

Cimino et al. developed an unbiased bioinformatics approach that identified homeobox A5 (HOXA5) as a gene whose expression correlated with gain of chromosome 7 and a more aggressive phenotype of the resulting glioma. HOXA5 overexpression promoted cellular proliferation and potentiated radioresistance.
Keywords: glioblastoma, HoxA, homeobox, RCAS, chromosome 7 gain
Abstract
Glioblastoma is the most frequently occurring and invariably fatal primary brain tumor in adults. The vast majority of glioblastomas is characterized by chromosomal copy number alterations, including gain of whole chromosome 7 and loss of whole chromosome 10. Gain of whole chromosome 7 is an early event in gliomagenesis that occurs in proneural-like precursor cells, which give rise to all isocitrate dehydrogenase (IDH) wild-type glioblastoma transcriptional subtypes. Platelet-derived growth factor A (PDGFA) is one gene on chromosome 7 known to drive gliomagenesis, but, given its location near the end of 7p, there are likely several other genes located along chromosome 7 that select for its increased whole-chromosome copy number within glioblastoma cells. To identify other potential genes that could select for gain of whole chromosome 7, we developed an unbiased bioinformatics approach that identified homeobox A5 (HOXA5) as a gene whose expression correlated with gain of chromosome 7 and a more aggressive phenotype of the resulting glioma. High expression of HOXA5 in glioblastoma was associated with a proneural gene expression pattern and decreased overall survival in both human proneural and PDGF-driven mouse glioblastoma. Furthermore, HOXA5 overexpression promoted cellular proliferation and potentiated radioresistance. We also found enrichment of HOXA5 expression in recurrent human and mouse glioblastoma at first recurrence after radiotherapy. Overall, this study implicates HOXA5 as a chromosome 7-associated gene-level locus that promotes selection for gain of whole chromosome 7 and an aggressive phenotype in glioblastoma.
Glioblastoma is the most common and aggressive primary brain tumor in the adult population (Ostrom et al. 2017). High-throughput technologies have considerably increased the knowledge of recurrent genetic and epigenetic alterations found in glioblastomas and other diffuse gliomas, helping to define molecularly distinct entities and subtypes, including gene expression-based subtypes designated proneural, classical, and mesenchymal types (The Cancer Genome Atlas Research Network 2008; Yan et al. 2009; Noushmehr et al. 2010; Verhaak et al. 2010; Sturm et al. 2012; The Cancer Genome Atlas Research Network et al. 2015; Olar et al. 2015; Weller et al. 2015; Ceccarelli et al. 2016; Louis et al. 2016). Specific point mutations in the isocitrate dehydrogenase-1 (IDH-1) and IDH-2 genes segregate glioblastoma into IDH mutant glioblastomas, characterized by epigenetic hypermethylation and proneural gene expression, and IDH wild-type glioblastomas, characterized by large chromosomal aberrations and proneural, mesenchymal, or classical gene expression patterns (Yan et al. 2009; Bleeker et al. 2010; The Cancer Genome Atlas Research Network et al. 2015; Eckel-Passow et al. 2015; Leeper et al. 2015; Olar et al. 2015; Reuss et al. 2015; Ceccarelli et al. 2016). IDH wild-type glioblastomas characteristically harbor gain of chromosome 7 and loss of chromosome 10 (Cimino et al. 2017), both of which arise from early nondisjunction events in gliomagenesis (Ozawa et al. 2014). Selection for neoplastic cells with gain of chromosome 7 is likely due to multiple genes that drive tumor aggressiveness. As one example, platelet-derived growth factor A (PDGFA) has been shown to be a main driver of chromosome 7 gain, and its overexpression is sufficient to induce proneural gliomas in mice (Ozawa et al. 2014). In this study, we sought to identify other gene-level loci on chromosome 7 that could provide a selective advantage for gain of whole chromosome 7 in IDH wild-type glioblastomas; moreover, we hypothesized that associations with survival will be specific for molecular glioblastoma subtypes. We used an unbiased bioinformatics approach to identify genes on chromosome 7 with the potential for driving gliomagenesis and found homeobox A5 (HOXA5) expression to be the most correlated with chromosome 7 copy number gain and survival in proneural glioblastomas. Mouse modeling supported this relationship in that HoxA5 overexpression enhances the aggressiveness of PDGF-driven proneural gliomas through cell cycle dysregulation and radioresistance. Further reinforcing its aggressive role in gliomagenesis, HOXA5 expression is selected for in glioblastomas following radiation therapy in mouse and human glioblastomas.
Results
Survival loci associated with chromosome 7 gain in IDH wild-type glioblastoma
As a first step to identifying potential genes that drive the selection of gain of whole chromosome 7, we queried the publically available The Cancer Genome Atlas (TCGA) glioblastoma data set (The Cancer Genome Atlas Research Network 2008; Ceccarelli et al. 2016). We identified genes with chromosomal copy number alterations showing differential expression across samples and whose expression was correlated with overall survival. Given the profound effect of IDH mutational status on patient survival and its relatively low mutational frequency in glioblastoma, IDH mutant glioblastomas were excluded from our analysis. Moreover, we focused on proneural glioblastomas because they represent the common progenitor of all IDH wild-type glioblastomas that may be genetically more uniform than classical or mesenchymal glioblastomas (Ozawa et al. 2014). Among the genes on chromosome 7, the gene expression changes of HOXA5, RNF32, and C7orf13 had the strongest association with patient survival (Fig. 1A). Expression of the known oncogenic drivers EGFR, MET, and CDK6 on chromosome 7 did not have correlation with chromosomal copy number and survival (P = 0.95, P = 0.35, and P = 0.71, respectively) (Fig. 1A). HOX genes encode for a family of 39 highly conserved transcription factors in four chromosomal clusters, including cluster A (HOXA) on chromosome 7p15. HOX gene products are critical regulators of embryonic morphogenesis (Holland et al. 2007), and dysregulation of HOX gene expression is associated with cancer (Shah and Sukumar 2010; Bhatlekar et al. 2014), but the functions of individual HOX genes in glioblastomas are incompletely defined. Biological functions of the long noncoding RNA encoded by C7orf13 and of a putative ubiquitin ligase encoded by RNF32 are widely elusive. Of the three genes associated with patient survival, HOXA5 was the only one that was overexpressed in association with chromosome 7 gain (Fig. 1A). Of note, HOXA3 and HOXA4 were the other HOXA cluster genes displaying an association with survival, albeit to a lesser extent than HOXA5 (Supplemental Table 1; Supplemental Fig. 1). Among glioblastoma transcriptional subtypes (Verhaak et al. 2010), HOXA5 gene expression was highest in proneural glioblastomas (Fig. 1B), which, moreover, was the only transcriptional subtype in which high versus low HOXA5 gene expression was associated with patient outcome (median overall survival: 10 vs. 22 mo; log rank P = 0.003) (Fig. 1C). RNF32 and C7orf13 were underexpressed in relation to chromosome 7 gain, and their expression was inversely associated with survival of patients with proneural glioblastomas (Fig. 1D,E). No association with survival was seen in the other transcriptional glioblastoma subtypes in relation to RNF32 and C7orf13 expression (P > 0.05). RNF32 and C7orf13 were the two genes having the most inverse correlation to HOXA5 expression in proneural glioblastomas (Fig. 1F).
Figure 1.
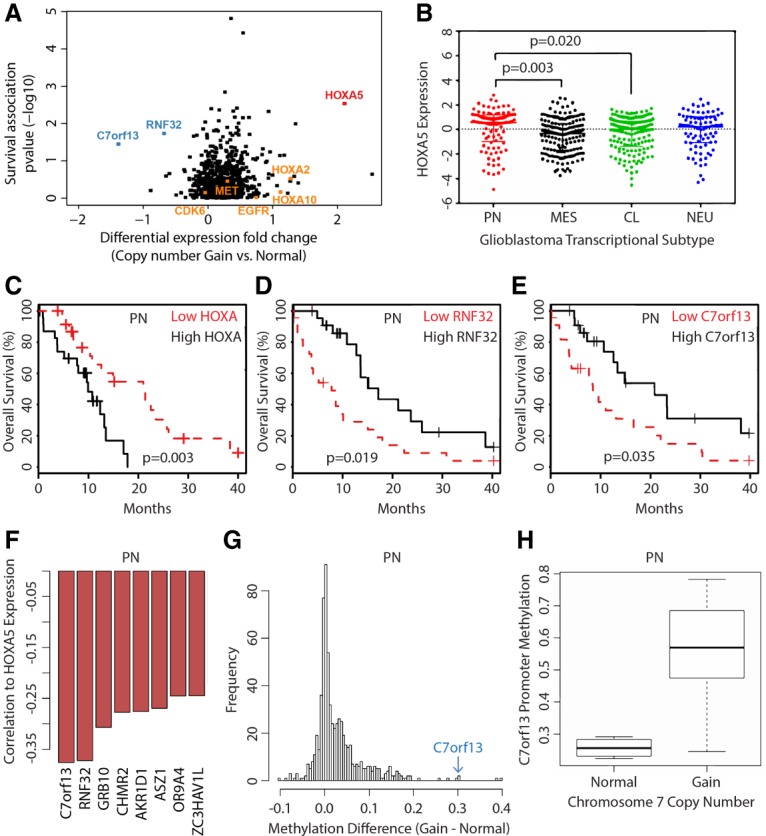
Genetic survival loci associated with chromosome 7 gain in human glioblastomas include HoxA5. (A) Genes on chromosome 7 ranked by association with overall survival in relation to differential gene expression between patients with normal and gained chromosome 7. The top-ranked genes include RNF32 (underexpressed), C7orf13 (underexpressed), and HOXA5 (overexpressed). (B) HOXA5 expression in proneural (PN; n = 97), classical (CL; n = 145), mesenchymal (MES; n = 156), and neural (NEU; n = 83) glioblastoma gene expression subtypes. Kaplan-Meier plots showing overall survival effects for proneural glioblastomas separated by quartile expression of chromosome 7 survival loci HOXA5 (C), RNF32 (D), and C7orf13 (E). (F) HOXA5 expression is inversely correlated to RNF32 and C7orf13 expression in proneural glioblastomas. (G) Methylation profiling in proneural glioblastomas comparing chromosomal 7 status (normal vs. gain) identifies C7orf13 as relatively highly methylated. (H) Methylation of the C7orf13 promoter increases with gain of chromosome 7, corresponding to decreased gene expression.
To explore a potential mechanism of gene silencing of RNF32 and C7orf13 in the setting of chromosome 7 gain, we investigated DNA methylation patterns across chromosome 7. The promoter for C7orf13, but not RNF32, had a high level of methylation in association with chromosome 7 gain in proneural glioblastomas (Fig. 1G,H). Furthermore, promoter methylation of C7orf13 was inversely correlated with gene expression, while RNF32 promoter methylation was positively correlated with gene expression (Supplemental Fig. 2). Of note, C7orf13 and RNF32 are neighboring genes, and potential coregulatory effects are not defined. Of these three genes associated with chromosome 7 gain and survival in glioblastomas, we chose to focus subsequent experimental mouse models on HOXA5, as (1) there is a lack of C7orf13 orthologs in mice; (2) there is no clear mechanism of gene silencing of RNF32 in glioblastomas, and there is a potential complex genomic relation with C7orf13; and (3) increased expression of HOXA5 was associated with chromosomal 7 gain, implying a simple mechanism of overexpression and providing a clear oncogenic candidate for glioblastomas.
Overexpression of HoxA5 decreases survival in murine glioblastomas
Our bioinformatic screen established a strong correlation of HOXA5 expression with gain of whole chromosome 7 and poor patient survival, implying that HOXA5 expression may provide a selective advantage for gain of chromosome 7 in IDH wild-type glioblastoma. To initially gain insight into the functional role of HOXA5 in glioblastomas and uncover a possible mechanism for its selective advantage in glioma cells, we introduced ectopic HoxA5 expression into an established mouse model of proneural glioblastomas. Because PDGFA is a chromosome 7-encoded driver of gliomagenesis, we used the RCAS/tva gene transfer system to induce PDGF-driven proneural glioblastomas in mice in order to determine whether elevated HOXA5 gene expression is causally related to aggressiveness of glioblastomas (Fig. 2A). The HoxA5-overexpressing group of tumor-bearing mice was created by infecting Nestin-positive (Ntva) cells in vivo with combined RCAS-PDGF and RCAS-HoxA5, while the control group was generated by combined RCAS-PDGF and RCAS-mCherry (Fig. 2B). Mice had a background of either homozygous (−/−) or heterozygous (+/−) deletion of Ink4a/Arf. After mice showed signs of disease, glioma tissue (Supplemental Fig. 3) was harvested, and tumor lysate confirmed the presence of RCAS-HoxA5 through Western blotting of protein tags (Fig. 2C). HoxA5 overexpression in mouse glioblastomas led to decreased symptom-free survival regardless of the deletion status of Ink4a/Arf (P < 0.001 for both genotypes), albeit this effect was more pronounced in mice with heterozygous deletion of Ink4a/Arf (hazard ratio [HR] 2.88, 95% confidence interval [CI] 1.58–5.25) compared with mice with homozygous deletion of Ink4a/Arf (HR 2.18, 95% CI, 1.50–3.17) (Fig. 2D–F).
Figure 2.
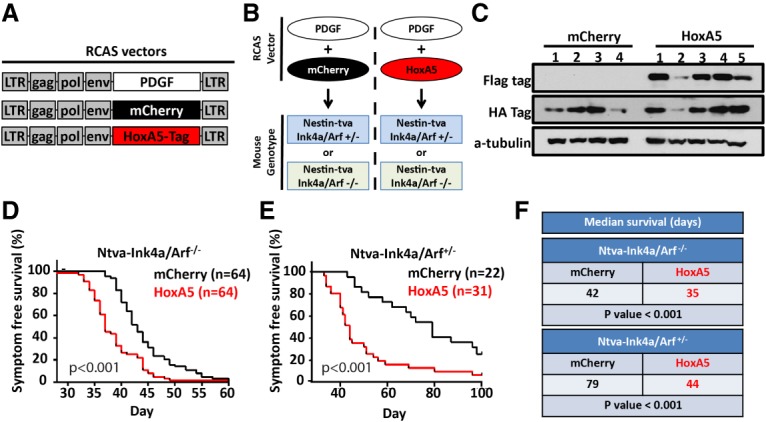
HoxA5 overexpression in mouse glioblastomas leads to decreased survival, reflective of human disease. (A) Schematic of RCAS vectors used for mouse glioblastoma production. (B) Strategy for RCAS vector coinjections in mice with background containing either homozygous or heterozygous loss of Ink4a/Arf. (C) Expression of tagged HoxA5 or mCherry in mouse PDGF-driven glioblastoma lysate as confirmed by Western blot. HoxA5-overexpressing mouse glioblastomas had decreased survival in mice with homozygous (D) or heterozygous (E) loss of Ink4a/Arf, with both having decreased median survival (log-rank test; F).
HoxA5 regulates cellular proliferation
In order to unravel the underlying biological processes yielding the association of HoxA5 gene expression with inferior survival, we began by performing a gene expression microarray focused on HoxA5 overexpression in bulk mouse glioblastomas. When compared with mCherry control tumors, the HoxA5-overexpressing tumors had 692 differentially expressed genes (190 up-regulated and 502 down-regulated) with false discovery rate (FDR) < 0.05 and absolute log2 fold change < 1 (Fig. 3A). To determine which of these 692 differentially expressed genes may be a result of direct HoxA5 DNA element binding rather than indirect compensatory effects, we queried ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) data of human carcinoma cells (Yan et al. 2013), which allows for filtering across species and cancer types. This analysis led to the identification of 136 common differentially expressed genes (46 up-regulated and 90 down-regulated) with known HOXA5 DNA-binding elements (Fig. 3B). Pathway enrichment analysis of the 46 up-regulated genes consisted of several biological processes involving cell cycle and DNA damage response pathways (Fig. 3C). Therefore, it is likely that HoxA5 promotes its aggressive glioma phenotype at least partially through direct up-regulation of cell cycle and DNA damage repair genes.
Figure 3.
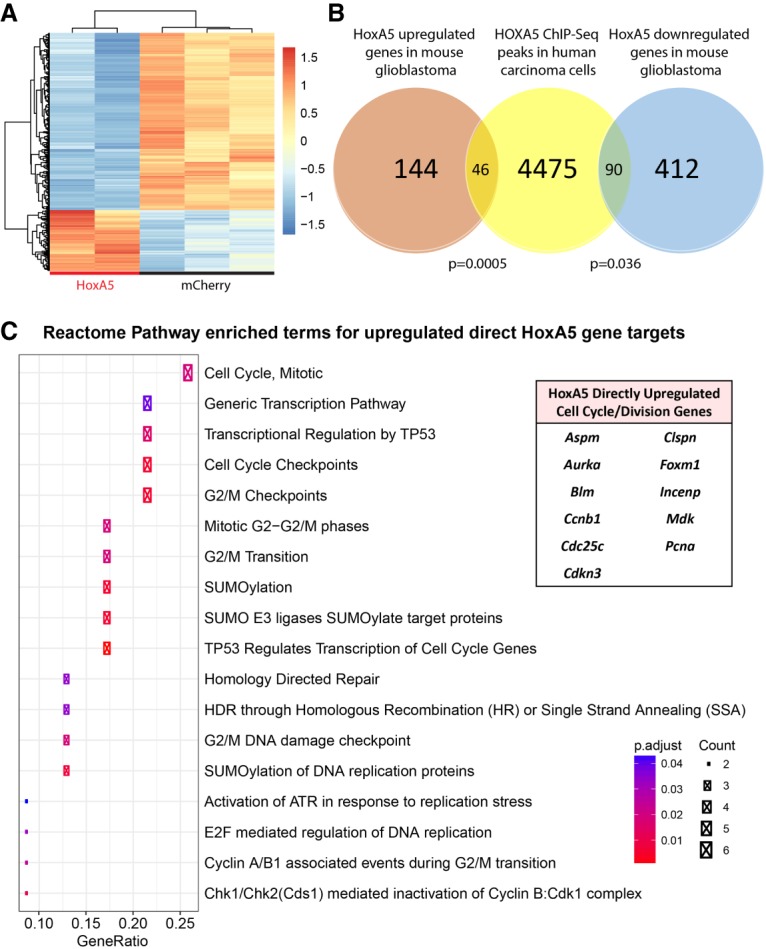
Gene expression profiling of HoxA5 mouse glioblastomas. (A) Microarray heat map highlighting 692 differentially expressed genes between HoxA5 and mCherry mouse glioblastomas. (B) Venn diagram comparing differentially expressed downstream HoxA5 genes in mouse glioblastomas with genes containing HOXA5 transcription factor DNA-binding elements by ChIP-seq analysis in human carcinoma cells. (C) Pathway enrichment analysis of the 46 common up-regulated genes contains several top-ranking biological processes related to cell cycle and DNA damage. The genes driving this pathway enrichment are listed in the table at the right. Statistical analysis of overlapping genes was performed using the hypergeometric test.
To investigate the functional roll of up-regulated cell cycle genes, we isolated mouse glioblastoma cells from bulk tumors and assayed cellular viability and growth in vitro. Cell doubling times of HoxA5-overexpressing versus control glioblastoma cells were 26.5 hr (HoxA5) versus 36.7 hr (mCherry; P = 0.0002) (Fig. 4A). HoxA5-overexpressing glioblastoma cells had a larger percentage (28.5%) of cells in S phase compared with controls (23.0% for mCherry; P < 0.0001) (Fig. 4B). Conversely, HoxA5 gene silencing had the opposite effect on cell doubling times; 59.8 hr (shHoxA5-1) and 38.2 hr (shHoxA5-2) versus 32.9 hr (shNT; nontarget control; P < 0.0001) (Fig. 4C; Supplemental Fig. 4). The percentage of cycling cells in S phase was decreased (P < 0.0001) with HoxA5 knockdown (11.6% shHoxA5-1 and 16.9% shHoxA5-2) compared with control nontarget shRNA (19.1%) (Fig. 4D). Consistent with the in vitro findings of growth and cell cycle analysis, immunohistochemistry confirmed that HoxA5 overexpression leads to a higher Ki-67 proliferative index in vivo for Ntva-Ink4a/Arf−/− (P = 0.007) and Ntva-Ink4a/Arf+/− (P = 0.002) mice (Fig. 4E). Last, the effect of HoxA5 expression on cellular proliferation was demonstrated through global gene expression analysis by RNA sequencing from bulk mouse glioblastoma tissue using gene set enrichment analysis (GSEA) with gene ontology (GO) terms. This approach resulted in the identification of gene expression sets in HoxA5-overexpressing tumors associated with biological processes linked to cell cycle regulation, such as “cell cycle,” “mitosis,” “DNA replication,” and “DNA metabolism” (Fig. 4F).
Figure 4.

HoxA5 promotes proliferation in glioblastoma cells. (A,B) Both cellular proliferation (A) and percentage of cells (B) in S phase increased over time with HoxA5 overexpression (as compared with control) in vitro in cultured mouse-derived glioblastoma cells. (C,D) Inversely, knockdown of HoxA5 by shRNA leads to decreased cell proliferation (C) and percentage of cells (D) in S phase in cultured mouse glioma cells. (E) The Ki-67 proliferative index (as determined by immunohistochemistry) was increased in HoxA5-overexpressing mouse gliomas in mice that had either homozygous or heterozygous background deletion of Ink4a/Arf. (F) GSEA from mouse glioma RNA demonstrates that HoxA5-overexpressing tumors are more enriched for GO gene sets corresponding to cell cycle and proliferation. Cellular proliferation assays underwent statistical analysis for genotype effect by two-way analysis of variance (ANOVA), and pairwise statistical analysis was performed using the Mann-Whitney U-test.
HoxA5 mediates resistance to ionizing irradiation
Ionizing irradiation is a mainstay in the standard of care for glioblastoma patients and preferentially induces apoptosis in fast cycling cells. In light of the role of HoxA5 for cellular proliferation and DNA damage response by gene expression microarray, we therefore sought to investigate whether HoxA5 expression influenced radiosensitivity. To analyze the impact of HoxA5 expression on radiation response in vivo, tumor-bearing mice underwent 10 Gy of irradiation at the time of symptomatology. HoxA5 overexpression in mouse glioblastomas was associated with decreased post-irradiation survival (P < 0.001), with a median survival of 22 d compared with a 33-d median survival in the mCherry control mice (Fig. 5A). In vitro, HoxA5-overexpressing mouse glioblastoma cells resumed proliferation earlier than tumor cells (7 d vs. 9 d; P = 0.032) (Fig. 5B). Clonal survival after irradiation was increased in HoxA5-overexpressing cells (Fig. 5C), indicating that resistance to irradiation-induced cell death contributed to earlier outgrowth. HoxA5 knockdown also decreased proliferation before (Fig. 5D) and after (Fig. 5E) irradiation in mouse glioblastoma-derived cells. To determine whether the delay in proliferation was due to the inherently higher proliferation rate of HoxA5-expressing cells or a differential to radiation response, a nonlinear regression of the proliferation data was performed and used to compare the times to reach half of the maximum proliferation (Supplemental Fig. 5). Radiation shifted the half-maximum proliferation from days 3, 5, and 7 to days 5, 8.5, and 11.5, resulting in proliferation delays of 2, 3.5, and 4.5 d for shNT (nontarget control), shHoxA5-1, and shHoxA5-2, respectively. After removing the effect of the inherently higher proliferation rate of HoxA5-expressing cells, HoxA5 knockdown still increased the time to reach the half-maximum after radiation by 1.5 and 2.5 d for shHoxA5-1 and shHoxA5-2, respectively. This finding suggests that HoxA5 expression allows cells to shorten the radiation-induced proliferation delay and may indicate that HoxA5 expression confers reduced sensitivity to radiation. Similar to mouse-derived glioblastoma cells, HOXA5 knockdown in U87 human glioblastoma cells had decreased proliferation after radiation (Fig. 5F). To investigate the sensitivity of U87 cells to irradiation in vitro after HoxA5 overexpression, H2AX phosphorylation (γ-H2AX) intensity was measured 2 h after irradiation by immunofluorescence. U87 cells with normal HoxA5 expression had a greater than twofold increase in y-H2AX staining after radiation, while HoxA5-overexpressing cells did not have any increased y-H2AX staining after radiation (Fig. 5G). Of note, HOXA5 overexpression induces an increased basal level of H2AX prior to radiation in mouse glioblastoma and human carcinoma cells (Supplemental Fig. 6; Ordonez-Moran et al. 2015). We also found that expression levels of the p21- and p53-activated proapoptotic genes BAX and PUMA in U87 cells were increased after radiation in U87 cells with normal HoxA5 expression (Fig. 5H). HoxA5-overexpressing U87 glioblastoma cells had increased PUMA and p21, but not BAX or p53, levels after radiation but to a far lesser extent than that of U87 cells with normal HoxA5 levels (Fig. 5H). These results suggest that cells overexpressing HoxA5 shut off DNA damage signaling pathways more rapidly following radiation and may be less likely to undergo apoptosis or maintain cell cycle arrest.
Figure 5.
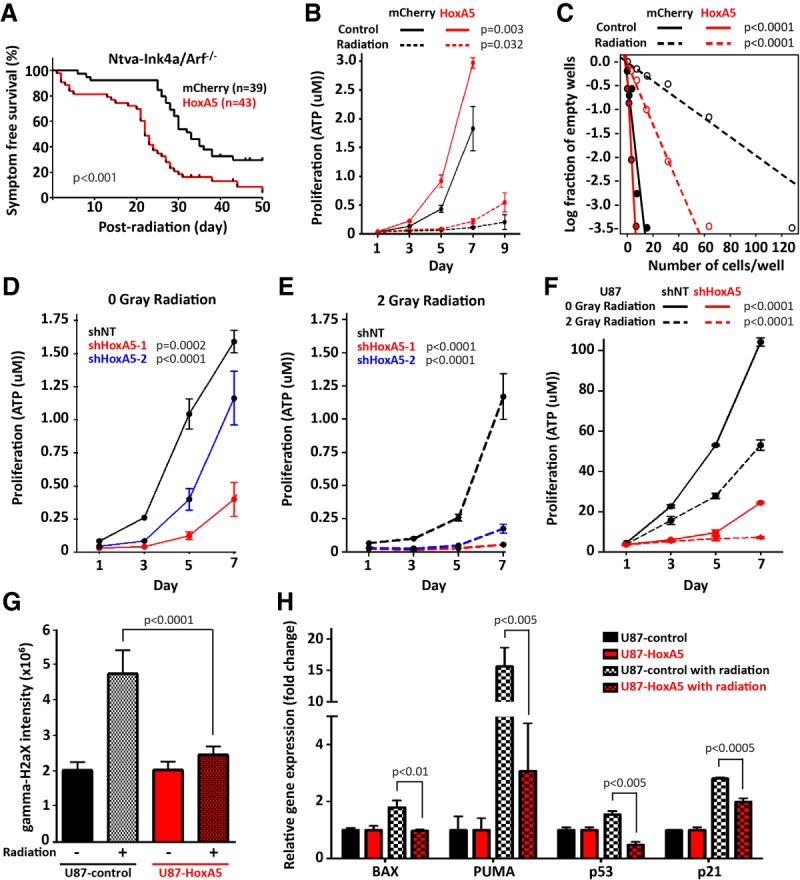
HoxA5 enhances radioresistance in glioblastoma cells. (A) Post-radiation survival is decreased for HoxA5-overexpressing glioblastomas in mice. (B) Proliferation in radiation-treated cultured mouse glioblastoma cells is increased with HoxA5 overexpression. (C) Limiting dilution clonal survival is increased with HoxA5 overexpression after radiation in glioblastoma cells. HoxA5 is associated with increased proliferation in vitro in mouse glioblastoma cells (D,E) and human U87 glioblastoma cells (F). (B–F) Cellular proliferation and clonality assays underwent statistical analysis for genotype effect by two-way ANOVA. (G) Radiation-induced DNA damage, as measured by γ-H2AX intensity, is inhibited by HOXA5 overexpression in human U87 glioblastoma cells. (H) HOXA5 mitigates radiation-induced up-regulation of cell cycle inhibitor- and apoptosis-related genes, including BAX, PUMA, p53, and p21 in human U87 glioblastoma cells. (G,H) Fluorescence intensity and gene expression assays underwent pairwise statistical analysis by the Mann-Whitney U-test.
HoxA5 expression is enriched in recurrent glioblastomas
Our data demonstrate that elevated HOXA5 expression is associated with increased cell proliferation and radioresistance in glioblastomas. These findings suggest that a subset of glioblastoma cells may be HOXA5-overexpressing and at least partially enriched in recurrent tumors due to therapeutic radioresistance. To investigate this observation further, we compared primary and recurrent post-radiation glioblastoma gene expression profiles for both mouse and human tumors. HoxA5 gene expression in standard PDGF-driven mouse glioblastomas (without RCAS-HoxA5 or RCAS-mCherry) was greater than twofold higher in post-radiation-treated recurrent mouse glioblastomas compared with primary tumors (P < 0.0001) (Fig. 6A). For the HoxA cluster overall, HoxA2, HoxA4, and HoxA5 were up-regulated in the recurrent mouse glioblastomas (Supplemental Fig. 7). To evaluate HoxA5 protein expression changes after radiation in vivo, immunohistochemistry of primary and recurrent mouse glioblastomas was performed to measure tumor cell percentages. The percentage of HoxA5-Myc-tagged-positive cells (41%) was approximately twofold higher in post-radiation recurrent tumors than primary mouse glioblastomas (22%) (Fig. 6B). These data indicate that radiation enriches for HoxA5-expressing cells in recurrent mouse glioblastomas.
Figure 6.
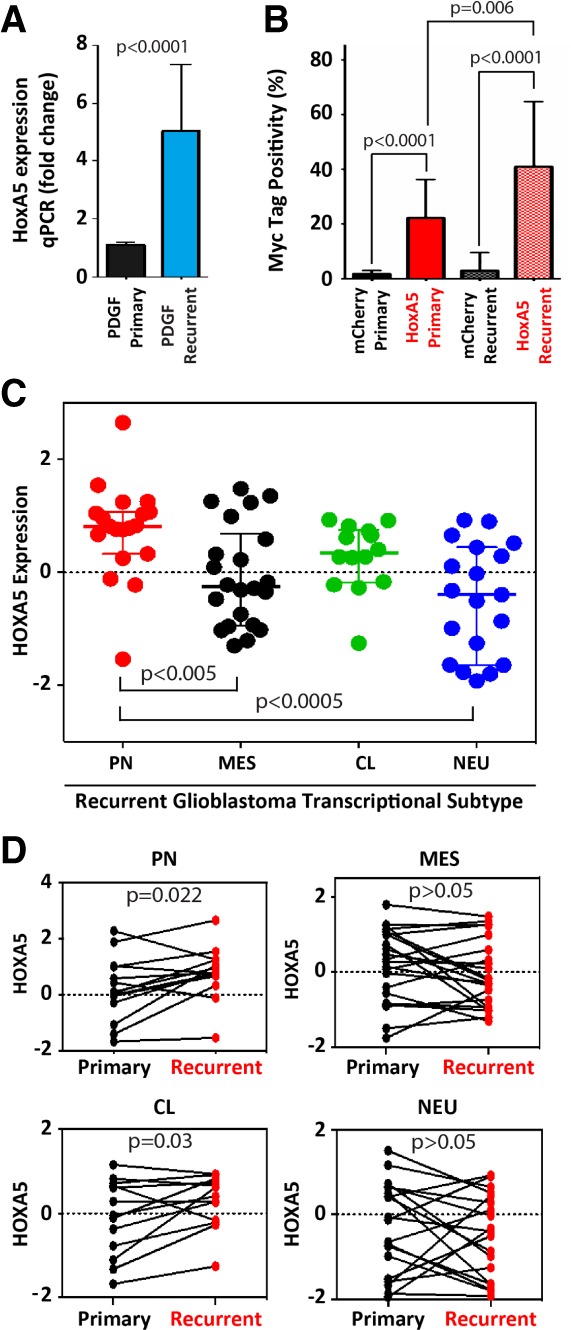
HoxA5 expression is enriched in mouse and human recurrent glioblastomas. (A) Post-radiation recurrent mouse PDGF-driven glioblastomas have approximately fivefold increased HoxA5 mRNA compared with primary glioblastomas. (B) Immunohistochemical quantification determines that Myc-tagged HoxA5, but not Myc-tagged mCherry, represents increased percentage of RCAS-infected cells in post-radiation recurrent mouse glioblastomas. (C) HOXA5 expression in recurrent human IDH wild-type glioblastomas is highest in the proneural (PN) transcriptional subtype (n = 19) compared with mesenchymal (MES; n = 22), classical (CL; n = 14), and neural (NEU; n = 19). (D) Recurrent human IDH wild-type glioblastomas exhibit HOXA5 mRNA enrichment in proneural and classical transcriptional subtypes compared with their matched primary tumors.
After determining that HoxA5 levels are increased in recurrent mouse glioblastomas, HOXA5 expression levels were also queried in recurrent human glioblastoma gene expression data (Wang et al. 2016). Consistent with our mouse results, the recurrent proneural subtype of IDH wild-type glioblastomas had higher levels of HOXA5 mRNA than the mesenchymal (P < 0.005) and neural (P < 0.0005) transcriptional subtypes (Fig. 6C). Furthermore, direct comparison of paired human primary and recurrent tumors determined that HOXA5 expression is enriched in recurrent proneural (P = 0.022) and classical (P = 0.03) transcriptional subtypes of glioblastomas (Fig. 6D).
Discussion
Whole-chromosome aneuploidy is a hallmark of cancer (Beroukhim et al. 2010), and, within IDH wild-type glioblastomas, gain of chromosome 7 is a frequent and early event. Stable aneuploidy implies a conferred selective advantage for having extra copies of chromosome 7 (Tang and Amon 2013). The number of genes located on chromosome 7 that drive its gain and selection has yet to be resolved. The main goal of this study was to identify genes whose expression may provide such a selection advantage for gain of whole chromosome 7. As a starting point, we took an unbiased bioinformatic approach to recognize HOXA5 overexpression as highly associated with gain of chromosome 7 and decreased overall survival in human proneural IDH wild-type glioblastomas. Proneural-like precursor cells give rise to all IDH wild-type transcriptional subtypes, and it is in these cells where gain of whole chromosome 7 occurs early in gliomagenesis. Genes on chromosome 7 that are regulated by mechanisms other than whole chromosome 7 copy number gain (i.e., extrachromosomal double minutes, pathway redundancy, etc.) would not have been expected to score highly in our initial screen. Such was the case for the known oncogenes on chromosome 7, including EGFR, MET, and CDK6. After establishing the relevance of increased HOXA5 expression in proneural glioblastomas, we went on to test the functional role by which HOXA5 overexpression might provide a selective advantage for gain of whole chromosome 7. The role of HOXA5 in promoting a more aggressive phenotype and selection in PDGF-driven glioblastomas was further supported through a combination of in vivo and in vitro studies that demonstrated HoxA5 overexpression causing increased cellular proliferation, cell cycle dysregulation, and radioresistance. Furthermore, HOXA5 expression was selected for in post-radiation human and mouse glioblastomas. In addition to having genes that confer increased tumor survival such as HOXA5, there are presumed genes on chromosome 7 whose overexpression could be detrimental to cellular fitness, and therefore their expression may be silenced by the neoplastic cells. Perhaps there is some amount of epigenetic suppression of RNF32 and C7orf13 (van Baren et al. 2002; Etcheverry et al. 2010) taking place in glioblastomas to counteract gain of chromosome 7. The reason and relevance of any such gene silencing on chromosome 7 have yet to be determined.
HOX genes encode for transcriptional factors that regulate cell fate decisions, and the dysregulation of expression of members of the HOX gene family initiates or supports tumorigenesis (Shah and Sukumar 2010; Bhatlekar et al. 2014). Examples of increased HOX expression in nonbrain neoplasms include acute myeloid leukemia, breast carcinoma, and prostate carcinoma. Elevated expression of HOX genes supports the immortalization of leukemic cells and formation of chimeric fusion genes (Armstrong et al. 2002; Ayton and Cleary 2003; Ghannam et al. 2004; Faber et al. 2009). HOXA and HOXB gene expression patterns in acute myeloid leukemia are regulated through epigenetic CpG island methylation (Spencer et al. 2015). In solid malignancies such as breast carcinoma, HOXA5 functions as a positive regulator of TP53, and loss of HOXA5 expression results in failure to block malignant transformation of epithelial cells (Raman et al. 2000; Chen et al. 2004). Similar findings of HOXA5 inducing apoptosis and a better clinical outcome have been observed in lung carcinoma (Chang et al. 2017) and liposarcoma (Lee et al. 2015). This previously described cooperative axis of HOXA5 and TP53 suppressing cellular proliferation in human carcinoma and liposarcoma is the inverse effect of what we observed in glioblastomas, where HOXA5 promotes cellular proliferation and inhibits TP53 expression. It is unclear why there is an apparent dichotomous effect of HOXA5 expression in glioblastomas compared with carcinomas and liposarcomas, but there are likely cell type-specific factors responsible for this behavior that have yet to be identified.
HOXA5 is located relatively centrally within the HOXA gene cluster, and it is unclear why HOXA5 in this study is specifically associated with gain of chromosome 7 and survival for proneural IDH wild-type glioblastomas. Given that the HOXA genes are colocated on chromosome 7, it remains unclear what the extent of involvement of HOXA genes other than HOXA5 is in glioblastoma biology. As a group, HOXA genes are overexpressed in glioblastomas compared with lower-grade astrocytomas and normal brain tissue (Abdel-Fattah et al. 2006; Costa et al. 2010). A subset of glioblastomas has increased HOXA transcription resultant from PI3K pathway-dependent regulation of H3K27me3 (Costa et al. 2010). HOXA9 was responsible for this phenotype, and HOXA9 overexpression in glioblastoma cells promoted proliferation and inhibited apoptosis. Increased HOXA9 expression was also associated with decreased progression-free and overall survival in two independent glioblastoma cohorts. However, these data are interpreted in the absence of knowing the IDH mutational status, so the relationship, if any, of HOXA9 to IDH status is unknown. A subgroup of glioblastomas with a transcriptional “stem cell signature” driven by HOX gene expression has been shown to have decreased survival following radiotherapy (Murat et al. 2008). Kurscheid et al. (2015) further showed that the HOX signature was associated with increased HOXA10 expression in glioblastomas and that these occurred in conjunction with complex epigenetic regulation and chromosome 7 gain. Gallo et al. (2013) provided additional insight into HOXA10 function in glioblastoma stem cells by showing that its expression is a direct result of activation of the Trithorax protein mixed-lineage leukemia (MLL) and that HOXA10 can then go on to directly activate other HOXA genes, such as HOXA7. Di Vinci et al. (2012) also demonstrated that methylation of HOXA9 and HOXA10 as wells as HOXA3 and HOXA7 increases in diffuse gliomas with increasing aggressiveness, as reflected by World Health Organization (WHO) grade. Outside of human tissue and cell line studies, one experimental investigation addressed several HoxA genes’ expression in a mouse glioma transplantation model (Schmid et al. 2016). Schmid et al. (2016) dedifferentiated cultured murine astrocytes into glioma stem cells through introduction of three mutations, including Rb knockout, Kras activation, and Pten deletion. These glioma stem cells were sufficient to form glioblastomas in their transplant mouse model. The dedifferentiation of those triple-mutant astrocytes resulted in an altered transcriptional profile, which had characteristic up-regulation of the HoxA locus (HoxA1–7, 9) through increased chromatin accessibility. Interestingly, of all of the up-regulated HoxA genes, HoxA5 had the largest magnitude of up-regulation with dedifferentiation. At this point, the magnitude of the role of HOXA genes in glioblastomas is not fully understood.
Because IDH wild-type glioblastomas have such a dismal prognosis, there is a need for increased targeted therapies for a precision medicine approach to this disease. Prospects for glioblastomas will likely include targeted therapies against molecular drivers of aggressive tumor behavior with respect to therapy resistance of the standard glioblastoma regimen of surgery, chemotherapy, and radiotherapy. Given the current data, it is possible that targeting HOXA genes may increase response to radiation therapy for glioblastomas and warrants further investigation.
Materials and methods
Human glioblastomas and gene-survival analysis
Gene expression data (level 3; n = 585; Agilent, G4502A) and DNA copy number GISTICS2 results (level 4; n = 571; Affymetrix Genome-Wide Human SNP srray 6.0) of glioblastoma samples from TCGA were used for analysis. Glioblastoma methylation and transcriptional subtype classifications (G-CIMP, classical, mesenchymal, neural, and proneural) and patient survival data were also obtained (Brennan et al. 2013). All analyses were done in R/Bioconductor (Gentleman et al. 2004). Transcriptional and clinical data for paired initial and recurrent glioblastomas were analyzed from a previously published glioblastoma cohort (Wang et al. 2016).
Proneural gene ranking and methylation promoter analysis
Selected genes on chromosome 7 were ranked by their association with overall survival and differences in expression by copy number for patients in the proneural subgroup as follows. Patients were divided into two groups: (1) glioblastoma diploid for chromosome 7 and (2) glioblastoma with gain of chromosome 7. Differential expression analysis was performed using Wilcoxon test for all genes on chromosome 7. For each gene, patients were sorted by gene expression, and the top and bottom 25% were selected to represent high and low gene expression subgroups. Cox proportional hazards regression was used to test for survival differences between the high and low gene expression subgroups for each gene. DNA promoter methylation levels were compared between the two groups separated by chromosome 7 copy number. In these analyses, the transcription start site ±1.5-kb regions were set as gene promoter regions.
HoxA5 RCAS vector construction
To prepare retroviral constructs for HoxA5 overexpression, pBABE-Puro retroviral vector (Cell Biolab, Inc.) and HoxA5 mouse cDNA (Open Biosystems) were used. HoxA5 cDNA was amplified using a forward primer containing a Flag tag sequence and a BamHI restriction site and a reverse primer containing a SalI restriction site. The amplified cDNA and the pBABE-Puro vector were incubated with the restriction enzymes BamHI and SalI, followed by gel purification. The HoxA5 cDNA and linear vector were ligated using T4 DNA ligase (New England Biolabs). Retroviral particles were generated by cotransfection of the pBABE HoxA5 vector and the pCL-10A1 retrovirus packaging vector. A forward primer with Flag tag or Myc tag sequences and BamHI restriction site and the reverse primer with a NotI restriction site were used to prepare the HoxA5 cDNA insert. The amplified cDNA and pENTR vector were digested using BamHI and NotI restriction enzymes and then ligated using T4 DNA ligase. The pENTR vector with the HoxA5 insert was subjected to Gateway LR ligation. After the HoxA5 insert was shuttled to the RCAS destination vector (RCAS-DV) using LR ligation, RCAS-HoxA5 was amplified.
Mouse glioma generation
Animal use was conducted with approval by the Institutional Animal Care and Use Committee of Fred Hutchinson Cancer Research Center, protocol 50842. To generate mCherry- and HoxA5-expressing gliomas, the RCAS/tva system was used as described previously (Holland et al. 1998; Holland and Varmus 1998; Shih and Holland 2006; Ozawa et al. 2014). Briefly, chicken fibroblast (DF1) cells were maintained with 10% fetal bovine serum (FBS) in Dulbecco's modified Eagle medium (DMEM). DF1 cells were transfected with each RCAS viral plasmid using Fugene 6 transfection reagent (Roche) following the manufacturer's protocol. Next, PDGFb-expressing DF1 cells were mixed with either mCherry- or HoxA5-expressing DF1 cells. This mixture of DF1 cells was injected into Ntva-Ink4a/Arf+/− and Ntva- Ink4a/Arf−/− mice. Adult mice anesthetized with isoflurane were positioned in a stereotactic device (Stoelting). RCAS transfected DF1 cells were injected using a 30-gauge needle inserted into the right frontal cortex (coordinates: 1.5 mm anterior to bregma, 0.5 mm lateral, and 1.5-mm depth). Next, mice were monitored daily until they developed signs of illness, such as lethargy, poor grooming, weight loss, dehydration, macrocephaly, seizure, jumping, and paralysis.
Mouse glioma irradiation
Following development of symptoms, DF1-injected mice were sedated with isoflurane, and their heads were irradiated with 10 Gy in one fraction using an X-RAD 320 from Precision X-Ray. The rest of the mouse was shielded with lead to minimize the radiation dose to normal tissue. Animals were sacrificed upon recurrence of neurological symptoms, as defined by the Institutional Animal Care and Use Committee. Any mice euthanized due to spontaneous tumors were removed from this experiment. Each survival arm was sufficiently powered to account for any baseline variability in response.
Immunohistochemistry
Tumor-bearing mouse brains were removed, fixed in 10% neutral-buffered formalin, and then embedded into paraffin blocks. The formalin-fixed paraffin-embedded (FFPE) tissue was serially sectioned 5 µm in depth and slide-mounted. Automated stain processing (Discovery, Ventana Medical Systems, Inc.) was used for immunohistochemical detection with the manufacturer's standard protocol. The following primary antibodies were used: Ki-67 (1:200, rabbit; Vector Laboratories, VP-RM04), Myc tag (1:500, rabbit; Cell Signaling Technology, 2272), and γ-H2A.X (Ser139) (1:500, rabbit; Cell Signaling Technology, 9718). For immunohistochemical staining quantification, tissue sections were imaged on a TissueFax slide scanner, and HistoQuest image analysis software was used to identify and count marker-positive and marker-negative cells (TissueGnostics GmbH). The number of marker-positive cells was divided by the total number of cells in each tumor region.
Glioma cell culture
Human U87 glioblastoma cells were maintained using 10% FBS (Invitrogen) in DMEM (American Type Culture Collection). Mouse glioma cells were derived from primary mouse tumors by tissue dissociation and single-cell isolation using papain enzyme (Leder et al. 2014). Mouse glioma cells were maintained in serum-free Neurocult NSC basal medium (mouse; Stem Cell Technologies) supplemented with 20 ng/mL EGF, 20 ng/mL bFGF, and heparin. Cell culture proliferation was assayed as described previously (Li et al. 2009; Lathia et al. 2010). Cells were plated in triplicate for each condition in 96-well culture dishes. ATP content was measured using CellTiter-Glo bioluminescence (Promega Corporation). For the in vitro limiting dilution assay, various dilutions of cells ranging from 400 cells to a single cell per well were plated into 96-well plates (Ploemacher et al. 1989). Ten days after plating, the number of wells without spheres was counted and analyzed using ELDA (http://bioinf.wehi.edu.au/software/elda).
Cell cycle analysis
The Click-iT EdU Alexa fluor 488 imaging kit (Invitrogen, C10337) was used for the cell cycle analysis using the manufacturer's protocol. Briefly, after cells were pretreated with retrovirus or lentivirus and puromycin, EdU was added, and cells were incubated for 30 min. Next, cells were trypsinized for a single-cell suspension and then fixed with 4% paraformaldehyde (PFA) for the Click-it Edu assay. Prepared cells were analyzed using fluorescence-activated cell sorting (FACS).
HoxA5 shRNA preparation
Lentiviral clones expressing human or mouse HoxA5-specific shRNAs (shHoxA5-1 for mice: 5′-CGCCGAAGAAGGATCGAAATA-3′; shHoxA5-2 for mice and humans: 5′-CCGGACTACCAGTTGCATAAT-3′) and control shNT (SHC002) were purchased from Sigma-Aldrich. Viral particles were produced in 293T cells with the pPACK set of helper plasmids (System Biosciences) in stem cell medium. Viral stocks were concentrated by precipitation with PEG-8000.
Western blotting
Western blots were performed as described previously (Ozawa et al. 2014). The following primary antibodies were used: β-Actin (1:2000, mouse; Sigma-Aldrich, A1978), α-tubulin (1:2000, mouse; Sigma-Aldrich, T5168), HA tag (1:1000, rabbit; Cell Signaling Technology, 3724), and anti-Flag (1:1000, rabbit; Sigma-Aldrich, F7425).
Quantitative RT–PCR
Total RNA was extracted from tumor tissue using RNeasy minikit (Qiagen) and was used to synthesize cDNA by using the SuperScript III first strand synthesis system for RT–PCR (Invitrogen) according to the manufacturer's protocol. SYBR Green real-time PCR was performed using primer sets, reagents, and protocols from Applied Biosystems in a 7900 HT Fast real-time PCR system (Applied Biosystems). Each sample was analyzed in triplicate. RT–PCR primer sequences are listed in Supplemental Table 2.
Mouse glioblastoma gene expression microarray
Total high-quality RNA was prepared and converted to biotin-labeled cDNA as described previously (Amankulor et al. 2017). Labeled cDNA was processed on a MouseWG-6v2 Expression BeadChip (Illumina, Inc.) and imaged using an Illumina iScan system. The Bioconductor package “lumi” was used to assess microarray data quality followed by quantile normalization (Du et al. 2008). Gene expression was initially filtered by retaining only probes above the signal “noise floor” threshold (established using the 75th percentile of the negative control probe signals within each array). The data set was subsequently filtered by using a variance filter using the “shorth” function of the Bioconductor package “genefilter” (Bourgon et al. 2010). Statistical analyses were performed using the Bioconductor package “limma” (Smyth et al. 2005), and a FDR method was applied to correct for multiple testing (Reiner et al. 2003). Differential expression was defined as log2 (ratio) ≥0.585 (±1.5-fold) with the FDR set to 5%. Microarray data are available at https://www.ncbi.nlm.nih.gov/gds as Gene Expression Omnibus (GEO) data set GSE89409. Mouse glioblastoma microarray results were further compared with gene expression of HOXA5-overexpressing human carcinoma cells (GEO data set GSE74862) and HOXA5 ChIP-seq of human carcinoma cells (GEO data set SM1239461) (Yan et al. 2013; Ordonez-Moran et al. 2015). Pathway enrichment analysis was done using R Bioconducter package “clusterProfiler” version 3.4.4 (Yu et al. 2012), and dotplots were made using R Bioconducter package DOSE (Yu et al. 2015).
Statistical analysis
Kaplan-Meier survival curves were prepared using Graphpad Prism 7 (Graphpad Software) and analyzed using the log-rank (Mantel-Cox) test. Time series data were analyzed in Graphpad Prism 7 using two-way analysis of variance (ANOVA) as designated. Microarray and ChIP-seq data sets were compared using the hypergeometric test in R (version 3.3.2, R Project for Statistical Computing, http://www.r-project.org). All other comparisons were determined by performing the Mann-Whitney U-test in Graphpad Prism 7.
Supplementary Material
Acknowledgments
Research reported in this work was supported by the National Cancer Institute (NCI) of the National Institutes of Health (NIH) under award numbers T32CA009657, U54CA193461, and U54CA193313. H.-G.W. was supported by a post-doctorate mobility grant of the Swiss National Science Foundation.
Author contributions: The study was conceived and designed by F.M., Y.K., and E.C.H. Acquisition of data was performed by P.J.C., Y.K., H.-J.W., J.A., H.-G.W., F.S., K.P., T.O., J.W., J.V., S.A., R.R., and R.L. Data were analyzed and interpreted by P.J.C., Y.K., H.-J.W., S.A., R.R., R.L., F.M., and E.C.H. Drafting of the manuscript was completed by P.J.C. and E.C.H. Critical review of the manuscript was additionally performed by F.M.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.312157.118.
References
- Abdel-Fattah R, Xiao A, Bomgardner D, Pease CS, Lopes MB, Hussaini IM. 2006. Differential expression of HOX genes in neoplastic and non-neoplastic human astrocytes. J Pathol 209: 15–24. [DOI] [PubMed] [Google Scholar]
- Amankulor NM, Kim Y, Arora S, Kargl J, Szulzewsky F, Hanke M, Margineantu DH, Rao A, Bolouri H, Delrow J, et al. 2017. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev 31: 774–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong SA, Staunton JE, Silverman LB, Pieters R, den Boer ML, Minden MD, Sallan SE, Lander ES, Golub TR, Korsmeyer SJ. 2002. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet 30: 41–47. [DOI] [PubMed] [Google Scholar]
- Ayton PM, Cleary ML. 2003. Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9. Genes Dev 17: 2298–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. 2010. The landscape of somatic copy-number alteration across human cancers. Nature 463: 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatlekar S, Fields JZ, Boman BM. 2014. HOX genes and their role in the development of human cancers. J Mol Med (Berl) 92: 811–823. [DOI] [PubMed] [Google Scholar]
- Bleeker FE, Atai NA, Lamba S, Jonker A, Rijkeboer D, Bosch KS, Tigchelaar W, Troost D, Vandertop WP, Bardelli A, et al. 2010. The prognostic IDH1(R132) mutation is associated with reduced NADP+-dependent IDH activity in glioblastoma. Acta Neuropathol 119: 487–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgon R, Gentleman R, Huber W. 2010. Independent filtering increases detection power for high-throughput experiments. Proc Natl Acad Sci 107: 9546–9551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. 2013. The somatic genomic landscape of glioblastoma. Cell 155: 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. 2008. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455: 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. 2015. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med 372: 2481–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, Morozova O, Newton Y, Radenbaugh A, Pagnotta SM, et al. 2016. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 164: 550–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CJ, Chen YL, Hsieh CH, Liu YJ, Yu SL, Chen JJW, Wang CC. 2017. HOXA5 and p53 cooperate to suppress lung cancer cell invasion and serve as good prognostic factors in non-small cell lung cancer. J Cancer 8: 1071–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Chung S, Sukumar S. 2004. HOXA5-induced apoptosis in breast cancer cells is mediated by caspases 2 and 8. Mol Cell Biol 24: 924–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimino PJ, Zager M, McFerrin L, Wirsching HG, Bolouri H, Hentschel B, von Deimling A, Jones D, Reifenberger G, Weller M, et al. 2017. Multidimensional scaling of diffuse gliomas: application to the 2016 World Health Organization classification system with prognostically relevant molecular subtype discovery. Acta Neuropathol Commun 5: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa BM, Smith JS, Chen Y, Chen J, Phillips HS, Aldape KD, Zardo G, Nigro J, James CD, Fridlyand J, et al. 2010. Reversing HOXA9 oncogene activation by PI3K inhibition: epigenetic mechanism and prognostic significance in human glioblastoma. Cancer Res 70: 453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Vinci A, Casciano I, Marasco E, Banelli B, Ravetti GL, Borzi L, Brigati C, Forlani A, Dorcaratto A, Allemanni G, et al. 2012. Quantitative methylation analysis of HOXA3, 7, 9, and 10 genes in glioma: association with tumor WHO grade and clinical outcome. J Cancer Res Clin Oncol 138: 35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du P, Kibbe WA, Lin SM. 2008. lumi: a pipeline for processing Illumina microarray. Bioinformatics 24: 1547–1548. [DOI] [PubMed] [Google Scholar]
- Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H, Pekmezci M, Rice T, Kosel ML, Smirnov IV, et al. 2015. Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N Engl J Med 372: 2499–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etcheverry A, Aubry M, de Tayrac M, Vauleon E, Boniface R, Guenot F, Saikali S, Hamlat A, Riffaud L, Menei P, et al. 2010. DNA methylation in glioblastoma: impact on gene expression and clinical outcome. BMC Genomics 11: 701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber J, Krivtsov AV, Stubbs MC, Wright R, Davis TN, van den Heuvel-Eibrink M, Zwaan CM, Kung AL, Armstrong SA. 2009. HOXA9 is required for survival in human MLL-rearranged acute leukemias. Blood 113: 2375–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo M, Ho J, Coutinho FJ, Vanner R, Lee L, Head R, Ling EK, Clarke ID, Dirks PB. 2013. A tumorigenic MLL-homeobox network in human glioblastoma stem cells. Cancer Res 73: 417–427. [DOI] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. 2004. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5: R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghannam G, Takeda A, Camarata T, Moore MA, Viale A, Yaseen NR. 2004. The oncogene Nup98-HOXA9 induces gene transcription in myeloid cells. J Biol Chem 279: 866–875. [DOI] [PubMed] [Google Scholar]
- Holland EC, Varmus HE. 1998. Basic fibroblast growth factor induces cell migration and proliferation after glia-specific gene transfer in mice. Proc Natl Acad Sci 95: 1218–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland EC, Hively WP, DePinho RA, Varmus HE. 1998. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev 12: 3675–3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland PW, Booth HA, Bruford EA. 2007. Classification and nomenclature of all human homeobox genes. BMC Biol 5: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurscheid S, Bady P, Sciuscio D, Samarzija I, Shay T, Vassallo I, Criekinge WV, Daniel RT, van den Bent MJ, Marosi C, et al. 2015. Chromosome 7 gain and DNA hypermethylation at the HOXA10 locus are associated with expression of a stem cell related HOX-signature in glioblastoma. Genome Biol 16: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathia JD, Gallagher J, Heddleston JM, Wang J, Eyler CE, Macswords J, Wu Q, Vasanji A, McLendon RE, Hjelmeland AB, et al. 2010. Integrin α6 regulates glioblastoma stem cells. Cell Stem Cell 6: 421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leder K, Pitter K, LaPlant Q, Hambardzumyan D, Ross BD, Chan TA, Holland EC, Michor F. 2014. Mathematical modeling of PDGF-driven glioblastoma reveals optimized radiation dosing schedules. Cell 156: 603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, Forscher C, Di Vizio D, Koeffler HP. 2015. Induction of p53-independent apoptosis by ectopic expression of HOXA5 in human liposarcomas. Sci Rep 5: 12580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeper HE, Caron AA, Decker PA, Jenkins RB, Lachance DH, Giannini C. 2015. IDH mutation, 1p19q codeletion and ATRX loss in WHO grade II gliomas. Oncotarget 6: 30295–30305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, Shi Q, Cao Y, Lathia J, McLendon RE, et al. 2009. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 15: 501–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. 2016. WHO classification of tumours of the central nervous system. International Agency for Research on Cancer, Lyon, France. [Google Scholar]
- Murat A, Migliavacca E, Gorlia T, Lambiv WL, Shay T, Hamou MF, de Tribolet N, Regli L, Wick W, Kouwenhoven MC, et al. 2008. Stem cell-related ‘self-renewal’ signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J Clin Oncol 26: 3015–3024. [DOI] [PubMed] [Google Scholar]
- Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, et al. 2010. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17: 510–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olar A, Wani KM, Alfaro-Munoz KD, Heathcock LE, van Thuijl HF, Gilbert MR, Armstrong TS, Sulman EP, Cahill DP, Vera-Bolanos E, et al. 2015. IDH mutation status and role of WHO grade and mitotic index in overall survival in grade II–III diffuse gliomas. Acta Neuropathol 129: 585–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordonez-Moran P, Dafflon C, Imajo M, Nishida E, Huelsken J. 2015. HOXA5 counteracts stem cell traits by inhibiting Wnt signaling in colorectal cancer. Cancer Cell 28: 815–829. [DOI] [PubMed] [Google Scholar]
- Ostrom QT, Gittleman H, Liao P, Vecchione-Koval T, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS. 2017. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro Oncol 19: v1–v88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa T, Riester M, Cheng YK, Huse JT, Squatrito M, Helmy K, Charles N, Michor F, Holland EC. 2014. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell 26: 288–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploemacher RE, van der Sluijs JP, Voerman JS, Brons NH. 1989. An in vitro limiting-dilution assay of long-term repopulating hematopoietic stem cells in the mouse. Blood 74: 2755–2763. [PubMed] [Google Scholar]
- Raman V, Martensen SA, Reisman D, Evron E, Odenwald WF, Jaffee E, Marks J, Sukumar S. 2000. Compromised HOXA5 function can limit p53 expression in human breast tumours. Nature 405: 974–978. [DOI] [PubMed] [Google Scholar]
- Reiner A, Yekutieli D, Benjamini Y. 2003. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics 19: 368–375. [DOI] [PubMed] [Google Scholar]
- Reuss DE, Mamatjan Y, Schrimpf D, Capper D, Hovestadt V, Kratz A, Sahm F, Koelsche C, Korshunov A, Olar A, et al. 2015. IDH mutant diffuse and anaplastic astrocytomas have similar age at presentation and little difference in survival: a grading problem for WHO. Acta Neuropathol 129: 867–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid RS, Simon JM, Vitucci M, McNeill RS, Bash RE, Werneke AM, Huey L, White KK, Ewend MG, Wu J, et al. 2016. Core pathway mutations induce de-differentiation of murine astrocytes into glioblastoma stem cells that are sensitive to radiation but resistant to temozolomide. Neuro Oncol 18: 962–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah N, Sukumar S. 2010. The Hox genes and their roles in oncogenesis. Nat Rev Cancer 10: 361–371. [DOI] [PubMed] [Google Scholar]
- Shih AH, Holland EC. 2006. Platelet-derived growth factor (PDGF) and glial tumorigenesis. Cancer Lett 232: 139–147. [DOI] [PubMed] [Google Scholar]
- Smyth GK, Michaud J, Scott HS. 2005. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics 21: 2067–2075. [DOI] [PubMed] [Google Scholar]
- Spencer DH, Young MA, Lamprecht TL, Helton NM, Fulton R, O'Laughlin M, Fronick C, Magrini V, Demeter RT, Miller CA, et al. 2015. Epigenomic analysis of the HOX gene loci reveals mechanisms that may control canonical expression patterns in AML and normal hematopoietic cells. Leukemia 29: 1279–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, Pfaff E, Tonjes M, Sill M, Bender S, et al. 2012. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22: 425–437. [DOI] [PubMed] [Google Scholar]
- Tang YC, Amon A. 2013. Gene copy-number alterations: a cost-benefit analysis. Cell 152: 394–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Baren MJ, van der Linde HC, Breedveld GJ, Baarends WM, Rizzu P, de Graaff E, Oostra BA, Heutink P. 2002. A double RING-H2 domain in RNF32, a gene expressed during sperm formation. Biochem Biophys Res Commun 292: 58–65. [DOI] [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. 2010. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17: 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Cazzato E, Ladewig E, Frattini V, Rosenbloom DI, Zairis S, Abate F, Liu Z, Elliott O, Shin YJ, et al. 2016. Clonal evolution of glioblastoma under therapy. Nat Genet 48: 768–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller M, Weber RG, Willscher E, Riehmer V, Hentschel B, Kreuz M, Felsberg J, Beyer U, Loffler-Wirth H, Kaulich K, et al. 2015. Molecular classification of diffuse cerebral WHO grade II/III gliomas using genome- and transcriptome-wide profiling improves stratification of prognostically distinct patient groups. Acta Neuropathol 129: 679–693. [DOI] [PubMed] [Google Scholar]
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, et al. 2009. IDH1 and IDH2 mutations in gliomas. N Engl J Med 360: 765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Enge M, Whitington T, Dave K, Liu J, Sur I, Schmierer B, Jolma A, Kivioja T, Taipale M, et al. 2013. Transcription factor binding in human cells occurs in dense clusters formed around cohesin anchor sites. Cell 154: 801–813. [DOI] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, He QY. 2012. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16: 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, Yan GR, He QY. 2015. DOSE: an R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics 31: 608–609. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.