

Abstract
The four dengue viruses (DENV1-4) are rapidly reemerging infectious RNA viruses. These positive-strand viral genomes contain structured 3′ untranslated regions (UTRs) that interact with various host RNA binding proteins (RBPs). These RBPs are functionally important in viral replication, pathogenesis, and defense against host immune mechanisms. Here, we combined RNA chromatography and quantitative mass spectrometry to identify proteins interacting with DENV1-4 3′ UTRs. As expected, RBPs displayed distinct binding specificity. Among them, we focused on quaking (QKI) because of its preference for the DENV4 3′ UTR (DENV-4/SG/06K2270DK1/2005). RNA immunoprecipitation experiments demonstrated that QKI interacted with DENV4 genomes in infected cells. Moreover, QKI depletion enhanced infectious particle production of DENV4. On the contrary, QKI did not interact with DENV2 3′ UTR, and DENV2 replication was not affected consistently by QKI depletion. Next, we mapped the QKI interaction site and identified a QKI response element (QRE) in DENV4 3′ UTR. Interestingly, removal of QRE from DENV4 3′ UTR abolished this interaction and increased DENV4 viral particle production. Introduction of the QRE to DENV2 3′ UTR led to QKI binding and reduced DENV2 infectious particle production. Finally, reporter assays suggest that QKI reduced translation efficiency of viral RNA. Our work describes a novel function of QKI in restricting viral replication.
Keywords: QKI, dengue virus, translation, host factors, RNA elements
INTRODUCTION
Dengue is a mosquito-borne viral disease caused by four dengue virus serotypes (DENV1-4). This disease is endemic in more than 100 countries, with a recent estimate of annual number of infections worldwide close to 400 million, which causes significant morbidity and negative economic impact (Bhatt et al. 2013). Recently, a tetravalent vaccine, Dengvaxia, was licensed in several dengue-endemic countries; however, its efficacy varies by age of the vaccine recipient, prior flaviviral or DENV infection at baseline, and by DENV serotype (Vannice et al. 2016). Hence, despite the availability of a dengue vaccine, novel strategies and targets are still urgently needed to develop therapeutics against dengue (Low et al. 2017).
DENVs are enveloped, positive-strand RNA viruses of the family Flaviviridae, which also includes the yellow fever (YFV), West Nile (WNV), Hepatitis C (HCV), and Zika (ZIKV) viruses. DENV genomes are ∼11 kb in length and contain one open reading frame flanked by 5′ and 3′ untranslated regions (UTRs). The 5′ end is modified with a type I cap structure while the 3′ end, unlike most other cellular mRNA, lacks a poly(A) tail. Translation of the viral genome produces a polyprotein which is co- and post-translationally cleaved into three structural proteins, capsid (C), premembrane (prM), and envelope (E), and seven nonstructural (NS) proteins, NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5. DENV initiates infection by binding to host cell receptors and undergoes endocytosis. Upon delivery of its genome into cells, the DENV genome acts as mRNA to produce viral proteins and serves as a template for the synthesis of complementary negative strands to generate new positive strand DENV genomes, which will be subsequently packaged into progeny virions. In addition to the full-length viral genome RNA, another abundant RNA species present during DENV infection is the subgenomic flaviviral RNA (sfRNA) (Pijlman et al. 2008; Liu et al. 2010). This sfRNA (∼300–600 nt [nucleotides]) is produced from the incomplete degradation of a viral genome by a cellular 5′–3′ exonuclease, and contains part of the 3′ UTR (Chapman et al. 2014). Production of sfRNA has been shown to inhibit antiviral interferon (IFN) response through protein interactions to promote viral replication (Bidet et al. 2014; Manokaran et al. 2015).
During this complex viral life cycle, DENV RNA molecules constantly engage and interact with various host proteins. The outcome of interplay between DENV RNAs and host RBPs is likely to determine whether viruses will succeed in establishing infection and thereby may impact the severity of disease. In order to understand the molecular mechanisms of DENV pathogenesis, earlier efforts using genome-wide screening have revealed key host factors that are required for DENV replication (Sessions et al. 2009; Marceau et al. 2016; Savidis et al. 2016; Zhang et al. 2016). Ribosomal proteins RPLP1 and RPLP2 are among those essential host factors and promote early viral protein accumulation (Campos et al. 2017). Interestingly, the RPLP1/2 heterodimer is not required for global cellular translation. This differential requirement of viral and cellular translation further highlights the complexity of virus-host interactions. Along with functional genomic studies, several approaches have been used to identify host factors that interact with DENV viral RNA in vivo and in vitro (Yocupicio-Monroy et al. 2003, 2007; Garcia-Montalvo et al. 2004; Paranjape and Harris 2007; Anwar et al. 2009; Polacek et al. 2009; Gomila et al. 2011; Lei et al. 2011; Ward et al. 2011; Phillips et al. 2016; Viktorovskaya et al. 2016). These identified DENV RBPs were shown to possess proviral or antiviral activities and could act at various stages of the viral life cycle, such as trafficking, translation, synthesis, and packaging of RNA genomes.
In this study, we combined RNA chromatography and quantitative mass spectrometry and sought to perform a comprehensive screen for host cell proteins that physically interact with the 3′ UTR of one clinical isolate from each of the four DENV serotypes. In this mass spectrometry screening, we identified several RBPs, including G3BP1, G3BP2, CAPRIN1, DDX6, and QKI. Each of these proteins displayed distinct binding preferences. For example, DDX6 was found to interact relatively equally with all DENV1-4 3′ UTRs, while QKI preferentially associated with the DENV4 3′ UTR; we focused on QKI because of this preference. QKI is a member of the STAR (signal transduction and activator of RNA) and an hnRNP K homology (KH)-type family of RBPs. Three main isoforms of QKI (QKI-5, QKI-6, and QKI-7) are produced from a single gene by alternative splicing (Ebersole et al. 1996; Kondo et al. 1999) and they are involved in multiple aspects of RNA metabolism (Darbelli and Richard 2016), such as alternative splicing (Hall et al. 2013; Fagg et al. 2017), translation (Saccomanno et al. 1999; Yamagishi et al. 2016), mRNA stability (Larocque et al. 2005), circular RNA production (Conn et al. 2015), and miRNA biogenesis (Wang et al. 2013). These processes are regulated by QKI binding to an RNA element, known as QKI response element (QRE), which includes ACUAAY and a half site UAAY separated by at least 1 nt (Galarneau and Richard 2005). Dysregulation of QKI-mediated biological activities has been implicated in several human diseases, such as atherosclerosis (de Bruin et al. 2016), cancer (Yang et al. 2010; Sebestyén et al. 2015; de Miguel et al. 2016), and schizophrenia (Aberg et al. 2006). Recently, QKI was shown to facilitate the expression of viral proteins and the production of viral particles in Herpesvirus infected cells (Sánchez-Quiles et al. 2011). A proviral role for QKI was also recently observed with ZIKV; however, there is no obvious QRE in the ZIKV UTRs (Ramanathan et al. 2018). Little is known about the molecular roles of QKI in viral replication.
Here, we show that QKI is a host restriction factor against a clinical isolate of DENV4 (DENV-4/SG/06K2270DK1/2005). RNA immunoprecipitation experiments demonstrated that DENV4 viral RNA physically associated with QKI in infected cells. The functional role of QKI was addressed using siRNA-mediated silencing. Knockdown of all QKI isoforms increased viral particle production in DENV4 infected cells but did not have a consistent impact on DENV2. Mapping of the QKI interaction site identified a QRE in DENV4 3′ UTR. Deletion of this putative QRE from DENV4 3′ UTR abolished this interaction and enhanced viral particle production. Consistent with the antiviral role of QKI, introduction of QRE to DENV2 3′ UTR conferred the ability to interact with QKI and reduced DENV2 infectious particle production. Mechanistically, QKI binding appears to reduce translation efficiency of DENV RNA. Taken together, our work describes a novel function of QKI in blocking DENV4 propagation.
RESULTS
Identification of DENV1-4 3′ UTR-associated proteins
The 3′ UTRs of DENVs play important roles in defining viral fitness in host cells (Manokaran et al. 2015; Ng et al. 2017; Pompon et al. 2017). Our group has previously identified several DENV2 3′ UTR interacting host proteins that regulate viral replication (Ward et al. 2011; Bidet et al. 2014; Manokaran et al. 2015). In order to achieve a more comprehensive understanding of the molecular mechanisms by which DENV 3′ UTRs impact viral propagation, we used a previously described approach and sought to comprehensively identify DENV1-4 3′ UTR interacting host RBPs. This method relies on the use of RNA affinity chromatography to isolate DENVs 3′ UTR binding RBPs from host cell lysates. First of all, we in vitro transcribed RNA molecules that contained two parts: tobramycin aptamer sequences and various DENV1-4 3′ UTRs sequences, the former of which allows reversible binding to tobramycin-conjugated sepharose beads. These in vitro transcribed RNA molecules were boiled, then cooled down to room temperature for secondary structure formation before they were mixed with tobramycin-conjugated sepharose beads. Following RNA binding to beads, immobilized RNA molecules were mixed with cell lysates for 1 h. After incubation and several washing steps, an excessive amount of tobramycin was used for elution. Resultant eluates were sent for mass spectrometry analysis for protein identification.
In order to facilitate identification of proteins that associate with DENV 3′ UTRs, we used stable isotopic labeling of amino acids in cell culture (SILAC) and quantitative mass spectrometry. This methodology is advantageous because it allows both identification and quantification of proteins in a complex mixture. For SILAC-based RNA pull-down experiments, HuH7 cells were grown in media containing normal isotopes of l-lysine and l-arginine (K0R0), l-lysine-(2D4) and l-arginine-(13C6) (K4R6), or l-lysine-(13C615N2) and l-arginine-(13C615N4) (K8R10). After six cell divisions, the cultures were labeled with K4R6 or K8R10 isotopes. Then these cells were propagated to generate a sufficient amount of cell lysates for pull-down experiments. Because phylogenetic analysis indicates that DENV1 and DENV3 are more closely related than DENV2 and DENV4 (Villordo et al. 2016), we divided DENV1-4 into two groups for RNA pull-down experiments. The first set includes a DENV2 New Guinea C strain (NGC) NS2A RNA as a negative control as well as the 3′ UTRs of DENV1 and DENV3 (Fig. 1A). In the second set, samples include the same negative control with the 3′ UTRs of DENV2 and DENV4. For binding reactions, they were mixed with K0R0 lysates, K4R6 lysates, and K8R10 lysates, respectively. Eluted samples were subjected to liquid chromatography mass spectrometry analysis (LC-MS). Two independent experiments for each set were performed. Results are presented as a ratio of proteins quantified in the experimental sample labeled with either K4R6 or K8R10 versus the control sample with K0R0. The complete list of identified proteins is presented in Supplemental Table S1. It should be noted that in replicate 2 of the DENV1 and DENV3 experiment, we carried out an extra wash before the tobramycin elution, resulting in less proteins being identified. This should be taken into account when comparing these two replicates. Protein candidates that have a ratio greater than 1.5 in either replicate (P-value <0.05) are considered as candidate DENV1-4 3′ UTR binding partners.
FIGURE 1.

Identification of DENV1-4 3′ UTR interacting proteins. (A,B) Schematic presentation of SILAC-based quantitative mass spectrometry following RNA affinity chromatography. Mass spectrometry data of selected proteins are shown. (C) Representative western blotting results, showing RBPs that interact with indicated DENV RNAs. In vitro transcribed RNA molecules containing tobramycin aptamer sequences and indicated DENV sequences were coupled with tobramycin conjugated sepharose beads. RNA bound beads were mixed with HuH7 cell lysates. After incubation, beads were washed and eluted. Resultant eluates were probed for indicated proteins using immunoblotting. (ND) Not detected.
The LC-MS analysis led to the identification of several novel DENV 3′ UTRs binding RBPs as well as those that have been reported before. Selected proteins are shown in Figure 1. As expected, these RBPs display distinct binding specificities. Mass spectrometry data suggests that G3BP1, G3BP2, and CAPRIN1 preferentially associated with DENV1 and DENV2 3′ UTRs (Fig. 1A,B): The enrichment of each protein relative to control is greater than 1.5-fold in either replicate (P-value <0.05). Consistently, higher amounts of G3BP1, G3BP2, and CAPRIN1 were observed using western blots in DENV1 and DENV2 3′ UTR pull downs (Fig. 1C). DDX6, on the other hand, bound to all DENV1-4 3′ UTRs (Fig. 1C; Ward et al. 2011). Interestingly, QKI preferentially interacted with DENV4 3′ UTR (∼7.62-fold enrichment in rep#1 and ∼6.49-fold enrichment in rep#2), possibly to a lesser extent with DENV1 3′ UTR, and it was not detectable in DENV2 and DENV3 3′ UTR pull downs (Fig. 1B,C). Agreement between mass spectrometry results and western blot analysis validated these interactions in vitro and increased our confidence in observing these physical associations in cells. These data suggest that each DENV 3′ UTR binds to different pools of host RBPs with different levels of binding affinity. We focused on QKI for the rest of this study because of its preferential binding to this DENV4 (DENV-4/SG/06K2270DK1/2005) 3′ UTR and its unknown function in DENV life cycle.
Interaction of QKI and DENV4 viral RNA in infected cells
In order to validate and extend the role for QKI suggested by the mass spectrometry screen, we utilized RNA immunoprecipitation (RIP) and RT-qPCR to test whether QKI interacts with DENV4 RNA during infection. We generated HEK293 cells expressing N terminus FLAG-tagged QKI-5 and QKI-6 isoforms under the control of a tetracycline-inducible promoter. QKI-5 and QKI-6 share identical amino-terminal RNA binding domains and differ at their extreme carboxy termini. A nuclear localization sequence at the carboxy-terminal of QKI-5 localizes it to the nucleus (Wu et al. 1999). QKI-6 resides both in the nucleus and cytoplasm (Hardy et al. 1996; Pilotte et al. 2001). Testing both isoforms in these RIP experiments would provide information about whether they have similar RNA binding specificity. Expression of FLAG-QKI-5 and FLAG-QKI-6 was induced by addition of tetracycline in culture media and these cells were infected with DENV4. Following infection, lysates were harvested and immunoprecipitated using a mouse IgG control antibody or a mouse FLAG antibody. Total RNA was purified from the immunoprecipitated material and the levels of DENV RNA precipitates were analyzed by RT-qPCR. The presence of an interaction between DENV4 RNA and QKI proteins was evaluated based on an enrichment of DENV4 RNA in the FLAG IP sample relative to the isotype control.
As seen in Figure 2A, only FLAG antibody efficiently immunoprecipitated FLAG-QKI-5 and FLAG-QKI-6 from lysates. Total RNA was isolated from a fraction of the same IP samples and RT-qPCR was performed to assess the levels of DENV4 RNA present. In addition, we also assessed the enrichment of two other abundant cellular RNAs with partial cytoplasmic localization, 18S rRNA and U1 spliceosomal RNA. As positive controls we tested hnRNPA1 and CTNNB1 RNAs, which have been shown to bind to QKI (Hafner et al. 2010; Yang et al. 2010; Zearfoss et al. 2011). Data are presented as the degree of enrichment of the indicated RNAs present in the FLAG IP sample relative to the isotype control. Results indicate that there was an approximately five- to 20-fold enrichment of DENV4 genome RNA (gRNA) in FLAG IP samples (Fig. 2B,C; Supplemental Fig. S1A,B). A similar trend was also observed using primers that amplify a specific region within the DENV4 3′ UTR, suggesting that both QKI-5 and QKI-6 proteins interacted with DENV4 viral RNA molecules in infected cells. Surprisingly, 18S rRNA was enriched in QKI pull-down samples (∼15-fold for QKI-5 and ∼40-fold for QKI-6). The significance of this association is unclear, but given QKI's role in protein synthesis (Saccomanno et al. 1999; Yamagishi et al. 2016), it is possible that QKI indirectly pulled down ribosomes associated with its target mRNAs. To confirm the specificity of QKI binding in our assay, we quantified SF3B1 mRNA as an additional negative control. SF3B1 was not enriched in FLAG IP pellets. (Fig. 2B,C; Supplemental Fig. S1A,B). As expected, both hnRNPA1 and CTNNB1 RNAs associated with QKI in cells. On the contrary, U1 RNA did not bind to QKI. These results suggest that DENV4 viral RNA interacts with QKI in cells during infection.
FIGURE 2.

DENV4 RNA interacts with QKI in infected cells. (A) Input samples and immunoprecipitated materials were analyzed by immunoblotting using antibodies against FLAG tag, showing immunoprecipitated FLAG-tagged QKI-5 and QKI-6, respectively. (B,C) Total RNA was isolated from immunoprecipitated materials and was analyzed by RT-qPCR using primers specific for indicated RNAs. Each experiment, in biological triplicates, was repeated three times. Statistical significance was determined using a two-tailed t-test: (*) P = 0.05; (**) P = 0.01; (***) P = 0.001. Error bars indicate SEM.
QKI depletion leads to enhanced viral particle production of DENV4
In order to determine whether QKI has a functional importance in viral replication, we performed siRNA-mediated knockdown of QKI followed by infection with DENVs. HuH7 cells were transfected with two independent siRNAs (siQKI-1 and siQKI-2) that targeted common regions in the 3′ UTR and in the open reading frame of all QKI transcript variants, respectively, or with a nontargeting control siRNA (NTC) as a negative control. Approximately 48 h after siRNA transfection, HuH7 cells were infected with the DENV4 or DENV2. Twenty-four or 48 h post-infection, tissue culture supernatants were harvested for focus forming assays to determine viral particle production. Total cellular RNA was harvested and analyzed for viral RNA levels by RT-qPCR with primers that amplify a region in the open reading frame. Relative levels of viral RNA were normalized to the geometric mean of two reference genes, SDHA and HPRT1, and are expressed relative to the NTC control. The QKI expression was efficiently knocked down by siQKI-1 and siQKI-2 in HuH7 cells, and this allowed us to test if QKI is functionally important in DENV infection (Fig. 3A).
FIGURE 3.

QKI depletion enhances viral particle production of DENV4. (A) Representative western blotting results, showing knockdown of QKI in HuH7 cells. (B–E) HuH7 were transfected with nontargeting control siRNA (NTC) or siRNAs targeting QKI (siQKI-1 and siQKI-2) and, two days later, cells were infected with DENV4 at MOI = 0.5 or with DENV2 at MOI = 10. Twenty-four hours post-DENV4 infection and 48 h post-DENV2 infection, supernatants were collected and viral titer was determined by focus formation assay. Cells were harvested for total RNA extraction, and levels of viral RNA accumulation were measured by RT-qPCR using primers that amplify a region in the middle of the viral coding sequence (gRNA). Each experiment, in biological triplicates, was repeated at least three times. Statistical significance was determined using a two-tailed t-test: (**) P = 0.01; (***) P = 0.001. Error bars indicate SEM.
First, we reported an enhanced production of infectious particles for DENV4 when QKI was depleted with two different siRNAs (approximately threefold increase in both siQKI-1 and siQKI-2 transfected cells) (Fig. 3B; Supplemental Fig. S2A). Next, when assessing viral RNA accumulation, we observed that siQKI-1 reduced DENV4 gRNA level while siQKI-2 did not, indicating that QKI knockdown did not impact DENV4 viral RNA accumulation (Fig. 3C; Supplemental Fig. S2B). Because DENV2 3′ UTR was not associated with QKI, we hypothesized that QKI depletion should not affect DENV2 replication. In support of this hypothesis, no consistent trends were observed in DENV2 viral particle production and DENV2 viral RNA accumulation using two independent siRNAs targeting QKI (Fig. 3D,E; Supplemental Fig. S2C,D). Taken together, these results indicate that siRNA-mediated silencing of QKI caused an increase of infectious particle production in DENV4 infected cells.
A QRE is important for QKI interaction in DENV 3′ UTR
Since our data showed QKI was a host restriction factor for DENV4, we mapped the QKI interaction site on the DENV4 3′ UTR. Modeling predicted that DENV4 has several relatively well-conserved secondary structures in its 3′ UTR, which includes a stem–loop (SL or exonuclease resistant RNA, known as xrRNA), two dumbbells (DB1 and DB2), and one 3′ end stem–loop (3′ SL) (Fig. 4A; Villordo et al. 2015). Unlike DENV4 3′ UTR, the DENV2 3′ UTR has one additional SL at its 5′ end and is slightly longer than the DENV4 3′ UTR (Fig. 4C; Chapman et al. 2014). To define the QKI binding site, we generated several DENV4 3′ UTR deletion mutants and performed RNA affinity chromatography experiments. Eluted samples were analyzed by western blotting using antibodies against QKI. DDX6 was used as a control because it has been shown to interact with the most well-conserved DB structures (Ward et al. 2011). D4 WT RNA bound to both QKI and DDX6, whereas D4 mut1 RNA, which contains only the SL, did not bind QKI and DDX6. D4 RNA mutants that include DBs (D4 mut2, 3, and 5) interacted with DDX6, as expected, but QKI only bound D4 mut4 and 5. QKI is thus likely to associate with a region that lies between the stop codon and the SL (Fig. 4D).
FIGURE 4.
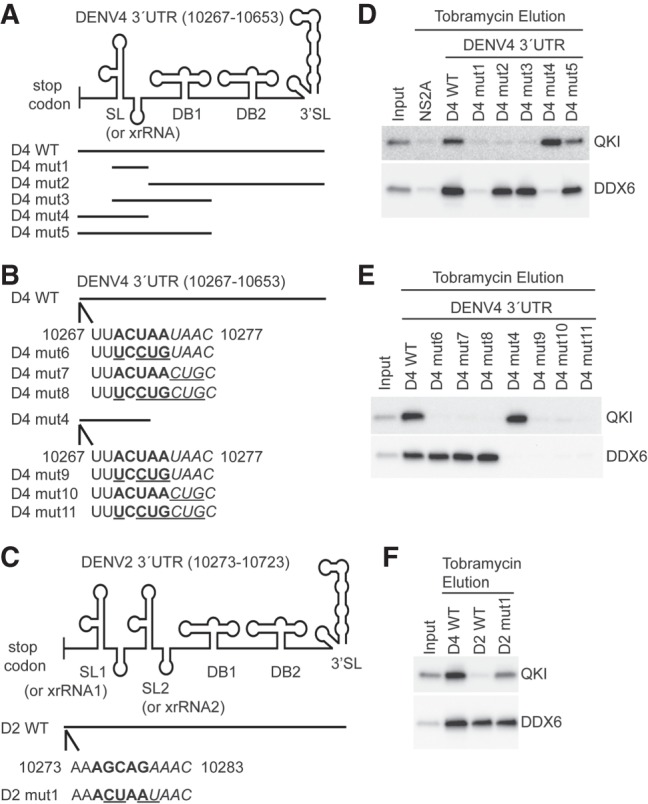
A QRE is important for QKI interaction in DENV 3′ UTR. (A–C) Schematic presentation of DENV 3′ UTR sequences used in RNA chromatography experiments. (D–F) Representative western blotting results showing interaction between indicated RNAs and QKI or DDX6. Indicated RNAs were immobilized on tobramycin sepharose beads, and RNA bound beads were mixed with HuH7 cell lysates. After incubation, beads were washed and eluted with excessive amount of tobramycin. Eluates were analyzed by immunoblotting.
QKI binds to a specific RNA sequence element, known as QRE, and it has been defined as a bipartite sequence with a core site ACUAAY and a half site UAAY (Galarneau and Richard 2005). Indeed, a QRE was identified 2 nt downstream from the stop codon in DENV4 3′ UTR (Fig. 4B). To test if this QRE is important for QKI binding in DENV4 3′ UTR, we mutated the core site alone, the half site alone, or both the core site and the half site in the context of full length DENV4 3′ UTR (D4-WT) or truncated DENV4 3′ UTR (D4 mut4). Mutated nucleotides are underlined in Figure 4B. All these mutants were tested in RNA chromatography experiments. In Figure 4E, mutations of QRE in DENV4 3′ UTR abolished its interaction with QKI but kept DDX6 association intact (D4 mut6-8). Consistently, some mutations of the QRE in a shorter version of DENV4 3′ UTR (D4 mut9-11) disrupted QKI-RNA interactions as well (Fig. 4E). These results suggest that QRE is required for QKI interaction in the DENV4 3′ UTR. It should be noted that although an identical 3′ UTR QRE was identified in an independent DENV4 clinical isolate (Bai et al. 2013), this QRE is not found in all DENV4 viruses.
Next, we determined if introduction of QRE to DENV2 3′ UTR is sufficient to render its interaction with QKI. We introduced the QRE 2 nt downstream from the stop codon in DENV2 3′ UTR, which is the same position as in the DENV4 3′ UTR (Fig. 4C). In support of our hypothesis, the introduction of a heterologous QRE in the DENV2 3′ UTR allowed its physical association with QKI (Fig. 4F). The QKI binding to D2mut1 was weaker than binding to DENV4 3′ UTR RNAs, suggesting that other proteins and/or RNA elements in the DENV4 3′ UTR might also contribute to QKI-RNA interactions. These results strongly suggest that QRE is critical in mediating QKI interaction in DENV 3′ UTRs.
Functional characterization of QRE in DENV 3′ UTRs
Since QRE was demonstrated to play critical roles in mediating QKI interaction in DENV4 3′ UTRs, we hypothesized that QRE would have functional significance in DENV4 replication. In order to test this hypothesis, we generated a mutant DENV4 virus that lacked the QRE in its 3′ UTR (DENV4 mut8). The sequence was mutated from ACUAAUAAC to UCCUGCUGC starting nucleotide 10,269 in DENV4 genome. For comparison, we also generated mutant DENV2 viruses with a heterologous QRE in their 3′ UTR (DENV2 mut1). The sequence was changed from AGCAGAAAC to ACUAAUAAC starting nucleotide 10,275 in DENV2 genome. HuH7 cells were infected with DENV4 WT viruses or DENV4 mut8 viruses at MOI = 0.5. Twenty-four hours post-infection, supernatants were harvested to determine the titer of infectious viral particles using focus forming assay. Total RNA was isolated from cells and viral RNA levels were assessed using RT-qPCR. In parallel experiments, cells were infected with DENV2 WT viruses or DENV2 mut1 viruses. Two days after infection, samples were harvested and analyzed as described above. Enhancement (∼10-fold) of infectious particle production was observed when cells were infected with DENV4 mut8 viruses as compared with DENV4 WT viruses (Fig. 5A; Supplemental Fig. S3A). Consistently, removal of QRE in DENV4 3′-UTR resulted in an approximately sevenfold increase of viral RNA accumulation (Fig. 5B; Supplemental Fig. S3B). For DENV2, introduction of QRE into DENV2 3′ UTR caused ∼50% reduction of viral titer in infected cells (Fig. 5C; Supplemental Fig. S3C). In line with this result, viral RNA accumulation was reduced by ∼50% when QRE was introduced into DENV2 3′ UTR (Fig. 5D; Supplemental Fig. S3D). Furthermore, we performed a time course experiment to monitor viral growth of DENV2 WT and mut1 viruses. In Figure 5E, inhibitory effect of the heterologous QRE was significant from 48 h post-infection and resulted in a 10-fold lower viral titer at 96 h post-infection (Supplemental Fig. S3E). Taken together, the data suggest that the presence of QRE in DENV 3′ UTR reduces viral multiplication in a serotype-independent manner.
FIGURE 5.

Functional characterization of QRE in DENV 3′ UTR. (A,B) HuH7 cells were infected with wild-type DENV4 viruses (DENV4-WT) or mutant DENV4 viruses (DENV4-mut8) at MOI = 0.5. Twenty-four hours post-infection, supernatants were collected and titer was measured by focus formation assay. Total RNA was isolated from infected cells and analyzed by RT-qPCR using primers that amplify a region in the middle of the viral coding sequence (gRNA). (C,D) HuH7 cells were infected with wild-type DENV2 viruses (DENV2-WT) or mutant DENV2 viruses (DENV2-mut1) at MOI = 10. Two days post-infection, samples were collected and analyzed as described above. (E) HuH7 cells were infected with DENV2 WT or mut1 viruses at MOI = 10. Supernatants were harvested at 1, 48, and 96 h post-infection and titer was measured by focus formation assay. Each experiment, in biological triplicates, was repeated at least two times. Statistical significance was determined using a two-tailed t-test: (**) P = 0.01; (***) P = 0.001. Error bars indicate SEM.
Removal of QRE in DENV4 3′ UTR increases relative translation efficiency
To investigate the molecular mechanisms by which QRE regulates viral replication, we generated Renilla luciferase (R Luc) reporters containing DENV4 3′ UTRs with or without QRE and sought to determine whether QRE affects the reporter expression at the translational level or at the transcriptional level. Three R Luc reporters were made: R Luc without DENV4 3′ UTR (R Luc), R Luc with DENV4 3′ UTR WT that has QRE (R Luc-D4 3′ UTR WT), and R Luc with DENV4 3′ UTR mut8 that does not have QRE (R Luc-D4 3′ UTR mut8). These R Luc reporter plasmids were transiently transfected into HuH7 cells together with a firefly luciferase (F Luc) transfection control. Approximately 24 h after transfection, cells were harvested and dual luciferase reporter assay was performed to assess the protein level of luciferase by measuring luciferase activity. In parallel experiments, total RNA was isolated from transfected cells for RT-qPCR analysis to measure the level of luciferase RNA accumulation. For both dual luciferase assay and RT-qPCR results, R Luc signal was normalized to F Luc signal, and data are presented as the fold change of indicated R Luc reporters with DENV4 3′ UTRs relative to R Luc reporter that does not have DENV4 3′ UTR (R Luc).
Similar to the observations on viral particle production, removal of QRE in DENV4 3′ UTR caused an increase (∼25%) of relative luciferase activity in dual luciferase assay (Supplemental Fig. S4A). The same mutation in DENV4 3′ UTR reduced the relative abundance of RNA levels (Supplemental Fig. S4B). To obtain a measure of relative translation efficiency, we divided the normalized relative luciferase activity by the normalized relative abundance of RNA levels. In Supplemental Figure S4C, a statistically significant approximately twofold increase of relative translation efficiency was observed in R Luc reporter with DENV4 3′ UTR that lacks QRE as compared to DENV4 3′ UTR WT that has QRE, suggesting that the presence of 3′ UTR QRE inhibits translation.
DISCUSSION
DENV RNA molecules are exposed to and physically interact with cellular RBPs throughout the course of infection. In particular, RBPs associated with DENV 3′ UTRs have been shown to play increasingly important roles in viral pathogenesis (Bidet and Garcia-Blanco 2014). To better understand RBP binding, we performed an interactome analysis of DENV 1-4 3′ UTRs using SILAC-based quantitative mass spectrometry following RNA affinity chromatography. This SILAC-based methodology facilitated the distinction of high confidence candidate host DENV 3′ UTRs interacting RBPs from false positive hits in a quantitative manner. From this mass spectrometry screening, we identified a list of putative host RBPs that interact with DENV1-4 3′ UTRs. The binding of selected protein candidates to DENV 3′ UTRs was confirmed using western blot analysis. Among these proteins, we focused on QKI because of its preferential binding to the DENV4 3′ UTR. Functional characterization revealed that QKI is a novel host restriction factor of DENV.
The preferential binding of QKI to DENV4 3′ UTR led us to discover its functional importance in inhibiting DENV4 propagation. Silencing of QKI caused an increase of viral particle production in DENV4 infected cells. On the contrary, no consistent changes were observed in DENV2 replication between control cells and two independent QKI knocked down treatments. These results suggest that QKI is not a universal host restriction factor against all DENVs and may have versatile roles in regulating virus replication. This notion is substantiated by the finding that QKI promotes Herpesvirus replication (Sánchez-Quiles et al. 2011) and ZIKV replication (Ramanathan et al. 2018). Although detailed molecular mechanisms of these QKI-mediated proviral and antiviral activities remain poorly understood, our experiments using mutant infectious clones suggest that QRE in the DENV 3′ UTR was the key for QKI antiviral activity. We demonstrated that removal of QRE from DENV4 3′-UTR abolished its interaction with QKI and resulted in a significant increase of viral particle production. Introduction of QRE into DENV2 3′ UTR confers the ability for QKI interaction and reduced infectious particle production. Intriguingly, the position of QRE in the viral genome is likely to determine the efficacy of QKI-mediated immunity. We examined the full-length DENV2 genome sequence manually and also identified putative QRE in the open reading frame; and yet, wild-type DENV2 viruses did not seem to encounter negative selective pressure from QKI and replicated efficiently in both control and QKI depleted cells. Insertion of QRE in the 3′ UTR of DENV2, instead, inhibited its replication. These results suggest that QRE in the 3′ UTR plays a pivotal role in regulating DENV propagation and that the QRE-mediated inhibitory effect is not serotype-dependent.
The 3′ UTRs are critical regions for regulating DENV RNA translation (Holden and Harris 2004; Chiu et al. 2005; Wei et al. 2009). The observed increase of DENV4 viral particle production in QKI knocked down cells while viral RNA level remained unaltered suggests that QKI can repress the translation of viral genomes by interacting with 3′ UTR QRE. In line with these results, our reporter assay showed that removal of QRE from reporters with DENV4 3′-UTR caused an increase of the relative luciferase activity but reduced the relative abundance of reporter RNA level. Although our plasmid-based system could not differentiate whether lower relative abundance of reporter RNA was caused by reduced RNA transcription or accelerated RNA decay, the measurement of relative translation efficiency indicated an approximately twofold increase in the absence of QRE, suggesting that QKI negatively regulates viral RNA translation. This scheme of regulation is consistent with a model where QKI sequesters viral RNA in a translationally quiescent but stable complex, generally known as stress granules (Wang et al. 2010). These stress granules could transport the bound viral RNA species to P-bodies where RNA decay takes place (Kedersha et al. 2005) and thereby viral replication is inhibited. Although stress granule formation has yet to be clearly defined during DENV infection (Emara and Brinton 2007; Ruggieri et al. 2012; Xia et al. 2015), partly because of the use of various markers in different cells, it is still possible that QKI transports viral RNAs to a translationally less active area, such as the nucleus (David et al. 2012), if not stress granules, to repress viral protein production. This QKI nucleus-cytoplasm shuttling has been observed in cells under various stress conditions (Wu et al. 1999; Sánchez-Quiles et al. 2011).
To conclude, we demonstrated that QKI is a novel host restriction factor inhibiting DENVs replication. By binding to QRE present in the 3′ UTR of viral genome, QKI can serve as a pattern recognition receptor and directly antagonize viral replication. Moreover, upon viral QRE recognition, it is plausible that QKI could also initiate a series of signaling pathways and contribute to the establishment of an antiviral state for host cells. Notably, how this QRE shapes viral evolution remains an open question. The loss of QRE increases the replication of the studied DENV viruses; however, the most cytopathic viruses in culture are not always the most efficient pathogens. Further investigation would shed light into the evolutionary role of QRE in DENVs.
MATERIALS AND METHODS
Cell culture and antibodies
HuH-7 and HEK293T cells were maintained in Dulbecco's modified Eagle medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (Pen Strep, Gibco) in a 37°C humidified incubator with 5% CO2. BHK-21 cells were propagated in RPMI Medium 1640 (Gibco) supplemented with 10% FBS and 1% Pen Strep at 37°C with 5% CO2. C6/36 cells were maintained in RPMI media supplemented with 2% fetal calf serum (FCS) at 28°C. Tetracycline-inducible cell lines were generated using the Flp-In T-REx system (Thermo Fisher Scientific) according to manufacturer's protocol. After introduction of transgenes, HEK-293 Flp-In T-REx cells were grown in medium with 100 µg/mL of hygromycin B and 15 µg/mL of blasticidin. The following primary antibodies were utilized during western blotting, RNA immunoprecipitation, and focus formation assay techniques: rabbit G3BP1 antibody (A302-033; Bethyl Laboratories), rabbit G3BP2 antibody (ab86135; Abcam), rabbit CAPRIN1 antibody (15112-1-AP; Proteintech Group), rabbit DDX6 antibody (A300-460A; Bethyl Laboratories), rabbit QKI antibody (A300-183A; Bethyl Laboratories), mouse FLAG antibody (F3165; Sigma), mouse IgG antibody (12–371; Millipore) as isotype control, mouse pan-actin (MAS-11869; Thermo Fisher Scientific), and mouse pan flaviviral envelope 4G2 antibody.
Small interfering RNA (siRNA) transfection
HuH-7 cells were seeded at 5 × 104 cells per well in 24-well plates immediately prior to siRNA transfection using Lipofectamine RNAiMAX reagent (Invitrogen) following manufacturer's protocol. Expression of QKI in HuH-7 cells was knocked down by reverse transfection with two independent siRNAs (Dharmacon), each at a final concentration of 50 nM: siQKI-1: 5′-CUAUUAACCCACAGCAUUAUU-3′ (Conn et al. 2015) and siQKI-2: 5′-GGACUUACAGCCAAACAACUU-3′ (Chen et al. 2007). The nontargeting control siRNA (NTC), ON-TARGETplus Nontargeting siRNA#2 (D-001810-02), was also transfected at a final concentration of 50 nM.
Western blotting
Samples were lysed in RIPA buffer (Cell Signaling Technology), and proteins were separated under denaturing conditions on 4%–15% polyacrylamide gels (Bio-Rad). After samples were transferred to polyvinylidene difluoride membranes (PVDF, Bio-Rad), blots were blocked and then incubated with primary antibodies overnight at 4°C. Goat anti-mouse HRP (115-035-003; Jackson ImmunoResearch) and goat anti-rabbit HRP (111-035-003; Jackson ImmunoResearch) were used for blot visualization on a chemiluminescence imaging system (Bio-Rad). Goat anti-mouse Dylight 680 (35518; Thermo Fisher Scientific) was used for detection of primary antibody on an Odyssey CLx imaging system (LI-COR).
Real-time quantitative PCR (RT-qPCR)
Total RNA for RNA immunoprecipitation experiments was isolated using RNAzol RT (Molecular Research Center). All other RNA was extracted using the EZNA Total RNA Kit I (OMEGA bio-tek). To assess the RNA levels of DENV genome (gRNA), DENV 3′ UTR, 18S, U1, and hnRNPA1, RT-qPCR was performed on a CFX96 RT thermal cycler (Bio-Rad) using iTaq Universal SYBR Green One-Step reagent (Bio-Rad) according to manufacturer's protocol. To measure RNA levels of beta catenin (CTNNB1), splicing factor 3b subunit 1 (SF3B1), luciferase reporters, hypoxanthine phosphoribosyl-transferase1 (HPRT1), and succinate dehydrogenase complex subunit A (SDHA), RNA samples were first reverse transcribed with the iScript cDNA Synthesis Kit (Bio-Rad). Then, RT-qPCR was accomplished using SensiFAST SYBR No-ROX reagent (BioLine) according to manufacturer's protocol. Primer sequences are listed in Supplemental Table S2.
RNA immunoprecipitation (RIP)
RNA immunoprecipitations were performed as described previously (Phillips et al. 2016), with slight modifications. One and a half million of Flp-In T-REx HEK-293 cells were seeded in 100 mm dishes. The day after seeding, cells were treated with 5 µg/mL tetracycline to induce protein expression. Twenty-four hours after tetracycline induction, Flp-In T-REx HEK-293 cells expressing FLAG-tagged QKI-5 or QKI-6 were infected with DENV4 at MOI = 10. One-day post-infection, cells were harvested, pelleted, and lysed in a buffer volume roughly equivalent to the cell pellet volume of RIP lysis buffer (200 mM KCl, 20 mM HEPES pH7.2, 2% N-dodecyl-β-D-maltoside, 1% Igepal CA-360, 100 U/mL Murine RNase inhibitor [NEB]). Subsequent lysates were cleared by centrifugation, and protein was normalized across samples to 400 µg in total volume of 500 µL RIP assay buffer (50 mM Tris HCl pH 7.5, 150 mM NaCl, 1 mM MgCl2, and 0.05% Igepal CA-360) per RIP reaction. To prepare RIP assay beads, Dynabeads protein G (Invitrogen) were washed with RIP assay buffer, blocked with BSA, and washed again with RIP assay buffer the day before cell harvest. The blocked beads were then incubated with 5 µg of mouse IgG control antibody or 5 µg FLAG antibody with head-to-tail rotation at 4°C overnight. Antibody-coupled beads were washed three times with RIP assay buffer and subsequently incubated with the prepared lysates on rotation at 4°C for 1 h. Complexes were washed four times in RIP assay buffer, and immunoprecipitated protein and RNA were analyzed by western blotting and RT-qPCR, respectively.
Plasmid construction and in vitro transcription
The plasmids containing tobramycin aptamer sequences and various DENV 3′ UTR sequences were generated in two steps. First, tobramycin aptamer sequences were amplified, using forward primer 5′-CGCGCTAGCGGGAGAAGACGACCGACCAGAATC-3′ and reverse primer 5′-ATAGGATCCAGACGCACATACCCGGCC-3′ from a synthesized DNA oligo. The PCR product was digested with NheI and BamHI and then ligated into pcDNA3 plasmids. The resultant plasmid (pcDNA3-Tobra) was used to generate all constructs containing DENV 3′ UTR sequences for in vitro transcription. DENV 3′ UTR fragments were amplified from infectious clone plasmids using primers indicated in the Supplemental Material. The PCR products were digested with BamHI and XhoI and then ligated into pcDNA3-Tobra plasmids.
The QKI-5 gene was amplified using forward primer 5′-CGCGGATCCATGGACTACAAAGACGATGACGACAAGATGGTCGGGGAAATGGAAACG-3′ and reverse primer 5′-CGCGCGGCCGCTTAGTTGCCGGTGGCGGCTCGGTC-3′ from an Origene plasmid (RC205779). QKI-6 was amplified using the same forward primer for QKI-5 and reverse primer 5′-CGCGCGGCCGCTTAGCCTTTCGTTGGGAAAGCCATAC-3′ from a Origene plasmid (RC224090). The PCR products were digested with BamHI and NotI and then ligated into pcDNA5 plasmids.
For in vitro transcription, plasmids were linearized with XhoI first. After purification, linearized plasmids were used for MEGAscript T7 RNA polymerase in vitro transcription reactions (Ambion) to generate RNA according to manufacturer's instructions.
Tobramycin RNA affinity chromatography
These experiments were performed as described previously (Ward et al. 2011, 2014). In brief, NHS-activated sepharose beads (GE Healthcare) were washed with 1 mM HCl four times and resuspended in coupling buffer (0.2 M NaHCO3, 0.5 M NaCl, pH 8.3) containing 5 mM Tobramycin. Following incubation overnight at 4°C, beads were spun down and resuspended in blocking buffer. After 1 h incubation at 4°C, beads were washed three times and resuspended in PBS. In vitro transcribed RNAs in RNA binding buffer (20 mM Tris pH 7.0, 1 mM CaCl2, 1 mM MgCl2, 75 mM NaCl, 145 mM KCl, 0.1 mg/mL yeast RNA, 0.2 mM DTT) were heated at 95°C for 5 min and cooled to room temperature. Renatured RNAs were mixed with 30 µL Tobramycin sepharose beads (50% slurry) at 4°C for 1 h. After incubation, RNA bound beads were washed two times and mixed with cell lysates at 4°C for 1 h with head-to-tail rotation. Beads were then washed with protein washing buffer (20 mM Tris pH 7.0, 1 mMCaCl2, 1 mM MgCl2, 145 mM KCl, 75 mM NaCl, 0.5% Igepal CA-360) and then incubated for 5 min at room temperature with elution buffer (20 mM Tris pH 7.0, 1 mM CaCl2, 1 mM MgCl2, 145 mM KCl, 5 mM Tobramycin, 0.2 mM DTT). Beads were centrifuged and supernatants were collected as eluate. Eluates were subjected to mass spectrometry analysis or analyzed by immunoblotting.
Mass spectrometry and data analysis
The eluates were separated by 4%–12% NuPage Novex Bis–Tris Gel (Invitrogen), stained using the Colloidal Blue Staining Kit (Invitrogen), and digested with trypsin as described previously (Swa et al. 2012). Samples were analyzed using Thermo Scientific Easy-nLC with 15 cm column (Acclaim PepMapTM RSLC, C18, 100 Å, 2 µm, 75 mm × 15 cm) coupled to an Orbitrap Classic (Thermo Scientific). Peptides were loaded with buffer A (0.1% formic acid, 2% acetonitrile) and eluted with a 120 min gradient at a flowrate of 250 nL/min from 8%–40% buffer B (0.1% formic acid, 80% acetonitrile). Mass spectra were acquired in positive ion mode. Survey full scan MS spectra (m/z 310–1400) were acquired with a resolution of r = 60,000, an AGC target of 1 × 106, and a maximum injection time of 1000 msec. The ten most intense peptide ions in each survey scan with an ion intensity of >2000 counts and a charge state ≥2 were isolated sequentially to a target value of 5 × 103, and fragmented in the linear ion trap by collision-induced dissociation using normalized collision energy of 35%. A dynamic exclusion was applied using a maximum exclusion list of 500 with one repeat count, and exclusion duration of 30 sec. SILAC peptide and protein quantification was performed with MaxQuant (Cox and Mann 2008) version 1.5.0.30 using default settings. Database searches of MS data were performed using Uniprot human fasta with tryptic specificity, allowing a maximum of two missed cleavages, two labeled amino acids, and an initial mass tolerance of 7 ppm for precursor ions and 0.5 kDa for fragment ions. Cysteine carbamidomethylation was searched as a fixed modification, and N-acetylation and oxidized methionine were searched as variable modifications. Labeled arginine and lysine were specified as fixed or variable modifications, depending on the prior knowledge about the parent ion. Maximum false discovery rates were set to 0.01 for both protein and peptide. Proteins were considered identified when supported by at least one unique peptide with a minimum length of seven amino acids.
Viruses and infections
DENV4 and DENV2 were initially isolated from patient sera and propagated as described previously (Low et al. 2006; Manokaran et al. 2015). Mutant viruses were constructed using approaches reported by Siridechadilok et al. (2013), with several modifications. In brief, first of all, wild-type viral genome RNA was extracted from culture media and cDNA was synthesized with the Superscript III First Strand Synthesis Kit (Invitrogen) using primers 5′-AGAACCTGTTGATTCAACAG-3′ for DENV2 and 5′-AGAACCTGTTGGATCAACAACACC-3′ for DENV4. Next, six PCR fragments of 1500–2000 nt were generated from cDNA by six pairs of primers (DENV2: primers# 35–46, DENV4: primers #47–58; Supplemental Material). These six fragments cover the whole viral genome (DENV2 and DENV4, respectively). To introduce mutations within the 3′ UTR of DENV2, the Site-Directed Mutagenesis Kit (NEB) was used as per manufacturer's protocol with primers 5′-GTGGTAGAAACTAATAACATCATGAGAC-3′ and 5′-AAGACTCCTGCCTCTTCCTCT-3′. The same protocol was used to introduce mutations in DENV4 3′ UTR using primers 5′-CTGTAATTTCCTGCTGCAAACACCAAA-3′ and 5′-AACTCCTTCACTTTCGGAAGGAGC-3′. After confirmation of site-directed mutagenesis by sequencing, six PCR fragments encompassing a complete DENV2 or DENV4 genome were amplified. These fragments were assembled with linearized expression vectors containing a CMV promoter, hepatitis D virus (HDV) ribozyme terminator, and SV40 poly(A) sequences using NEBuilder HiFi DNA Assembly Master Mix (NEB), according to manufacturer's instructions. Resultant ligated products were transfected into HEK 293T cells using lipofectamine 2000 (Invitrogen). Supernatants containing viruses were harvested 3 d after transfection, and, subsequently, viruses were further propagated in C6/36 cells.
For infection in RIP experiments, Flp-In T-Rex HEK-293 cells expressing FLAG-tagged QKI-5 or QKI-6 were infected with DENV4 in serum-free media for 1 h using an MOI of 10. The inoculum was then substituted with reduced-serum media (DMEM containing 2% FBS and 1% Pen Strep). Twenty-four hours post-infection, cells were washed with PBS and processed as described above. Infection of HuH-7 cells was carried out at an MOI of 0.5 for DENV4. Twenty-four hours post-infection, supernatants were collected for titer determination using focus formation assay. Total RNA was isolated from cells and analyzed by RT-qPCR. DENV2 infections were performed at an MOI of 10. Forty-eight hours post-infection, both supernatants and cells were harvested and analyzed as described above. Regarding infections comparing wild-type and mutant DENVs, DENV4 infections were performed at an MOI of 0.5 and samples were collected for analysis 24 h post-infection. Wild-type and mutant DENV2 viruses were used for infection at an MOI of 10 and samples were analyzed 48 h post-infection unless indicated otherwise.
Focus formation assay
BHK-21 cells were seeded at 1.5 × 105 cells per well in 24-well plates the day prior to infection. Cell monolayers were washed with serum-free RPMI 1640 media and infected with 10-fold serially diluted supernatant media from previously infected HuH-7 cells. After 1 h, inoculum was substituted with overlay media containing RPMI 1640, 2% FBS, and 1% Aquacide II (Calbiochem). Three days later, media was disposed and BHK-21 cells were fixed for 30 min with 3.75% formaldehyde in 1× PBS. Fixed cells were permeabilized and blocked for 30 min with 1× PBS containing 0.5% Triton X-100 and 2% FBS. Viral foci were detected using the 4G2 antibody and anti-mouse Dylight 680 secondary antibody. After visualization on an Odyssey CLx imaging system, viral foci were counted and expressed as FFU/mL.
Accession numbers
DENV1(EU081230.1), DENV2(EU081177.1), DENV3(EU081190.1), DENV4(GQ398256.1), QKI-5(NM_006775.2), QKI-6(NM_ 206853.2), DENV2 NGC NS2A (AF038403.1: 3739–4224 with three different nucleotides observed at C3995T, T4001C, and A4156G).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank our colleagues from the Pompon/Garcia-Blanco and Ooi laboratories, Duke-NUS Medical School, for their support and materials. We also thank colleagues from Bradrick/Garcia-Blanco laboratory, University of Texas Medical Branch for comments on the manuscript, and Asfa Alli Shaik and Siok Ghee Ler from Gunaratne's laboratory, IMCB, A*STAR for technical support in mass spectrometry analysis. Support for this research came from a CBRG (NMRC/CBRG/0074/2014) and from the Duke-NUS Signature Research Programme funded by the Agency for Science, Technology and Research (A*STAR), Singapore, and the Ministry of Health, Singapore.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.064006.117.
Freely available online through the RNA Open Access option.
REFERENCES
- Aberg K, Saetre P, Jareborg N, Jazin E. 2006. Human QKI, a potential regulator of mRNA expression of human oligodendrocyte-related genes involved in schizophrenia. Proc Natl Acad Sci 103: 7482–7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anwar A, Leong KM, Ng ML, Chu JJ, Garcia-Blanco MA. 2009. The polypyrimidine tract-binding protein is required for efficient dengue virus propagation and associates with the viral replication machinery. J Biol Chem 284: 17021–17029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Z, Liu Q, Jiang LY, Liu LC, Cao YM, Xu Y, Jing QL, Luo L, Yang ZC, Jiang YQ, et al. 2013. Complete genome sequence of dengue virus serotype 4 from Guangzhou, China. Genome Announc 1: e00299–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, et al. 2013. The global distribution and burden of dengue. Nature 496: 504–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidet K, Garcia-Blanco MA. 2014. Flaviviral RNAs: weapons and targets in the war between virus and host. Biochem J 462: 215–230. [DOI] [PubMed] [Google Scholar]
- Bidet K, Dadlani D, Garcia-Blanco MA. 2014. G3BP1, G3BP2 and CAPRIN1 are required for translation of interferon stimulated mRNAs and are targeted by a dengue virus non-coding RNA. PLoS Pathog 10: e1004242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos RK, Wong B, Xie X, Lu YF, Shi PY, Pompon J, Garcia-Blanco MA, Bradrick SS. 2017. RPLP1 and RPLP2 are essential flavivirus host factors that promote early viral protein accumulation. J Virol 91: e01706–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman EG, Moon SL, Wilusz J, Kieft JS. 2014. RNA structures that resist degradation by Xrn1 produce a pathogenic dengue virus RNA. Elife 3: e01892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Tian D, Ku L, Osterhout DJ, Feng Y. 2007. The selective RNA-binding protein quaking I (QKI) is necessary and sufficient for promoting oligodendroglia differentiation. J Biol Chem 282: 23553–23560. [DOI] [PubMed] [Google Scholar]
- Chiu WW, Kinney RM, Dreher TW. 2005. Control of translation by the 5′- and 3′-terminal regions of the dengue virus genome. J Virol 79: 8303–8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn SJ, Pillman KA, Toubia J, Conn VM, Salmanidis M, Phillips CA, Roslan S, Schreiber AW, Gregory PA, Goodall GJ. 2015. The RNA binding protein quaking regulates formation of circRNAs. Cell 160: 1125–1134. [DOI] [PubMed] [Google Scholar]
- Cox J, Mann M. 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26: 1367–1372. [DOI] [PubMed] [Google Scholar]
- Darbelli L, Richard S. 2016. Emerging functions of the Quaking RNA-binding proteins and link to human diseases. Wiley Interdiscip Rev RNA 7: 399–412. [DOI] [PubMed] [Google Scholar]
- David A, Dolan BP, Hickman HD, Knowlton JJ, Clavarino G, Pierre P, Bennink JR, Yewdell JW. 2012. Nuclear translation visualized by ribosome-bound nascent chain puromycylation. J Cell Biol 197: 45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin RG, Shiue L, Prins J, de Boer HC, Singh A, Fagg WS, van Gils JM, Duijs JM, Katzman S, Kraaijeveld AO, et al. 2016. Quaking promotes monocyte differentiation into pro-atherogenic macrophages by controlling pre-mRNA splicing and gene expression. Nat Commun 7: 10846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Miguel FJ, Pajares MJ, Martínez-Terroba E, Ajona D, Morales X, Sharma RD, Pardo FJ, Rouzaut A, Rubio A, Montuenga LM, et al. 2016. A large-scale analysis of alternative splicing reveals a key role of QKI in lung cancer. Mol Oncol 10: 1437–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebersole TA, Chen Q, Justice MJ, Artzt K. 1996. The quaking gene product necessary in embryogenesis and myelination combines features of RNA binding and signal transduction proteins. Nat Genet 12: 260–265. [DOI] [PubMed] [Google Scholar]
- Emara MM, Brinton MA. 2007. Interaction of TIA-1/TIAR with West Nile and dengue virus products in infected cells interferes with stress granule formation and processing body assembly. Proc Natl Acad Sci 104: 9041–9046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagg WS, Liu N, Fair JH, Shiue L, Katzman S, Donohue JP, Ares M Jr. 2017. Autogenous cross-regulation of Quaking mRNA processing and translation balances Quaking functions in splicing and translation. Genes Dev 31: 1894–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galarneau A, Richard S. 2005. Target RNA motif and target mRNAs of the Quaking STAR protein. Nat Struct Mol Biol 12: 691–698. [DOI] [PubMed] [Google Scholar]
- Garcia-Montalvo BM, Medina F, del Angel RM. 2004. La protein binds to NS5 and NS3 and to the 5′ and 3′ ends of dengue 4 virus RNA. Virus Res 102: 141–150. [DOI] [PubMed] [Google Scholar]
- Gomila RC, Martin GW, Gehrke L. 2011. NF90 binds the dengue virus RNA 3′ terminus and is a positive regulator of dengue virus replication. PLoS One 6: e16687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr, Jungkamp AC, Munschauer M, et al. 2010. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141: 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MP, Nagel RJ, Fagg WS, Shiue L, Cline MS, Perriman RJ, Donohue JP, Ares M Jr. 2013. Quaking and PTB control overlapping splicing regulatory networks during muscle cell differentiation. RNA 19: 627–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy RJ, Loushin CL, Friedrich VL Jr, Chen Q, Ebersole TA, Lazzarini RA, Artzt K. 1996. Neural cell type-specific expression of QKI proteins is altered in quakingviable mutant mice. J Neurosci 16: 7941–7949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden KL, Harris E. 2004. Enhancement of dengue virus translation: role of the 3′ untranslated region and the terminal 3′ stem-loop domain. Virology 329: 119–133. [DOI] [PubMed] [Google Scholar]
- Kedersha N, Stoecklin G, Ayodele M, Yacono P, Lykke-Andersen J, Fritzler MJ, Scheuner D, Kaufman RJ, Golan DE, Anderson P. 2005. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J Cell Biol 169: 871–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo T, Furuta T, Mitsunaga K, Ebersole TA, Shichiri M, Wu J, Artzt K, Yamamura K, Abe K. 1999. Genomic organization and expression analysis of the mouse qkI locus. Mamm Genome 10: 662–669. [DOI] [PubMed] [Google Scholar]
- Larocque D, Galarneau A, Liu HN, Scott M, Almazan G, Richard S. 2005. Protection of p27Kip1 mRNA by quaking RNA binding proteins promotes oligodendrocyte differentiation. Nat Neurosci 8: 27–33. [DOI] [PubMed] [Google Scholar]
- Lei Y, Huang Y, Zhang H, Yu L, Zhang M, Dayton A. 2011. Functional interaction between cellular p100 and the dengue virus 3′ UTR. J Gen Virol 92: 796–806. [DOI] [PubMed] [Google Scholar]
- Liu R, Yue L, Li X, Yu X, Zhao H, Jiang Z, Qin E, Qin C. 2010. Identification and characterization of small sub-genomic RNAs in dengue 1–4 virus-infected cell cultures and tissues. Biochem Biophys Res Commun 391: 1099–1103. [DOI] [PubMed] [Google Scholar]
- Low JG, Ooi EE, Tolfvenstam T, Leo YS, Hibberd ML, Ng LC, Lai YL, Yap GS, Li CS, Vasudevan SG, et al. 2006. Early dengue infection and outcome study (EDEN)—study design and preliminary findings. Ann Acad Med Singapore 35: 783–789. [PubMed] [Google Scholar]
- Low JG, Ooi EE, Vasudevan SG. 2017. Current status of dengue therapeutics research and development. J Infect Dis 215: S96–S102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manokaran G, Finol E, Wang C, Gunaratne J, Bahl J, Ong EZ, Tan HC, Sessions OM, Ward AM, Gubler DJ, et al. 2015. Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science 350: 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marceau CD, Puschnik AS, Majzoub K, Ooi YS, Brewer SM, Fuchs G, Swaminathan K, Mata MA, Elias JE, Sarnow P, et al. 2016. Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature 535: 159–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng WC, Soto-Acosta R, Bradrick SS, Garcia-Blanco MA, Ooi EE. 2017. The 5′ and 3′ untranslated regions of the flaviviral genome. Viruses 9: E137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paranjape SM, Harris E. 2007. Y box-binding protein-1 binds to the dengue virus 3′-untranslated region and mediates antiviral effects. J Biol Chem 282: 30497–30508. [DOI] [PubMed] [Google Scholar]
- Phillips SL, Soderblom EJ, Bradrick SS, Garcia-Blanco MA. 2016. Identification of proteins bound to dengue viral RNA in vivo reveals new host proteins important for virus replication. mBio 7: e01865–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pijlman GP, Funk A, Kondratieva N, Leung J, Torres S, van der Aa L, Liu WJ, Palmenberg AC, Shi PY, Hall RA, et al. 2008. A highly structured, nuclease-resistant, noncoding RNA produced by flaviviruses is required for pathogenicity. Cell Host Microbe 4: 579–591. [DOI] [PubMed] [Google Scholar]
- Pilotte J, Larocque D, Richard S. 2001. Nuclear translocation controlled by alternatively spliced isoforms inactivates the QUAKING apoptotic inducer. Genes Dev 15: 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polacek C, Friebe P, Harris E. 2009. Poly(A)-binding protein binds to the non-polyadenylated 3′ untranslated region of dengue virus and modulates translation efficiency. J Gen Virol 90: 687–692. [DOI] [PubMed] [Google Scholar]
- Pompon J, Manuel M, Ng GK, Wong B, Shan C, Manokaran G, Soto-Acosta R, Bradrick SS, Ooi EE, Missé D, et al. 2017. Dengue subgenomic flaviviral RNA disrupts immunity in mosquito salivary glands to increase virus transmission. PLoS Pathog 13: e1006535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan M, Majzoub K, Rao DS, Neela PH, Zarnegar BJ, Mondal S, Roth JG, Gai H, Kovalski JR, Siprashvili Z, et al. 2018. RNA-protein interaction detection in living cells. Nat Methods 15: 207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggieri A, Dazert E, Metz P, Hofmann S, Bergeest JP, Mazur J, Bankhead P, Hiet MS, Kallis S, Alvisi G, et al. 2012. Dynamic oscillation of translation and stress granule formation mark the cellular response to virus infection. Cell Host Microbe 12: 71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccomanno L, Loushin C, Jan E, Punkay E, Artzt K, Goodwin EB. 1999. The STAR protein QKI-6 is a translational repressor. Proc Natl Acad Sci 96: 12605–12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Quiles V, Mora MI, Segura V, Greco A, Epstein AL, Foschini MG, Dayon L, Sanchez JC, Prieto J, Corrales FJ, et al. 2011. HSV-1 Cgal+ infection promotes quaking RNA binding protein production and induces nuclear-cytoplasmic shuttling of quaking I-5 isoform in human hepatoma cells. Mol Cell Proteomics 10: M111 009126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savidis G, McDougall WM, Meraner P, Perreira JM, Portmann JM, Trincucci G, John SP, Aker AM, Renzette N, Robbins DR, et al. 2016. Identification of Zika virus and dengue virus dependency factors using functional genomics. Cell Rep 16: 232–246. [DOI] [PubMed] [Google Scholar]
- Sebestyén E, Zawisza M, Eyras E. 2015. Detection of recurrent alternative splicing switches in tumor samples reveals novel signatures of cancer. Nucleic Acids Res 43: 1345–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessions OM, Barrows NJ, Souza-Neto JA, Robinson TJ, Hershey CL, Rodgers MA, Ramirez JL, Dimopoulos G, Yang PL, Pearson JL, et al. 2009. Discovery of insect and human dengue virus host factors. Nature 458: 1047–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siridechadilok B, Gomutsukhavadee M, Sawaengpol T, Sangiambut S, Puttikhunt C, Chin-inmanu K, Suriyaphol P, Malasit P, Screaton G, Mongkolsapaya J. 2013. A simplified positive-sense-RNA virus construction approach that enhances analysis throughput. J Virol 87: 12667–12674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swa HL, Blackstock WP, Lim LH, Gunaratne J. 2012. Quantitative proteomics profiling of murine mammary gland cells unravels impact of annexin-1 on DNA damage response, cell adhesion, and migration. Mol Cell Proteomics 11: 381–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannice KS, Durbin A, Hombach J. 2016. Status of vaccine research and development of vaccines for dengue. Vaccine 34: 2934–2938. [DOI] [PubMed] [Google Scholar]
- Viktorovskaya OV, Greco TM, Cristea IM, Thompson SR. 2016. Identification of RNA binding proteins associated with dengue virus RNA in infected cells reveals temporally distinct host factor requirements. PLoS Negl Trop Dis 10: e0004921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villordo SM, Filomatori CV, Sanchez-Vargas I, Blair CD, Gamarnik AV. 2015. Dengue virus RNA structure specialization facilitates host adaptation. PLoS Pathog 11: e1004604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villordo SM, Carballeda JM, Filomatori CV, Gamarnik AV. 2016. RNA structure duplications and flavivirus host adaptation. Trends Microbiol 24: 270–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lacroix G, Haines J, Doukhanine E, Almazan G, Richard S. 2010. The QKI-6 RNA binding protein localizes with the MBP mRNAs in stress granules of glial cells. PLoS One 5: e12824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Vogel G, Yu Z, Richard S. 2013. The QKI-5 and QKI-6 RNA binding proteins regulate the expression of microRNA 7 in glial cells. Mol Cell Biol 33: 1233–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward AM, Bidet K, Yinglin A, Ler SG, Hogue K, Blackstock W, Gunaratne J, Garcia-Blanco MA. 2011. Quantitative mass spectrometry of DENV-2 RNA-interacting proteins reveals that the DEAD-box RNA helicase DDX6 binds the DB1 and DB2 3′ UTR structures. RNA Biol 8: 1173–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward AM, Gunaratne J, Garcia-Blanco MA. 2014. Identification of dengue RNA binding proteins using RNA chromatography and quantitative mass spectrometry. Methods Mol Biol 1138: 253–270. [DOI] [PubMed] [Google Scholar]
- Wei Y, Qin C, Jiang T, Li X, Zhao H, Liu Z, Deng Y, Liu R, Chen S, Yu M, et al. 2009. Translational regulation by the 3′ untranslated region of the dengue type 2 virus genome. Am J Trop Med Hyg 81: 817–824. [DOI] [PubMed] [Google Scholar]
- Wu J, Zhou L, Tonissen K, Tee R, Artzt K. 1999. The quaking I-5 protein (QKI-5) has a novel nuclear localization signal and shuttles between the nucleus and the cytoplasm. J Biol Chem 274: 29202–29210. [DOI] [PubMed] [Google Scholar]
- Xia J, Chen X, Xu F, Wang Y, Shi Y, Li Y, He J, Zhang P. 2015. Dengue virus infection induces formation of G3BP1 granules in human lung epithelial cells. Arch Virol 160: 2991–2999. [DOI] [PubMed] [Google Scholar]
- Yamagishi R, Tsusaka T, Mitsunaga H, Maehata T, Hoshino S. 2016. The STAR protein QKI-7 recruits PAPD4 to regulate post-transcriptional polyadenylation of target mRNAs. Nucleic Acids Res 44: 2475–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Fu H, Zhang J, Lu X, Yu F, Jin L, Bai L, Huang B, Shen L, Feng Y, et al. 2010. RNA-binding protein quaking, a critical regulator of colon epithelial differentiation and a suppressor of colon cancer. Gastroenterology 138: 231–240 e231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yocupicio-Monroy RM, Medina F, Reyes-del Valle J, del Angel RM. 2003. Cellular proteins from human monocytes bind to dengue 4 virus minus-strand 3′ untranslated region RNA. J Virol 77: 3067–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yocupicio-Monroy M, Padmanabhan R, Medina F, del Angel RM. 2007. Mosquito La protein binds to the 3′ untranslated region of the positive and negative polarity dengue virus RNAs and relocates to the cytoplasm of infected cells. Virology 357: 29–40. [DOI] [PubMed] [Google Scholar]
- Zearfoss NR, Clingman CC, Farley BM, McCoig LM, Ryder SP. 2011. Quaking regulates Hnrnpa1 expression through its 3′ UTR in oligodendrocyte precursor cells. PLoS Genet 7: e1001269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Miner JJ, Gorman MJ, Rausch K, Ramage H, White JP, Zuiani A, Zhang P, Fernandez E, Zhang Q, et al. 2016. A CRISPR screen defines a signal peptide processing pathway required by flaviviruses. Nature 535: 164–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.