

Abstract
Circular RNAs (circRNAs) are a relatively new class of RNA molecules, and knowledge about their biogenesis and function is still in its infancy. It was recently shown that alternative splicing underlies the formation of circular RNAs (circRNA) arising from the Titin (TTN) gene. Since the main mechanism by which circRNAs are formed is still unclear, we hypothesized that alternative splicing, and in particular exon skipping, is a major driver of circRNA production. We performed RNA sequencing on human and mouse hearts, mapped alternative splicing events, and overlaid these with expressed circRNAs at exon-level resolution. In addition, we performed RNA sequencing on hearts of Rbm20 KO mice to address how important Rbm20-mediated alternative splicing is in the production of cardiac circRNAs. In human and mouse hearts, we show that cardiac circRNAs are mostly (∼90%) produced from constitutive exons and less (∼10%) from alternatively spliced exons. In Rbm20 KO hearts, we identified 38 differentially expressed circRNAs of which 12 were produced from the Ttn gene. Even though Ttn appeared the most prominent target of Rbm20 for circularization, we also detected Rbm20-dependent circRNAs arising from other genes including Fan1, Stk39, Xdh, Bcl2l13, and Sorbs1. Interestingly, only Ttn circRNAs seemed to arise from Rbm20-mediated skipped exons. In conclusion, cardiac circRNAs are mostly derived from constitutive exons, suggesting that these circRNAs are generated at the expense of their linear counterpart and that circRNA production impacts the accumulation of the linear mRNA.
Keywords: circRNAs, splicing, gene expression, heart, Rbm20
INTRODUCTION
Circular RNAs (circRNAs) have only recently been added to the family of regulatory RNAs, despite their discovery over 20 years ago (Nigro et al. 1991). This species of RNA molecules was largely ignored due to their unusual splicing behavior in which exons are joined at consensus splice sites, but in a scrambled order relative to the primary transcript, and due to their low expression levels compared to mRNAs. Decades later, with the advent of next-generation sequencing, thousands of endogenous circRNAs were found to be expressed in various tissues and cells types (Salzman et al. 2012; Memczak et al. 2013; Jeck and Sharpless 2014), including the heart (Jakobi et al. 2016; Khan et al. 2016; Werfel et al. 2016; Tan et al. 2017). Recent studies have revealed that circRNAs can regulate gene expression by different mechanisms; specifically, by serving as miRNA sponges (Hansen et al. 2013; Memczak et al. 2013; Zheng et al. 2016), facilitating transcription of their host gene by directly associating with RNA polymerase II (Zhang et al. 2013) or forming platforms for protein interactions (Du et al. 2016). Emerging evidence also suggests that some circRNAs can act as templates for protein synthesis (Legnini et al. 2017; Pamudurti et al. 2017; Yang et al. 2017).
The biogenesis of circRNAs is linked to pre-mRNA splicing, a process carried out by the spliceosome (Ashwal-Fluss et al. 2014; Starke et al. 2015). Pre-mRNA splicing represents the process of removal of introns and joining of exons in a linear fashion to produce a mature mRNA. CircRNAs, however, are formed by a back-splicing event, in which a donor site of an exon is not connected with an acceptor site of a downstream exon as observed in linear splicing, but rather with an acceptor site of an upstream exon (or with the acceptor site of the same exon in case of a single-exon circRNA). This gives rise to a single-stranded RNA loop with a unique exon–exon junction not present in the linear transcript. Mechanistically, back-splicing requires that the donor and acceptor site of the back-spliced exons are brought in close proximity to each other, which can be accomplished by direct RNA base-pairing of reverse complementary sequences (RCS) in the introns flanking the back-spliced exons, or by the interaction of RNA-binding proteins (RBPs) that dock on these flanking introns (Ashwal-Fluss et al. 2014; Zhang et al. 2014; Conn et al. 2015).
Ashwal-Fluss et al. (2014) demonstrated that there is strong competition between linear splicing and circRNA biogenesis. By introducing strong 5′ and 3′ splice sites in the exons that flank the circRNAs, they observed a dramatic reduction in circularization efficiency in minigene experiments in HEK293 cells. Moreover, flies carrying a mutation in RNA polymerase II, which has been shown to increase cotranscriptional splicing efficiency, produced significantly lower numbers of circRNAs. Both interventions decreased circRNA abundance in a context where linear splicing was more efficient, indicating that inclusion of exons in the linear transcript competes with back-splicing events of the same exons, thereby inhibiting circRNA production. Thus circRNA production seems to have a negative effect on linear splicing and therefore on gene expression (Ashwal-Fluss et al. 2014). Furthermore, Liang et al. (2017) recently showed that the steady-state output of protein-coding genes shifts to circular RNAs when canonical pre-mRNA splicing events are inhibited.
How RNA circularization is mechanistically connected to “alternative” splicing remains largely unknown, but splicing factors and RBPs, such as Muscleblind, Quaking (Ashwal-Fluss et al. 2014; Conn et al. 2015), RNA-binding domain containing immune factors NF90/NF110, hnRNPs, and SR proteins (Kramer et al. 2015; Li et al. 2017), have been shown to regulate back-splicing and circRNA biogenesis. RNA-Binding Motif protein 20 (RBM20) is yet another splicing factor that has recently been implicated in circRNA production, specifically from the TTN gene (Khan et al. 2016). RBM20 is highly expressed in the heart and regulates alternative splicing of a large number of genes, including titin (TTN): a structural component of sarcomeres (Labeit and Kolmerer 1995; Guo et al. 2012). Mutations in the gene encoding RBM20 have been implicated in a clinically aggressive form of dilated cardiomyopathy (DCM), characterized by progressive dilation and dysfunction of the left ventricle (Brauch et al. 2009; Beqqali et al. 2016). The missplicing of TTN is considered an important reason why RBM20 mutation carriers develop DCM. We recently discovered that a remarkably large number of circRNAs are produced from the TTN gene and that RBM20 is required for the production of a subset of these TTN circRNAs (Khan et al. 2016).
In the current study, we hypothesized that alternative splicing, and in particular exon skipping, drives the formation of cardiac circRNAs, and that Rbm20-mediated alternative splicing drives the formation of circRNAs that arise from Rbm20 target genes. To investigate to what extent alternative splicing underlies circRNA formation in the heart, we performed RNA sequencing on human and mouse hearts, and on the hearts of Rbm20 knockout (KO) mice. We integrated in silico analyses of alternative splicing and circRNA production in human heart and revealed that ∼90% of cardiac circRNAs are produced from constitutive exons and ∼10% from alternatively spliced exons. Strikingly, of the circRNAs that were formed from alternatively spliced exons, a large fraction (∼26%) stemmed from the TTN gene. In addition, we show that the strong correlation observed between exon skipping and circular RNA formation in the Ttn gene does not appear as a common mechanism for other Rbm20 target genes, suggesting that circRNA formation is not a general function of Rbm20. In conclusion, we show that alternative splicing drives the formation of only a small subset of cardiac circRNAs, as most cardiac circRNAs arise from exons that are constitutively spliced.
RESULTS
Identification of circRNAs in mouse hearts
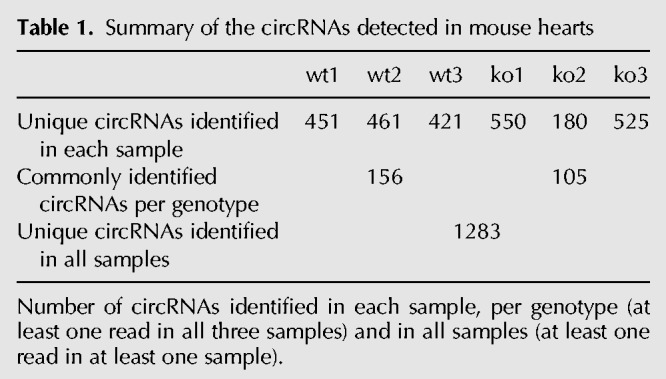
To detect circRNAs in the mouse heart, ribosomal-depleted RNA obtained from left ventricles of three wild-type and three Rbm20 KO mice was used for whole transcriptome sequencing (Fig. 1A). The MapSplice tool was used to identify back-splice junctions in the RNA-seq data (Wang et al. 2010; Khan et al. 2016). We detected a total of 1283 unique circRNAs in these six mouse samples (Table 1; Supplemental Table S1). However, the expression of the circRNAs was rather variable between hearts, as there were only 156 circRNAs commonly detected in all three wild-type hearts (Table 1). Most circRNAs were predicted to comprise between two and six exons within the back-spliced exons, but we also detected circRNAs potentially harboring as many as 30–100 exons within the back-spliced exons, albeit less frequently (Supplemental Fig. S1A). We confirmed the previous observation (Salzman et al. 2012) that particularly exon 2 of the host gene is overrepresented in circRNAs (Supplemental Fig. S1B). Furthermore, half of the host genes were predicted to produce a single circRNA, while the other half generated between two and nine different circRNAs, with the exception of Ttn, which generated a total of 38 putative circRNAs (Supplemental Fig. S1C). These Ttn specific circRNAs were mostly formed from the I-band region of the gene; a region known to undergo complex alternative splicing. This is in line with our previous report showing robust circRNA production from titin's I-band region in human hearts (Fig. 1B; Khan et al. 2016).
FIGURE 1.

Characterization of circRNAs in mouse hearts. (A) Strategy used to elucidate the relation between the circRNAs formation and alternative splicing. (B) Schematic representation of two splice isoforms of the Ttn gene (N2BA-G and N2B) and the location of the 38 identified circRNAs in the mouse hearts and the previously identified TTN circRNAs in human hearts (Khan et al. 2016) (C) Table showing conservation of cardiac circRNAs derived from mouse and human after filtering the human circRNAs for the expression level. Between brackets, the number of circRNAs passing the filter are indicated. (D) Scatter plot showing the relation between the percentage of complementarity and the length of the identified reverse complementary sequences (RCS) passing the P-value cutoff (FDR ≤ 0.05). RCS sharing high sequence similarity with the mouse B1 elements are depicted in black.
Table 1.
Summary of the circRNAs detected in mouse hearts
Only a small subset of circRNAs expressed in the heart are evolutionarily conserved
We have previously identified a total of 7130 unique circRNAs in human hearts, of which 3478 were found in hearts of healthy individuals. In the current study, using the exact same methodology, we detected a total of 1283 circRNAs in the mouse hearts. Overall, we found that ∼9% and ∼3% of the genes expressed in the human and mouse hearts produced circRNAs, respectively. To investigate to what extent circRNAs in human and mouse hearts are evolutionarily conserved, we converted mouse circRNA back-splice genomic coordinates to human coordinates and compared the genomic positions of circRNAs expressed in human and mouse hearts. We found that only 5% of human circRNAs were conserved in the mouse (Fig. 1C; Supplemental Table S2). Interestingly, when filtering human circRNAs for high expression levels (≥70 reads), the overlap with mouse circRNAs increases from 5% to 19% (Fig. 1C). This indicates that the highly expressed human circRNAs show a higher degree of conservation. Vice versa, up to 50% of the mouse circRNAs were also identified in human hearts (Supplemental Table S3).
Mouse cardiac circRNAs are flanked by reverse complementary sequences (RCS)
There is a consensus that the presence of RCS in introns flanking back-spliced exons is associated with the formation of circRNAs (Dubin et al. 1995; Zhang et al. 2014; Ivanov et al. 2015). To investigate the presence of these RCS, we retrieved 1000 bp of intronic sequence flanking each of the 1283 mouse circRNAs and investigated complementarity (Supplemental Fig. S2A). We selected the best local alignment found between each circRNA-flanking intron pair and compared it with randomly selected introns to assess the statistical significance (see Materials and Methods). All the introns flanking the detected circRNAs had RCS ranging from 9 to 332 bp, but only 802 were statistically significant (FDR ≤ 0.05, at least 67% identity) (Supplemental Table S4). Analysis of the identified RCS showed that ∼13% of them share sequence similarity with inverted B1 elements (mouse equivalent of Alu-repeats in human) (Fig. 1D), ∼10% with other elements like short interspersed sequence elements (SINEs) B2–B4, long interspersed sequence elements (LINEs) and simple repeats, and ∼77% with none of the known elements (Supplemental Fig. S2B). The identification of the above repeats is in line with other groups who revealed that intronic repeats are necessary and sufficient for circularization (Liang and Wilusz 2014; Zhang et al. 2014; Ivanov et al. 2015; Starke et al. 2015). The sequences of our identified RCS are shown in Supplemental Table S4. Experimental studies are needed to provide evidence of whether base-pairing events of the identified sequences are necessary and/or sufficient for circularization of these mouse circRNAs.
Thirty-eight circRNAs are differentially expressed in hearts of RBM20 knockout mice
To identify Rbm20-dependent circRNAs expressed in the heart, we performed differential expression analysis by comparing circRNAs in Rbm20 KO with wild-type mice. We identified 38 out of 1283 circRNAs to be differentially expressed, of which 26 were down-regulated and 12 were up-regulated in Rbm20 KO mice compared to wild-type mice (Fig. 2A; Supplemental Table S1). Notably, our RNA-seq data showed that a subset of 19 circRNAs was completely absent in Rbm20 KO mouse hearts, while they were readily expressed in wild-type hearts. Eleven of those circRNAs were generated from the Ttn gene and the remaining from the host genes Unc13b, Stk39, Arhgap10, Tfdp2, Sorbs1, Fan1, Insr, and Xdh (Table 2). A subset of seven up-regulated circRNAs was uniquely expressed in the hearts of Rbm20 KO mice, and they were generated from the host genes Sorbs1, D19Ertd386e, Ehmt1, Gcom1, Bcl2l13, Smad1, and Ttn (Table 2).
FIGURE 2.

A subset of circRNAs are differentially expressed in hearts of Rbm20 knockout mice. (A) Volcano plot showing differentially expressed circRNAs in Rbm20 KO hearts compared to wild-type hearts. Differentially expressed circRNAs (−1 ≥ Log2 fold change ≥ 1 and P-value ≤ 0.05) are marked in gray. (B) RT-PCR for circRNAs on wild-type (WT), heterozygous (HET), and Rbm20 knockout (KO) mouse hearts. GAPDH was used as input control.
Table 2.
Normalized read counts of differentially expressed circRNAs in hearts of Rbm20 knockout mice

To validate these differentially expressed circRNAs experimentally, we selected six circRNA candidates, based on P-value and expression level, that were down-regulated (circTtn_2, circTtn_3, circTtn_4, circFan1, circStk39, and circXdh) and four circRNA candidates that were up-regulated (circTtn_7, circSorbs1, circBcl2l13, circEhmt1) in Rbm20 KO hearts (Fig. 2B; Table 2). We designed divergent primers flanking the circRNA-specific back-spliced junctions and performed RT-PCR on RNA isolated from hearts of six wild-type, six heterozygous, and six Rbm20 KO mice. Sanger sequencing confirmed the presence of back-splice junctions in the amplicons, and RT-PCR on poly(A)-negative and -positive RNA, and on RNase R-treated RNA and mock samples revealed that we detected bona fide circRNAs (Supplemental Fig. S3A; Supplemental Table S5). In line with our previous report (Khan et al. 2016), we confirmed the loss of the three Ttn circRNAs in the Rbm20 KO hearts, but also the down-regulation of circRNAs produced from Fan1 and Stk39. Interestingly, the circRNA derived from the Fan1 gene seemed to depend critically on Rbm20 expression, as we found that circFan1 was substantially reduced in the heterozygotes and completely absent in the Rbm20 KO hearts (Fig. 2B). Of the circRNAs that were up-regulated in the Rbm20 KO hearts, we could experimentally validate circTtn_7, circSorbs1, and circBcl2l13. Based on RT-PCR, expression of circXdh and circEhmt1 did not seem to be affected in the absence of Rbm20 in this set of 18 mouse hearts. Finally, we performed quantitative RT-PCR for circFan1, circStk39, circXdh, circEhmt1, circTtn3, and circTtn7 and further confirmed the differential expression of these circRNAs, except for circEhmt1 (Supplemental Fig. S3B; Supplemental Table S5). Overall, nine out of 10 differentially expressed circRNAs could be experimentally validated.
Furthermore, expression analysis of the host genes of the 38 differentially expressed circRNAs revealed that there were only three genes differently expressed (when considering an absolute fold change ≥1.5), indicating that the differential expression of the circRNAs was not due to dysregulation of the corresponding host genes (Supplemental Table S6).
Rbm20-binding site enrichment analysis showed that introns flanking the 38 differentially expressed circRNAs are twofold enriched for Rbm20-binding sites compared to a random set of introns; with the most significant enrichment occurring within a flanking distance of 100 bp (P ≤ 0.001) (Supplemental Fig. S4). Interestingly, even after removing Ttn circRNAs from the analysis, a significant enrichment remained. For instance, we identified a total of four potential Rbm20-binding sites in the introns flanking circFan1 (Supplemental Fig. S5A). In conclusion, the RNA-seq in wild-type and Rbm20 KO hearts in combination with the Rbm20-binding site prediction revealed that the Ttn gene seems to be the most prominent target of Rbm20-regulated circRNA production. Nevertheless, of the 38 differentially expressed circRNAs, 26 originated from other genes, suggesting that circRNAs arising from Ttn gene are not the only ones that are regulated by Rbm20.
Rbm20-dependent alternative splicing and circRNA production
We have previously shown that Rbm20-dependent exon skipping within Ttn's I-band region is associated with circRNA formation (Khan et al. 2016). To assess whether Rbm20-controlled alternative splicing and circRNA formation are connected beyond the Ttn gene, we calculated the percentage spliced in (PSI) (Kakaradov et al. 2012; Schafer et al. 2015) of the back-spliced exons of all 38 differentially expressed circRNAs. The PSI indicates the efficiency of splicing of a specific exon into the transcript population of a gene. It ranges from 0 to 1, indicating an exon is never (0) to always (1) spliced into the expressed transcripts of a gene. In Figure 3A and B, we plotted the PSI of back-spliced exons of the 38 differentially expressed circRNAs against the expression of the corresponding circRNAs. Figure 3A shows that particularly the circRNAs from the Ttn gene arise from exons that are spliced out of the linear transcripts. The back-spliced exons of almost all other differentially expressed circRNAs, except those arising from Arhrgap10 and 2310046A06Rik host genes, have a PSI close to 1, indicating that these exons are always included in the linear transcripts. In Figure 3B, where the PSI is plotted against the expression of the circRNAs in the Rbm20 KO hearts, it can be appreciated that PSI levels of the Ttn back-spliced exons increase to 1, indicating that these exons are now included in the linear Ttn transcripts. Alternative splicing changes for Ttn and Arhgap10 in wild-type, heterozygous, and Rbm20 KO hearts were confirmed by semi-quantitative RT-PCR (Supplemental Figure S6; Supplemental Table S5). To further quantify the difference in exon skipping between Rbm20 KO and wild-type mice, we calculated the delta-PSI of the back-spliced exons and plotted these against the expression changes of the circRNAs (Fig. 3C; Supplemental Table S7). Again, this shows that the exons used for circularization are mostly constitutive exons since the exons that are differentially back-spliced upon Rbm20 loss are not differentially spliced in the linear transcript. Also, in this approach, the most notable exceptions are Ttn circRNAs.
FIGURE 3.

Rbm20-dependent alternative splicing and circRNA production. Scatterplots showing the relation between the PSI of the back-spliced exons and the expression of the circRNAs (Log2 bsj reads) in (A) wild-type mice and (B) Rbm20 KO mice. Only the 38 differentially expressed circRNAs are depicted. The names of the circRNA host genes are plotted. (C) Scatterplot showing the relation between the dPSI of the back-spliced exons of the 38 differentially expressed circRNAs (dPSI back-spliced exons) and the expression fold changes (Log2 bsj reads FC) of the corresponding circRNAs between Rbm20 KO hearts and WT hearts.
In addition to these PSI calculations, we used an alternative strategy to assess whether circRNA formation and alternative splicing are concurrently regulated. We used MapSplice to retrieve the linear junction reads (lj reads) stemming from the back-spliced exons in the Rbm20 KO and wild-type mice. We then plotted the fold changes of the linear junction reads against the fold changes of back-spliced junction reads (bsj reads) in Rbm20 KO compared to wild-type mice (Supplemental Fig. S7). This also shows that reduced numbers of Ttn circRNAs in Rbm20 KO hearts associates with increased linear junction read counts, indicative of a negative correlation between circRNA production and linear exon usage.
Besides regulating Ttn splicing, Rbm20 is known to regulate linear splicing of many other cardiac genes, including Camk2d, Ldb3, and Myh7 (Maatz et al. 2014). We next performed a dedicated search for circRNAs that may have arisen by exon skipping events in these other Rbm20 target genes. Genome-wide differential exon usage analysis (using the R Bioconductor package DEXSeq) in our Rbm20 mouse model confirmed that loss of Rbm20 leads to missplicing of several genes, including Ttn, Myh7, Obscn, Slc8a1, Sorbs2, Camk2d, Ldb3, and Rtn4 (Supplemental Table S8). We retrieved the exons that are alternatively spliced by Rbm20 and overlapped these with the back-spliced exons to assess whether these exons generated circRNAs. Interestingly, the exons that were found to be differentially spliced in the Rbm20 KO mice do not produce detectable circRNAs, except the ones arising from the Ttn gene (Supplemental Table S9). As described in the previous section, we identified circFan1 as a circRNA that also critically depends on Rbm20 expression. To investigate whether Rbm20-dependent alternative splicing may underlie circFan1 production, we generated a splicing plot for the Fan1 gene showing exon usage in Rbm20 KO and wild-type mice. As shown in Supplemental Figure S5B, several exons in the Fan1 mRNA are alternatively spliced but not the two exons that circularize (Supplemental Table S10). Therefore, it remains unclear at this point how Rbm20 regulates the production of circFan1. In conclusion, Rbm20-mediated exon skipping results in circRNA production from the Ttn gene, but not from other bona fide splicing targets of Rbm20.
Genome-wide analysis of alternative splicing and circRNA production in the human heart
With the availability of RNA data sets of the mouse and human heart, we have the opportunity to investigate the connection between alternative splicing and circRNAs formation, in an unbiased and genome-wide manner. Therefore, we calculated the PSI of the back-spliced exons of all circRNAs passing the filtering step (read counts ≥2 in both samples) in two control human hearts from our previous study (Khan et al. 2016). Figure 4A shows the PSI of back-spliced exons of the 1211 circRNAs detected in human hearts, plotted against the expression level of the corresponding circRNAs. We considered exons to be constitutive if they were included in the linear mRNA in at least 90% of the transcripts (PSI > 0.90) (Schafer et al. 2017). The results show that only ∼10% of the circRNAs had back-spliced exons with a PSI ≤ 0.90 and thus may have been derived from exon skipping events (Supplemental Table S11). Of note, when we use a more stringent filtering step (read counts ≥8 in both samples) for circRNA expression, the percentage of alternative back-spliced exons and constitutive back-spliced exons does not change, suggesting that our PSI-findings are not biased by lowly expressed circRNAs or false-positive circRNA predictions (Supplemental Table S12).
FIGURE 4.

CircRNAs in the human heart are derived from exons that are typically not alternatively spliced. (A) Scatterplot showing the relation between the PSI of the back-spliced exons with the expression of the corresponding circRNAs (Log2 bsj reads) in the human hearts. Gray dots represent back-spliced exons with PSI > 0.90. Blue dots represent back-spliced exons from TTN gene. Black dots represent back-spliced exons with PSI ≤ 0.90. (B–E) The PSI of the four host genes (PALM2, YAF2, CUX1, and ZEB1), indicated in orange in A are plotted, in which exon skipping is connected with circRNA production. Black dots represent all exon bins of the corresponding genes, and orange dots represent the exon bins that give rise to the circRNAs (e.g., back-spliced exons). (F) Proposed model showing two mechanisms for circRNA production: competition-based and exon skipping-based circRNA production.
We also calculated the ratio of constitutive vs. alternative exons in the complete human and mouse transcriptome, and found that, after correcting for the ratio of constitutive vs alternative exons in the transcriptome, a 3.3–2.1-fold enrichment of constitutive exons in the group of back-spliced exons was still present (Supplemental Table S13). This confirms our observation that alternative splicing is not a major driver of circRNA production.
Thus, these findings indicate that most circRNAs are not derived from alternatively spliced exons but from constitutive exons. Of the 10% of the circRNAs that we found to associate with alternative splicing, a large proportion (26%) were derived from the TTN gene (Fig. 4A, blue dots). Interestingly, we also found several other genes where exon skipping was associated with circRNA production. To visualize the relation between exon skipping and circRNA production in greater detail, we plotted the PSI of four of these genes, PALM2, YAF2, CUX1, and ZEB1, and marked the exons used for circularization in orange (Fig. 4B–E). In the mouse hearts, ∼13% of the circRNAs were associated with exon skipping events, when considering a PSI of 0.90 as a cutoff (Supplemental Fig. S8; Supplemental Table S14). In conclusion, many of the cardiac circRNAs that coincide with alternative splicing are derived from the Ttn gene. However, particularly in the human heart, we found several examples of non-TTN circRNAs that were produced from regions that undergo extensive alternative splicing.
Based on these results, we propose a model in which there are two mechanisms for circRNA production: competition-based and exon skipping-based circRNA production (Fig. 4F). In the competition-based mechanism, which is the most frequently observed mechanism (∼90% of the circRNAs), a pre-mRNA is used to produce a circRNA, but not a mature linear mRNA. In this case, circRNA biogenesis may be an important regulator in the control of gene expression; and depending on the gene in question, there might be a favored production of linear mRNAs or circRNAs. In the exon skipping-based mechanism, which is less frequently observed (∼10%), pre-mRNA undergoes alternative splicing and results in the formation of both a linear exon-skipped mRNA and a circRNA.
DISCUSSION
It is known that the splicing machinery can circularize exons and that linear splicing is directly involved in circRNA production (Zaphiropoulos 1996). However, to what extent alternative splicing, and in particular exon skipping, drives circRNA formation is currently unknown. Here, we analyzed RNA-seq data from mouse and human hearts on alternative splicing and circRNA production and show in humans that ∼90% of the circRNAs are derived from exons that are typically not alternatively spliced (i.e., constitutive exons). This suggests that these circRNAs are mostly generated at the expense of linear mRNA isoforms, and that circRNA production competes with the accumulation of linear mRNA. Conversely, ∼10% of the human circRNAs may have been produced from exon skipping events, as these circRNAs are derived from exons that are subjected to alternative splicing (Fig. 4F). Interestingly, a large proportion (∼26%) of the circRNAs that are associated with exon skipping events in the human heart is derived from the TTN gene.
Our finding that circRNAs can be derived from exon-skipping events is in line with other studies (Salzman et al. 2012; Jeck et al. 2013; Barrett et al. 2015; Kelly et al. 2015), although the described frequency of these events differs. Jeck et al. (2013) determined exon-skipped linear transcripts from human fibroblasts and found that 45% of the expressed circRNAs are derived from linear splicing reads with characteristics of an exon-skipping event. Salzman et al. (2012) provided circumstantial evidence that in a variety of normal and malignant human cells, 2% of the circRNAs are derived from exons predicted to undergo alternative splicing. In the current study, we used an integrated approach to evaluate alternative splicing and circRNA production by calculating PSI, back-spliced junction reads, and linear junction reads and provide evidence that at most 10% of the circRNAs are derived from exon skipping events. This difference in frequencies between the studies may be explained by differences in examined tissue, or the difference in methods/cutoffs to detect exon skipping. Unfortunately, we were unable to examine the underlying differences since it is unclear what methods and cutoffs were exactly used for the detection of alternative splicing in the other two studies. We were rather strict in setting the cutoff for the detection of alternative splicing (PSI ≤ 0.90); with a more lenient cutoff for exon skipping (PSI ≤ 0.98), we find that up to 27% of the circRNAs can be derived from exon skipping. In contrast to the studies mentioned above and our current study, Kelly et al. (2015) presented an extraordinarily high correlation between exon skipping and circRNA production. However, two fundamental problems seem to underlie their analyses. Firstly, they express the abundance of back-spliced exons within circRNAs as expression ratios (back-spliced junction reads in circRNAs/back-spliced exon aligning reads in linear mRNA). The expressions of the back-spliced exons in the linear mRNA were subsequently plotted against these ratios and used to calculate correlations. However, since a common factor (i.e., reads aligning to the back-spliced exons in linear mRNA) is present in both correlation variables, this will automatically, but falsely result in a high correlation. Secondly, to assess exon skipping, Kelly and colleagues only measured the abundance of exonic reads in linear RNA. However, simply measuring reduced exonic reads does not provide evidence for exon skipping. To be included as a possible product of exon skipping, they should have selected only those linear reads that completely traverse the back-spliced exons. In our study, we calculated the PSI, in which the efficiency of splicing of a specific exon within a transcript population of a gene is calculated based on exon-skipped junctional reads, which to our opinion is a more appropriate approach for this purpose.
A limitation of the current study is that we cannot rule out that post-transcriptional degradation processes, such as nonsense-mediated decay (NMD), clear away mRNAs that have skipped exons that yield circRNAs (and hence the alternatively spliced mRNAs would not be detected in RNA-seq). Furthermore, Conn et al. (2017) showed that low-abundance splice variants may be missed in RNA-seq experiments, at least in plants. Both events may have led to an underestimation of the proportion of exon skipping-based circRNAs. In regard to the first point, it would be interesting to block NMD, for example pharmacologically with drugs like emitine, and then examine the expression of linear mRNA transcripts arising from the circRNA host genes to ascertain the proportion of exon skipping-based circRNAs that lead to the degradation of their linear counterpart. A related point is that current circRNA profiling studies typically examine steady-state levels of circRNAs and mRNAs and do not provide information on nascent RNA. It has recently been shown that back-splicing is less favorable than canonical splicing, and that the expression of circRNAs for a large part depends on accumulation of synthesized circRNAs rather than increased synthesis (Zhang et al. 2016), and in that sense, the effect of circRNA biogenesis on linear mRNA expression can be only minor. Therefore, experiments in which newly transcribed RNAs are labeled metabolically (for instance using 4-thioridine [Fuchs et al. 2015]) to study the kinetics of circRNA biogenesis and nascent RNA splicing in cardiac cells are awaited with great interest.
We recently discovered a causal link between exon skipping regulated by the splice factor RBM20 and circRNA formation in the human TTN gene (Khan et al. 2016). These results led us to hypothesize that this phenomenon may not be restricted to a single gene but that RBM20 may also regulate the formation of circRNAs arising from other RBM20 target genes. Therefore, we designed this study to investigate whether Rbm20 is required for the production of circRNAs beyond those of the Ttn gene. We identified 38 differentially expressed circRNAs in Rbm20 KO hearts, and the Ttn gene appeared as the most prominent target of Rbm20 for circRNA production, as we found that 11 Ttn circRNAs were completely absent in Rbm20 KO hearts. We next investigated whether the set of 38 circRNAs that were regulated in the Rbm20 KO hearts could have been formed by exon skipping events. We used two different strategies (PSI and linear junction read count of back-spliced exons) to interrogate alternative splicing of linear transcripts, and both methods similarly showed that predominantly the circRNAs from the Ttn gene are associated with exon skipping events. This is a remarkable finding since Rbm20 is known to act as a splicing repressor of many cardiac genes, including Camk2d, Ldb3, Sorbs1, and Myh7 (Maatz et al. 2014). Dedicated searches for circRNAs in the mapped reads of these well-described Rbm20 target genes did not yield any evidence of back-spliced junctions in the exons that are targeted by Rbm20 for splicing. Also, the Sorbs1 and Ryr2 circRNAs that we found to be differentially expressed in the Rbm20 KO hearts were derived from different exons than the ones that are normally repressed for splicing by Rbm20. A differential exon usage plot of the Fan1 gene in wild-type and Rbm20 KO hearts revealed that Rbm20 regulates alternative splicing (i.e., exon inclusion) in the region of the back-spliced exons, but not in the back-spliced exons specifically. The presence of four Rbm20-binding sites in the introns flanking the back-spliced exons of circFan1 further suggest that Rbm20 underlies circRNA formation from this gene directly; however, the precise mechanism remains elusive. Overall, the tight correlation between exon skipping and circRNA formation observed in the Ttn gene does not appear to be a common mechanism for Rbm20 target genes, indicating that circRNA formation is not a general function of Rbm20.
We performed a comparative analysis and showed that the normal human heart expresses four times more circRNAs than the mouse heart (3478 circRNAs in humans and 848 in mice). This is in line with a study by Tan et al. (2017), where five times more circRNAs were detected in the human heart compared to the mouse heart. Werfel et al. (2016) only found 1.6 times more circRNAs in human hearts compared to the mouse hearts, which is probably caused by the relatively high number of circRNAs they detected in the mouse hearts. We further interrogated evolutionary conservation of the circRNAs by converting the mouse genomic coordinates of the back-spliced junctions to the human genome. We show that only 184 (i.e., 5%) of the human circRNAs are also expressed in mouse hearts. When discarding the lowly expressed human circRNAs, the overlap with mouse circRNAs increased from 5% to 19%. This level of evolutionary conservation is in agreement with the study of Werfel et al. (2016), who showed that approximately 13% of the human cardiac circRNAs are conserved to mouse and rats. Vice versa, due to the lower numbers of circRNAs expressed in the mouse heart, a much higher percentage (between 20% and 50%) of the mouse circRNAs are conserved to human. This suggests that during evolution, an elusive mechanism has led to an astounding increase in the number of circRNAs in the human transcriptome. The observation that the expression of circRNAs is highly variable between hearts from the same species suggests that occasional circularization of exons is easy to evolve and may provide a mechanism for rapid evolution of stably expressed circRNAs. It has been shown that inverted Alu repeats in the introns flanking back-spliced exons are associated with the formation of circRNAs (Jeck et al. 2013). Alu repeats are primate-specific SINE retrotransposons and thus not present in the mouse genome (Kriegs et al. 2007). In the mouse genome, the B1 element is the most abundant class of repetitive elements. They belong to the SINE family and, like Alu repeats in human, originated from an initial duplication of the 7SL RNA and amplified and duplicated independently in the two genomes while accumulating specific mutations (Quentin 1994). The discrepancy in the numbers of identified circRNAs in human and mouse hearts may relate to the fact that regulatory sequences for circularization (for instance Alu and B1 elements) are not well conserved (Tsirigos and Rigoutsos 2009). Analysis of the introns flanking the back-spliced exons of the mouse circRNAs revealed besides the B1 elements, also reverse complementary sequences that do not share any sequence similarity with transposable elements. Further investigation is required to evaluate whether the identified sequences are necessary for circularization.
In conclusion, this study revealed that the splice factor Rbm20 is not a global regulator of circRNA formation in the heart. The tight correlation between exon skipping and circRNA formation that we observed for the Ttn gene does not appear to be a common mechanism for other Rbm20 target genes. In addition, we have shown that cardiac circRNAs are mostly (∼90%) produced from constitutive exons, indicating that these circRNAs are generated at the expense of their linear counterpart. Future studies investigating how circRNA production from these constitutive exons affect gene expression and protein production of the host gene will be awaited with great interest.
MATERIALS AND METHODS
Rbm20 knockout mice
Generation of the Rbm20 KO mice has been described previously (Khan et al. 2016). In line with other Rbm20 KO models (Guo et al. 2012; Methawasin et al. 2014), homozygous Rbm20 knockout mice revealed abnormal Ttn splicing and a DCM phenotype, which is manifested by LV dilatation and impaired cardiac function. Total RNA was extracted from LV tissue samples of 6-mo-old mice (three wild-type and three Rbm20-KO) with TRI Reagent (Sigma-Aldrich) according to manufacturer's protocol.
All animal studies were approved by the Institutional Animal Care and Use Committee of the University of Amsterdam and in accordance with the guidelines of this institution and the Directive 2010/63/EU of the European Parliament.
Library preparation and whole transcriptome RNA sequencing
RNA quality was assessed with the Agilent 2100 Bioanalyzer. All samples had a RIN score of ∼8. Total RNA samples (500 ng) were treated with biotin-streptavidin-based bead systems (Exiqon) to minimize ribosomal contamination. Ribosomal-depleted RNA libraries were sequenced on an Illumina NextSeq 500 platform in paired-end mode and with a read length of 101 bp at Exiqon A/S. Sequencing depth was approximately 70 million raw reads per sample (Supplemental Table S15). Base-calling was performed using the bcl2fastq 2.0 Conversion Software from Illumina.
Quality control, processing and mapping of RNA-seq data
Quality control of fastq files was performed using FASTQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Trimmomatic version 0.351 (Bolger et al. 2014) was used to remove Illumina adapters and low quality bases, using a Phred score cutoff of 30 while discarding reads with a length below 25 bases. The paired-end RNA-seq reads passing the quality controls from the three Rbm20 KO mice and wild-type mice were then aligned against the mouse genome, Gencode annotation release M1 (NCBI37), using TopHat2 (Kim et al. 2013) version 2.0.14 with default values.
Conservation analysis between human and mouse circRNAs
To determine homologous exon groups, we used UCSC liftOver (Karolchik et al. 2004) to convert mouse splice coordinates (mm9) to human coordinates (hg19). For the analysis, we used the 848 unique circRNAs detected in wild-type mice (one read count in at least one mouse) and the 3487 unique circRNAs detected in human controls (one read count in at least one control) from our previous work (Khan et al. 2016). For a more stringent analysis, we filtered the human and mouse circRNAs for the expression level using the cutoff of at least two, 10, 40, and 70 normalized read counts in at least two samples.
RCS analysis in the flanking introns of the predicted circRNAs
For each circRNA, we aligned the downstream and reversed complement of the upstream intron, using the pairwiseAlignment function in the R package Biostrings and retrieved the score of the best local alignment. To calculate a P-value associated with each match, we compared the actual score to the distribution of scores obtained by aligning the sequence of the best local alignment (without gap) with 1000 random introns. The P-value was then adjusted for multiple testing using the FDR methods. The following alignment parameters were used: match = 2; mismatch = −3, gap opening = −5, gap extension = −2. To calculate the percent sequence identity for a pairwise sequence alignment we used the following formula: 100*(identical positions)/(aligned positions + internal gap positions). We repeated the procedure for each pair intron flanking circRNA. As the last step, we blasted the identified RCS with Repeat Masker Database using ABBlast algorithm (Smit et al. 2013–2015).
Identification of back-spliced and linear junction reads—MapSplice
Back-spliced and linear junction reads were identified using MapSplice version 2.2.0 (Wang et al. 2010). For circRNAs detection we set the following options: --min-fusion-distance 200 (as suggested by the authors), --filtering 1, and --min-map-len 25. The reported linear and back-spliced junction coverage for each sample were then formatted into matrices, where the value in the ith row and the jth column of each matrix represented the total number of reads successfully mapped to the back-splice or linear junctions of the host gene i in sample j. The raw counts were then normalized using the R Bioconductor package DESeq2 (Love et al. 2014). GenomicRanges function and custom R scripts were used to overlay linear splicing events with back-splicing events.
Differential circRNA expression analysis—DESeq2
Differential expression analysis of circRNAs identified by MapSplice in the previous step was performed using the R Bioconductor package, DESeq2 (Love et al. 2014). To investigate circRNAs expression in Rbm20 KO and wild-type mice, two sets of differential expression analyses were conducted. The first analysis was performed using the total number of circRNAs detected in the hearts of wild-type mice and Rbm20 KO mice. To increase sensitivity, the second analysis was performed considering only circRNAs detected in at least two wild-type or two Rbm20 KO mice with normalized reads counts ≥2. In the end, only those circRNAs with an absolute log2 fold change cutoff ≥1 together with a P-value cutoff ≤0.05 were deemed significantly differentially expressed. Owing to the limited number of samples (n = 3 per group), the adjusted P-value was not used as a criterion to select candidates and the final set comprised of 38 circRNAs, which were selected for downstream analyses.
Differential gene expression analysis—DESeq2
Differential gene expression analysis was performed using the R Bioconductor package, DESeq2 (Love et al. 2014). GenomicRanges infrastructure was used to count the number of aligned reads overlapping with each gene. The expression of the host genes of the 38 differentially expressed circRNAs was retrieved. Only those circRNA host genes with an absolute log2 fold change cutoff ≥1 together with an adjusted P-value cutoff ≤0.05 were deemed significantly differentially expressed.
Rbm20-binding site enrichment analysis
Genomic sequences of introns flanking the differentially expressed circRNAs were obtained from the UCSC Genome Table Browser. Gregexpr2 function in the R package Biostrings and custom R scripts were then used to determine for each candidate circRNA the genomic positions of the Rbm20 motif (UCUU) occurring within introns flanking 100 bp upstream of and downstream from the back-splicing acceptor and donor sites, respectively. Rbm20 motifs occurring in flanking introns of the circRNAs were then tallied. An equal number of randomly selected introns was utilized as a control set. The number of Rbm20 motifs occurring within introns flanking 100 bp upstream of and downstream from these “control” intron pairs were tallied.
Differential exon usage analysis—DEXSeq
To test for exon usage differences between Rbm20 KO and wild-type mice we used the R Bioconductor package DEXSeq version 1.20.0 (Anders et al. 2012), using GenomicRanges infrastructure to count the number of aligned reads overlapping with each exonic region. Only those exons with an absolute log2 fold change cutoff ≥1 together with an adjusted P-value cutoff ≤0.05 were deemed significantly differentially expressed.
Percentage spliced in of the back-spliced exons—PSI
The “percentage spliced in” index (PSI) was computed by executing the script PSI.sh of Schafer et al. (2015) on exonic parts derived from the Gencode annotation release M1 (NCBIM37) for mouse, and Gencode annotation release 19 (GRCh37.p13) for human. GenomicRanges function and custom R scripts were used to overlay the coordinates of the back-spliced exons with the coordinates of the exon bins identified during the PSI calculation. In mice we found the corresponding PSI of back-spliced exons for 1148 on a total of 1283 detected circRNAs. In human, of the 3487 detected circRNAs, we found the corresponding PSI of back-spliced exons for 3401 of them. Back-spliced exons of circRNAs were detected in at least two samples, wild-type or Rbm20 KO mice, and two human controls with normalized reads counts ≥2 or ≥8 (high-confidence expressed circRNAs [Zhang et al. 2014] with RPM ≥0.1 corresponding to eight reads in our sample with the lowest number of mapped reads) were used in the analysis. If an exon's boundary was not the same in all transcripts, the exon was cut into two or more parts, referred to as exon bins. We considered exons to be constitutive if they were included in the linear mRNA in at least 90% of the transcripts (PSI 0.9) (Schafer et al. 2017).
Data visualization and statistical analysis
All results generated from differential circRNA analysis were visualized using the R Bioconductor package, ggplot2. The graphical representations of the TTN gene and circRNAs were constructed using the R package, GenomicRanges and custom R scripts, respectively. Differential exon usage and differential expression analyses of circRNAs and circRNA host genes across conditions were statistically analyzed using the negative binomial test in the R Bioconductor package, DESeq2. All correlation tests were performed in R, using the Spearman's correlation as the method of choice. Differences in the enrichment of Rbm20 sites between circRNAs and control sequences were compared using the Fisher exact test. To be able to analyze circRNAs that were not expressed in the RBM20 KO mice or wild-type mice, a count of 1 was added to avoid problems with infinite values when log transforming. The P-value for PSI differences was calculated using the Student's t-test. Scatter plots were used to plot the mean values of the samples. qRT-PCR data were analyzed using LinRegPCR quantitative PCR data analysis software (Ruijter et al. 2009) and normalized with Hprt. Values are expressed as mean ± standard error. Differences between groups were compared using the Student's t-test.
Preparation of poly(A)-positive and poly(A)-negative RNA fractions
Poly(A)-positive and poly(A)-negative RNA fractions were generated as described previously (Khan et al. 2016). Briefly, 6 μg total RNA from three wild-type mouse hearts and 200 μL oligo d(T)25 Magnetic Beads (NEB) were used to generate poly(A) enriched and depleted fractions. The beads were washed first and subsequently incubated with the RNA in a 100 μL reaction for 10 min with gentle agitation. Afterwards, the poly(A) negative fraction was collected as supernatant by pulling the beads to the side of the tube. After three times washing, beads were incubated in 100 μL elution buffer at 50°C for 2 min to recover the poly(A) positive RNA fraction. The poly(A) negative RNA fraction was also treated at 50°C for 2 min.
RNase R treatment
RNase R treatment was performed as described previously (Khan et al. 2016). Briefly, 1 μg total RNA from three wild-type mouse hearts was treated with or without five units of RNase R (Epicentre) at 37°C for 10 min in a 20 μL reaction followed by heat inactivation at 95°C for 3 min.
RT-PCR and qRT-PCR
RT-PCR and qRT-PCR were performed as described previously (Khan et al. 2016). Briefly, 1 μg of poly(A) enriched/depleted or RNase R treated/nontreated RNA was DNA depleted with DNase I (Invitrogen) and subsequently used to generate cDNA using SuperScript II Reverse Transcriptase (Invitrogen) and random hexamers (Invitrogen). Two microliters of five-times diluted cDNA was amplified for 30–35 cycles with HOT FIREpol DNA polymerase together with divergent primers for circRNA detection or normal convergent primers for mRNA detection. Primers are designed to amplify the smallest band depicted on the gels. Sanger sequencing confirmed the presence of the expected back-splice junctions in these bands. For qRT-PCR, 2 μL of five-times diluted cDNA was amplified for 45 cycles with SYBR Green I Master (Roche) on a LightCycler480 system II (Roche). Primer sequences are included in Supplemental Table S5.
DATA DEPOSITION
Raw RNA sequencing data are available at NCBI BioProject, under accession number PRJNA417769.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO-836.12.002, NWO-821.02. 021) and by the Cardiovasculair Onderzoek Nederland (CVON): CVON-ARENA-2011-11.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.064394.117.
REFERENCES
- Anders S, Reyes A, Huber W. 2012. Detecting differential usage of exons from RNA-seq data. Genome Res 22: 2008–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashwal-Fluss R, Meyer M, Pamudurti NR, Ivanov A, Bartok O, Hanan M, Evantal N, Memczak S, Rajewsky N, Kadener S. 2014. circRNA biogenesis competes with pre-mRNA splicing. Mol Cell 56: 55–66. [DOI] [PubMed] [Google Scholar]
- Barrett SP, Wang PL, Salzman J. 2015. Circular RNA biogenesis can proceed through an exon-containing lariat precursor. eLife 4: e07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beqqali A, Bollen IAE, Rasmussen TB, van den Hoogenhof MM, van Deutekom HWM, Schafer S, Haas J, Meder B, Sørensen KE, van Oort RJ, et al. 2016. A mutation in the glutamate-rich region of RNA-binding motif protein 20 causes dilated cardiomyopathy through missplicing of titin and impaired Frank-Starling mechanism. Cardiovasc Res 112: 452–463. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauch KM, Karst ML, Herron KJ, de Andrade M, Pellikka PA, Rodeheffer RJ, Michels VV, Olson TM. 2009. Mutations in RNA binding protein gene cause familial dilated cardiomyopathy. J Am Coll Cardiol 54: 930–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn SJ, Pillman KA, Toubia J, Conn VM, Salmanidis M, Phillips CA, Roslan S, Schreiber AW, Gregory PA, Goodall GJ. 2015. The RNA binding protein quaking regulates formation of circRNAs. Cell 160: 1125–1134. [DOI] [PubMed] [Google Scholar]
- Conn VM, Hugouvieux V, Nayak A, Conos SA, Capovilla G, Cildir G, Jourdain A, Tergaonkar V, Schmid M, Zubieta C, et al. 2017. A circRNA from SEPALLATA3 regulates splicing of its cognate mRNA through R-loop formation. Nat Plants 3: 17053. [DOI] [PubMed] [Google Scholar]
- Du WW, Yang W, Liu E, Yang Z, Dhaliwal P, Yang BB. 2016. Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res 44: 2846–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubin RA, Kazmi MA, Ostrer H. 1995. Inverted repeats are necessary for circularization of the mouse testis Sry transcript. Gene 167: 245–248. [DOI] [PubMed] [Google Scholar]
- Fuchs G, Voichek Y, Rabani M, Benjamin S, Gilad S, Amit I, Oren M. 2015. Simultaneous measurement of genome-wide transcription elongation speeds and rates of RNA polymerase II transition into active elongation with 4sUDRB-seq. Nat Protoc 10: 605–618. [DOI] [PubMed] [Google Scholar]
- Guo W, Schafer S, Greaser ML, Radke MH, Liss M, Govindarajan T, Maatz H, Schulz H, Li S, Parrish AM, et al. 2012. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat Med 18: 766–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J. 2013. Natural RNA circles function as efficient microRNA sponges. Nature 495: 384–388. [DOI] [PubMed] [Google Scholar]
- Ivanov A, Memczak S, Wyler E, Torti F, Porath HT, Orejuela MR, Piechotta M, Levanon EY, Landthaler M, Dieterich C, et al. 2015. Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep 10: 170–177. [DOI] [PubMed] [Google Scholar]
- Jakobi T, Czaja-Hasse LF, Reinhardt R, Dieterich C. 2016. Profiling and validation of the circular RNA repertoire in adult murine hearts. Genomics Proteomics Bioinformatics 14: 216–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeck WR, Sharpless NE. 2014. Detecting and characterizing circular RNAs. Nat Biotechnol 32: 453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, Sharpless NE. 2013. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 19: 141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakaradov B, Xiong HY, Lee LJ, Jojic N, Frey BJ. 2012. Challenges in estimating percent inclusion of alternatively spliced junctions from RNA-seq data. BMC Bioinformatics 13(Suppl 6): S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ. 2004. The UCSC Table Browser data retrieval tool. Nucleic Acids Res 32: D493–D496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly S, Greenman C, Cook PR, Papantonis A. 2015. Exon skipping is correlated with exon circularization. J Mol Biol 427: 2414–2417. [DOI] [PubMed] [Google Scholar]
- Khan MAF, Reckman YJ, Aufiero S, van den Hoogenhof MMG, van der Made I, Beqqali A, Koolbergen DR, Rasmussen TB, van der Velden J, Creemers EE, et al. 2016. RBM20 regulates circular RNA production from the titin gene. Circ Res 119: 996–1003. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer MC, Liang D, Tatomer DC, Gold B, March ZM, Cherry S, Wilusz JE. 2015. Combinatorial control of Drosophila circular RNA expression by intronic repeats, hnRNPs, and SR proteins. Genes Dev 29: 2168–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegs JO, Churakov G, Jurka J, Brosius J, Schmitz J. 2007. Evolutionary history of 7SL RNA-derived SINEs in Supraprimates. Trends Genet 23: 158–161. [DOI] [PubMed] [Google Scholar]
- Labeit S, Kolmerer B. 1995. Titins: giant proteins in charge of muscle ultrastructure and elasticity. Science 270: 293–296. [DOI] [PubMed] [Google Scholar]
- Legnini I, Di Timoteo G, Rossi F, Morlando M, Briganti F, Sthandier O, Fatica A, Santini T, Andronache A, Wade M, et al. 2017. Circ-ZNF609 is a circular RNA that can be translated and functions in myogenesis. Mol Cell 66: 22–37.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Liu CX, Xue W, Zhang Y, Jiang S, Yin QF, Wei J, Yao RW, Yang L, Chen LL. 2017. Coordinated circRNA biogenesis and function with NF90/NF110 in viral infection. Mol Cell 67: 214–227.e7. [DOI] [PubMed] [Google Scholar]
- Liang D, Wilusz JE. 2014. Short intronic repeat sequences facilitate circular RNA production. Genes Dev 28: 2233–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang D, Tatomer DC, Luo Z, Wu H, Yang L, Chen LL, Cherry S, Wilusz JE. 2017. The output of protein-coding genes shifts to circular RNAs when the pre-mRNA processing machinery is limiting. Mol Cell 68: 940–954.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maatz H, Jens M, Liss M, Schafer S, Heinig M, Kirchner M, Adami E, Rintisch C, Dauksaite V, Radke MH, et al. 2014. RNA-binding protein RBM20 represses splicing to orchestrate cardiac pre-mRNA processing. J Clin Invest 124: 3419–3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, et al. 2013. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495: 333–338. [DOI] [PubMed] [Google Scholar]
- Methawasin M, Hutchinson KR, Lee E-J, Smith JE, Saripalli C, Hidalgo CG, Ottenheijm CAC, Granzier H. 2014. Experimentally increasing titin compliance in a novel mouse model attenuates the Frank-Starling mechanism but has a beneficial effect on diastole. Circulation 129: 1924–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigro JM, Cho KR, Fearon ER, Kern SE, Ruppert JM, Oliner JD, Kinzler KW, Vogelstein B. 1991. Scrambled exons. Cell 64: 607–613. [DOI] [PubMed] [Google Scholar]
- Pamudurti NR, Bartok O, Jens M, Ashwal-Fluss R, Stottmeister C, Ruhe L, Hanan M, Wyler E, Perez-Hernandez D, Ramberger E, et al. 2017. Translation of circRNAs. Mol Cell 66: 9–21.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quentin Y. 1994. Emergence of master sequences in families of retroposons derived from 7sl RNA. Genetica 93: 203–215. [DOI] [PubMed] [Google Scholar]
- Ruijter JM, Ramakers C, Hoogaars WMH, Karlen Y, Bakker O, van den Hoff MJB, Moorman AFM. 2009. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res 37: e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO. 2012. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE 7: e30733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer S, Miao K, Benson CC, Heinig M, Cook SA, Hubner N. 2015. Alternative splicing signatures in RNA-seq data: percent spliced in (PSI). Curr Protoc Hum Genet 87: 11.16.1–11.16.14. [DOI] [PubMed] [Google Scholar]
- Schafer S, de Marvao A, Adami E, Fiedler LR, Ng B, Khin E, Rackham OJL, van Heesch S, Pua CJ, Kui M, et al. 2017. Titin-truncating variants affect heart function in disease cohorts and the general population. Nat Genet 49: 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P. 2013–2015. RepeatMasker Open-4.0. http://www.repeatmasker.org/.
- Starke S, Jost I, Rossbach O, Schneider T, Schreiner S, Hung LH, Bindereif A. 2015. Exon circularization requires canonical splice signals. Cell Rep 10: 103–111. [DOI] [PubMed] [Google Scholar]
- Tan WLW, Lim BTS, Anene-Nzelu CGO, Ackers-Johnson M, Dashi A, See K, Tiang Z, Lee DP, Chua WW, Luu TDA, et al. 2017. A landscape of circular RNA expression in the human heart. Cardiovasc Res 113: 298–309. [DOI] [PubMed] [Google Scholar]
- Tsirigos A, Rigoutsos I. 2009. Alu and B1 repeats have been selectively retained in the upstream and intronic regions of genes of specific functional classes. PLOS Comput Biol 5: e1000610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, He X, Mieczkowski P, Grimm SA, Perou CM, et al. 2010. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res 38: e178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werfel S, Nothjunge S, Schwarzmayr T, Strom TM, Meitinger T, Engelhardt S. 2016. Characterization of circular RNAs in human, mouse and rat hearts. J Mol Cell Cardiol 98: 103–107. [DOI] [PubMed] [Google Scholar]
- Yang Y, Fan X, Mao M, Song X, Wu P, Zhang Y, Jin Y, Yang Y, Chen L, Wang Y, et al. 2017. Extensive translation of circular RNAs driven by N6-methyladenosine. Cell Res 27: 626–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaphiropoulos PG. 1996. Circular RNAs from transcripts of the rat cytochrome P450 2C24 gene: correlation with exon skipping. Proc Natl Acad Sci 93: 6536–6541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhang XO, Chen T, Xiang JF, Yin QF, Xing YH, Zhu S, Yang L, Chen LL. 2013. Circular intronic long noncoding RNAs. Mol Cell 51: 792–806. [DOI] [PubMed] [Google Scholar]
- Zhang XO, Wang HB, Zhang Y, Lu X, Chen LL, Yang L. 2014. Complementary sequence-mediated exon circularization. Cell 159: 134–147. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xue W, Li X, Zhang J, Chen S, Zhang JL, Yang L, Chen LL. 2016. The biogenesis of nascent circular RNAs. Cell Rep 15: 611–624. [DOI] [PubMed] [Google Scholar]
- Zheng Q, Bao C, Guo W, Li S, Chen J, Chen B, Luo Y, Lyu D, Li Y, Shi G, et al. 2016. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun 7: 11215. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.