

Abstract
Prp8 is an essential protein that regulates spliceosome assembly and conformation during pre-mRNA splicing. Recent cryo-EM structures of the spliceosome model Prp8 as a scaffold for the spliceosome's catalytic U snRNA components. Using a new amino acid probing strategy, we identified a dynamic region in human Prp8 that is positioned to stabilize the pre-mRNA in the spliceosome active site through interactions with U5 snRNA. Mutagenesis of the identified Prp8 residues in yeast indicates a role in 5′ splice site recognition. Genetic interactions with spliceosome proteins Isy1, which buttresses the intron branch point, and Snu114, a regulatory GTPase that directly contacts Prp8, further corroborate a role for the same Prp8 residues in substrate positioning and activation. Together the data suggest that adjustments in interactions between Prp8 and U5 snRNA help establish proper positioning of the pre-mRNA into the active site to enhance 5′ splice site fidelity.
Keywords: pre-mRNA splicing, spliceosome, Prp8, U5 snRNA, 5′ exon, chemical probing
INTRODUCTION
Pre-mRNA splicing is an essential step of eukaryotic gene expression, which results in the removal of intron sequences from pre-mRNA transcripts. This RNA processing event is orchestrated by a dynamic ribonucleoprotein (RNP) complex named the spliceosome. During spliceosome assembly, specific landmark sequences within introns must be properly recognized in order to distinguish intron sequences from coding exon sequences. Following early assembly, the spliceosome structures its active site in a process referred to as activation. Activation allows first step chemistry of splicing to proceed, in which the spliceosome severs the 5′ splice site while simultaneously generating an intron lariat intermediate. This is followed by second step chemistry, where the 3′ splice site is cleaved concomitant with exon ligation.
Recent cryo-electron microscopy (cryo-EM) structures of the spliceosome provide an unprecedented view of the organization of spliceosome components before and after active site formation (Galej et al. 2016; Bertram et al. 2017a,b; Zhang et al. 2017). Differences between the stable conformations represented by the cryo-EM models indicate that a number of large and small structural rearrangements are required to generate the catalytic core of the spliceosome, which is primarily composed of three uridine-rich small nuclear RNAs (U snRNAs). Specifically, U2 snRNA positions the intron branch point, U6 snRNA holds the 5′ end of the intron and catalytic metals, and U5 snRNA interacts with the 5′ exon. These snRNAs are all supported by the large scaffold protein Prp8. Prp8 also interacts with several proteins that are known to regulate different steps of spliceosome assembly and catalysis, placing it at the nexus of structural rearrangements that allows the spliceosome to specifically position splice site consensus sequences at intron boundaries with high fidelity (Umen and Guthrie 1996; Liu et al. 2007; Mayerle and Guthrie 2016). Changes in Prp8 structure correlate with spliceosome progression, but additional biochemical analysis is required to delineate which Prp8 interactions are critical for snRNA active site formation and pre-mRNA splice site identification. Using chemical probing, we have identified a dynamic interaction between Prp8 and U5 snRNA, and functionally link the residues involved to specific splicing factors in yeast. Combined, our data serve as a platform for additional mechanistic interpretation of spliceosome structural features.
RESULTS AND DISCUSSION
Two-stage chemical probing pinpoints structural changes between spliceosome conformations
In this study, we sought to investigate interactions that regulate spliceosome transitions involved in activation and catalysis by identifying changes in solvent accessibility of lysine side chains. Because of their positive charge, lysine side chains often play important structural roles in RNPs by helping stabilize specific conformations of the phosphate backbone of RNA. We devised a new two-stage amino acid probing strategy combined with tandem-mass spectrometry (MS/MS) to assess the reactivity of lysine side chains of the proteins in spliceosome complexes (Fig. 1A). First, we acetylated primary amines of lysine residues that are solvent accessible and reactive in affinity-purified spliceosomes assembled in vitro. We then denatured proteins via SDS-PAGE and modified the remaining nonacetylated lysine residues with a deuterated acetylation reagent. The additional mass units of the deutero-acetyl group are sufficient to differentiate the modification state as acetyl or deutero-acetyl by MS/MS (Fig. 1B,C). The second modification step is important because trypsin cleaves after arginine and lysine residues, but not after acetyl-lysine. By ensuring complete acetylation of all lysines, the two-stage modification strategy guarantees that trypsin digestion will generate comparable peptides from spliceosomes in different conformations for MS/MS analysis. Accordingly, nearly all peptides from spliceosome proteins that we sequenced by MS/MS ended in arginine (Fig. 1D). Furthermore, we found the majority of individual lysine residues have the same acetylation state when comparing replicate samples of two different spliceosome assembly states, indicating that the modification patterns are reproducible across multiple preparations (Fig. 1E).
FIGURE 1.

Two-step lysine probing of spliceosomes. (A) Schematic of probing strategy: First lysines in native spliceosomes are modified by acetylation (yellow stars), and then following denaturation are modified with a deutero-acetylating reagent (cyan stars). (B) Sample MS spectra containing two doubly charged peptides of m/z 555.37 and m/z 556.88 corresponding to the peptide VLIGVGKLLR from splicing factor 3B subunit 3. In the m/z 555.37 version, the lysine is modified with an acetyl group, whereas in the m/z 556.88 version, the lysine is modified with a deuteron-acetyl group, leading to a 3 Dalton mass difference. (C) Theoretical modeling of the predicted isotope pattern for the lighter version using MS-Isotope (Chalkley et al. 2005) indicates a fourth isotope peak would contribute 5% peak intensity to the first isotope of the deuterated monoisotopic peak signal, and have an insignificant effect on overall quantification. (D) Frequency of C-terminal amino acid identity for all unique MS/MS measured peptides from precatalytic and catalytic spliceosomes. (E) Frequency of individual lysines identified with the same, mixed, or opposite modification state between replicate samples of preactivated and catalytic spliceosomes.
We applied the probing method to purified human preactivated spliceosomes containing all five snRNPs (B-like complex) (Bertram et al. 2017a) and human spliceosomes captured after the first step of catalysis (C/C* complex) (Jurica et al. 2002) to assess structural differences before and after the RNA catalytic core is established (Fig. 2A; Supplemental Fig. 1). We interpreted lysines modified with an acetyl group as being in a solvent accessible position in native spliceosomes. In contrast, lysines modified with a deutero-acetyl group were presumably protected during the first round of chemical probing by molecular interactions. Although the spliceosome undergoes significant gain and loss of proteins along the transition from the preactivation state (B complex) through activation (Bact complex) to the catalytic states (C and C* complexes), we were able to compare the modification state of 91 lysine residues from twelve proteins identified in both samples. Over 70% of these lysines maintained the same modification state, while 17% showed a distinct change. Modification of the remaining portion was variable among replicates.
FIGURE 2.

Prp8 lysine residues surrounding U5 snRNA change accessibility between preactivation and catalysis. (A) Schematic of spliceosome assembly highlighting involvement of U snRNPs in the formation of the catalytic core during activation, and the two steps of splicing chemistry. (B, left) Schematic of Prp8 domain structure and relative positions of four differentially modified lysine residues that spatially located near U5 snRNA in the spliceosome. RT/EN indicates the large reverse transcriptase/endonuclease domain. (Right) Secondary structure of U5 snRNA highlighting SLI interaction with the 5′ exon of pre-mRNA. Red indicates the region of U5 snRNA shown in C. (C) Position of Prp8 (gray) side chains K595, K597, K1300, and K1306 (cyan) interactions with U5 SLI (blue) contacting the 5′ exon (gold) U5 SLI heteroatoms are colored red. (D) View of Prp8 K595 and K597 in close proximity to the phosphate backbone of U5 SLI. Distances (Å) from lysine terminal nitrogens and nonbridging oxygens of phosphates of U5 snRNA C45, U46, and A30 are indicated by the dotted red lines. Panels C and D are derived from the cryo-EM model of the human catalytic (C*) spliceosome (Zhang et al. 2017).
Aiming to relate changes in lysine reactivity to structure, we examined the location of differentially modified lysine residues shared in the two available cryo-EM models of human spliceosomes in a preactivation state (B complex [Bertram et al. 2017a]) and a catalytic state remodeled for exon ligation (C* complex [Zhang et al. 2017]). Based on the presence of both Isy1, a C complex factor, and Prp22, a C* complex factor, in the MS/MS peptides, the catalytic complex that we purified likely represents a mixture of spliceosomes in both catalytic states. The lysine residues are from U5 snRNP proteins that remain with the spliceosome from preactivation through catalysis: seven in Prp8 and one each in EFTUD2 and SNRNP40. Interestingly, they all were identified as solvent accessible in preactivated spliceosomes, and as protected in catalytic spliceosomes. The change in reactivity suggests that side chains acquire an interaction partner at some point between the preactivation state and splicing catalysis. The cryo-EM structures clearly bear out this prediction for Prp8 residues K68, which is located in interaction interfaces that Prp8 acquires with Bud31 during activation. Likewise, SNRNP40 residue K286 sits in an interaction interface with Prp8, which is present in the catalytic C* spliceosome, but not the preactivation B complex.
The structural basis for the change in modification state for the five other differentially modified lysine side chains is not as clear-cut. Four conserved Prp8 lysines, K595, K597, K1300, and K1306, are notable in that they are located within 16 Å of each other in the region of Prp8 surrounding stem loop I of U5 snRNA (U5 SLI) as it interacts with the 5′ exon of pre-mRNA (Fig. 2B,C; Supplemental Fig. 2). Consistent with our interpretation of the probing data, the four lysine residues appear in contact with RNA or protein in the cryo-EM reconstruction of the human catalytic spliceosome (C* complex [Zhang et al. 2017]), which includes side chains in the Prp8 model. Residues K595 and K597 are situated in a turn between two α helices in the amino-terminal domain of Prp8, and in position to stabilize a highly negative groove created by a bend in U5 SLI toward the 5′ splice site (Fig. 2D). In the C* spliceosome structure the terminal nitrogen of K595 is modeled as 2.9 and 3.1 Å, respectively, from C45 and A30 phosphate groups of U5 snRNA, which are only 4.0 Å apart. K597 is closer to the phosphate groups of U46 and A30 (3.3 and 3.9 Å, respectively [Fig. 2D]). K1300 and K1306 are in the reverse transcriptase/endonuclease (RT/EN) domain of PRP8. K1306 sits between the phosphates of U5 snRNA residue C39 and a nucleotide in the 5′ exon. K1300 is surrounded by other amino acids in Prp8 with no clear access to solvent.
Based on the chemical probing results, we anticipated the same four Prp8 lysine residues to appear in a solvent accessible conformation in the cryo-EM structure of human preactivated spliceosome (Bertram et al. 2017a). Unfortunately, the limited resolution of the cryo-EM density precluded modeling of side chains and their potential interactions. Compared with the catalytic structure, residues K1300 and K1306 are significantly shifted relative to U5 snRNA due to a conformational change between the RT/EN and amino-terminal domains of Prp8. K595 and K597 remain in the same orientation relative to U5 snRNA and are positioned to stabilize a similar bend in U5 SLI (Supplemental Fig. 3). The discordance between chemical probing results and the position of these lysine residues in the preactivated spliceosome structure may indicate that the solvent accessible conformation is too flexible or too brief to be observed by cryo-EM. Likewise, our probing method cannot discern lysine side chains that are continuously solvent accessible from those that shift between solvent accessible and protected conformations over the time scale of the probing experiment. It is also worth noting that the two pre-mRNAs sequences used to assemble preactivated spliceosomes differed, which may result in different conformations of the complexes (Bertram et al. 2017a).
Differentially modified lysine residues genetically interact with multiple factors involved in 5′ splice site recognition
If Prp8 residues K595 and K597 modulate U5 SLI structure, then neutralizing their positive charge will alter the conformation of U5 SLI and affect splicing. To test this hypothesis, we mutated the homologous residues (K670 and K672) to alanine in Saccharomyces cerevisiae. In yeast spliceosome cryo-EM structures, these residues interact with a similar bend in U5 SLI in a preactivated B complex (Plaschka et al. 2017) and both C and C* catalytic complexes (Galej et al. 2016; Wan et al. 2016; Yan et al. 2016; Fica et al. 2017), suggesting that any functional role is evolutionarily conserved (Supplemental Fig. 3). Yeast harboring the prp8-K670/672A allele are viable, and show no differences in growth relative to wild type PRP8 (Supplemental Fig. 4A,B). However, prp8-K670/672A mutants do exhibit increased intron retention of two endogenous pre-mRNAs as measured by RT-qPCR, indicating the lysine to alanine mutations modestly perturb splicing (Fig. 3A).
FIGURE 3.
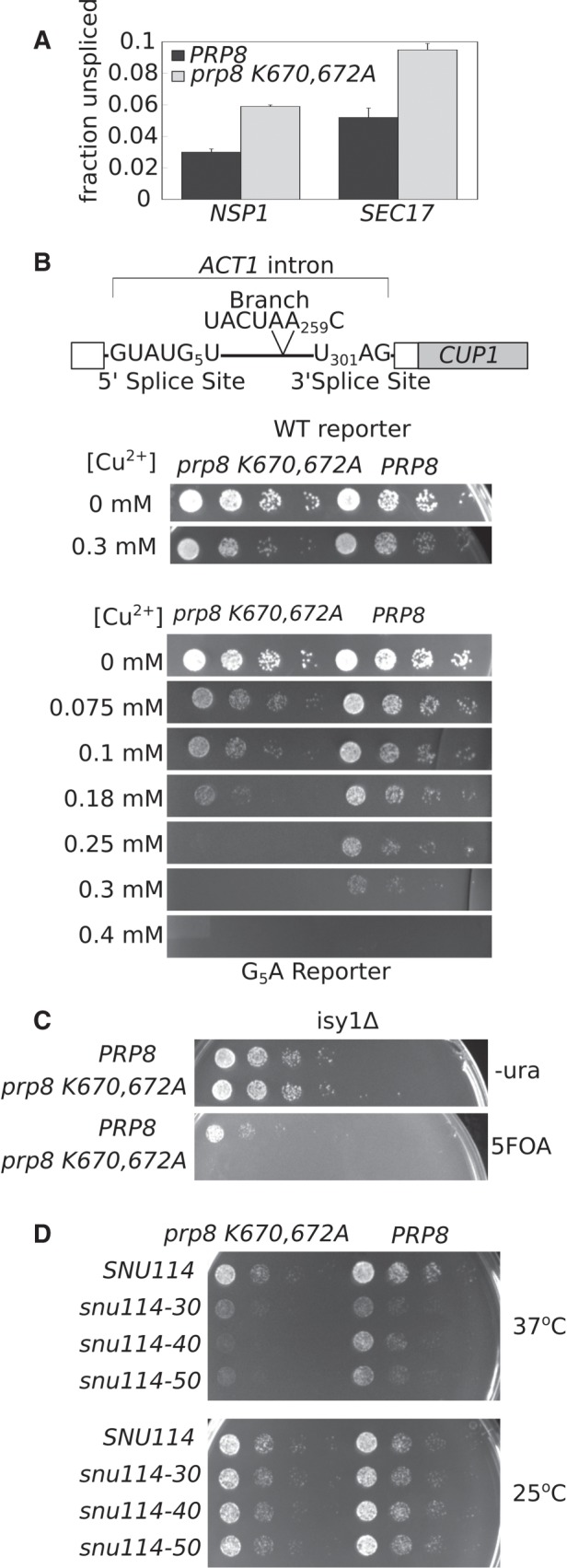
prp8-K670/672A genetically interacts with 5′ splice site and splicing factors involved in activation and first step chemistry. (A) qRT-PCR analysis of relative NSP1 and SEC17 pre-mRNA transcript abundance in prp8-K670/672A (gray bars) and PRP8 (black bars). Primers that span the intron-exon junction were used to quantify pre-mRNA abundance, while primers that amplify a portion of the 3′ exon present in both the pre- and mature mRNA were used to quantify total RNA levels. Fraction unspliced is calculated as Fpre-mRNA/Ftotal. The average of three biological replicates is shown. Error bars represent standard error of the mean. (B) The ACT1–CUP1 splicing reporter. The ACT1 intron is fused to the CUP1 coding sequence such that splicing efficiency can be directly assessed by relative yeast growth on copper-containing media. The 5′ and 3′ and branch site consensus sequences are shown. Numbers indicate position of specific nucleotide where the first nucleotide of the intron is defined as one. Prp8-K670/672A and PRP8 yeast strains carrying either a WT or G5A ACT1–CUP1 reporter were serially diluted and spotted onto plates containing varying concentrations of Cu2+ and allowed to grow at 30°C for 2 d. (C) Double mutant isy1Δ prp8Δ strains carrying either prp8-K670/672A or PRP8 on a HIS plasmid and PRP8 on a URA plasmid were serially diluted and spotted onto 5FOA or –ura plates and grown at 30°C. 5FOA prohibits growth of URA+ strains. prp8-K670/672A is lethal in an isy1Δ background. (D) Double mutant snu114Δ prp8Δ complemented with prp8-K670/672A or PRP8 in combination with SNU114, or snu114-30, snu114-40, or snu114-50 C-terminal truncation alleles were serially diluted and spotted onto rich media and allowed to grow at 37°C and 25°C.
To assess whether the prp8-K670/672A allele impacts splicing fidelity and/or efficiency, we used an ACT1–CUP1 reporter system that directly links intron removal to the ability of yeast to grow in the presence of copper (Fig. 3B; Lesser and Guthrie 1993). With the reporter, the prp8-K670/672A strain grew identically to wild type PRP8 yeast, except when the reporter contained a guanine to adenosine substitution at the 5th position of the 5′ splice site consensus (G5A). RT-PCR confirmed a small decrease in reporter splicing (Supplemental Fig. 4C). By weakening interactions between the intron and U6 snRNA, the G5A mutation has been suggested to make the 5′ splice site “slippery,” thereby destabilizing the intron's position in the active site of the spliceosome (Parker and Guthrie 1985). Exacerbation of the G5A phenotype by prp8-K670/672A may be due to additional “slack” in the position of the upstream exon when U5 SLI conformation is disturbed. The effect appears to be specific to 5′ splice site recognition because Prp8-K670/672A yeast have no phenotype when the branch point or 3′ splice site is mutated in the ACT1–CUP1 reporter (Supplemental Fig. 4).
To further explore the role of the Prp8 lysine residues in spliceosome function, we searched for genetic interactions between prp8-K670/672A and proteins involved in splicing activation and first step chemistry. Isy1 is a nonessential protein that is implicated in stabilizing the first step active site conformation of the spliceosome to help maintain fidelity during transition to second step chemistry (Villa and Guthrie 2005). Consistent with this model, Isy1 contacts intron residues flanking the branch point in structures of S. cerevisiae C spliceosome caught immediately after first step chemistry (Supplemental Fig. 5; Galej et al. 2016). Like prp8-K670/672A, isy1Δ yeast are viable with no growth phenotype, but show reduced splicing efficiency in vivo (Dix et al. 1999). Strikingly, when Isy1 is deleted, the prp8-K670/672A mutation becomes lethal (Fig. 3C). This synthetic lethality suggests that the spliceosome as a whole cannot compensate for a combined loss of interactions supporting the first step active site conformation.
The U5 snRNP protein Snu114 is a GTPase that is proposed to regulate early snRNA rearrangements involved in spliceosome activation (Brenner and Guthrie 2005; Frazer et al. 2009). Its influence on those rearrangements is likely indirect considering that Snu114 sits at a considerable distance from the RNA catalytic core in cryo-EM structures of the spliceosome, where it primarily contacts the amino-terminal domain of Prp8 (Supplemental Fig. 5). Our prp8-K670/672A allele exacerbates temperature sensitivity of snu114-40 (M842R) and snu114-50 (E910G, C928R) alleles (Fig. 3D; Brenner and Guthrie 2005), suggesting that Snu114's influence on splicing is mediated, at least in part, by these Prp8 lysine residues. The same Snu114 alleles also show genetic interactions with Prp8 mutations linked to activation (Brenner and Guthrie 2005), as well as with deletions in U5 internal loop I (U5 ILI) at the base of U5 SLI (ΔC79-A81 and ΔA78-A81, ΔC11, ΔC112-G113) (Frazer et al. 2009). Considering these data in conjunction with the position of the lysines relative to SLI in the cryo-EM structures, we suggest that the role of Snu114 in spliceosome activation is linked to Prp8's hold on U5 snRNA.
Implications for Prp8's role in 5′ splice site positioning
Cryo-EM structures of human spliceosomes reveal the central role that Prp8 plays in supporting RNA interactions at the heart of the splicing machinery (Bertram et al. 2017a,b; Zhang et al. 2017). It augments U2 and U6 snRNA interactions with the intron and with each other as the branch point region is juxtaposed to the 5′ splice site. Prp8 also cradles much of U5 snRNA, and encloses the 5′ exon as it interacts with U5 SLI. U5 snRNA contacts with the exon are thought to assist in docking the 5′ splice site into the spliceosome catalytic core (Turner et al. 2004). Consistent with that model, mutations that alter the alignment of U5 SLI and the 5′ exon inhibit the second step of splicing (O'Keefe and Newman 1998).
Differences between the structures of human preactivated and catalytic spliceosomes imply large-scale rearrangements in U2 and U6 snRNA conformations and their interactions with the intron to create the active sites for first and second step chemistries (Bertram et al. 2017a; Zhang et al. 2017). Although more subtle, U5 snRNA interactions with the 5′ exon are also different before and after catalytic activation. Specifically, the last nucleotide of the exon has a different U5 snRNA base-pairing partner (U41 instead of U40) in the human catalytic versus preactivation complex, and the number of base interactions decreases from five to three (Fig. 4B). Chemical probing supports a transient loss and gain of interactions for both RNA and protein in this region during spliceosome activation. Our protein probing indicates Prp8 lysine residues in the area surrounding the loop of U5 SLI are accessible to an acetylating reagent, and then become protected by first step catalysis. Bases in the loop of U5 snRNA SLI also show increased protection to chemical probing following preactivation (Anokhina et al. 2013). To explain these changes in reactivity, and the difference in U5 snRNA interactions with pre-mRNA visualized by cryo-EM, we hypothesize that a transient conformation of the preactivated spliceosome exists. In our model, Prp8 partially disengages the U5 SLI, allowing it to reposition relative to the 5′ exon, which aids in docking the 5′ splice site into the first step active site.
FIGURE 4.
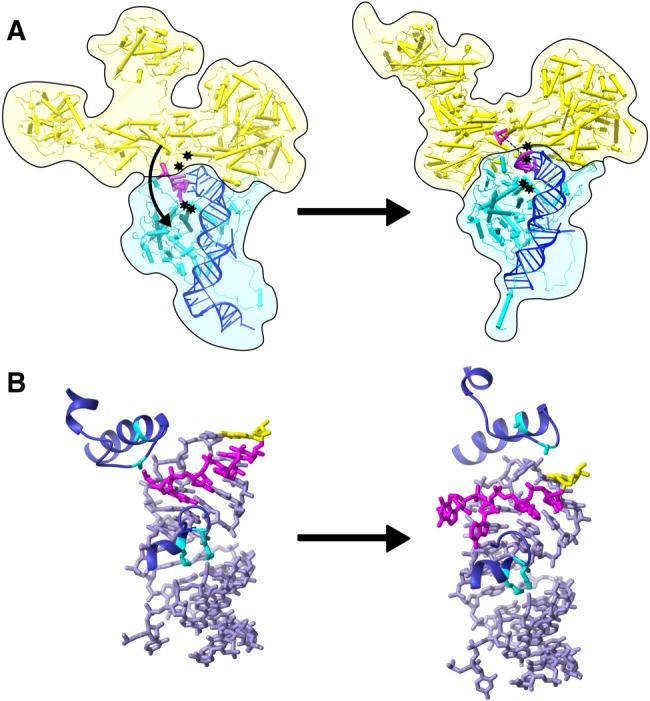
Model of Prp8, U5 SLI, and 5′ exon interactions as the human spliceosome undergoes activation and first step chemistry. (A) Global domain movements of Prp8 body (yellow) relative to N-terminal domain (cyan) in human preactivation (Bertram et al. 2017b) (left) and human C* (Zhang et al. 2017) (right) spliceosome models; showing U5 snRNA (blue) and 5′ exon (magenta). The positions of differentially modified lysine residues K1300 and K1306 (upper) and K595 and K597 (lower) are indicated by black stars. (B) Close-up view of the relative positions of U5 SLI (lavender), 5′ exon (magenta), and the same lysine side chains (cyan) of Prp8 (purple) in preactivation (left) and catalytic (right) spliceosome conformations. The last nucleotide of the 5′ exon is highlighted in yellow.
Given this model, it is not surprising that lysines mediating U5 SLI contacts genetically interact with factors previously implicated in spliceosome catalytic core formation, i.e., activation. Individually, the 5′ splice site G5A mutation impairs 5′ splice site positioning by removing a pre-mRNA/U6 contact, and Isy1 null yeast lose contacts between Isy1 and the intron branch. The Snu114 mutants exhibit temperature sensitive growth, presumably from problems activating the spliceosome. In combination with our Prp8 lysine mutations these phenotypes are more severe, indicating that cooperation of these discrete molecular interactions supports the first step active site and is a piece of the larger mechanism used by the spliceosome to control accurate positioning of the pre-mRNA.
MATERIALS AND METHODS
In vitro assembly of preactivated and catalytic spliceosomes
Catalytic and preactivated pre-mRNA substrates are derivatives of the AdML transcript, and tagged with three MS2 sites in the intron or the 3′ end, respectively. T7 runoff transcription was used to generate G(5′)ppp(5′)G-capped radiolabeled pre-mRNA, which was gel purified and preincubated with a 50-fold excess of MS2-MBP fusion protein. In vitro splicing reactions contained 10 nM pre-mRNA, 80 mM potassium glutamate, 2 mM magnesium acetate, 2 mM ATP, 5 mM creatine phosphate, 0.05 mg/mL tRNA, and 40% HeLa cell (Biovest International) nuclear extract.
Preactivated spliceosomes were generated in vitro by first forming the exon definition complex containing U1/U2/U4/U5/U6 snRNAs on a substrate that consists of an exon proceeded by a branch point and 3′ splice site, and then adding 100-fold excess 5′ splice site oligo (AAGGUAAAGUAU) as previously described (Schneider et al. 2010), but with the following changes: (i) the exon definition complex was formed for 8 min prior to 5′ splice site oligo addition, and then incubated an additional 7 min, and (ii) the 3′ exon of the pre-mRNA lacked a 5′ splice site. Catalytic spliceosomes arrested after first step chemistry and containing U2/U5/U6 snRNAs were accumulated in vitro using a pre-mRNA with a mutant GG 3′ splice site, and excess unspliced pre-mRNA from catalytic splicing reactions were depleted by DNA oligo directed RNase H digestion as previously described (Jurica et al. 2002).
pre-mRNA sequences (intron sequence is underlined)
Catalytic pre-mRNA
5′-GGGAGACCGGCAGATCAGCTTGGCCGCGTCCATCTGGTCATCTAGGATCTGATATCATCGATGAATTCGAGCTCGGTACCCCGTTCGTCCTCACTCTCTTCCGCATCGCTGTCTGCGAGGGCCAGCTGTTGGGGTGAGTACTCCCTCTCAAAAGCGGGCATGACTTCTGCCCTCGAGCGATATCCGTACACCATCAGGGTACGAGCTAGCCCATGGCGTACACCATCAGGGTACGACTAGTAGATCTCGTACACCATCAGGGTACGGAATTCTCTAGACTCGAGTTATTAACCCTCACTAAAGGCAGTAGTCAAGGGTTTCCTTGAAGCTTTCGTACTAACCCTTTCCCTTTTTTTTCCTCTCTCTCTGGCCATGGGTCGACGTTGAGGACAAACTCTTCGCGGTCTTTCCAGTACTCTTGGA-3′
Preactivated pre-mRNA
5′-GGGCAAGGGTTTCCTTGAAGCTTTCGTACTAACCCTTTCCCTTTTTTTTCCTCTCTCTCTGGCCATGGGTCGACGTTGAG GACAAACTCTTCGCGGTCTTTCCAGTACTCTTGGATCCGATATCCGTACACCATCAGGGTACGAGCTAGCCCATGGCGTACACCATCAGGGTACGACTAGTAGATCTCGTACACCATCAGGGTACGGAATTCTCT-3′
Spliceosome purification
Catalytic spliceosomes were fractionated by size exclusion chromatography using Sephacryl S-400 resin (GE Healthcare) in SCB2H-N buffer (20 mM HEPES pH 7.9, 150 mM KCl, 5 mM EDTA, 0.05% v/v NP-40). Preactivated spliceosomes were fractionated in 10%–30% v/v glycerol gradients in G-75 buffer (20 mM HEPES pH 7.9, 75 mM KCl, and 1.5 mM MgCl2) for 12 h at 4°C in a SW-41 rotor at 25,000 RPM in a Beckman ultracentrifuge. Corresponding fractions of catalytic and preactivated spliceosomes were pooled and bound to amylose resin via MS2:MBP. Preactivated spliceosomes were washed with G75 buffer, and catalytic spliceosomes were washed with SCB2H (20 mM HEPES pH 7.9, 150 mM KCl, 5 mM EDTA). Spliceosome complexes were then eluted with their respective wash buffers containing 10 mM maltose.
Chemical probing by two-step acetylation
In the first acetylation reaction, one picomole of purified spliceosomes in elution buffer was incubated in 5 mM sulfo-NHS-acetate (Pierce Technology) at room temperature for 1 h. Reactions were quenched with 1/10th volume 1 M Tris pH 7.9, and then subjected to SDS-PAGE. After staining with Coomassie-G (5% w/v aluminum sulfate 14–18 hydrate, 10% v/v ethanol, 0.02% w/v CBB G-250, 2% v/v phosphoric acid), the entire lane containing spliceosome proteins was excised into six–nine slices. For the second acetylation reaction, gel slices were washed with shaking agitation in (i) water for 10 min at 37°C, (ii) 10 mM ammonium bicarbonate at room temperature, (iii) 50 mM ammonium bicarbonate:acetonitrile (1:1) twice for 45 min at 37°C, (iv) water twice, and (v) 100% acetonitrile twice at 37°C for 5 min. After complete removal of the acetonitrile, 20 µL of D6-acetic anhydride (Acros Organics) mixed with 40 µL of 100 mM ammonium bicarbonate was added and completely absorbed by the gel. The gel slices were then submerged in 100 mM ammonium bicarbonate, and the pH adjusted to 7–8 with 1 M ammonium bicarbonate. After 60 min at 37°C, the gel slices were washed with water three times and submitted for MS/MS analysis.
Protein digestion and mass spectrometric analysis
For protein digestion, gel pieces were washed in 25 mM ammonium bicarbonate/60% v/v acetonitrile. Disulfide bonds were reduced using 10 mM dithiothreitol in 25 mM ammonium bicarbonate for 30 min at 60°C, then free sulfhydryls were alkylated using 20 mM iodoacetamide in 25 mM ammonium bicarbonate for an hour. Gel pieces were washed again using 25 mM ammonium bicarbonate/60% v/v acetonitrile, and then digested overnight using 200 ng TPCK-treated porcine trypsin (Promega) in 25 mM ammonium bicarbonate. Peptides were extracted using 50% v/v acetonitrile/1% v/v formic acid. Extracted peptides were dried down by vacuum centrifugation, and then resuspended in 0.1% v/v formic acid prior for mass spectrometric analysis.
Peptides were analyzed by LC-MS/MS using a NanoAcquity (Waters) UPLC system interfaced with either an LTQ Orbitrap Velos or QExactive Plus (both Thermo) mass spectrometer. Peptides were separated using an integrated 75 µm × 15 cm PEPMAP reverse phase column and spray tip (EasySpray source) using a gradient from 2% to 27% v/v solvent B (0.1% v/v formic acid in acetonitrile) over solvent A (0.1 % acetonitrile in water) at a flow rate of 400 nL/min for 27 min. Using the Velos, survey scans were measured at a resolution of 30,000 full width at half maximum (FWHM). The six most intense precursor ions were automatically selected for HCD fragmentation analysis by data-dependent acquisition and measured at a resolution of 7500 FWHM. For QExactive data, survey scans were measured a resolution of 70,000 FWHM, and were followed by data-dependent acquisition of the top ten most intense precursors at a resolution of 17,500 FWHM.
Raw data was converted to mgf format peak list files using in-house software based on the Raw_Extract script in Xcalibur version 2.4, and then searched using Protein Prospector version 5.18.22 against a database of all human proteins in SwissProt downloaded on May 9th 2016 supplemented with entries for E. coli maltose-binding periplasmic protein, enterobacteria phage MS2 coat protein and pig trypsin, plus decoy versions of all of these sequences (a total of 20,204 entries searched). Carbamidomethylation of cysteine residues was searched as a constant modification, and variable modifications considered were acetylation or deuteroacetylation of uncleaved lysines, methionine oxidation, pyroglutamate formation from N-terminal glutamines, and protein N-terminal methionine removal and/or acetylation. Results were thresh-holded to an estimated 1% protein FDR according to target:decoy database searching (Elias and Gygi 2007).
Intensities of peaks were extracted from the raw data using Protein Prospector by summing together signal over a period −10 to +20 sec from the time the peak was selected for MSMS.
Yeast plasmids
Plasmids used in this study are indicated in Table 1. Plasmid pRS313-PRP8-WT, pRS314-PRP16 plasmids, ACT-CUP reporter plasmids, and pTB-SNU114 plasmids have been previously described (Lesser and Guthrie 1993; Query and Konarska 2004; Brenner and Guthrie 2005; Mayerle and Guthrie 2016). Plasmid pRS313-prp8-K670,672A was made from pRS313-PRP8-WT using Quikchange Mutagenesis (Agilent Technologies). All plasmids have been deposited in the Addgene Vector Database, www.addgene.org.
TABLE 1.
Plasmids used for yeast experiments
Strains and growth assays
Strains used in this study are listed in Table 2. All strains were constructed using standard genetic methods. Double mutant strains were constructed via 5FOA shuffling, as described previously (Guthrie and Fink 2002). Relative growth was assessed by diluting an overnight culture to an OD600 of 0.1 and allowing it to grow to mid-log phase, an approximate OD600 of 0.5. Strains were then serially diluted 1:5 from a starting OD600 concentration of 0.1 and spotted onto rich media, minimal media lacking Ura, or minimal media containing 5FOA and then allowed to grow at 37°C, 30°C, 25°C, and 17°C before being photographed.
TABLE 2.
Yeast strains used for the studies

ACT-CUP reporter assays
Prp8 and ACT-CUP reporter plasmids (WT, G1A, U2A, G5A, C256A, BrC, BrG, U301G, A302G, G303-4C) were transformed into strain yMeg030 using standard methods (Guthrie and Fink 2002). Saturated overnight cultures grown in –Leu media were diluted to an approximate OD600 of 0.1 and allowed to grow again to an OD600 of approximately 0.5 before being diluted to an OD600 of 0.1 and spotted onto –Leu plates supplemented with 0 to 1.5 mM CuSO4. Plates were placed at 30°C and allowed to grow for 2–3 d and photographed.
RNA isolation and RT-qPCR
RNA isolation and detection were carried out as previously described (Mayerle and Guthrie 2016). Briefly, cultures were grown at 30°C overnight to saturation and then diluted to an OD600 of approximately 0.1. Diluted cultures were allowed to grow to an approximate OD600 of 0.5, pelleted, washed briefly with water, and then snap frozen at −80°C for further processing. Total cellular RNA was isolated using hot acid phenol followed by ethanol precipitation, and 20 µg total RNA was treated with 15 µL RNase free DNase I (NEB) in a total volume of 250 µL for 1 h at 37°C, and then re-extracted with phenol–chloroform. Superscript III Reverse Transcription System (Invitrogen) dN9 primers (Life Technologies) were used in all reverse transcription reactions. qPCR was performed using NEB Taq Polymerase and the gene-specific primers listed in Table 3, or previously described NSP1 and SEC17 primers. qPCR cycle conditions were 95°C for 3 min, 39 cycles of 95°C for 15 sec, 55°C for 30 sec, 72°C for 15 sec, followed by one cycle at 72°C for 3 min. Three biological replicates using total RNA isolated from separate biological cultures were assayed in technical triplicate.
TABLE 3.
Primer sequences for RT-qPCR experiments
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant R01GM72649 to M.S.J. A.J.M. was supported by the National Institutes of Health training grant T32GM08646. M.M. and C.G. were supported by National Institutes of Health R01GM2119 and C.G. is an American Cancer Society Research Professor of Molecular Genetics. Support for R.J.C. and A.L.B. was provided by National Center for Research Resources grants RR001614, RR015804, and RR019934 and National Institute of General Medical Sciences grant P41 GM103481.
Author contributions: A.J.M., M.M., R.J.C., C.G., A.L.B., and M.S.J. conceived and designed the experiments; A.J.M., M.M. E.H.-R., and R.J.C. performed the experiments; A.J.M., M.M., R.J.C., and M.S.J. analyzed the data; A.J.M., M.M., and M.S.J. wrote the paper.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.065458.117.
REFERENCES
- Anokhina M, Bessonov S, Miao Z, Westhof E, Hartmuth K, Lührmann R. 2013. RNA structure analysis of human spliceosomes reveals a compact 3D arrangement of snRNAs at the catalytic core. EMBO J 32: 2804–2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram K, Agafonov DE, Dybkov O, Haselbach D, Leelaram MN, Will CL, Urlaub H, Kastner B, Lührmann R, Stark H. 2017a. Cryo-EM structure of a pre-catalytic human spliceosome primed for activation. Cell 170: 701–713 e711. [DOI] [PubMed] [Google Scholar]
- Bertram K, Agafonov DE, Liu WT, Dybkov O, Will CL, Hartmuth K, Urlaub H, Kastner B, Stark H, Lührmann R. 2017b. Cryo-EM structure of a human spliceosome activated for step 2 of splicing. Nature 542: 318–323. [DOI] [PubMed] [Google Scholar]
- Brenner TJ, Guthrie C. 2005. Genetic analysis reveals a role for the C terminus of the Saccharomyces cerevisiae GTPase Snu114 during spliceosome activation. Genetics 170: 1063–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalkley RJ, Hansen KC, Baldwin MA. 2005. Bioinformatic methods to exploit mass spectrometric data for proteomic applications. Methods Enzymol 402: 289–312. [DOI] [PubMed] [Google Scholar]
- Dix I, Russell C, Yehuda SB, Kupiec M, Beggs JD. 1999. The identification and characterization of a novel splicing protein, Isy1p, of Saccharomyces cerevisiae. RNA 5: 360–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, Gygi SP. 2007. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods 4: 207–214. [DOI] [PubMed] [Google Scholar]
- Fica SM, Oubridge C, Galej WP, Wilkinson ME, Bai XC, Newman AJ, Nagai K. 2017. Structure of a spliceosome remodelled for exon ligation. Nature 542: 377–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer LN, Lovell SC, O'Keefe RT. 2009. Analysis of synthetic lethality reveals genetic interactions between the GTPase Snu114p and snRNAs in the catalytic core of the Saccharomyces cerevisiae spliceosome. Genetics 183: 497–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galej WP, Wilkinson ME, Fica SM, Oubridge C, Newman AJ, Nagai K. 2016. Cryo-EM structure of the spliceosome immediately after branching. Nature 537: 197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie C, Fink GR. 2002. Guide to yeast genetics and molecular and cell biology. Academic Press, Amsterdam. [Google Scholar]
- Jurica MS, Licklider LJ, Gygi SR, Grigorieff N, Moore MJ. 2002. Purification and characterization of native spliceosomes suitable for three-dimensional structural analysis. RNA 8: 426–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesser CF, Guthrie C. 1993. Mutational analysis of pre-mRNA splicing in Saccharomyces cerevisiae using a sensitive new reporter gene, CUP1. Genetics 133: 851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Query CC, Konarska MM. 2007. Opposing classes of prp8 alleles modulate the transition between the catalytic steps of pre-mRNA splicing. Nat Struct Mol Biol 14: 519–526. [DOI] [PubMed] [Google Scholar]
- Mayerle M, Guthrie C. 2016. Prp8 retinitis pigmentosa mutants cause defects in the transition between the catalytic steps of splicing. RNA 22: 793–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Keefe RT, Newman AJ. 1998. Functional analysis of the U5 snRNA loop 1 in the second catalytic step of yeast pre-mRNA splicing. EMBO J 17: 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker R, Guthrie C. 1985. A point mutation in the conserved hexanucleotide at a yeast 5′ splice junction uncouples recognition, cleavage, and ligation. Cell 41: 107–118. [DOI] [PubMed] [Google Scholar]
- Plaschka C, Lin PC, Nagai K. 2017. Structure of a pre-catalytic spliceosome. Nature 546: 617–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Query CC, Konarska MM. 2004. Suppression of multiple substrate mutations by spliceosomal prp8 alleles suggests functional correlations with ribosomal ambiguity mutants. Mol Cell 14: 343–354. [DOI] [PubMed] [Google Scholar]
- Schneider M, Will CL, Anokhina M, Tazi J, Urlaub H, Lührmann R. 2010. Exon definition complexes contain the tri-snRNP and can be directly converted into B-like precatalytic splicing complexes. Mol Cell 38: 223–235. [DOI] [PubMed] [Google Scholar]
- Turner IA, Norman CM, Churcher MJ, Newman AJ. 2004. Roles of the U5 snRNP in spliceosome dynamics and catalysis. Biochem Soc Trans 32: 928–931. [DOI] [PubMed] [Google Scholar]
- Umen JG, Guthrie C. 1996. Mutagenesis of the yeast gene PRP8 reveals domains governing the specificity and fidelity of 3′ splice site selection. Genetics 143: 723–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa T, Guthrie C. 2005. The Isy1p component of the NineTeen complex interacts with the ATPase Prp16p to regulate the fidelity of pre-mRNA splicing. Genes Dev 19: 1894–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan R, Yan C, Bai R, Huang G, Shi Y. 2016. Structure of a yeast catalytic step I spliceosome at 3.4 Å resolution. Science 353: 895–904. [DOI] [PubMed] [Google Scholar]
- Yan C, Wan R, Bai R, Huang G, Shi Y. 2016. Structure of a yeast step II catalytically activated spliceosome. Science 355: 149–155. [DOI] [PubMed] [Google Scholar]
- Zhang X, Yan C, Hang J, Finci LI, Lei J, Shi Y. 2017. An atomic structure of the human spliceosome. Cell 169: 918–929 e914. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.