial activity follow a temporal pattern similar to that of CMRGlc and closely track changes in synaptic number during childhood brain development, providing further evidence that the general pattern of brain metabolism during childhood is likely correct (21).
An alternative approach to determine the glucose requirements for brain development would be to consider the effects of hypoglycemia on the brain. This approach, however, is complicated by the fact that in the setting of acute hypoglycemia, the brain and body have multiple mechanisms to compensate. Compensatory mechanisms might include increased cerebral blood flow, lactate and ketone production and utilization, and decreasing the demand for glucose by lowering overall neuronal activity (23). In neonatal hypoglycemia, multiple other systemic insults, such as sepsis or hypoxic-ischemic injury, may further confound determination of the lower limits of brain-glucose requirements. Further, the duration and severity of the hypoglycemia both influence the risk of brain injury, complicating efforts to define a threshold below which hypoglycemia should be treated (24,25). Nonetheless, severe acute hypoglycemia is well documented to result in areas of brain injury. The areas of injury typically occur a regionally specific manner that corresponds to the age-dependent regional differences in brain glucose metabolism (24) (Figure 1). The detailed effects of acute hypoglycemia on the neonatal and child’s brain are beyond the scope of this short review, but have been discussed at length elsewhere (23–25).
Figure 1. Regional brain vulnerability to hypoglycemia is predicted by baseline glucose demand.
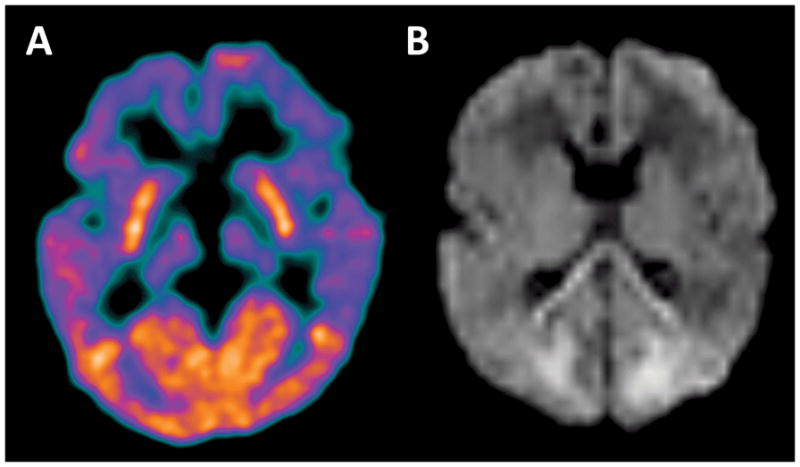
(A) FDG-PET brain imaging was performed in a 7 month old for the purposes of epilepsy source localization. This shows the well documented finding of elevated FDG uptake in the occipital lobes during early infancy. (B) Brain MRI including diffusion weighted imaging (DWI) was performed on an infant a few days after experiencing severe refractory hypoglycemia. This DWI image shows areas of diffusion restriction, representing focal brain injury, predominantly in the medial occipital lobes. An additional area of diffusion restriction in the splenium of the corpus callosum was interpreted to represent developing secondary Wallerian degeneration arising from the occipital lobe injury.
Malnutrition and the developing brain
More prolonged periods of inadequate nutrition, such as those that might occur in the setting of gut malabsorptive states or socio-economically driven malnutrition, might also constrain the developing brain’s access to adequate amounts of glucose. As noted above, an intriguing hypothesis is that if malnutrition delays/inhibits the normal trajectory of brain development, it might also then increase the latent demand for aerobic glycolysis in the brain. If true, brain aerobic glycolysis should be increased during the early nutritional rehabilitation phase in malnourished children. Mehta and colleagues provide some evidence to support this hypothesis using Kety Schmidt based methods to measure whole brain glucose and oxygen metabolism in young malnourished children (26). They found that the mean oxygen/glucose index (OGI), which is an inverse measure of aerobic glycolysis, was lower in a cohort of healthy and mildly malnourished children (n = 10, age 11±2 months, OGI = 65.8%) as compared to prior reported values in adults (approximately 90%). In severely malnourished children, aerobic glycolysis was substantially higher (n = 6, age 23±5 months, OGI = 34.7%), despite similar levels of oxidative glycolysis as compared to the healthy/mildly malnourished cohort. Though this study has not yet been replicated in humans, it supports our contention that the developing brain has high demands for aerobic glycolysis, and possibly even more so in the context of malnutrition.
An open question is to what degree is the developing brain most vulnerable to the effects of under- and malnutrition, and if so, in what manner and during what period(s) of development. Is there truly a ‘time window’ of greater concern, as has been proposed for the first 1000 days, for example, or does the brain remain highly vulnerable to malnutrition beyond this period (27)? Understanding the compensatory mechanisms in this setting, the time and degree of vulnerability for brain development, and the long term effects of malnutrition represent a critical goal for future investigations aimed at understanding the effects of malnutrition on brain development.
Equally important will be understanding the role of gut physiology, including its microbiota in securing adequate glucose and other nutrient resources for brain development. The tools to begin such investigations from the gut to the brain in small animal models and humans are now available (28). A multidisciplinary approach will be required to understand these relationships thoroughly, but could ultimately lead to targeted and/or more efficient approaches to alleviating the untoward effects of childhood malnutrition.
What is known
The human brain has high demands for glucose throughout the lifespan.
Glucose requirements in the human brain peak during childhood, reaching approximately twice that of the adult brain, per gram of brain tissue.
However, data is limited due to ethical constraints in applying current brain metabolism methodologies to study healthy children
What is new
Aerobic glycolysis might be particularly relevant to brain development in the context of malnutrition.
The high brain glucose requirements during childhood warrant further investigations into the effects of under- and malnutrition on brain development.
Acknowledgments
Sources of support that require acknowledgment: MSG is supported by a grant from the RSNA R&E Foundation. MER receives support from the NIH (P01-NS080675).
Footnotes
Author Contributions
Both MSG and MR contributed to the conception and writing of the manuscript. Both reviewed and approved the final draft.
Disclosure of funding: Above.
References
- 1.Raichle ME, Posner JB, Plum F. Cerebral blood flow during and after hyperventilation. Arch Neurol. 1970;23(5):394–403. doi: 10.1001/archneur.1970.00480290014002. [DOI] [PubMed] [Google Scholar]
- 2.Mink JW, Blumenschine RJ, Adams DB. Ratio of central nervous system to body metabolism in vertebrates: its constancy and functional basis. Am J Physiol. 1981;241(3):R203–12. doi: 10.1152/ajpregu.1981.241.3.R203. [DOI] [PubMed] [Google Scholar]
- 3.Fonseca-Azevedo K, Herculano-Houzel S. Metabolic constraint imposes tradeoff between body size and number of brain neurons in human evolution. Proc Natl Acad Sci USA. 2012;109(45):18571–6. doi: 10.1073/pnas.1206390109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Herculano-Houzel S. The remarkable, yet not extraordinary, human brain as a scaled-up primate brain and its associated cost. Proc Natl Acad Sci USA. 2012;109(Suppl 1):10661–8. doi: 10.1073/pnas.1201895109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aiello LC, Wheeler P. The expensive-tissue hypothesis - the brain and the digestive-system in human and primate evolution. Curr Anthropol. 1995;36(2):199–221. [Google Scholar]
- 6.Navarrete A, van Schaik CP, Isler K. Energetics and the evolution of human brain size. Nature. 2011;480(7375):91–U252. doi: 10.1038/nature10629. [DOI] [PubMed] [Google Scholar]
- 7.Chugani HT. Positron emission tomography: principles and applications in pediatrics. Mead Johnson Symp Perinat Dev Med. (25) 1987:15–8. [PubMed] [Google Scholar]
- 8.Chugani HT, Phelps ME, Mazziotta JC. Positron emission tomography study of human brain functional development. Ann Neurol. 1987;22(4):487–97. doi: 10.1002/ana.410220408. [DOI] [PubMed] [Google Scholar]
- 9.Madsen PL, Hasselbalch SG, Hagemann LP, et al. Persistent resetting of the cerebral oxygen glucose-uptake ratio by brain activation - Evidence obtained with the Kety-Schmidt technique. J Cereb Blood Flow Metab. 1995;15(3):485–91. doi: 10.1038/jcbfm.1995.60. [DOI] [PubMed] [Google Scholar]
- 10.Kinnala A, Suhonen-Polvi H, Aarimaa T, et al. Cerebral metabolic rate for glucose during the first six months of life: an FDG positron emission tomography study. Arch Dis Child Fetal Neonat Ed. 1996;74(3):F153–7. doi: 10.1136/fn.74.3.f153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Powers WJ, Rosenbaum JL, Dence CS, et al. Cerebral glucose transport and metabolism in preterm human infants. J Cereb Blood Flow Metab. 1998;18(6):632–8. doi: 10.1097/00004647-199806000-00005. [DOI] [PubMed] [Google Scholar]
- 12.De Volder A, Bol A, Michel C, et al. Brain glucose metabolism in children with the autistic syndrome: positron tomography analysis. Brain Dev. 1987;9(6):581–7. doi: 10.1016/s0387-7604(87)80089-x. [DOI] [PubMed] [Google Scholar]
- 13.Shan ZY, Leiker AJ, Onar-Thomas A, et al. Cerebral glucose metabolism on positron emission tomography of children. Hum Brain Mapp. 2014;35(5):2297–309. doi: 10.1002/hbm.22328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goyal MS, Hawrylycz M, Miller JA, et al. Aerobic glycolysis in the human brain is associated with development and neotenous gene expression. Cell Metab. 2014;19(1):49–57. doi: 10.1016/j.cmet.2013.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Ann Rev Cell Dev Biol. 2011;27:441–64. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- 16.Baquer NZ, Hothersall JS, McLean P, et al. Aspects of carbohydrate metabolism in developing brain. Dev Med Child Neurol. 1977;19(1):81–104. doi: 10.1111/j.1469-8749.1977.tb08027.x. [DOI] [PubMed] [Google Scholar]
- 17.Bauernfeind AL, Barks SK, Duka T, et al. Aerobic glycolysis in the primate brain: reconsidering the implications for growth and maintenance. Brain Struct Funct. 2014;219(4):1149–67. doi: 10.1007/s00429-013-0662-z. [DOI] [PubMed] [Google Scholar]
- 18.Chugani HT. A critical period of brain development: studies of cerebral glucose utilization with PET. Prev Med. 1998;27(2):184–8. doi: 10.1006/pmed.1998.0274. [DOI] [PubMed] [Google Scholar]
- 19.Huttenlocher PR, Dabholkar AS. Regional differences in synaptogenesis in human cerebral cortex. J Comp Neurol. 1997;387(2):167–78. doi: 10.1002/(sici)1096-9861(19971020)387:2<167::aid-cne1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 20.Petanjek Z, Judas M, Simic G, et al. Extraordinary neoteny of synaptic spines in the human prefrontal cortex. Proc Natl Acad Sci U S A. 2011;108(32):13281–6. doi: 10.1073/pnas.1105108108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goyal MS, Raichle ME. Gene expression-based modeling of human cortical synaptic density. Proc Natl Acad Sci USA. 2013;110(16):6571–6. doi: 10.1073/pnas.1303453110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kang HJ, Kawasawa YI, Cheng F, et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478(7370):483–9. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hay WW, Jr, Raju TN, Higgins RD, et al. Knowledge gaps and research needs for understanding and treating neonatal hypoglycemia: workshop report from Eunice Kennedy Shriver National Institute of Child Health and Human Development. J Pediatr. 2009;155(5):612–7. doi: 10.1016/j.jpeds.2009.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barkovich AJ, Ali FA, Rowley HA, et al. Imaging patterns of neonatal hypoglycemia. AJNR Am J Neuroradiol. 1998;19(3):523–8. [PMC free article] [PubMed] [Google Scholar]
- 25.Burns CM, Rutherford MA, Boardman JP, et al. Patterns of cerebral injury and neurodevelopmental outcomes after symptomatic neonatal hypoglycemia. Pediatrics. 2008;122(1):65–74. doi: 10.1542/peds.2007-2822. [DOI] [PubMed] [Google Scholar]
- 26.Mehta S, Kalsi HK, Nain CK, et al. Energy Metabolism of Brain in Human Protein-Calorie Malnutrition. Pediat Res. 1977;11:290–3. doi: 10.1203/00006450-197704000-00006. [DOI] [PubMed] [Google Scholar]
- 27.Leroy JL, Ruel M, Habicht JP, et al. Linear Growth Deficit Continues to Accumulate beyond the First 1000 Days in Low- and Middle-Income Countries: Global Evidence from 51 National Surveys. J Nutr. 2014;144(9):1460–66. doi: 10.3945/jn.114.191981. [DOI] [PubMed] [Google Scholar]
- 28.Goyal MS, Venkatesh S, Milbrandt J, et al. Feeding the brain and nurturing the mind: Linking nutrition and the gut microbiota to brain development. Proc Natl Acad Sci USA. 2015;112(46):14105–12. doi: 10.1073/pnas.1511465112. [DOI] [PMC free article] [PubMed] [Google Scholar]